

Abstract
The cerebral cortex is characterized by multiple layers and many distinct cell-types that together as a network are responsible for many higher cognitive functions including decision making, sensory-guided behavior or memory. To understand how such intricate neuronal networks perform such tasks, a crucial step is to determine the function (or electrical activity) of individual cell types within the network, preferentially when the animal is performing a relevant cognitive task. Additionally, it is equally important to determine the anatomical structure of the network and the morphological architecture of the individual neurons to allow reverse engineering the cortical network. Technical breakthroughs available today allow recording cellular activity in awake, behaving animals with the valuable option of post hoc identifying the recorded neurons. Here, we demonstrate the juxtasomal biocytin labeling technique, which involves recording action potential spiking in the extracellular (or loose-patch) configuration using conventional patch pipettes. The juxtasomal recording configuration is relatively stable and applicable across behavioral conditions, including anesthetized, sedated, awake head-fixed, and even in the freely moving animal. Thus, this method allows linking cell-type specific action potential spiking during animal behavior to reconstruction of the individual neurons and ultimately, the entire cortical microcircuit. In this video manuscript, we show how individual neurons in the juxtasomal configuration can be labeled with biocytin in the urethane-anaesthetized rat for post hoc identification and morphological reconstruction.
Keywords: Bioengineering, Issue 84, biocytin, juxtasomal, morphology, physiology, action potential, structure-function, histology, reconstruction, neurons, electrophysiological recordings
Introduction
Neuronal networks consist of multiple cell types, characterized by highly specific morphological and physiological properties1-7. As a consequence, individual cell types perform specialized tasks within the network (see for instance Gentet et al.8 and Burgalossi et al.9). We are only beginning to understand cell type-specific functions across neuronal networks and much is still to be discovered. To this end, many labs are implementing experimental approaches that allow the analysis of morphological properties of the same neuronal population from which physiological parameters have been obtained1,10-15. Here, we demonstrate the juxtasomal labeling technique16,17 which involves electrophysiological recordings using conventional patch pipettes in the extracellular (thus noninvasive) configuration in combination with electroporation of the recorded neuron with biocytin. The major advantage of this approach is that the noninvasive nature ensures that action potential spiking of individual neurons is recorded without altering (e.g. dialyzing) the intracellular content of the cell. Followed by electroporation, the juxtasomal approach provides the option of post hoc cell identification and reconstruction to link function (physiology) to structure (morphology). Typically, morphological reconstruction involves reconstruction of dendritic and axonal morphology which can be extended to quantification of spine and/or bouton densities or even reconstruction of neuronal morphology at nanometer resolution using electron microscopy. The juxtasomal recording technique can be used for in vivo recordings of various cell-types across cortical layers or in sub-cortical areas in a range of species, although most studies have applied the technique in small rodents such as mice or rats. Our research is focused on recording and labeling neurons from rat primary somatosensory cortex (S1) and involves visual identification of recorded neurons18, dendritic reconstructions in combination with precise registration in a standardized reference frame to reverse engineer cortical networks4,19 and detailed reconstruction of axonal architecture to characterize cell type-specific local and long-range projection targets20.
Compared to alternative in vivo recording techniques (intracellular or whole-cell), juxtasomal recordings are relatively stable and can therefore be applied across behavioral states including anesthetized21,22, sedated14, awake head-fixed23, or even freely-moving animals9. Here, we show juxtasomal labeling in S1 of a urethane-anesthetized rat, although we emphasize the general applicability of this technique to many preparations of choice.
Protocol
1. Preparation of the Animal
All experimental procedures are carried out in accordance with the Dutch law and after evaluation by a local ethical committee at the VU University Amsterdam, The Netherlands.
Anesthetize a Wistar rat (P25-P45, ♂/♀) with isoflurane (2-3% in oxygen) and subsequently with urethane (20% in 0.9% NaCl, 1.6-1.7 g/kg) by intraperitoneal injection. Assess the depth of the anesthesia by monitoring pinch withdrawal, eyelid reflexes, and vibrissae movements.
Administer an additional dose of urethane (10% of initial dose) in case the animal does not reach sufficient anesthetic depth or whenever vibrissae twitches are observed during the course of the experiment.
Place the anesthetized animal on the stereotaxic frame equipped with a heating pad, insert rectal temperature probe and maintain the animal’s body temperature at 37.5±0.5 °C by a heating pad.
Position the animal’s head in the stereotaxic frame; trim the hair on top of the head using scissors to render the skin visible.
Inject 100 µl 1% lidocaine (in 0.9% NaCl) subcutaneously for local anesthesia at the operation site and wait 3-5 min. Remove the skin covering the surface of the skull by cutting in the caudal-to-rostral direction and clean the remaining tissue.
Clean the skull extensively with 0.9% NaCl and absorbent swabs.
Determine the coordinates of the target area on the left hemisphere (for primary somatosensory cortex (S1): 2.5 mm posterior and 5.5 mm lateral to bregma). Mark the location on the skull with a surgical skin marker pen.
Scrape off the bone gently by a dental drill from the region of interest until the bone becomes transparent and blood vessels are clearly visible.
Make a 0.5 mm x 0.5 mm craniotomy by cutting a window in the thinned skull with a scalpel (#11), avoiding damage to the dura mater and blood vessels. Optional: mark the edges of the craniotomy using a surgical skin marker pen to improve visibility.
Apply a thin layer of superglue on the dry skull and then dental cement to construct a bath (i.e. recording chamber) enclosing the craniotomy.
Trim the whiskers at the contralateral side to exactly 5 mm from the whisker follicle. Optional: highlight the trimmed whiskers with black mascara.
2. Juxtasomal Recordings and Biocytin Labeling
Make patch pipettes of borosilicate glass with inside tip diameter of ~1 μm to obtain electrodes with 3-5 MΩ resistance. Note: The ideal pipette morphology for the juxtasomal recording is a gradual slender taper, a low cone angle, and a ~1 μm tip (Figure 1).
Depending on the recording depth (with respect to the pial surface), the taper dimensions of the recording electrode should be adjusted. Critically important is that the taper diameter should be less than 75 μm at point of entry in the brain to avoid mechanical damage or stress to the recording area. This indicates that for superficial cortical recordings, a taper of 300-500 μm with an outer diameter of maximally 75 μm is required (Figure 1A) whereas recordings from relatively deep cortical layers require a longer taper of 1,500-1,800 μm, again with maximal outer diameter of 75 μm (at 1,800 μm from the electrode tip).
Load the patch pipette with normal rat ringer (NRR) supplemented with 2% biocytin (see Table 1 for solutions and reagents) and mount the patch pipette on a head stage fixed to a micromanipulator.
Set the angle of the electrode holder to 34° with respect to the sagittal plane to specifically target the D2 column of rat S1.
Connect the head stage to an amplifier in bridge-mode (i.e. current clamp).
Position the patch pipette in close proximity to the craniotomy. Fill the bath with 0.9% NaCl and determine the electrode resistance which should be between 3-5 MΩ, assessed by applying a square pulse with positive current injection (1 nA, 200 msec on/off).
Establish overpressure of 100-150 mbar and advance the patch pipette in 1 µm steps while applying positive current as square pulses (1 nA, 200 msec on/off). Monitor the robust resistance change upon establishing contact with the dura mater. At this point, set the coordinates of the micromanipulator to ‘zero’ to allow accurate depth measurements.
Advance in 1 µm step-mode until the patch pipette penetrates the dura mater indicated by a sudden drop in electrode resistance. Remove the holding pressure of the pipette.
Search for single units while advancing in 1 µm steps. Monitor the electrode resistance continuously by applying 200 msec on/off pulses. An increase in electrode resistance typically indicates the proximity of a single neuron. Advance the electrode until positive action potential (AP) waveforms of ~2 mV are recorded (Figures 2A and 2B).
Optional: Record the spontaneous spiking pattern of the neuron and determine whisker-evoked spiking by, for instance, caudally deflecting individual whiskers for 200 msec at an angle of 3.3° using a piezoelectric device18.
Advance the electrode until the resistance is 25-35 MΩ and spikes have amplitudes of 3-8 mV to obtain optimal conditions of juxtasomal filling. Start the juxtasomal filling by applying square pulses of positive current (1 nA, 200 msec on/off). Slowly and gradually increase the current by steps of 0.1 nA while monitoring the AP waveform and frequency (Figures 2A and 2B).
Monitor the membrane opening as a clear increase in AP frequency during the on-phase of the block pulse (Figure 2C). The spike waveform during filling shows an increased width and reduced after-hyperpolarization (Figures 2C and 2D). Additional parameters include increased noise or a small (1-5 mV) negative DC shift.
Increase or decrease the current (1 nA) while filling to maintain stable biocytin infusion. Reduce or stop the current pulses upon sudden increase of the AP frequency to avoid toxicity by excess influx of extracellular ions, such as sodium ions. Note: every neuron will respond differently to the current injection and juxtasomal labeling parameters need to be adjusted depending on individual recording conditions.
Closely monitor the signal after stopping the current injection. The spike waveform after a filling session is usually broadened and shows a strongly reduced after hyperpolarization. Wait for recovery of the neuron, which is apparent when the spike waveform returns to its original properties (i.e. presence of normal after-hyperpolarization, Figure 2).
Repeat biocytin filling sessions after complete recovery of the neuron to increase biocytin load for improved staining quality.
Retract the patch pipette in steps of 1 µm until the spike amplitude decreases to reduce any mechanical stress to the cell. Wait at least 1 hr for the biocytin to diffuse intracellularly while closely monitoring the animal’s body temperature and breathing.
3. Perfusing the Animal and Removing the Brain
Prepare the perfusion setup, rinse, and preload the tubing with 0.9% NaCl.
Position the animal on a surgical tray and secure with standard labeling tape. Ensure sufficient depth of the anesthesia; foot pinch and eyelid reflexes should be absent.
Make a medial to lateral incision (5-6 cm) through the abdominal wall just beneath the rib cage and proceed in posterior-anterior direction to expose the sternum. Pull the sternum anteriorly and carefully separate the liver from the diaphragm.
Make a small incision in the diaphragm, cut through the lower ribs and continue the incision along the entire length of the abdominal cavity to expose the heart.
Remove the pericardium.
Insert the needle into the left ventricle and make an incision in the right atrium. Perfuse with 0.9% NaCl (~8 ml/min) until decoloration of the liver is complete.
Switch the infusion to 4% paraformaldehyde (PFA) until stiffness of front paw and lower jaw is apparent.
Decapitate the rat using a pair of scissors
Remove the remaining neck muscles and expose the skull completely.
Position the scissors in the brain stem on the dorsal side and cut the bone carefully along the sagittal suture, maintaining the dorsal position.
Remove the bones from both sides of the sagittal suture to expose the brain using forceps. Carefully remove the dura to avoid damage.
Carefully insert a blunt spatula to the ventral side of the brain and remove the brain gently.
Post-fix the entire brain overnight in PFA at 4 °C. Switch the brain to 0.05 M phosphate buffer (PB) and store at 4 °C.
4. Slicing the Brain in Tangential Sections
Take the brain out of the 0.05 M PB and put it on a filter paper facing anterior. Use a sharp razor blade to cut off the cerebellum along the coronal plane and separate the hemispheres by cutting along the mid sagittal plane.
Apply superglue on the mounting platform and mount the left hemisphere on its sagittal plane with anterior facing right. Secure the mounting platform at an angle of 45° on a vibratome and submerge the brain in 0.05 M PB.
Secure a razor blade on the vibratome and make sure that the first contact with the brain surface is in the middle of the anterior-posterior plane of the hemisphere. Cut 24 100 µm sections and collect them in a 24-well plate containing 0.05 M PB.
5. Histological Procedures
Perform histological protocols for the cytochrome oxidase staining24 and the avidin-biotin-peroxidase method according to previously described methods25. Optional: visualize biocytin using fluorescent avidin/streptavidin-Alexa conjugates. This additionally allows double-staining with retrograde or anterograde tracing techniques.
Wash sections 5 x 5 min with 0.05 M PB and prepare the 3,-3’-diaminobenzidine tetrahydrochloride (DAB)-containing solution for the cytochrome oxidase staining to visualize barrels in layer 4 (L4) of S1 (see Table 2 for solutions and reagents). Incubate sections 6-12 from the pia in the preheated solution for 30-45 min at 37 °C.
Rinse sections with 0.05 M PB for 6 x 5 min and quench endogenous peroxidase activity by incubating all sections in 3% H2O2 in 0.05 M PB for 20 min at room temperature (RT).
Rinse sections with 0.05 M PB for 5 x 10 min. Incubate sections in ABC-solution (see Table 3 for solutions and reagents) overnight at 4 °C.
Rinse sections with 0.05 M PB for 5 x 10 min and prepare the DAB-solution to visualize the biocytin-filled neuron (see Table 4 for solutions and reagents). Incubate sections in filtered solution for 45-60 min at RT.
Rinse sections with 0.05 M PB for 5 x 10 min. Mount sections on microscope slides and cover slip with Mowiol (see Table 5 for solutions and reagents).
Determine labeling quality using light microscopy.
Representative Results
Detailed knowledge on 3D structure of individual neurons is crucial for elucidating organizational principles of neuronal networks. Our method involves a pipeline to achieve high quality biocytin labeling from an in vivo preparation, thereby allowing post hoc neuronal classification and detailed reconstruction of dendritic and axonal architecture of single neurons at high resolution. Depending on the quality of juxtasomal labeling, neurons are recovered with different DAB-intensities ranging from faint to intense DAB signals at positions that very accurately correspond to the recording location19,26. In our lab, a trained experimenter operates at a success-rate of 30-40% in urethane anaesthetized rodents. This indicates that one neuron is selected for dendritic and axonal reconstruction in one out of three experiments which then meets the following criteria: 1) only one neuron filled in the brain, 2) excellent labeling quality, 3) biocytin signal is constant along axonal projections and does not decrease at distal endings, 4) Cyt C counterstaining is successful to allow neuronal registration, and 5) no damage to brain slices during histology. The limiting factor is typically insufficient biocytin labeling, meaning that in ~80% of all experiments (anaesthetized and/or awake), the recorded neuron will be recovered (soma and dendrites). This is sufficient for cell type classification but not for reliable and complete reconstruction of the axonal architecture. In Figure 3, we show two examples of biocytin-labeled neurons with DAB signal after appropriate juxtasomal labeling; one spiny pyramidal neuron (Figure 3A) and one interneuron (Figure 3B). Reconstruction of adjacent tangential sections allows serial reconstruction to obtain full neuronal morphology18,22,23,27,28. Additionally, histological staining of anatomical reference frames (for instance in primary somatosensory cortex) allows registering single neurons to standardized reference frames4,19. The two examples presented here reflect optimal conditions to classify and subsequently reconstruct the morphological properties with a manual or automated system (Figure 4)15,20,29-31.
 Figure 1. Electrode characteristics for juxtasomal biocytin labeling. (A) Electrode for juxtasomal recording of cortical neurons at 300-500 μm depth with respect to pial surface. (B) Electrode for juxtasomal recording of cortical neurons at 1,500-1,800 μm recording depth. Note the difference in taper length between panels (A) and (B). (C) High magnification image to illustrate electrode tip shape which remains constant across various recording depths and/or cell types. Click here to view larger image.
Figure 1. Electrode characteristics for juxtasomal biocytin labeling. (A) Electrode for juxtasomal recording of cortical neurons at 300-500 μm depth with respect to pial surface. (B) Electrode for juxtasomal recording of cortical neurons at 1,500-1,800 μm recording depth. Note the difference in taper length between panels (A) and (B). (C) High magnification image to illustrate electrode tip shape which remains constant across various recording depths and/or cell types. Click here to view larger image.
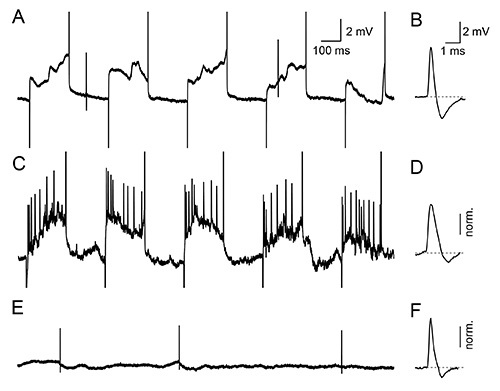 Figure 2. Electrophysiological properties during biocytin filling. (A, B) Spontaneous action potentials are observed during the application of square pulses (200 msec, on/off) with positive current but with insufficient amplitude to load the neuron with biocytin. (C) Increasing the current amplitude results in opening of the neuronal membrane allowing biocytin-loading, visible in the trace as phase-locked burst-spiking during the on-phase of the pulse. (D) The spike waveform during filling shows an increased width and reduced after hyperpolarization. Spike amplitude is normalized to spike amplitude in B for comparison. (E, F) After successful recovery of the neuron subsequent to the filling session a normal spike width and frequency can be observed. Spike amplitude is normalized to spike amplitude in B for comparison. Click here to view larger image.
Figure 2. Electrophysiological properties during biocytin filling. (A, B) Spontaneous action potentials are observed during the application of square pulses (200 msec, on/off) with positive current but with insufficient amplitude to load the neuron with biocytin. (C) Increasing the current amplitude results in opening of the neuronal membrane allowing biocytin-loading, visible in the trace as phase-locked burst-spiking during the on-phase of the pulse. (D) The spike waveform during filling shows an increased width and reduced after hyperpolarization. Spike amplitude is normalized to spike amplitude in B for comparison. (E, F) After successful recovery of the neuron subsequent to the filling session a normal spike width and frequency can be observed. Spike amplitude is normalized to spike amplitude in B for comparison. Click here to view larger image.
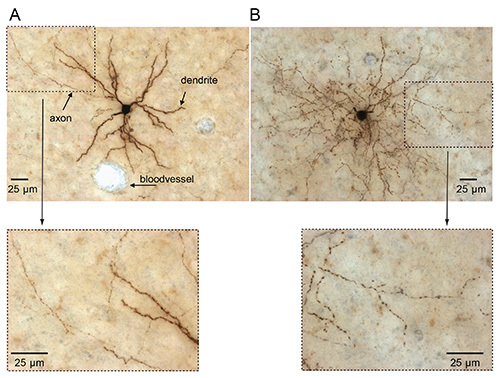 Figure 3. Biocytin-DAB labeling from juxtasomally filled neurons in primary somatosensory cortex of urethane anaesthetized rats. (A) Pyramidal neuron in tangential view. Note the prominent difference in diameter size of dendrites and axons. (B) Fast-spiking interneuron in tangential view. Note the beaded structure of the dendrites and axons. Click here to view larger image.
Figure 3. Biocytin-DAB labeling from juxtasomally filled neurons in primary somatosensory cortex of urethane anaesthetized rats. (A) Pyramidal neuron in tangential view. Note the prominent difference in diameter size of dendrites and axons. (B) Fast-spiking interneuron in tangential view. Note the beaded structure of the dendrites and axons. Click here to view larger image.
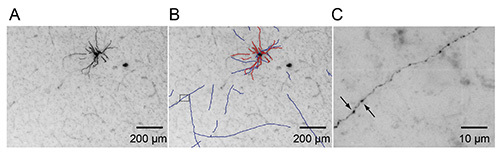 Figure 4. Digital reconstruction of juxtasomally labeled neuron. (A) Neuron projection image at high resolution (but low magnification) obtained from 100 µm tangential slice of layer 6 neuron of rat primary somatosensory cortex using semi-automated imaging pipeline. (B) The same image as in (A) with automated digital reconstruction overlaid (axon in blue, dendrite in red, respectively). (C) Bracket box from (B) zoomed in to illustrate reliability of axonal reconstruction using juxtasomal labeling. Note the axon boutons throughout the length of the axon; two boutons are highlighted by black arrows. Click here to view larger image.
Figure 4. Digital reconstruction of juxtasomally labeled neuron. (A) Neuron projection image at high resolution (but low magnification) obtained from 100 µm tangential slice of layer 6 neuron of rat primary somatosensory cortex using semi-automated imaging pipeline. (B) The same image as in (A) with automated digital reconstruction overlaid (axon in blue, dendrite in red, respectively). (C) Bracket box from (B) zoomed in to illustrate reliability of axonal reconstruction using juxtasomal labeling. Note the axon boutons throughout the length of the axon; two boutons are highlighted by black arrows. Click here to view larger image.
| mM | g/ltr | |
| 135 | NaCl | 7.8894 |
| 5.4 | KCl | 0.40257 |
| 1.8 | CaCl2 | 0.26464 |
| 1 | MgCl2 | 0.2033 |
| 5 | HEPES | 1.1915 |
| Adjust pH to 7.2 with NaOH | ||
| Add 20 mg/ml-1 biocytin |
Table 1. Normal Rat Ringer Supplemented with Biocytin.
| 8 mg | Cytochrome C from equine heart |
| 8 mg | Catalase from bovine liver |
| 265 µl | 75 mg/ml DAB |
| Dissolve in 40 ml 0.05M PB | |
| Filter and preheat solution at 37 °C |
Table 2. Solution for Cytochrome C oxidase staining.
| 1 drop | Reagent A |
| 1 drop | Reagent B |
| 0.5 ml | 10% Triton X100 |
| Dissolve in 9.5 ml 0.05M PB (45 min in advance) | |
| Add 410 µl solution/well | |
| Protocol for ABC-solution for one 24-well plate. |
Table 3. Solution with Vectastain standard ABC-kit.
| 265 µl | 75 mg/ml DAB |
| 13.4 µl | 30% H2O2 |
| Dissolve in 40 ml 0.05 M PB and filter solution before use |
Table 4. Solution for DAB staining.
| 6 g | Analytical grade glycerol |
| 2.4 g | Mowiol 4-88 |
| Manually stir 10 min to coat Mowiol with glycerol | |
| 6 ml | Distilled H2O |
| Incubate at RT for 2 hr | |
| 12 ml | 0.2 M Tris pH 8.5 |
| Incubate in 50 °C waterbath for 1hr - O/N, stir occasionally | |
| Centrifuge 15 min at 5,000 x g, aliquot and store at -20 °C |
Table 5. Mowiol solution.
Discussion
The juxtasomal method allows recording in vivo action potential spiking from single units across behavioral conditions (anesthetized, awake head-fixed or freely moving) with the option of biocytin-labeling the recorded neuron for post hoc cell type classification and/or 3D reconstruction. The major advantage is to obtain physiological parameters in the extracellular (thus noninvasive) configuration, yet being able to label the neuron intracellularly with biocytin16,17,32. In addition to biocytin labeling, this technique can be used to inject neurons with DNA, RNA, proteins, or fluorescent dyes33,34. The most obvious disadvantage of this approach is perhaps the lack of visual control of the labeling quality, and thus no means of checking the labeling quality during the experiment. However, recording conditions and particularly action potential shape can be used to assess the success of the labeling procedure. For instance, the likelihood of recovering a neuron with dense biocytin-DAB label increases when the spiking frequency during current pulses dramatically increases, accompanied with disappearance of the after-hyperpolarization and broadening of the action potential waveform (see Figure 2). Additionally, the quality of biocytin-labeling is directly correlated to the summed length of the separate labeling sessions, such that several long filling sessions (>60 sec) will result in better histological quality compared to an individual short filling session (<30 sec). Finally, the survival time for tracer diffusion will critically determine the labeling intensity of distal compartments. In general, survival time of 1 hr after high-quality electroporation ensures sufficient labeling of long-range intracortical axonal projections. One example of such long-range projections are neurons from primary somatosensory cortex projecting to secondary somatosensory cortex or dysgranular zones thus projecting to areas which are several millimeters away from the labeling site4,20. When axonal projections of even larger dimensions are investigated (such as thalamocortical projections), survival time should be increased15. Important to realize is that electroporation of the neuron will have a profound effect on the physiological condition of the neuron due to excessive sodium influx from the electrode solution. Thus, it is highly recommended to intermingle filling episodes with quiescent episodes to allow full recovery of action potential spiking and monitoring recording conditions after filling sessions when biocytin is allowed to diffuse along the dendritic and axonal arborizations12,31.
Disclosures
The authros have nothing to disclose.
Acknowledgments
We would like to thank Profs. Huibert Mansvelder and Bert Sakmann for extensive support, Dr. Marcel Oberlaender for fruitful discussions and providing neuronal tracing, and Brendan Lodder for technical assistance. Data was acquired using the ntrode VI for LabView, generously provided by R. Bruno (Columbia Univ., NY, USA). This research was supported by the Max Planck Society and the Bernstein Center for Computational Neuroscience, Tuebingen (funded by the German Federal Ministry of Education and Research (BMBF; FKZ: 01GQ1002))( R.T.N.), Center for Neurogenomics and Cognitive Research (CNCR), Neuroscience Campus Amsterdam (NCA), funding to C.P.J.d.K. (NWO-ALW #822.02.013 and ENC-Network #p3-c3) and the VU University Amsterdam.
References
- Brown SP, Hestrin S. Cell-type identity: a key to unlocking the function of neocortical circuits. Curr. Opin. Neurobiol. 2009;19(4):415–421. doi: 10.1016/j.conb.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFelipe J, et al. New insights into the classification and nomenclature of cortical GABAergic interneurons. Nat. Rev. Neurosci. 2013;14(3):202–216. doi: 10.1038/nrn3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean P, Porrill J, Ekerot CF, Jorntell H. The cerebellar microcircuit as an adaptive filter: experimental and computational evidence. Nat. Rev. Neurosci. 2010;11(1):30–43. doi: 10.1038/nrn2756. [DOI] [PubMed] [Google Scholar]
- Oberlaender M, et al. Cell type-specific three-dimensional structure of thalamocortical circuits in a column of rat vibrissal cortex. Cereb. Cortex. 2012;22(10):2375–2391. doi: 10.1093/cercor/bhr317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer MA, Cepko CL. Regulating proliferation during retinal development. Nat. Rev. Neurosci. 2001;2(5):333–342. doi: 10.1038/35072555. [DOI] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 2008;321(5885):53–57. doi: 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban N, Tripathy S. Neuroscience: Circuits drive cell diversity. Nature. 2012;488(7411):289–290. doi: 10.1038/488289a. [DOI] [PubMed] [Google Scholar]
- Gentet LJ, et al. Unique functional properties of somatostatin-expressing GABAergic neurons in mouse barrel cortex. Nat. Neurosci. 2012;15(4):607–612. doi: 10.1038/nn.3051. [DOI] [PubMed] [Google Scholar]
- Burgalossi A, et al. Microcircuits of functionally identified neurons in the rat medial entorhinal cortex. Neuron. 2011;70(4):773–786. doi: 10.1016/j.neuron.2011.04.003. [DOI] [PubMed] [Google Scholar]
- Bock DD, et al. Network anatomy and in vivo physiology of visual cortical neurons. Nature. 2011;471(7337):177–182. doi: 10.1038/nature09802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggman KL, Helmstaedter M, Denk W. Wiring specificity in the direction-selectivity circuit of the retina. Nature. 2011;471(7337):183–188. doi: 10.1038/nature09818. [DOI] [PubMed] [Google Scholar]
- Herfst L, et al. Friction-based stabilization of juxtacellular recordings in freely moving rats. J. Neurophysiol. 2012;108(2):697–707. doi: 10.1152/jn.00910.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx M, Gunter RH, Hucko W, Radnikow G, Feldmeyer D. Improved biocytin labeling and neuronal 3D reconstruction. Nat. Protoc. 2012;7(2):394–407. doi: 10.1038/nprot.2011.449. [DOI] [PubMed] [Google Scholar]
- Bruno RM, Sakmann B. Cortex is driven by weak but synchronously active thalamocortical synapses. Science. 2006;312(5780):1622–1627. doi: 10.1126/science.1124593. [DOI] [PubMed] [Google Scholar]
- Oberlaender M, Ramirez A, Bruno RM. Sensory experience restructures thalamocortical axons during adulthood. Neuron. 2012;74(4):648–655. doi: 10.1016/j.neuron.2012.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Hawken MJ. Loose-patch-juxtacellular recording in vivo-a method for functional characterization and labeling of neurons in macaque V1. J. Neurosci. Methods. 2006;156(1-2):37–49. doi: 10.1016/j.jneumeth.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Pinault D. A novel single-cell staining procedure performed in vivo under electrophysiological control: morpho-functional features of juxtacellularly labeled thalamic cells and other central neurons with biocytin or Neurobiotin. J. Neurosci. Methods. 1996;65(2):113–136. doi: 10.1016/0165-0270(95)00144-1. [DOI] [PubMed] [Google Scholar]
- de Kock CP, Bruno RM, Spors H, Sakmann B. Layer and cell type specific suprathreshold stimulus representation in primary somatosensory cortex. J. Physiol. 2007;581(1):139–154. doi: 10.1113/jphysiol.2006.124321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger R, Narayanan RT, Helmstaedter M, de Kock CP, Oberlaender M. 3D reconstruction and standardization of the rat vibrissal cortex for precise registration of single neuron morphology. PLoS Comput. Biol. 2012;8(12) doi: 10.1371/journal.pcbi.1002837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlaender M, et al. Three-dimensional axon morphologies of individual layer 5 neurons indicate cell type-specific intracortical pathways for whisker motion and. Proc. Natl. Acad. Sci. U.S.A. 2011;108(10):4188–4193. doi: 10.1073/pnas.1100647108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata S, Harris KD. Laminar structure of spontaneous and sensory-evoked population activity in auditory cortex. Neuron. 2009;64(3):404–418. doi: 10.1016/j.neuron.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kock CP, Sakmann B. High frequency action potential bursts (>or= 100 Hz) in L2/3 and L5B thick tufted neurons in anaesthetized and awake rat primary somatosensory cortex. J. Physiol. 2008;586(14):3353–3364. doi: 10.1113/jphysiol.2008.155580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kock CP, Sakmann B. Spiking in primary somatosensory cortex during natural whisking in awake head-restrained rats is cell-type specific. Proc. Natl. Acad. Sci. U.S.A. 2009;106(38):16446–16450. doi: 10.1073/pnas.0904143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Riley M. Changes in the visual system of monocularly sutured or enucleated cats demonstrable with cytochrome oxidase histochemistry. Brain Res. 1979;171(1):11–28. doi: 10.1016/0006-8993(79)90728-5. [DOI] [PubMed] [Google Scholar]
- Horikawa K, Armstrong WE. A versatile means of intracellular labeling: injection of biocytin and its detection with avidin conjugates. J. Neurosci. Methods. 1988;25(1):1–11. doi: 10.1016/0165-0270(88)90114-8. [DOI] [PubMed] [Google Scholar]
- O'Connor DH, Peron SP, Huber D, Svoboda K. Neural activity in barrel cortex underlying vibrissa-based object localization in mice. Neuron. 2010;67(6):1048–1061. doi: 10.1016/j.neuron.2010.08.026. [DOI] [PubMed] [Google Scholar]
- Veinante P, Deschenes M. Single-cell study of motor cortex projections to the barrel field in rats. J. Comp. Neurol. 2003;464(1):98–103. doi: 10.1002/cne.10769. [DOI] [PubMed] [Google Scholar]
- Boudewijns ZS, et al. Layer-specific high-frequency action potential spiking in the prefrontal cortex of awake rats. Front. Cell. Neurosci. 2013;7 doi: 10.3389/fncel.2013.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlaender M, Bruno RM, Sakmann B, Broser PJ. Transmitted light brightfield mosaic microscopy for three-dimensional tracing of single neuron morphology. J. Biomed. Opt. 2007;12(6) doi: 10.1117/1.2815693. [DOI] [PubMed] [Google Scholar]
- Boudewijns ZS, et al. Semi-automated three-dimensional reconstructions of individual neurons reveal cell type-specific circuits in cortex. Commun. Integr. Biol. 2011;4(4):486–488. doi: 10.4161/cib.4.4.15670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno RM, Hahn TT, Wallace DJ, de Kock CP, Sakmann B. Sensory experience alters specific branches of individual corticocortical axons during development. J. Neurosci. 2009;29(10):3172–3181. doi: 10.1523/JNEUROSCI.5911-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D. Observing without disturbing: how different cortical neuron classes represent tactile stimuli. J. Physiol. 2007;581(1) doi: 10.1113/jphysiol.2007.132225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann E, Kakorin S, Toensing K. Fundamentals of electroporative delivery of drugs and genes) Bioelectrochem. Bioenerg. 1999;48(1):3–16. doi: 10.1016/s0302-4598(99)00008-2. [DOI] [PubMed] [Google Scholar]
- Haas K, Sin WC, Javaherian A, Li Z, Cline HT. Single-cell electroporation for gene transfer in vivo. Neuron. 2001;29(3):583–591. doi: 10.1016/s0896-6273(01)00235-5. [DOI] [PubMed] [Google Scholar]