

Abstract
Nonalcoholic fatty liver disease (NAFLD) is a common liver disease, and the incidence increases year by year. The pathogenesis of NAFLD is correlated with insulin resistant (IR), and oxidative stress which induces varied inflammatory cytokines (TNF-α, IL-1, IL-6, etc). Different signal transductions such as MAPK, NF-κB, AMPK, JAK2/STAT3, PPAR, PI3K/Akt, TLR were activated by the pathogenic factors to regulate correlative reactions. Thus, in-depth study of the signal transductions will probably provide new suitable solutions for the prevention and therapy of NAFLD.
Keywords: Nonalcoholic fatty liver disease, pathogenesis, signal transduction
“Nonalcoholic Fatty Liver Disease” which was coined by Ludwing in 1980 early is a clinical syndrome characterized by the excessive fat accumulation in the liver and hepatic cellular degeneration even in the absence of alcohol consumption [1]. Defined as a genetic-environmental-metabolic stress-related disease, NAFLD encompasses a spectrum of disease from simple steatosis to nonalcoholic steatohepatitis (NASH), which can progress to cirrhosis and hepatocellular carcinoma. NAFLD affects 10% to 24% of the general population in various countries and the prevalence has even been up to 75 percent in obese people [2]. In United States it translates to approximately 30.1 million obese people affected with steatosis and 8.6 million with steatohepatitis [3]. Hyperlipidemia (hypertriglyceridemia and/or hypercholesterolemia), which is frequently associated with both obesity and type 2 diabetes, has been reported in 20% to 80% of patients with NASH [4]. There is increasing evidence that NAFLD represents the hepatic component of a metabolic syndrome characterized by obesity, hyperinsulinemia, peripheral insulin resistance, diabetes, hypertriglyceridemia, and hypertension. In recent years, with the improvement of living standard and lifestyle, especially the change of the diet structure and decrease in physical activity, the NAFLD patients increase year by year, and their ages are to be younger, therefore, NAFLD has become a global public health problem. So far the pathogenesis of NAFLD has not been fully clarified, and currently, a variety of factors based on the insulin-oxidative stress injury are considered to be involved in it, but its intracellular signal transduction mechanisms are not clear yet. As is known that the intracellular signaling transduction is an important way for a numerous factors to stimulate cell changes, in other words, the relevant causative factors play a role by the signal transmission within the liver cells. Currently many factors such as free fatty acids (FFAs), reactive oxidative stress (ROS), tumor necrosis factor-α (TNF-α), interleukin (IL)-6 that cause insulin resistance (IR), hepatocyte fat accumulation and cellular injury are involved in the pathologic processes of NAFLD by directly activating the c-Jun N-terminal kinase (JNK) [5]. Otherwise, signaling pathways related with insulin resistance, oxidative stress and inflammation fibrosis include: nuclear factor κB (NF-κB), AMP-activated protein kinase (AMPK), Janus kinase/signal transducers and activators of transcription (JAK/STAT), peroxisome proliferator-activated-receptors (PPARs, phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt), Toll-like receptor (TLR). Blocking any one of the above pathways would not be effective in the prevention and treatment of NAFLD [6-11].
Pathogenesis of NAFLD
The pathogenesis of NAFLD is not a simple mechanism, the most widespread and prevailing theory is the so-called “two-hit” model (Figure 1). The first hit is insulin resistant(IR) leading to hepatic fat accumulation; on this basis a large number of adipokines (leptin, adiponectin, resistin) regulate free fatty acids (FFAs) to induce ROS injury is to be the second hit [3,12-15]; ROS could activate Fas ligand/Fas system, and progress to lead to structural protein of the Fas death zone to raise the downstream caspase family members to form the protease procascade reaction and then results in cellular disorganization and apoptosis; What’s more, the apoptotic hepatocytes could form the aggregation of inflammatory cells, thereby inducing a variety of inflammatory cytokines (TNF-α, IL-1, IL-6, IL-8, IL-18, monocyte chemoattractant protein (MCP-1), etc.) directly mediate fibrosis to induce inflammation of the liver and steatohepatitis, the process above is inflammatory-necrotic circulation [16,17]; One more hit by fibrosis factors (such as TGF-β) results in the synthesis of hepatocyte extracellular matrix is greater than the degradation, thus forming a progressive fibrosis [18]. Although a variety of factors are involved in the pathogenesis of NAFLD, the development of fatty liver is a highly integrated process. Now it’s clear that intracellular signaling transduction is an important way for a variety of factors to stimulate cell changes. Namely, the relevant causative factors play an important role by the signal transmission within the liver cells, so it will be helpful for the prevention and cure of NAFLD by in-depth study of the signal transductions in liver cells about the pathogenesis of NAFLD.
Figure 1.
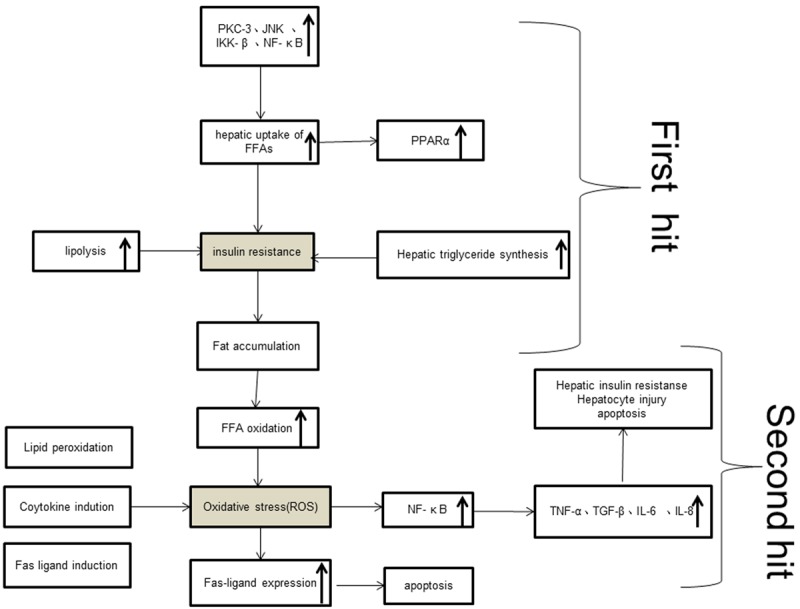
The pathogenesis of NAFLD: “two-hit” model.
Signal transductions and NAFLD
MAPK and NAFLD
Mitogen-activated protein kinase (MAPK) is a class of serine/threonine protein kinase widely existed in mammalian cells and mediates signal conduction from the cell surface to the endonuclear, which includes cell extracellular signal-regulated kinases (ERKs), c-Jun amino-terminal kinase (JNK) and p38MAPK. The ERKs play a major role in the cellular response induced by growth factor stimulation, and JNK and p38 are related with stress and inflammation [19]. MAPK activation is involved in regulation of cell proliferation, differentiation, transformation and apoptosis through phosphorylation of nuclear transcription factors, cytoskeletal proteins and enzymes, which is closely related with mechanisms of inflammation, cancer and many other diseases [20]. In recent years, many researches have indicated that JNK was closely related with IR; And inflammatory cytokines such as tumor necrosis factor α (TNF-α), free fatty acids (FFAs), oxidative stress that could lead to IR, hepatocyte fat accumulation and cell injury were involved in the pathogenesis of NAFLD rat model by activating the JNK [5,21]. Dynamic observation of the JNK signaling pathway protein expression in rat liver tissue and the generated impact and mechanism of IR in the process of the formation of high-fat-diet induced NAFLD from the first week, drawing that the high-fat-diet rat liver tissue JNK1 protein expression levels were higher compared with the control group over the same period, and JNK1 protein expression and IR level was positively correlated from the end of the second week, which indicated that high-fat- diet activated JNK1, diminished insulin signaling, thus causing IR [22]. p38 signaling pathways are involved in the cell inflammatory response and apoptosis process under stress conditions, which are related with the release of a variety of inflammatory cytokines (such as IL-1, TNF-α and IL-6, etc.) after activation [23]. To study the action mechanism of an antioxidant in the high-fat-diet rats, Sinha-Hikim et al [24] analyzed the phosphorylation of JNK and p38 signal level in the high-fat-diet-induced NAFLD rats, which confirmed a significant (P<0.05) increase in both phospho-JNK and phospho-p38MAPK levels than the normal control group and significantly reduced after antioxidants treatment, and drew that oxidative stress can promote activation of both p38MAPK and JNK, which through mitochondria-dependent intrinsic pathway signaling promotes apoptosis in various cell types. The pathogenesis of NAFLD is related with IR and a variety of inflammatory cytokines, so it is known that the JNK and p38 signaling pathways are involved in the pathological process of NAFLD.
NF-κB and NAFLD
Nuclear factor-κB (NF-κB) is a nuclear transcription factor widely present in varied cells, which could regulate a variety of cytokines involved in inflammation, adhesion molecules and protease gene transcription in vivo, and it is closely related with inflammation. NF-κB activation can promote the expression of inflammatory cytokines, while inflammatory factors, in turn, can further enhance the activity of NF-κB, to make the inflammation worse [25]. Cytokines (TNF-α, TNF-β, IL-1, etc.), growth factors (such as insulin), immune receptors, the medium, stress response (oxygenation, etc.), bacteria and products (lipopolysaccharide, etc.), viruses and the product, biological xenobiotics, environmental hazards, and other factors could induce NF-κB activation [25], initiating transcription of many genes such as TNF-α, IL-1, IL-6, IL-8 after activation. These gene products regulated by NF-κB are involved in inflammation of the liver, liver fibrosis, liver regeneration and apoptosis. In addition, FFAs, ROS initiate the activation of the transcription factor NF-κB, which leads to increased pro-inflammatory cytokines (TNF-α, TGF-β, IL-1β, IL-6, IL-8) production, and insulin resistance in the liver [4]. Several pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin (IL)-1, IL-6, various adipocytokines, and several transcription factors and kinases such as c-Jun N-terminal kinase (JNK) and a kinase located proximal of nuclear factor-κB (NF-κB) participate in the occurrence and development of the IR [6]. The IR and inflammatory cytokines are related with the pathogenesis of NAFLD. There were also experiments confirmed that NAFLD rat liver tissue NF-κB expression was significantly enhanced than the normal group [26]. Therefore, the NF-κB signaling pathway are probably involved in the pathological process of NAFLD.
AMPK and NAFLD
AMP-activated protein kinase (AMPK) is a heterologous trimeric protein kinase activated by adenosine monophosphate (AMP) and widely exists in eukaryotic cells, which is a central regulator of cellular energy balance and plays an important role in fatty acid metabolism through the fatty acid biosynthetic pathway. AMPK is composed of α, β and γ subunit, α is the catalytic subunit controlled by AMP/ATP ratio, which could be activated by stress stimuli caused by any reduction in intracellular ATP that include metabolic product, the oxidation stress, hypoxia and low sugar [27]; when the AMP/ATP ratio increases, AMPK phosphorylation activates a large number of downstream target molecule acetyl coenzyme A carboxylase (ACC), which reduce the use of ATP (inhibition of glycogen, fat and cholesterol synthesis) and increase ATP production (promotion of fatty acid oxidation and glucose transporter) [28], so that the cell catabolism increases. Sterol regulatory element-binding proteins (SREBPs) are a family of transcription factors that control the expression of genes required for the biosynthesis of cholesterol, fatty acids, triglycerides, and phospholipids, and SREBP-1c preferentially controls the expression of genes involved in triglyceride synthesis and accumulation, such as fatty acid synthase (FAS) and ACC [29]. Therefore, AMPK activation suppresses the expression of ACC and FAS via down-regulation of SREBP-1c [30]. AMPK activators, metformin, have been shown to inhibit the expression of the SREPB-1c gene and to prevent the development of hepatic steatosis [31]. Adiponectin is a recently discovered hormone against diabetes insulin resistance, which comes from the fat cells, has function of regulating energy balance, glucose and fat metabolism, and it is closely related with insulin resistance and atherosclerosis hardening. Adiponectin is inhibited by TNF-α, IL-6, resistin, insulin, and stimulates mitochondrial β-oxidation by activating AMPK and inhibits lipogenesis by down-regulating SREBP-1c [28]. Yamauchi [32] et al. also believed that its signal transduction mechanisms were activating AMPK function. In conclusion, AMPK is closely related with insulin resistance and liver lipid content. A number of experimental studies also confirmed that phosphorylated AMPK protein expression level in high-fat-diet-induced NAFLD was significantly lower than the normal control group [7,24], which suggested that AMPK was involved in the pathological process of NAFLD.
JAK2/STAT3 and NAFLD
Leptin is the main regulator of fat in the organism. It is released from the fat tissue into blood, then dispensed into the tissues (musculi skeleti, adipose tissue, peripheral lymphoid tissue, central nervous system, gastrointestinal tract, and liver) over the body by circulation and combines with its receptors, which then interact with Janus family protein tyrosine kinase/signal transduction and activates transcription factors (JAK/STAT), mainly JAK2/STAT3 [33], to cause the related biological effects, and to exhibit the function of diet control, energy metabolism regulation and interfere with the role of insulin in the liver, thus decreasing triglyceride (TG), elevating the insulin sensitivity of liver and peripheral tissues and reducing fat deposition [8]; When the receptor expression is in dysfunction, JAK2/STAT3 signal transduction disorder occurs, thus leptin could not play the biological effects to cause the leptin resistance. The NAFLD patients commonly have leptin resistance [34], thereby promoting lipid synthesis in the liver and leading to the development of fatty liver. Bartek [35] et al. found that leptin and free leptin receptor in the serum of patients with fatty liver were significantly increased, suggesting that leptin bound to its receptor may exist obstacles to cause leptin resistance. The experimental results showed that leptin receptor mRNA and phosphorylation of JAK2/STAT3 level in NAFLD rat liver were lower than that in the control group [8]. To sum up, leptin JAK2/STAT3 signal pathway is probably involved in the pathological process of NAFLD.
PPARs and NAFLD
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors of nuclear receptor family that exist three subtypes, namely α, β, and γ. PPARα mainly distributes in the tissue with a high efficiency of mitochondrial fatty acid oxidation, which highly expresses in the liver, and PPARγ highly presents in adipose tissue and the immune system. PPARs regulate not only the expression of genes involved in fatty acid synthesis, oxidation, and storage, but also participate in the molecular mechanism of altered metabolic homeostasis, such as is found in type 2 diabetes or obesity [9,36]. PPARα agonist and PPARγ agonist are involved in diabetes mellitus and dyslipidemia, and they play an important role in insulin resistance [37]. Seo [38] et al found PPAR agonists, especially a PPARα agonist, improved the histological and biochemical parameters in the NAFLD rat model by inducing fatty-acid metabolic enzymes. PPARα is considered to be the main regulator of fatty acid oxidation: PPARα is activated in combination with polyunsaturated fatty acids, thereby it can improve insulin resistance, reduce blood lipids, promote β-oxidation of fatty acids, inhibit lipogenesis gene expression, inhibit genes transcription related with inflammatory response, which are conducive to control NAFLD [39]. In addition, PPARα is activated by adiponectin and could inhibit NF-kB pathway [28]. PPARγ enhances insulin action, FFA oxidation, adiponectin secretion, and inhibits secretion of proinflammatory cytokines [28], so PPARγ could improve NAFLD. However hepatic PPARγ 2 expression is increased in high-fat-diet fed mice due to elevated rates of lipogenesis via the upregulation of de novo lipogenic genes FAS and ACC [40]. So it is still not conclusive as to whether the PPARγ is beneficial or detrimental. Thus, we could know PPARs play an important role in the pathogenesis of fatty liver.
PI3K/Akt and NAFLD
Phosphatidylinositol 3-kinase (PI3K) pathway is one of the main signal transduction pathways of insulin action, and PKB/Akt (protein kinase B) as PI3K downstream kinase is the important serine/threonine kinase in this pathway which is primarily responsible for the conduction of the initial biological information by PI3K. PI3K/Akt signaling pathway as insulin downstream molecular pathway, plays an important role in a variety of biological processes such as cell metabolism, cell cycle regulation, cell growth, apoptosis, glucose transporter, and it is closely related with the development of IR [10]. In addition, the p85 subunit of PI3K could be combined with the insulin receptor substrate, so close to the insulin receptor and is anchored in the cell membrane, thereby activating the p110 subunit to regulate fat cells and liver cells in the uptake of glucose through a series of signal transduction [41]. Yuan J [42] et al. found that long-term insulin-stimulated HepG2 cells could lower insulin signal transduction through the PI3K signaling pathway to inhibit insulin signaling, leading to insulin resistance, meanwhile it has been confirmed that insulin signal could not pass in the direction of glucose uptake through the PI3K pathway and to cause IR if the PI3K expression and activity reduce. Akt could suppress fatty acid oxidation gene expression, thereby regulating the process of hepatic glucose and lipid metabolism [43]. There were some study results showed that NAFLD rat liver PI3K and Akt protein were significantly lower than the normal groups [44,45].
TLR and NAFLD
TLR (Toll-like receptor) family members are characterized by highly evolutionarily conserved TIR domain (Toll/IL-1R domain) in the intracellular region, which could recognize varies of pathogen-associated molecular patterns (PAMP) by the leucine repeat(leucine-rich repeat LRR) Ribbon in the extracellular region. Various TLR could induce a series of gene activation through the same or a different signal transduction pathway in pathogen invasion minutes, resulting in cell secretion of pro-inflammatory cytokines and chemokines, upregulation of co-stimulatory factor, increase of antigen-presenting ability, which cause systemic inflammatory response and the occurrence of innate immune response [46]. Knockout rats results showed that TLR4, TLR9 promoted the development of NAFLD [47]. TLR4 is LPS receptor, and LPS elevation in most animal models of NAFLD causes liver steatosis, hepatic insulin resistance, increased liver weight, so the TLR4-LPS is the key pathway to promote NAFLD development [48]. In addition, FFAs could also activate TLR4 [47]. TLR9 signal is related with the development of NASH, TLR9 missing rats present less steatohepatitis, insulin resistance, and fiber reaction [49]. MyD88 is an adapter protein of all TLRs except TLR3, which is related with expression of a variety of inflammatory cytokines and inflammatory chemokines, and TLR-MyD88 signaling pathway could also activate JNK and NF-κB signaling [47], while JNK and NF-κB signals are involved in the pathogenesis of NAFLD. TLR signal mediates the occurrence of steatosis, inflammation and fibrosis, which is closely related with the incidence of NAFLD.
Conclusion
NAFLD is increasingly common in the crowd, the “two-hit” theory based on IR and oxidative stress is the main theoretical basis to explain its pathogenesis. Based on the pathogenesis of NAFLD, it could be found that MAPK, NF-κB, AMPK, JAK2/STAT3, PPARs, PI3K/Akt, TLR signaling pathways are closely related with the incidence of NAFLD. In the NAFLD pathogenesis, the same kind of stimulation could activate different signal pathways, whereas different stimuli could activate a signal pathway (Figure 2): Inflammatory cytokines (TNF-α, TGF-β, IL-1, IL-6, etc.) could activate JNK, p38 and NF-κB signaling pathways, in turn, the activation of these signaling pathways could increase the expression of inflammatory cytokines; adiponectin could reduce fatty acid synthesis and enhance fatty acid β-oxidation via activation of AMP kinase, and then resist steatosis and increase insulin sensitivity [50], but also could activate PPARα and inhibit NF-kB pathway [13]; resistin could inhibit AMPK activity in liver and skeletal muscle, impede insulin signaling PI3K/Akt pathway and induce NF-κB nuclear transcription [28,51,52]; leptin plays biological effects through JAK2/STAT3, may also stimulate AMPK to increase in β-oxidation [28]. These signaling pathways are interconnected to form a network of signaling pathways, so blocking any one of pathway could not be effective to prevent and treat the NAFLD. Therefore, in-depth study of liver cells signaling in the NAFLD pathogenesis process will contribute to the prevention and treatment of NAFLD.
Figure 2.
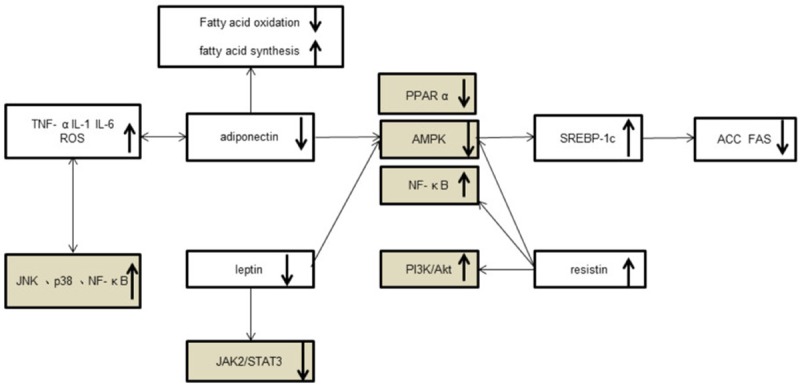
The signal transductions in NAFLD.
References
- 1.Yeh MM, Brunt EM. Pathology of nonalcoholic fatty liver disease. Am J Clin Pathol. 2007;128:837–847. doi: 10.1309/RTPM1PY6YGBL2G2R. [DOI] [PubMed] [Google Scholar]
- 2.Miele L, Forgione A, Hernandez AP, Gabrieli ML, Vero V, Di Rocco P, Greco AV, Gasbarrini G, Gasbarrini A, Grieco A. The natural history and risk factors for progression of non-alcoholic fatty liver disease and steatohepatitis. Eur Rev Med Pharmacol Sci. 2005;9:273–277. [PubMed] [Google Scholar]
- 3.Yan E, Durazo F, Tong M, Hong K. Nonalcoholic fatty liver disease: pathogenesis, identification, progression, and management. Nutr Rev. 2007;65:376–384. doi: 10.1301/nr.2007.aug.376-384. [DOI] [PubMed] [Google Scholar]
- 4.Duvnjak M, Lerotic I, Barsic N, Tomasic V, Virovic JL, Velagic V. Pathogenesis and management issues for non-alcoholic fatty liver disease. World J Gastroenterol. 2007;13:4539–4550. doi: 10.3748/wjg.v13.i34.4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kodama Y, Brenner DA. c-Jun N-terminal kinase signaling in the pathogenesis of nonalcoholic fatty liver disease: Multiple roles in multiple steps. Hepatology. 2009;49:6–8. doi: 10.1002/hep.22710. [DOI] [PubMed] [Google Scholar]
- 6.Tilg H, Moschen AR. Inflammatory mechanisms in the regulation of insulin resistance. Mol Med. 2008;14:222–231. doi: 10.2119/2007-00119.Tilg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park KG, Min AK, Koh EH, Kim HS, Kim MO, Park HS, Kim YD, Yoon TS, Jang BK, Hwang JS, Kim JB, Choi HS, Park JY, Lee IK, Lee KU. Alpha-lipoic acid decreases hepatic lipogenesis through adenosine monophosphate-activated protein kinase (AMPK)-dependent and AMPK-independent pathways. Hepatology. 2008;48:1477–1486. doi: 10.1002/hep.22496. [DOI] [PubMed] [Google Scholar]
- 8.Zheng P, Ji G, Ma Z, Liu T, Xin L, Wu H, Liang X, Liu J. Therapeutic effect of puerarin on non-alcoholic rat fatty liver by improving leptin signal transduction through JAK2/STAT3 pathways. Am J Chin Med. 2009;37:69–83. doi: 10.1142/S0192415X09006692. [DOI] [PubMed] [Google Scholar]
- 9.Aleman G, Torres N, Tovar AR. [Peroxisome proliferator-activated receptors (PPARs) in obesity and insulin resistance development] . Rev Invest Clin. 2004;56:351–367. [PubMed] [Google Scholar]
- 10.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 11.Mencin A, Kluwe J, Schwabe RF. Toll-like receptors as targets in chronic liver diseases. Gut. 2009;58:704–720. doi: 10.1136/gut.2008.156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol. 2010;5:145–171. doi: 10.1146/annurev-pathol-121808-102132. [DOI] [PubMed] [Google Scholar]
- 13.Polyzos SA, Kountouras J, Zavos C, Tsiaousi E. The role of adiponectin in the pathogenesis and treatment of non-alcoholic fatty liver disease. Diabetes Obes Metab. 2010;12:365–383. doi: 10.1111/j.1463-1326.2009.01176.x. [DOI] [PubMed] [Google Scholar]
- 14.Gentile CL, Frye MA, Pagliassotti MJ. Fatty acids and the endoplasmic reticulum in nonalcoholic fatty liver disease. Biofactors. 2011;37:8–16. doi: 10.1002/biof.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tilg H. Adipocytokines in nonalcoholic fatty liver disease: key players regulating steatosis, inflammation and fibrosis. Curr Pharm Des. 2010;16:1893–1895. doi: 10.2174/138161210791208929. [DOI] [PubMed] [Google Scholar]
- 16.Kneeman JM, Misdraji J, Corey KE. Secondary causes of nonalcoholic fatty liver disease. Therap Adv Gastroenterol. 2012;5:199–207. doi: 10.1177/1756283X11430859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harmon RC, Tiniakos DG, Argo CK. Inflammation in nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol. 2011;5:189–200. doi: 10.1586/egh.11.21. [DOI] [PubMed] [Google Scholar]
- 18.De Minicis S, Svegliati-Baroni G. Fibrogenesis in nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol. 2011;5:179–187. doi: 10.1586/egh.11.28. [DOI] [PubMed] [Google Scholar]
- 19.Chakrabarty S, Kondratick L. Insulin-like growth factor binding protein-2 stimulates proliferation and activates multiple cascades of the mitogen-activated protein kinase pathways in NIH-OVCAR3 human epithelial ovarian cancer cells. Cancer Biol Ther. 2006;5:189–197. doi: 10.4161/cbt.5.2.2333. [DOI] [PubMed] [Google Scholar]
- 20.Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tarantino G, Caputi A. JNKs, insulin resistance and inflammation: A possible link between NAFLD and coronary artery disease. World J Gastroenterol. 2011;17:3785–3794. doi: 10.3748/wjg.v17.i33.3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–12101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 23.Pearson G, Robinson F, Beers GT, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 24.Sinha-Hikim I, Sinha-Hikim AP, Shen R, Kim HJ, French SW, Vaziri ND, Crum AC, Rajavashisth TB, Norris KC. A novel cystine based antioxidant attenuates oxidative stress and hepatic steatosis in diet-induced obese mice. Exp Mol Pathol. 2011;91:419–428. doi: 10.1016/j.yexmp.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaidashev IP. [NF-kB activation as a molecular basis of pathological process by metabolic syndrome] . Fiziol Zh. 2012;58:93–101. [PubMed] [Google Scholar]
- 26.Leclercq IA, Farrell GC, Sempoux C, Dela PA, Horsmans Y. Curcumin inhibits NF-kappaB activation and reduces the severity of experimental steatohepatitis in mice. J Hepatol. 2004;41:926–934. doi: 10.1016/j.jhep.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 27.Zhang BB, Zhou G, Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009;9:407–416. doi: 10.1016/j.cmet.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 28.Musso G, Gambino R, Cassader M. Non-alcoholic fatty liver disease from pathogenesis to management: an update. Obes Rev. 2010;11:430–445. doi: 10.1111/j.1467-789X.2009.00657.x. [DOI] [PubMed] [Google Scholar]
- 29.Yuan H, Shyy JY, Martins-Green M. Second-hand smoke stimulates lipid accumulation in the liver by modulating AMPK and SREBP-1. J Hepatol. 2009;51:535–547. doi: 10.1016/j.jhep.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kohjima M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, Fujino T, Yada M, Yada R, Harada N, Enjoji M, Takayanagi R, Nakamuta M. SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int J Mol Med. 2008;21:507–511. [PubMed] [Google Scholar]
- 31.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 33.Saxena NK, Ikeda K, Rockey DC, Friedman SL, Anania FA. Leptin in hepatic fibrosis: evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology. 2002;35:762–771. doi: 10.1053/jhep.2002.32029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chitturi S, Abeygunasekera S, Farrell GC, Holmes-Walker J, Hui JM, Fung C, Karim R, Lin R, Samarasinghe D, Liddle C, Weltman M, George J. NASH and insulin resistance: Insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology. 2002;35:373–379. doi: 10.1053/jhep.2002.30692. [DOI] [PubMed] [Google Scholar]
- 35.Bartek J, Bartos J, Galuska J, Galuskova D, Stejskal D, Ivo K, Ehrmann J, Ehrman JJ, Chlup R. Expression of ob gene coding the production of the hormone leptin in hepatocytes of liver with steatosis. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2001;145:15–20. doi: 10.5507/bp.2001.003. [DOI] [PubMed] [Google Scholar]
- 36.Ferre P. The biology of peroxisome proliferator-activated receptors: relationship with lipid metabolism and insulin sensitivity. Diabetes. 2004;53(Suppl 1):S43–S50. doi: 10.2337/diabetes.53.2007.s43. [DOI] [PubMed] [Google Scholar]
- 37.Choi KC, Ryu OH, Lee KW, Kim HY, Seo JA, Kim SG, Kim NH, Choi DS, Baik SH, Choi KM. Effect of PPAR-α and -γ agonist on the expression of visfatin, adiponectin, and TNF-α in visceral fat of OLETF rats. Biochem Bioph Res Co. 2005;336:747–753. doi: 10.1016/j.bbrc.2005.08.203. [DOI] [PubMed] [Google Scholar]
- 38.Seo YS, Kim JH, Jo NY, Choi KM, Baik SH, Park JJ, Kim JS, Byun KS, Bak YT, Lee CH, Kim A, Yeon JE. PPAR agonists treatment is effective in a nonalcoholic fatty liver disease animal model by modulating fatty-acid metabolic enzymes. J Gastroenterol Hepatol. 2008;23:102–109. doi: 10.1111/j.1440-1746.2006.04819.x. [DOI] [PubMed] [Google Scholar]
- 39.Videla LA, Pettinelli P. Misregulation of PPAR Functioning and Its Pathogenic Consequences Associated with Nonalcoholic Fatty Liver Disease in Human Obesity. PPAR Res. 2012;2012:107434. doi: 10.1155/2012/107434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ables GP. Update on ppargamma and nonalcoholic Fatty liver disease. PPAR Res. 2012;2012:912351. doi: 10.1155/2012/912351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foster FM, Traer CJ, Abraham SM, Fry MJ. The phosphoinositide (PI) 3-kinase family. J Cell Sci. 2003;116:3037–3040. doi: 10.1242/jcs.00609. [DOI] [PubMed] [Google Scholar]
- 42.Yuan J, Gao H, Sui J, Duan H, Chen WN, Ching CB. Cytotoxicity evaluation of oxidized single-walled carbon nanotubes and graphene oxide on human hepatoma HepG2 cells: an iTRAQ-coupled 2D LC MS/MS proteome analysis. Toxicol Sci. 2012;126:149–161. doi: 10.1093/toxsci/kfr332. [DOI] [PubMed] [Google Scholar]
- 43.Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature. 2007;447:1012–1016. doi: 10.1038/nature05861. [DOI] [PubMed] [Google Scholar]
- 44.Liu Y, Han X, Bian Z, Peng Y, You Z, Wang Q, Chen X, Qiu D, Ma X. Activation of liver X receptors attenuates endotoxin-induced liver injury in mice with nonalcoholic fatty liver disease. Dig Dis Sci. 2012;57:390–398. doi: 10.1007/s10620-011-1902-9. [DOI] [PubMed] [Google Scholar]
- 45.Matsuda S, Kobayashi M, Kitagishi Y. Roles for PI3K/AKT/PTEN Pathway in Cell Signaling of Nonalcoholic Fatty Liver Disease. ISRN Endocrinol. 2013;2013:472432. doi: 10.1155/2013/472432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48:322–335. doi: 10.1002/hep.22306. [DOI] [PubMed] [Google Scholar]
- 47.Miura K, Seki E, Ohnishi H, Brenner DA. Role of toll-like receptors and their downstream molecules in the development of nonalcoholic Fatty liver disease. Gastroenterol Res Pract. 2010;2010:362847. doi: 10.1155/2010/362847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soares JB, Pimentel-Nunes P, Roncon-Albuquerque R, Leite-Moreira A. The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol Int. 2010;4:659–672. doi: 10.1007/s12072-010-9219-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. 2010;139:323–334. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Das K, Lin Y, Widen E, Zhang Y, Scherer PE. Chromosomal localization, expression pattern, and promoter analysis of the mouse gene encoding adipocyte-specific secretory protein Acrp30. Biochem Biophys Res Commun. 2001;280:1120–1129. doi: 10.1006/bbrc.2001.4217. [DOI] [PubMed] [Google Scholar]
- 51.Hegarty BD, Turner N, Cooney GJ, Kraegen EW. Insulin resistance and fuel homeostasis: the role of AMP-activated protein kinase. Acta Physiol (Oxf) 2009;196:129–145. doi: 10.1111/j.1748-1716.2009.01968.x. [DOI] [PubMed] [Google Scholar]
- 52.Sheng CH, Di J, Jin Y, Zhang YC, Wu M, Sun Y, Zhang GZ. Resistin is expressed in human hepatocytes and induces insulin resistance. Endocrine. 2008;33:135–143. doi: 10.1007/s12020-008-9065-y. [DOI] [PubMed] [Google Scholar]