

Abstract
Diabetes mellitus was originally conceived as a renal disorder. In the last decade, however, there has been renewed interest in role of the kidney in the development and maintenance of high glucose levels. This has led to the development of novel agents to inhibit sodium glucose transporter-2 (SGLT2) as a means to better control glucose levels and at the same time augment calorie wasting and lower insulin, blood pressure and uric acid levels. Such actions, indirectly, may also have benefits for the prevention of diabetic complications including renal disease. However, there are also data to support the potential for direct renoprotective actions arising from inhibition of SGLT2, including actions to attenuate diabetes-associated hyperfiltration and tubular hypertrophy, as well as reduce the tubular toxicity of glucose. Some studies have demonstrated significant reductions in albumin excretion in various experimental models, independent of its effects on blood pressure or glucose control. Although promising, such actions remain to be established by comprehensive clinical trials with a renal focus, many of which are currently in progress. This article reviews the clinical and experimental data pertaining to the renal effects of SGLT2 inhibition with a particular focus on dapaglifozin.
Keywords: dapagliflozin, diabetes, diabetic nephropathy, hyperfiltration, SGLT2, SGLT2 inhibition
Background
Diabetes mellitus was originally conceived as a renal disorder, as the ‘wasting of flesh into urine’. The discovery of insulin changed the focus of diabetes management to the pancreas. In the last decade, however, there has been renewed interest in role of the kidney in the development and maintenance of high glucose levels [Gerich and Bastien, 2011; Tahrani and Barnett, 2012]. In healthy adults, approximately 180 litres of plasma containing 5.5 mM glucose is filtered every day through the kidneys. This means that approximately 180 grams of glucose are filtered daily through the glomeruli and lost into the primary urine. Almost of this filtered load (~ 99.9%) is then actively reabsorbed by sodium-coupled transport across the brush border membrane of the proximal tubule and then returned to the circulation by glucose transporters [Gerich and Bastien, 2011; Tahrani and Barnett, 2012]. More than 90% of the glucose that is initially filtered is reabsorbed by a low affinity, high capacity system controlled by sodium glucose transporter-2 (SGLT2) in the early convoluted segment of the proximal tubule (s1) [Gerich and Bastien, 2011]. Reabsorption of almost all remaining filtered glucose is performed by sodium glucose transporter-1 (SGLT1), a low affinity, high capacity transporter further down in the straight segment of the descending proximal tubule (s3) [Gerich and Bastien, 2011; Tahrani and Barnett, 2012]. So efficient is this glucose reabsorption pathway that, under normal circumstances, less than 0.1 g will ever find its way into the urine of nondiabetic individuals [Gerich and Bastien, 2011; Kim and Babu, 2012; Tahrani and Barnett, 2012; Whaley et al. 2012].
Yet, however efficient the renal reabsorption of glucose is, its capacity is limited. If plasma glucose levels rise, as in patients with diabetes, the filtered load may exceed the capacity for glucose reabsorption. At which time, glucose (and the water held with it) will spill over into the urine. This produces the classical symptoms of polyuria, frequency and polydipsia that characterizes uncontrolled hyperglycaemia. The threshold at which elevated blood glucose levels results in glycosuria is 10–11 mM on average, but this can vary significantly from person to person. In some people, blood glucose levels greater than 7 mM may produce glycosuria, especially in children and pregnant women. In others, spillover of glucose into the urine may not occur until much higher plasma glucose levels up to 15 mM. This is more often the case in individuals with diabetes where, perhaps as a way to conserve energy, the glucose reabsorption capacity from the proximal tubule is upregulated. This is partly due to tubular hypertrophy and increased of Na/K/ATPase activity in the diabetic kidney. In addition, the expression and activity of apical SGLT2 and basolateral glucose transporter proteins are also increased.
These changes act to increase the threshold for glycosuria and retain extra glucose, paradoxically at a time when glucose levels are already elevated. Moreover, all the extra glucose retained by the kidneys means extra insulin needs to made (or given) to keep the plasma glucose levels under control. And instead of losing the calories attributable to glucose into the urine, (glucose) energy is retained, chiefly as body fat. These changes in renal glucose reabsorption are considered to significantly contribute to the maintenance of hyperglycaemia in patients with diabetes [Defronzo et al. 2012] and provide a strong rationale for inhibition of SGLT2 as a means to better control glucose levels, and at the same time augment calorie wasting and lower blood pressure. Such actions may also have benefits for the prevention of diabetic complications including renal disease. But in addition, there are data to support the potential for direct renoprotective actions arising from inhibition of SGLT2 (Figure 1) [Gilbert, 2013]. This article reviews the clinical and experimental data pertaining to the renal effects of SGLT2 inhibition with a particular focus on dapagliflozin.
Figure 1.
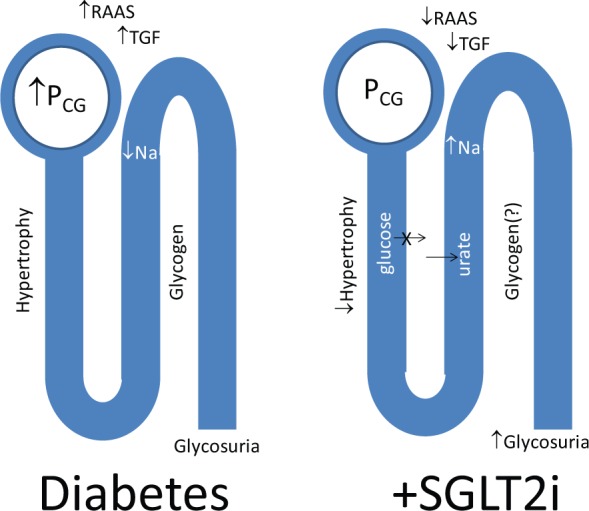
Direct effects of sodium glucose transporter-2 (SGLT2) inhibition in the kidney.
PCG, intraglomerular capillary pressure; RAAS, renin–angiotensin aldosterone system; SGLT2i, sodium glucose transporter-2 inhibitor; TGF, tubuloglomerular feedback.
What does dapagliflozin do to the kidney?
The molecular actions of dapagliflozin
Dapagliflozin is a potent selective reversible inhibitor of SGLT2 in the kidney. The selectivity of dapagliflozin for SGLT2 is at least 1200-fold greater than that for SGLT1 [Riser Taylor and Harris, 2013]. SGLT2 is exclusively expressed in the proximal tubule of the kidney [Chen et al. 2010]. In contrast, SGLT1, with which SGLT2 shares 59% identity, is also expressed in the brain, the heart, and in the intestinal epithelium, where it is involved in water and glucose uptake. Indeed, it has been estimated that almost half the daily uptake of water from the small intestine occurs via SGLT1 dependent pathways [Meinild et al. 1998]. Moreover, SGLT1 also serves as the intestinal glucose sensor for glucose-induced incretin secretion [Moriya et al. 2009], a key component in glycaemic control. These characteristics potentially make SGLT1 a less acceptable clinical target, and selectivity for SGLT2 a desirable property.
Inhibition of SGLT2 following treatment with dapagliflozin reduces the capacity for tubular glucose reabsorption by approximately 30–50%. Why a greater level of inhibition is not achieved in vivo, despite being a potent and comprehensive inhibitor in vitro is unclear. It may be that pharmacokinetics at the proximal tubule, including shuttling of transporters, means that at any one time most SGLT2 are not occupied [Abdul-Ghani et al. 2013]. In addition the upregulation of SGLT1 expression and activity also acts to reduce the excretion of filtered glucose following inhibition of SGLT2 [Rieg et al. 2014]. Nonetheless, this level of inhibition is sufficient to lower the threshold for spillover and result in urinary glucose wasting in both healthy volunteers and patients with diabetes. However, total urinary glucose losses are proportional to the ambient plasma glucose level, so that absolute losses are greater in patients with diabetes and especially those with poor control. In the short term, on average, a 10 mg daily dose of dapagliflozin will increase the amount of glucose excreted in the urine of a patients with type 2 diabetes by 50–80 g per day. However, in the long term it may be less, as improvements in glycaemic control leading to reduction in plasma glucose act to attenuate glycosuria.
There are no clear data that dapagliflozin has actions beyond inhibition of SGLT2 and the induction of glycosuria. Its parent compound, the plant-derived dihydrochalcone phlorizin has been associated with actions on other targets including histone deacetylase, the sodium iodide symporter, as well as antioxidant activities [Ehrenkranz, 2009]. However, these may reflect its other actions on SGLT1 or other receptors.
The renal phenotype of benign familial glycosuria
To obtain some understanding of the long-term renal effects of inhibition of SGLT2, it is possible to extrapolate from the experience of patients with familial glycosuria. The most common genetic forms involve a mutation in the SLCA2 gene that reduces the expression and/or activity of SGLT2. In type O glycosuria, the reabsorption of glucose is severely reduced or absent, meaning that otherwise normoglycaemic individuals can waste over a 100 g of glucose every day. Despite this, familial glycosuria is a generally benign condition, although dehydration and ketosis may sometimes develop during starvation or pregnancy. Such data suggest that long-term inhibition of SGLT2 by selective pharmacological, at the very least, may represent a safe strategy for the management of patients with type 2 diabetes.
The renal effects of glycosuria
The presence of glucose in the urine beyond the straight segment of the descending proximal tubule has a number of effects on the kidney function. The most obvious is that urine flow is increased as water is held in the tubular lumen due to osmotic effects of unabsorbed glucose. This ultimately results in an increased urine output of about 300–400 ml/day. Glycosuria also results in the rapid accumulation of intracellular glycogen granules in the distal tubules, including the thick ascending limbs in the cortex and the outer stripe of the outer medulla (so-called Armanni–Ebstein lesion [Holck and Rasch, 1993]). But while glycogen accumulation may be associated with tubular apoptosis, the pathological significance of intracellular glycogen accumulation remains to be established. Certainly, the Fanconi–Bickel syndrome, a primary tubular disorder due to mutation of the GLUT-2 transporter, is associated with tubular glycogen accumulation as well as progressive diabetic-like changes in the kidney [Berry et al. 1995]. Hyperglycaemia also results in the rapid induction of tubular hypertrophy. Interestingly, in the absence of hyperglycaemia, the lack of SGLT2 in mice is associated with a modest increase in kidney weight [Vallon et al. 2013]. This possibly reflects the impact of increased distal sodium and glucose delivery and storage as glycogen. Interestingly, although hyperglycaemia is associated with a compensatory increase in glomerular filtration rate (GFR) (discussed below), GFR is not affected genetic deletion or inhibition of SGLT2 in nondiabetic mice [Vallon et al. 2013].
Indirect actions of SGLT2 inhibitors on the kidney
Improving glucose control
By reducing the capacity for tubular glucose reabsorption and augmenting urinary glucose wasting, inhibition of SGLT2 results in improved glycaemic control (including improvement in HbA1c, fasting plasma glucose and postprandial glucose levels) in patients with type 2 diabetes [Wilding et al. 2009, 2010; List et al. 2009; Bailey et al. 2010; Ferrannini et al. 2010; Strojek et al. 2011]. There is consistent evidence across many trials that simply improving control, regardless of how it is achieved, is associated with a lower risk of new onset microalbuminuria and a lower rate of progression to overt nephropathy [Boussageon et al. 2011; Coca et al. 2012]. Moreover, these benefits appear to be sustained even if optimal glycaemic control is subsequently lost. In addition, both the United Kingdom Prospective Diabetes Study (UKPDS) and the Action in Diabetes and Vascular Disease: Preterax and Diamicron MR Controlled Evaluation (ADVANCE) study reported a significant reduction in patients requiring renal replacement therapy following intensification of glycaemic control. However, progressive declines in GFR and doubling of serum creatinine are not modified following improvement in glycaemic control [Boussageon et al. 2011].
Reduced insulin levels and improved insulin sensitivity
By promoting glycosuria and lowering the plasma glucose concentration in individuals with diabetes, insulin-stimulated tissue glucose disposal in skeletal muscle is substantially enhanced [Merovci et al. 2014]. This is probably due to the amelioration of glucotoxicity and reduction in body fat. In addition, reduction in glucose levels also leads to a compensatory fall in insulin secretion. Both result in a significant decline in insulin levels in patients with type 2 diabetes. This reduction may contribute to beneficial effects in many organs subject to the ravages of hyperinsulinism, including the kidneys [Han and Susztak, 2014]. It is now clear that insulin has direct actions in the microvasculature that influence the development and progression of microvascular disease. In type 2 diabetes, selective resistance to insulin mediated activation of the phosphatidylinositol-3-kinase signalling pathway leads to reduced synthesis of nitric oxide, impaired metabolic control and compensatory hyperinsulinemia. By contrast, insulin signalling via extracellular signal-regulated kinase dependent pathways is relatively unaffected in diabetes, tipping the balance of insulin’s actions so that they favour abnormal vasoreactivity, angiogenesis, fibrogenesis and other pathways implicated in progressive renal disease [Groop et al. 2005].
Improving blood pressure control
In several studies, a mild naturetic effect has been observed following treatment with dapagliflozin associated with a reduction in systolic blood pressure of ~4 mmHg [Musso et al. 2012], although greater reductions may be observed in hypertensive patients. Although modest, these effects on blood pressure may contribute to the potential renoprotective actions of SGLT2 inhibition. It is well established that improvements in blood pressure control in hypertensive patients are associated with improved renal outcomes. For example, in the UKPDS study of patients with type 2 diabetes, a reduction of blood pressure from 154 to 144 mm Hg was associated with a 30% reduction in microalbuminuria [UK Prospective Diabetes Study Group, 1998]. In the large ADVANCE study, treatment with the angiotensin-converting-enzyme (ACE) inhibitor, perindopril and the naturetic, indapamide (4 mg/1.25 mg daily) lowered the systolic blood pressure by ~7 mmHg and reduced new onset microalbuminuria by 26% compared with conventional antihypertensive treatment [Patel, 2007].
Worsening renal perfusion
Diabetic nephropathy is associated with impaired autoregulation of renal blood flow, meaning that drops in blood pressure will be more likely to be associated with reduction in renal perfusion in the diabetic kidney. This is particularly the case in patients with established renal impairment. In some cases, volume depletion and blood pressure lowering with dapagliflozin as well as other SGLT2 inhibitors having been associated with acute-on-chronic renal impairment [Scheen, 2014]. For this reason, as well as the lack of glucose lowering efficacy in this setting, SGLT2 inhibitors are not used in patients with moderate to severe renal impairment. Interestingly, effects on blood pressure and weight remain preserved in the setting of renal impairment [Kohan et al. 2014]. What may happen in patients with undiagnosed bilateral renal artery stenosis or very vulnerable renal perfusion at lesser degrees of renal impairment is unclear. Monitoring of renal function is currently justified when using renin–angiotensin system (RAS) blockers in patients with diabetes. Similar considerations may also be appropriate in selected (volume sensitive) patients receiving SGLT2 inhibitors.
Improving weight control
In most clinical trials, dapagliflozin has been associated with modest reductions in body weight (approximately 2–3 kg over 12–24 weeks of therapy). This appears to be attributable to the loss of glucose energy, lower insulin levels and increased oxidative metabolism of fat [Cefalu, 2014]. Obesity is recognized as a risk factor for the development of kidney disease and may directly contribute to glomerulopathy [Kambham et al. 2001]. However, it is less clear whether weight loss prevents or attenuates the development of chronic kidney disease (CKD). In a meta-analysis of 5 controlled and 8 uncontrolled trials in which urinary protein was examined among obese or overweight adults, weight loss interventions including dietary restriction, exercise, anti-obesity medications and bariatric surgery decreased urinary albumin excretion, such that each 1 kg weight loss was associated with 110 mg decrease in proteinuria and 1.1 mg decrease in microalbuminuria [Afshinnia et al. 2010]. The Prevention of Renal and Vascular End-Stage Disease (PREVEND) study also reported that weight loss was associated with a reduction in urinary albumin excretion independent of blood pressure, albeit in the general population [Bello et al. 2007].
Lowering uric acid levels
Uric acid levels are elevated in many people with diabetes, in whom their levels are highly correlated with the risk of renal damage in diabetes [Hovind et al. 2011]. More than being a marker, it has been argued that uric acid and its metabolic effects may contribute to microvascular injury, especially in the kidney [Kang et al. 2002]. Consequently, recent attention has turned to whether lowering uric acid levels may have direct renoprotective effects in diabetes. In two small clinical studies, inhibition of xanthine oxidase with achieved a modest reduction in albuminuria [Goicoechea et al. 2010; Momeni et al. 2010]. Whether this is due to the antioxidant effects of inhibition of xanthine oxidase or secondary to lowering uric acid is unclear. A nonpurine alternative to allopurinol, febuxostat, is currently recruiting for a specific clinical trial in diabetic CKD [ClinicalTrials.gov identifier: NCT01350388]. Inhibition of SGLT2 with dapagliflozin also has modest but significant effects on uric acid elimination and lowers plasma uric acid levels [Musso et al. 2012], which may contribute to its renal effects. Uricosuria following SGLT2 inhibition is thought to be due to the actions of SLC2A9b which secretes urate back into the urine in exchange for luminal glucose [Cheeseman, 2009].
Potential direct renoprotective actions of SGLT2 inhibitors?
Glomerular hyperfiltration
Uncontrolled hyperglycaemia is associated with an increase in GFR, driven by increases in glomerular capillary pressure and single-nephron GFR (SNGFR). According to the ‘tubulocentric’ hypothesis, this glomerular hyperfiltration is caused by increased proximal sodium reabsorption associated with tubular hypertrophy and augmented sodium glucose cotransport which leads to reduced distal sodium delivery and subsequent activation of tubuloglomerular feedback and a proportionally greater decrease in afferent versus efferent arteriolar resistance. Intraglomerular hypertension has been strongly implicated in the development and progression of diabetic kidney disease, and has been used to explain the role of nephron endowment in determining the predisposition for diabetic nephropathy as well as the renoprotective effects following from blockade of RAS. Similar arguments are now being used to explain the potential renoprotective actions of SGLT2 inhibition. Certainly, genetic deletion or inhibition of SGLT2 is able to attenuate hyperfiltration association with experimental diabetes [Vallon et al. 2013]. This is thought to be mediated by the reduction is proximal sodium absorption when SGLT2 is inhibited, leading to increased distal sodium delivery to the macula densa, suppression of tubuloglomerular feedback pathways and a compensatory and persistent reduction in intraglomerular pressure [Thomson et al. 2012; Vallon et al. 2013].
Activation of RAS
One of the key pathways suppressed by increasing the distal delivery of sodium is the renin–angiotensin–aldosterone system (RAAS). RAAS activation is, at its most basic, a system for vascular homeostasis that promotes salt and water reabsorption in states of volume depletion. The less sodium that is delivered to the macula densa, which provides the signal of salt/volume depletion, the greater the activation of RAAS to retain salt. Equally, the more sodium that is delivered to the macula densa, the greater the suppression of RAAS to facilitate natriuresis. Consequently, natriuresis induced following inhibition of SGLT2 should effectively inhibit the intrarenal RAAS. At the same time, volume depletion associated with osmotic diuresis will activate the systemic RAAS. In a recent study in patients with type 1 diabetes, aldosterone levels were increased following treatment with empagliflozin both in the presence and absence of hyperglycaemia [Cherney et al. 2014]. What the net effect is and how this may impact the RAAS dependent lesion of diabetic nephropathy is unclear. Certainly, RAAS activation is critical for diabetes associated hyperfiltration, tubular hypertrophy, fibrogenesis and many other pathogenic features of diabetic nephropathy. Indeed it is widely believed that the most potent means currently to inhibit kidney damage is inhibition of RAAS. Such changes may suggest that a combination of RAAS blockade and SGLT2 inhibition, as is the inadvertent norm in patients with type 2 diabetes, may have advantages for the diabetic kidney. Indeed in one experimental study, the combination of RAAS blockade with SGLT2 inhibition was associated with additive renoprotective effects compared to either drug alone [Kojima et al. 2013].
Tubular hypertrophy
One of the earliest structural changes in the diabetic kidney is the hypertrophy and hyperplasia of the cortical tubuli and concomitant renal enlargement. Early studies have demonstrated that diabetic rats exhibit a 15% increase in whole kidney weight within 72 hours of exposure to hyperglycaemia [Osterby and Gundersen, 1975]. Although there are increases in glomerular and, in particular glomerular capillary volume, most of the increase in cortical mass may be accounted for by hypertrophy of the proximal tubule. Tubular epithelial cells in experimental diabetes undergo both proliferation and an increase in cellular size leading to overall increase in tubular length [Rasch and Dorup, 1997]. These abnormalities appear to be largely confined to the cortex and the outer stripe of the outer medulla, with little initial change in the inner stripe of the outer medulla or in the collecting ducts. It had been thought that tubular glucose reabsorption associated with hyperglycaemia is a dominant driving force, leading to increased expression of growth factors like transforming growth factor β (TGF-β), vascular endothelial growth factor (VEGF) and insulin-like growth factor (IGF). In support, studies with the nonspecific SGLT inhibitor, phlorizin, and more recently with selective SGLT2 inhibitor, empaglifozin, have shown that inhibition of glucose reabsorption was able to attenuate renal hypertrophy associated with experimental diabetes [Malatiali et al. 2008; Vallon et al. 2014]. In contrast, gene-targeted knockout of SGLT2 did not prevent renal hypertrophy [Vallon et al. 2013]. However, this may be explained by the fact that knockout of SGLT2 did not increase glycosuria in streptozotocin diabetes, as the profound reduction in plasma glucose in knockout mice completely offset their reduced threshold for urinary glucose retention [Vallon et al. 2013], meaning that the glucose load reaching the tubule was identical in wildtype and SGLT2 knockout mice with diabetes. Whether hypertrophy is a mediator of long-term dysfunction, for example through accelerating the senescence of tubular cells, or is a marker of the induction of pathogenic mediators like TGF-β, VEGF and IGF remains to be established. At least in experimental models, inhibition of tubular hypertrophy appears to have renoprotective effects [Thomson et al. 2001].
Tubulotoxicity of glucose
Exposure of proximal tubular cells to increased levels of glucose results in functional changes, including increased expression of pro-inflammatory cytokines, growth factors and profibrotic mediators, as well as increased formation of advanced glycation endproducts and reactive oxygen species. Each of these has been implicated in the development and progression of diabetic nephropathy. In so far as the induction of these pathways is mediated by glucose entry into the tubular cell and the major mechanism of entry is mediated by SGLT2, inhibition of SGLT2 would appear a useful way of reducing intracellular exposure to glucose in the proximal tubule and consequently its adverse actions in it. Indeed, in cell culture, inhibition of SGLT2 may antagonize many of the deleterious actions of high glucose exposure [Panchapakesan et al. 2013]. Whether this is also the case in vivo, where effective SGLT2 inhibition is far less complete, is unclear.
Nephrolithiasis
Renal stones occur in at least 1 in every 10 patients with diabetes. They remain a major cause of morbidity and can only be modestly reduced by lifestyle interventions. Although inhibition of SGLT2 increases the excretion of uric acid (detailed above), the osmotic effect of glucose considerably dilutes the urine and increases urine flow rate making nidus formation less likely. Clinical studies with SGLT2 inhibitors have not shown an increase in nephrolithiasis. Interestingly AstraZeneca holds a patent for preventing and treating kidney stones by inhibiting SGLT2, although no data validating this strategy are currently available.
Summary
SGLT2 inhibitors offer a complementary approach to reduce glucose levels by promoting urinary glucose wasting. This approach has the added advantage of losing calories that may be otherwise be deposited as fat and sodium that may otherwise support blood pressure elevation. This will have particular advantages in obese and/or hypertensive patients. Beyond these actions, there are sound physiological reasons why inhibition of SGLT2 may have beneficial effects in the kidney (Table 1), in particular by reducing hyperfiltration and renal hypertrophy in patients with incipient nephropathy. But while actions on hyperfiltration are clear, any direct effects of SGLT2 inhibition on actual renal injury has been harder to detect. Gene-targeted knockout of SGLT2, though it prevents hyperfiltration associated with diabetes, does not alter markers of renal injury, inflammation or fibrosis in streptozotocin diabetes [Vallon et al. 2013]. However, studies with SGLT2 inhibitors have demonstrated significant reductions in albumin excretion in various experimental models independent of its effects on blood pressure or blood glucose control. Moreover, a recent clinical trial suggests the regression of albuminuria may be more common in patients treated with dapagliflozin than placebo, despite having little or no effect on glycaemic control [Kohan et al. 2014]. Although promising, such actions remain to be established by comprehensive clinical trials with a renal focus, many of which are currently in progress.
Table 1.
Actions of dapagliflozin with potential to directly or indirectly protect the kidney in diabetes.
| Indirect effects | Direct effects |
|---|---|
| Improved glycaemic control | Prevent hyperfiltration |
| Reduced insulin levels | Renin angiotensin system (?) |
| Improved insulin sensitivity | Prevent tubular hypertrophy |
| Weight loss | Reduced tubular toxicity of glucose |
| Reduced blood pressure | Reduced nephrolithiasis? |
| Reduced uric acid |
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: The author has received honoraria from companies involved in the development and marketing of SGLT2 inhibitors including Bristol Meyers Squibb, AstraZeneca, Boehringer Ingelhiem, Lilly, Takeda, and Merck Sharpe and Dohme.
References
- Abdul-Ghani M., Defronzo R., Norton L. (2013) Novel hypothesis to explain why SGLT2 inhibitors inhibit only 30–50% of filtered glucose load in humans. Diabetes 62: 3324–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afshinnia F., Wilt T., Duval S., Esmaeili A., Ibrahim H. (2010) Weight loss and proteinuria: systematic review of clinical trials and comparative cohorts. Nephrol Dial Transplant 25: 1173–1183 [DOI] [PubMed] [Google Scholar]
- Bailey C., Gross J., Pieters A., Bastien A., List J. (2010) Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with metformin: a randomised, double-blind, placebo-controlled trial. Lancet 375: 2223–2233 [DOI] [PubMed] [Google Scholar]
- Bello A., De Zeeuw D., El Nahas M., Brantsma A., Bakker S., De Jong P., et al. (2007) Impact of weight change on albuminuria in the general population. Nephrol Dial Transplant 22: 1619–1627 [DOI] [PubMed] [Google Scholar]
- Berry G., Baker L., Kaplan F., Witzleben C. (1995) diabetes-like renal glomerular disease in Fanconi–Bickel syndrome. Pediatr Nephrol 9: 287–291 [DOI] [PubMed] [Google Scholar]
- Boussageon R., Bejan-Angoulvant T., Saadatian-Elahi M., Lafont S., Bergeonneau C., Kassai B., et al. (2011) Effect of intensive glucose lowering treatment on all cause mortality, cardiovascular death, and microvascular events in type 2 diabetes: meta-analysis of randomised controlled trials. Br Med J 343: d4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cefalu W. (2014) Paradoxical insights into whole body metabolic adaptations following SGLT2 inhibition. J Clin Invest 124: 485–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman C. (2009) Solute carrier family 2, member 9 and uric acid homeostasis. Curr Opin Nephrol Hypertens 18: 428–432 [DOI] [PubMed] [Google Scholar]
- Chen J., Williams S., Ho S., Loraine H., Hagan D., Whaley J., et al. (2010) Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther 1: 57–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherney D., Perkins B., Soleymanlou N., Maione M., Lai V., Lee A., et al. (2014) Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation 129: 587–597 [DOI] [PubMed] [Google Scholar]
- Coca S., Ismail-Beigi F., Haq N., Krumholz H., Parikh C. (2012) Role of intensive glucose control in development of renal end points in type 2 diabetes mellitus: systematic review and meta-analysis intensive glucose control in type 2 diabetes. Arch Intern Med 172: 761–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defronzo R., Davidson J., Del Prato S. (2012) The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab 14: 5–14 [DOI] [PubMed] [Google Scholar]
- Ehrenkranz J. (2009) Glucose control, the kidney, and potential treatments for diabetes: more to the story. Am J Kidney Dis 54: 583. [DOI] [PubMed] [Google Scholar]
- Ferrannini E., Ramos S., Salsali A., Tang W., List J. (2010) Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care 33: 2217–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerich J., Bastien A. (2011) Development of the sodium-glucose co-transporter 2 inhibitor dapagliflozin for the treatment of patients with type 2 diabetes mellitus. Expert Rev Clin Pharmacol 4: 669–683 [DOI] [PubMed] [Google Scholar]
- Gilbert R. (2013) Sodium-glucose linked transporter-2 inhibitors: potential for renoprotection beyond blood glucose lowering? Kidney Int 10.1038/ki.2013.451 [DOI] [PubMed] [Google Scholar]
- Goicoechea M., De Vinuesa S., Verdalles U., Ruiz-Caro C., Ampuero J., Rincon A., et al. (2010) Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin J Am Soc Nephrol 5: 1388–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groop P., Forsblom C., Thomas M. (2005) Mechanisms of disease: pathway-selective insulin resistance and microvascular complications of diabetes. Nat Clin Pract Endocrinol Metab 1: 100–110 [DOI] [PubMed] [Google Scholar]
- Han S., Susztak K. (2014) The hyperglycemic and hyperinsulinemic combo gives you diabetic kidney disease immediately. Focus on ‘combined acute hyperglycemic and hyperinsulinemic clamp induced profibrotic and proinflammatory responses in the kidney’. Am J Physiol Cell Physiol 306: C198–C199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holck P., Rasch R. (1993) Structure and segmental localization of glycogen in the diabetic rat kidney. Diabetes 42: 891–900 [DOI] [PubMed] [Google Scholar]
- Hovind P., Rossing P., Johnson R., Parving H. (2011) Serum uric acid as a new player in the development of diabetic nephropathy. J Ren Nutr 21: 124–127 [DOI] [PubMed] [Google Scholar]
- Kambham N., Markowitz G., Valeri A., Lin J., D’Agati V. (2001) Obesity-related glomerulopathy: an emerging epidemic. Kidney Int 59: 1498–1509 [DOI] [PubMed] [Google Scholar]
- Kang D., Nakagawa T., Feng L., Watanabe S., Han L., Mazzali M., et al. (2002) A role for uric acid in the progression of renal disease. J Am Soc Nephrol 13: 2888–2897 [DOI] [PubMed] [Google Scholar]
- Kim Y., Babu A. (2012) Clinical potential of sodium-glucose cotransporter 2 inhibitors in the management of type 2 diabetes. Diabetes Metab Syndr Obes 5: 313–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan D., Fioretto P., Tang W., List J. (2014) Long-term study of patients with type 2 diabetes and moderate renal impairment shows that dapagliflozin reduces weight and blood pressure but does not improve glycemic control. Kidney Int 85: 962–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima N., Williams J., Takahashi T., Miyata N., Roman R.J. (2013) Effects of a new SGLT2 inhibitor, luseogliflozin, on diabetic nephropathy in T2DN rats. J Pharmacol Exp Ther 345: 464–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- List J., Woo V., Morales E., Tang W., Fiedorek F. (2009) Sodium-glucose cotransport inhibition with dapagliflozin in type 2 diabetes. Diabetes Care 32: 650–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malatiali S., Francis I., Barac-Nieto M. (2008) Phlorizin prevents glomerular hyperfiltration but not hypertrophy in diabetic rats. Exp Diabetes Res 2008: 305403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinild A., Klaerke D., Loo D., Wright E., Zeuthen T. (1998) The human Na+-glucose cotransporter is a molecular water pump. J Physiol 508: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merovci A., Solis-Herrera C., Daniele G., Eldor R., Fiorentino T., Tripathy D., et al. (2014) Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest 124: 2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momeni A., Shahidi S., Seirafian S., Taheri S., Kheiri S. (2010) Effect of allopurinol in decreasing proteinuria in type 2 diabetic patients. Iran J Kidney Dis 4: 128–132 [PubMed] [Google Scholar]
- Moriya R., Shirakura T., Ito J., Mashiko S., Seo T. (2009) Activation of sodium-glucose cotransporter 1 ameliorates hyperglycemia by mediating incretin secretion in mice. Am J Physiol Endocrinol Metab 297: E1358–E1365 [DOI] [PubMed] [Google Scholar]
- Musso G., Gambino R., Cassader M., Pagano G. (2012) A novel approach to control hyperglycemia in type 2 diabetes: sodium glucose co-transport (SGLT) inhibitors: systematic review and meta-analysis of randomized trials. Ann Med 44: 375–393 [DOI] [PubMed] [Google Scholar]
- Osterby R., Gundersen H. (1975) Glomerular size and structure in diabetes mellitus. I. Early abnormalities. Diabetologia 11: 225–229 [DOI] [PubMed] [Google Scholar]
- Panchapakesan U., Pegg K., Gross S., Komala M., Mudaliar H., Forbes J., et al. (2013) Effects of SGLT2 inhibition in human kidney proximal tubular cells—renoprotection in diabetic nephropathy? PLoS One 8: e54442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A., Advance Collaborative Group (2007) Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus (the ADVANCE trial): a randomised controlled trial. Lancet 370: 829–840 [DOI] [PubMed] [Google Scholar]
- Rasch R., Dorup J. (1997) Quantitative morphology of the rat kidney during diabetes mellitus and insulin treatment. Diabetologia 40: 802–809 [DOI] [PubMed] [Google Scholar]
- Rieg T., Masuda T., Gerasimova M., Mayoux E., Platt K., Powell D., et al. (2014) Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am J Physiol Renal Physiol 306: F188–F193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riser Taylor S., Harris K. (2013) The clinical efficacy and safety of sodium glucose cotransporter-2 inhibitors in adults with type 2 diabetes mellitus. Pharmacotherapy 33: 984–999 [DOI] [PubMed] [Google Scholar]
- Scheen A. (2014) Evaluating SGLT2 inhibitors for type 2 diabetes: pharmacokinetic and toxicological considerations. Expert Opin Drug Metab Toxicol 10: 647–663 [DOI] [PubMed] [Google Scholar]
- Strojek K., Yoon K., Hruba V., Elze M., Langkilde A., Parikh S. (2011) Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with glimepiride: a randomized, 24-week, double-blind, placebo-controlled trial. Diabetes Obes Metab 13: 928–938 [DOI] [PubMed] [Google Scholar]
- Tahrani A., Barnett A. (2012) Dapagliflozin: A sodium glucose cotransporter 2 inhibitor in development for type 2 diabetes. DiabetesTher 1: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson S., Deng A., Bao D., Satriano J., Blantz R., Vallon V. (2001) Ornithine decarboxylase, kidney size, and the tubular hypothesis of glomerular hyperfiltration in experimental diabetes. J Clin Invest 107: 217–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson S., Rieg T., Miracle C., Mansoury H., Whaley J., Vallon V., et al. (2012) Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am J Physiol Regul Integr Comp Physiol 302: R75–R83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- UK Prospective Diabetes Study Group (1998) Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. Br Med J 317: 703–713 [PMC free article] [PubMed] [Google Scholar]
- Vallon V., Gerasimova M., Rose M., Masuda T., Satriano J., Mayoux E., et al. (2014) SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic akita mice. Am J Physiol Renal Physiol 306: F194–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallon V., Rose M., Gerasimova M., Satriano J., Platt K., Koepsell H., et al. (2013) Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am J Physiol Renal Physiol 304: F156–F167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whaley J., Tirmenstein M., Reilly T., Poucher S.M., Saye J., Parikh S., et al. (2012) Targeting the kidney and glucose excretion with dapagliflozin: preclinical and clinical evidence for SGLT2 inhibition as a new option for treatment of type 2 diabetes mellitus. Diabetes Metab Syndr Obes 5: 135–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilding J., Norwood P., T’Joen C., Bastien A., List J., Fiedorek F. (2009) A study of dapagliflozin in patients with type 2 diabetes receiving high doses of insulin plus insulin sensitizers: applicability of a novel insulin-independent treatment. Diabetes Care 32: 1656–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilding J., Woo V., Pahor A., Sugg J., Langkilde A., Parikh S. (2010) Effect of dapagliflozin, a novel insulin-independent treatment, over 48 weeks in patients with type 2 diabetes poorly controlled with insulin. Diabetologia 53(Suppl.): abstract S348 [Google Scholar]