

ABSTRACT
The intestinal brush border Na+/H+ exchanger NHE3 is tightly regulated through changes in its endocytosis and exocytosis. Myosin VI, a minus-end-directed actin motor, has been implicated in endocytosis at the inter-microvillar cleft and during vesicle remodeling in the terminal web. Here, we asked whether myosin VI also regulates NHE3 movement down the microvillus. The basal NHE3 activity and its surface amount, determined by fluorometry of the ratiometric pH indicator BCECF and biotinylation assays, respectively, were increased in myosin-VI-knockdown (KD) Caco-2/Bbe cells. Carbachol (CCH) and forskolin (FSK) stimulated NHE3 endocytosis in control but not in myosin VI KD cells. Importantly, immunoelectron microscopy results showed that NHE3 was preferentially localized in the basal half of control microvilli but in the distal half in myosin VI KD cells. Treatment with dynasore duplicated some aspects of myosin VI KD: it increased basal surface NHE3 activity and prevented FSK-induced NHE3 endocytosis. However, NHE3 had an intermediate distribution along the microvillus (between that in myosin VI KD and untreated cells) in dynasore-treated cells. We conclude that myosin VI is required for basal and stimulated endocytosis of NHE3 in intestinal cells, and suggest that myosin VI also moves NHE3 down the microvillus.
KEY WORDS: Na+/H+ exchanger, Na+ absorption, Endocytosis, Trafficking, Myosin VI, Brush border
INTRODUCTION
Acute regulation of the activity of the intestinal brush border Na+/H+ exchanger NHE3 during digestion contributes to intestinal water and Na+ homeostasis. This regulation occurs primarily through changes in plasma membrane NHE3 expression, which is mediated by modulation of the rates of its endocytosis and exocytosis (Sarker et al., 2008; Cha et al., 2006; Donowitz and Li, 2007; Collazo et al., 2000; Bobulescu et al., 2005). The great majority of NHE3 resides in the intestinal microvillus under basal conditions and its activity is downregulated early in the post-prandial state by stimulated endocytosis. The steady-state distribution of NHE3 along the microvillus is not known, nor is the machinery regulating it under basal and stimulated conditions.
In membrane trafficking, motor proteins moving along cytoskeletal tracks play major roles in transporting cargo or vesicles between donor and acceptor compartments. There are three classes of mammalian motors: myosins, which move along actin filaments, and kinesins and dyneins, which use microtubules. Only myosin motors are relevant to membrane protein movements within or on the microvillus because each microvillus contains a core of cross-linked actin filaments but no microtubules (Bretscher et al., 1982; Coudrier et al., 1982; Matsudaira, 1983; Mooseker et al., 1982; Mooseker, 1983; Mooseker et al., 1984). The myosin superfamily is made up of ∼20 different classes, and the intestinal brush border, which comprises the microvillus, the inter-microvillar region and underlying terminal web, contains at least 14 myosin members (Hodge and Cope, 2000; Berg et al., 2001; McConnell et al., 2011). However, myosin VI is the only negatively directed myosin isoform in this group capable of moving material from the microvillus to the inter-microvillar region (Wells et al., 1999).
Myosin VI has been suggested to take part in at least three distinct steps of apical endocytosis in polarized epithelial cells (reviewed by Hasson, 2003). These include movement of cargo from the microvillus to the clathrin-coated pit area, clathrin-coated vesicle formation and movement of uncoated vesicles through the actin–Arp2/3-complex-rich terminal web to early endosomes (reviewed by Hasson, 2003). There is evidence supporting the second and third steps, but evidence for myosin-VI-dependent movement of cargo from the microvillus to inter-microvillar region remains indirect (Gottlieb et al., 1993; Biemesderfer et al., 2002; Birn et al., 1997; Christensen and Nielsen, 1991; Aschenbrenner et al., 2003; Yang et al., 2005; Ameen and Apodaca, 2007; Yang et al., 2008; Riquier et al., 2009; Collaco et al., 2010; Kravtsov et al., 2012).
In this study, we provide direct evidence that myosin VI is required to move NHE3 along the microvillus length to the inter-microvillar region of intestinal epithelial cells, and confirm its role in basal and stimulated endocytosis. Our approach was to knockdown myosin VI in polarized Caco-2/Bbe cells and assay the activity, protein abundance and surface distribution of NHE3 using biochemical and morphological methods.
RESULTS
Myosin VI is present in the microvilli of intestinal epithelial cells
The expression of myosin VI in the brush border of intestinal epithelial cells is well documented (Hegan et al., 2012; Buss et al., 2001; Biemesdorfer et al., 2002). We confirmed the presence of myosin VI in the brush border of Caco-2 cells and mouse ileum, and determined the extent to which it overlapped with villin, a brush border marker, in wild-type Caco-2/Bbe cells, and aquaporin 7, a microvillus marker, in mouse ileum. Approximately 45% of myosin VI overlapped with brush border villin in Caco-2/Bbe cells (Fig. 1Aa1–a4, Fig. 1B), illustrating that it is abundant throughout the brush border. The overlap of myosin VI with aquaporin, which is restricted to the microvillus membrane, was ∼9% (Fig. 1Ab1–b4, Fig. 1B). There was no signal in the ileum of myosin-VI-knockout (KO) mice (Fig. 1Ad) or in the myosin-VI-knockdown (KD) Caco-2/Bbe cells (Fig. 1Af). These results demonstrate that myosin VI is present throughout the brush border (including the microvillus) in both Caco-2/Bbe cells and mouse small intestine.
Fig. 1.

A pool of myosin VI localizes on the microvillus of intestinal epithelial cells. (A) Immunofluorescence (confocal microscopic) detection of myosin VI expression and localization in Caco-2/Bbe cells and mouse ileum. In Caco-2/Bbe cells, myosin VI is present throughout the brush border, including in the microvillus and in a vesicular pattern in the terminal web and other intracellular compartments (a1–a4). In mouse ileum, myosin VI is present to a lesser extent on the microvillus and in the terminal web (b1–b4). As a negative control, anti-myosin VI antibody was used to show there is no myosin VI staining in ileum from myosin VI KO mice (d) and myosin VI KD Caco-2/Bbe cells (f). (c) wild-type (WT) control; (e) empty-vector-infected control. Scale bar: 20 µm. (B) Analysis of the overlap of myosin VI and protein markers of microvillus by MetaMorph colocalization software. Please note this software shows presence of two proteins in the same intracellular compartment but does not establish physical association. In Caco-2/Bbe cells, ∼46% of myosin VI overlaps with villin, and in mouse ileum ∼9% of myosin VI overlaps with aquaporin 7. For both Caco-2/Bbe cells and mouse ileum, results are mean±s.e.m. of three separate experiments with five or six images analyzed in each experiment (the total number of images analyzed was 16).
Knocking down myosin VI does not dramatically change Caco-2/Bbe cell morphology
To study the regulatory role of myosin VI in NHE3 localization and transport activity, we knocked down the protein in Caco-2/Bbe cells. Myosin VI expression was reduced by ∼90%, as judged by western blotting (Fig. 2A), and was not detected by immunofluorescence (Fig. 1Af) in myosin VI KD cells. Light microscopy images showed that control and KD cells had similar morphologies (data not shown), and transmission electron microscopic (TEM) studies confirmed this view (Fig. 2B,C). It should be noted that individual microvilli in both control and KD cells were variable in length and surface density, as has been seen in other studies (Marzesco et al., 2009; Wilson, et al., 1990). Nevertheless, the microvilli lengths were comparable in these two cell types (Fig. 2D). The KD cells did not show an apparent lifting of the plasma membrane in the inter-microvillar region, as reported for intestinal cells in vivo (Hegan, et al., 2012; T.C. and M.D., unpublished observations). Finally, the localization of the tight junction proteins ZO-1 (also known as TJP1) (supplementary material Fig. S1a,b), occludin (supplementary material Fig. S1c,d) and claudin-1 (supplementary material Fig. S1e,f) were similar in control and KD cells, as were the trans-epithelial resistances (Fig. 2E).
Fig. 2.
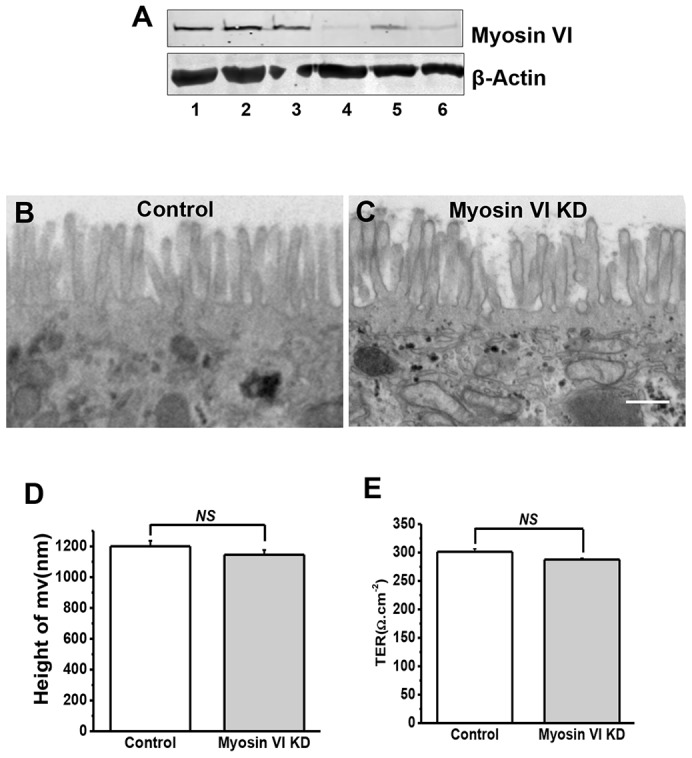
Efficient KD of myosin VI in Caco-2/Bbe cells did not significantly alter the ultrastructure of their microvilli. (A) Myosin VI expression in lenti-shRNA virus-infected Caco-2/Bbe cells. Caco-2/Bbe cells were infected with empty vector (lane 1), lentiviral vector encoding shRNA against GFP (lanes 2 and 3), and three different shRNAs against myosin VI (shMyoVI) [no. 1 (lane 4), no. 2 (lane 5) and no. 3 (lane 6) (sequences are shown in supplementary material Table S1)]. After 48 h of incubation, the cells were harvested for western blot analysis. This experiment was repeated three times with similar results. (B,C) TEM of microvillus in control (B) and myosin VI KD (C) cells grown on Transwell filters for 14 days. Representative images are shown. No obvious morphological changes were seen in the myosin VI KD cells, such as pulling away of microvillar membrane from the base of microvillus, as previously reported in myosin VI KO mouse intestine (Ameen and Apodaca, 2007). Scale bar: 500 nm. (D) The lengths of microvilli in myosin VI KD cells were not significantly (NS) different from those in control cells. Results are mean±s.e.m. Results are from a single experiment using conventional (i.e. glutaraldehyde) processing (>100 microvilli analyzed) and six experiments using immunoelectron microscopy processing (>200 microvilli analyzed). (E) Measurement of transepithelial electrical resistance (TER) in Caco-2/Bbe cells ∼14 days post confluency. The value obtained is expressed in Ω.cm−2. Results are mean±s.e.m. (n = 3). There was no significant difference of TER between control cells and myosin VI KD cells.
Knocking down myosin VI increases NHE3 activity and the amount of NHE3 at the surface
NHE3 activity is increased upon myosin VI knockdown
To test the effect of knocking down myosin VI on NHE3 function, NHE3 basal and stimulated activities were measured by fluorometry of the ratiometric pH indicator BCECF [it should be noted that NHE3 activity measured with this indicator is not influenced by changes in cell surface area (Levine et al., 1993)]. Basal NHE3 activity was increased >60% after myosin VI KD (Fig. 3A,B). If myosin VI were necessary for NHE3 endocytosis, we would predict that myosin VI KD would greatly reduce the inhibition of NHE3 activity caused by carbachol (CCH) or forskolin (FSK), both of which stimulate NHE3 endocytosis. The results shown in Fig. 3 confirm this prediction: whereas CCH and FSK treatment reduced NHE3 activity in control Caco-2/Bbe cells neither had any effect on NHE3 activity in myosin VI KD cells (Fig. 3C,D; P<0.05). We then asked whether myosin VI was necessary for the stimulatory effect of epidermal growth factor (EGF) on NHE3 activity, which we have previously shown is due to an increase in surface NHE3 (Sarker et al., 2011). Unlike the effects of CCH or FSK, EGF treatment resulted in similar increases in NHE3 activity in control and myosin VI KD cells (Fig. 3E). A summary of the role of myosin VI in NHE3 basal and regulated activity is shown in Table 1. Taken together, these data indicate that myosin VI is necessary for setting basal NHE3 activity in the microvillus and for regulating the endocytosis of the transporter after exposure of cells to CCH or FSK. Importantly, our results show that myosin VI is not required for exocytosis of NHE3 (Janecki et al., 2000; Li et al., 2001).
Fig. 3.

Knocking down myosin VI increases basal NHE3 activity and prevents reduction of NHE3 activity by CCH and FSK in Caco-2/Bbe cells but does not affect EGF stimulation of NHE3. (A,B) Myosin VI KD in Caco-2/Bbe cells increased basal NHE3 activity by more than 60%. Basal transport activity was measured in Ad-HA-NHE3-infected Caco-2/Bbe cells. NHE3 activity was measured from the initial rates of Na+-dependent intracellular alkalinization. A is a single experiment and B is mean±s.e.m. (µM/s) of four experiments. (C–E) Knocking down myosin VI in Caco-2/Bbe cells prevents the inhibition of NHE3 activity by FSK (C) (P<0.05) and CCH (D) (P<0.05), but not its stimulation by EGF (E) (P<0.05). Results are mean±s.e.m. of three independent experiments. NS, not significant.
Table 1. Disruption of myosin VI in Caco-2/Bbe/HA-NHE3 cells prevents downregulation of NHE3 activity by CCH and FSK but not its stimulation by EGF.
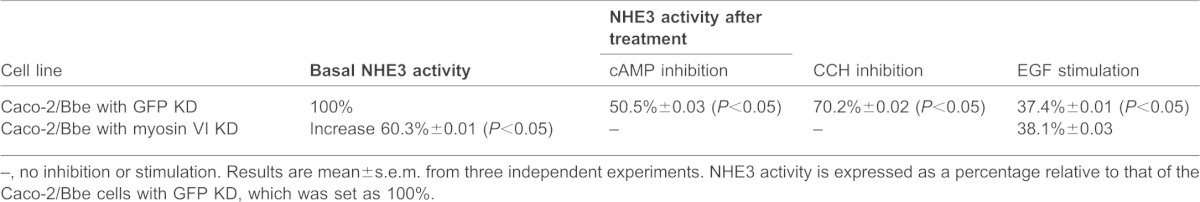
–, no inhibition or stimulation. Results are mean±s.e.m. from three independent experiments. NHE3 activity is expressed as a percentage relative to that of the Caco-2/Bbe cells with GFP KD, which was set as 100%.
NHE3 surface amount is increased upon myosin VI knockdown
We next asked whether myosin VI KD altered the surface expression of NHE3 protein. We compared several parameters of NHE3 expression in control and myosin VI KD cells: total NHE3 expression level by immunoblot analysis and quantitative immunofluorescence, and apical membrane NHE3 amounts by cell-surface biotinylation and quantitative immunoblot analysis. There was a significant increase in total NHE3 expression in KD versus control cells (Fig. 4A,B,D). We also measured the extent of overlap between NHE3 and fluorophore-labeled wheat germ agglutinin (WGA), which was applied to fixed cells at 4°C, prior to their permeabilization and staining for NHE3 (Fig. 4C). The extent of overlap between NHE3 and WGA, which represents microvillar NHE3, was increased in the myosin VI KD cells (Fig. 4E). Finally, we compared the amount of microvillar NHE3 in control and myosin VI KD cells by measuring surface biotinylation after exposure to CCH or FSK. In control cells, the amount of surface NHE3 was significantly reduced after treatment with CCH or FSK (Fig. 5A,C). However, myosin VI KD cells, which we already showed had increased amounts of NHE3 on the cell surface, showed no subsequent decrease in surface NHE3 upon exposure to either CCH or FSK (Fig. 5B,D). Again, these results suggest that myosin VI sets the amount of surface NHE3 expression and activity under basal conditions and conditions that stimulate endocytosis.
Fig. 4.

Total and surface NHE3 levels were increased under basal conditions in myosin VI KD Caco-2/Bbe cells. (A,B) Western blot detection and quantification of total NHE3 in myosin VI KD cells. Cells were grown in six-well plates and infected with Ad-HA-NHE3 at day 12. At 48 h after infection, cells were processed for western blotting. Total NHE3 amount was normalized with β-actin. Results are mean±s.e.m. of three independent experiments. In myosin VI KD cells, the total NHE3 amount was significantly increased (P<0.05). (C) NHE3 expression in myosin VI KD cells as detected by immunofluorescence (confocal microscopical) analysis. Cells were filter grown for 12 days, infected with Ad-HA-NHE3 and 2 days later processed for antibody staining. Cells were not initially permeabilized and were surface labeled with Alexa-Fluro-568-conjugated WGA at 4°C; then cells were permeabilized and labeled with anti-HA antibody. (C) Compared to control cells (a1, a2, a3), NHE3 expression was dramatically increased in myosin VI KD cells (b1, b2, b3). Scale bar: 20 µm. (D) Statistical analysis of total NHE3 intensity by Volocity software. The total NHE3 amount was increased (P<0.05) in myosin VI KD cells. Results are mean±s.e.m. of ten images from three independent experiments. (E) Quantification of the percentage of NHE3 on the surface by Volocity software. The percentage of surface NHE3 was increased (P<0.05) in myosin VI KD cells. Results are mean±s.e.m. of 15 images from three independent experiments.
Fig. 5.

The surface NHE3 amount was not changed by FSK and CCH in myosin VI KD Caco-2/Bbe cells but was decreased in control cells. Cells were grown in six-well plates and infected with Ad-HA-NHE3 at day 12. After 48 h, cells were serum-starved for 4–5 h then treated with 25 µM CCH for 5 min or 10 µM FSK for 15 min at 37°C before assessing the amount NHE3 by a cell surface biotinylation assay. Detection and quantification of total and surface amounts of NHE3 are shown in A and B (single experiments), and C and D (mean±s.e.m. of three independent experiments). Two dilutions of total, surface and cytosol fractions were loaded and the surface expression of NHE3 was determined as a percentage of total NHE3 and normalized to β-actin; the values for the controls were set to 100 in each experiment. As shown in A and C, CCH- and FSK-treated control cells expressed ∼40% less surface NHE3 than non-treated cells (P<0.05). In myosin VI KD cells, there was no significant (NS) change of surface NHE3 expression after treatment with CCH or FSK compared to non-treated cells (B,D).
Myosin VI regulates the distribution of NHE3 along the microvillus length
Endocytosis of apical membrane proteins in polarized epithelial cells with brush borders, such as the intestine, can be separated into three steps: (1) movement to and accumulation of microvillus membrane proteins in the inter-microvillus region; (2) internalization in clathrin-coated vesicles (and also non-clathrin-dependent processes) that form in the inter-microvillus region; and (3) movement of vesicles through the actin-rich terminal web to early endosomal compartments (Aschenbrenner et al., 2003; Hasson, 2003). Evidence for the involvement of myosin VI in the second and third steps has been reported by others (Gottlieb et al., 1993; Biemesderfer et al., 2002; Birn et al., 1997; Christensen and Nielsen, 1991; Aschenbrenner et al., 2003; Yang et al., 2005; Ameen et al., 2007; Yang et al., 2008; Riquier et al., 2009; Collaco et al., 2010; Kravtsov et al., 2012), and our results described so far support its requirement for both basal and agonist-stimulated endocytosis of NHE3 in Caco-2/Bbe cells. However, the localization of NHE3 along the microvillus in control and myosin VI KD cells has not been determined; consequently, we used immunoelectron microscopy to address this question. After paraformaldehyde fixation of filter-grown control and myosin VI KD cells, surface HA–NHE3 was labeled with anti-HA antibodies, then gold-labeled secondary antibodies, after which the cells were fixed with glutaraldehyde before being processed for electron microscopy. As shown in Fig. 6, ∼85% of the microvillar NHE3 in control cells was located in the first 600 nm of the microvillus length and the rest was >600 nm away from the microvillus base (Fig. 6). In contrast, <50% of the microvillar NHE3 in myosin VI KD cells was located in the first 600 nm and >50% was >600 nm away from the microvillus base (Fig. 6). This shift in NHE3 distribution along the microvillus length is important and suggests that the increase in NHE3 surface activity that we consistently detected in myosin VI KD cells (Fig. 3A–E) could be represented by the NHE3 present in the outer microvillus half (see Discussion).
Fig. 6.

More NHE3 distributes on the outer half of the microvillus in myosin VI KD Caco-2/Bbe cells compared to controls. (A,B) HA-NHE3 was detected by immunoelectron microscopy using gold-labeled secondary antibodies. HA–NHE3 was detected on paraformaldehyde-fixed cells by labeling first with mouse anti-HA antibodies followed by anti-mouse-IgG conjugated to 12-nm gold particles (red circles). The cells were then fixed with glutaraldehyde. The circles surround gold particles on the microvillus of the epithelial cells. mv, microvillus. Ctrl, empty vector infected Caco-2/Bbe cells. Scale bar: 0.5 µm. (C) A diagram of a microvillus shows how the HA–NHE3 distribution on the microvillus was quantified. The whole microvillus from the baseline to tip (∼1200 nm) was divided into 10–12 sections, each 100-nm wide, and the number of gold particles in each section was counted. (D) Histograms show the fraction of microvillar NHE3 distributed 0–600 nm (dark gray) and >600nm (light gray) away from the microvillus base in control and myosin VI KD cells from all individual data points of three separate experiments. Number in brackets represent the total gold particles counted in each condition. The total number of cells with gold particles counted are 79 in control and 125 in myosin VI KD. Two-sample Wilcoxon rank-sum (Mann–Whitney) tests showed that NHE3 was significantly differently distributed in control and myosin VI KD cells, <600 nm (*P<0.0001) and ≧600 nm (†P<0.0001). <600 nm and ≧ 600 nm refer to distance from baseline.
Finally, we used dynasore to determine whether the effect of myosin VI KD on basal NHE3 localization was duplicated by blocking endocytosis only at the level of the inter-microvillus region. Dynasore is a potent inhibitor of dynamin, a large GTPase that mediates scission of clathrin-coated pits and consequent formation of clathrin-coated vesicles (Macia et al., 2006). We treated control and myosin VI KD cells with or without 80 µM dynasore for 30 min and then assayed NHE3 activity using fluorometry. As shown in Fig. 7A, the NHE3 activity was increased by dynasore treatment in control cells but not in myosin VI KD cells, which already exhibited increased NHE3 activity. Importantly, treatment of myosin VI KD cells with dynasore did not show an additive effect, indicating that the change in NHE3 activity with dynasore treatment and KD of myosin VI were both due to an increase in surface NHE3 (supplementary material Fig. S2A–C). These results are consistent with dynasore and myosin VI acting in the same pathway under basal conditions, but not necessarily at the same steps. In further studies, the effect of dynasore on FSK inhibition of NHE3 was determined. As with myosin VI KD, FSK-mediated inhibition of NHE3 was blocked by dynasore in control cells (Fig. 8).
Fig. 7.

Dynasore treatment increases microvillar NHE3 activity and alters the distribution of NHE3 along the microvillus in control but not myosin VI KD cells. (A) Dynasore treatment (Dyn, 80 µM, 30 min) stimulated basal Caco-2/Bbe NHE3 activity by 40% in control (Ctrl) but not myosin VI KD cells (P<0.05). Results are mean±s.e.m. from three independent experiments. (B) Dynasore treatment shifts the microvillus NHE3 distribution in control but not myosin VI KD cells. See Fig. 6 legend for details of immunoelectron microscopy analysis. Histograms show the fraction of microvillar NHE3 distributed 0–600 nm (dark gray) and >600 nm (light gray) away from the microvillus base for each of the four treatments. Individual data points from three separate experiments were grouped and analyzed. The number in brackets represents the total gold particles counted in each condition. The total number of cells with gold particles counted are 99 in control, 185 in control plus dynasore, 140 in myosin VI KD and 198 in myosin VI KD plus dynasore. Two-sample Wilcoxon rank-sum (Mann–Whitney) tests showed that there was significantly more NHE3 on the outer half of the microvillus in dynasore-treated compared to the untreated control (*P<0.0001), and that the distribution of NHE3 was significantly closer to the microvillus base compared to myosin VI KD (†P<0.0001). The combination of myosin VI KD and dynasore exposure had a similar distribution to myosin VI KD. (C) The proposed distribution patterns of microvillar NHE3 in Caco-2/Bbe cells with or without myosin VI KD and/or dynasore are presented together with a summary of results of NHE3 surface activities and endocytic and exocytic behaviors in the different conditions. (D) FRAP analysis shows changes in the NHE3 mobile fractions in Caco-2/Bbe cells with or without myosin VI KD and dynasore (80 μM, 30 min) treatment. The mobile fraction was increased by ∼160% in myosin VI KD versus control cells (P<0.05). Dynasore treatment of control cells did not significantly change the microvillar NHE3 mobile fraction, but significantly reduced the mobile fraction in myosin VI KD cells (P<0.05). Results are mean±s.e.m. from three independent experiments. NS, not significant.
Fig. 8.
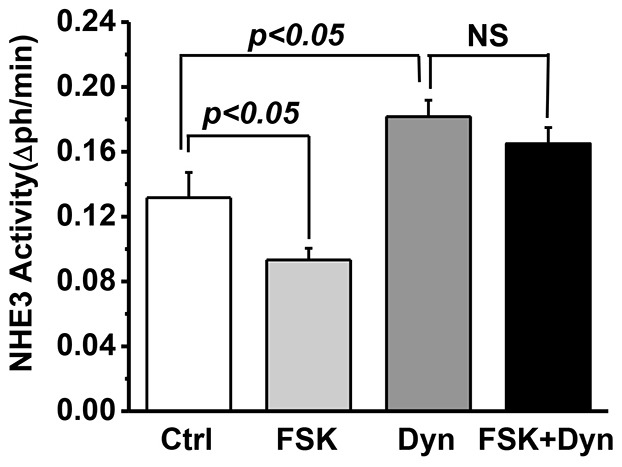
Dynasore inhibits NHE3 endocytosis stimulated by FSK in Caco-2/Bbe cells. Basal NHE3 transport activity was measured in Ad-HA-NHE3-infected Caco-2/Bbe cells with or without dynasore (80 µM, 30 min) or with or without FSK (25 µM, 30 min) pre-incubation. Dynasore significantly increased NHE3 activity, and FSK decreased NHE3 activity (P<0.05). The inhibition of NHE3 activity by FSK was blocked by dynasore. Results are mean±s.e.m. from three independent experiments. NS, not significant.
Given that dynasore and myosin VI KD had similar effects on NHE3 activity, we asked whether they caused similar changes in the NHE3 distribution along microvillus. Again immunoelectron microscopy was used. As shown in Fig. 7B, dynasore treatment of control cells resulted in more NHE3 located >600 nm from the microvillus base than in untreated control cells (∼45% compared to <20%, respectively; P<0.001). However, untreated myosin VI KD cells still showed more NHE3 in the outer half of microvillus than did control cells treated with dynasore (Fig. 7, ∼63% compared to ∼45%, respectively; P<0.001). Finally, dynasore treatment of myosin VI KD cells had no further effect on the microvillus distribution of NHE3 than did myosin VI KD alone (Fig. 7B). A diagram illustrating the different distributions of NHE3 under the four conditions is shown in Fig. 7C. Given that the mechanism of action of dynasore is thought to involve only the inter-microvillar region, the difference in the distribution of NHE3 in the microvillus in myosin VI KD cells versus control cells treated with dynasore suggests that myosin VI KD results in the loss of the NHE3 tip-to-base microvillus movement.
Effect of myosin VI KD and dynasore on NHE3 brush border mobility
Fluorescence recovery after photobleaching (FRAP) was performed on NHE3 in the apical domain of Caco-2/Bbe cells to determine whether dynasore and myosin VI KD affected NHE3 differently (Fig. 7D). Compared to the relatively immobile NHE3 observed under basal conditions, NHE3 mobility was greatly increased in the myosin VI KD cells, consistent with myosin VI anchoring a substantial amount of microvillar NHE3. In contrast, NHE3 mobility was slightly, but not significantly, reduced after dynasore treatment. The combination of dynasore with myosin VI KD increased NHE3 mobility to a level between that of dynasore treatment alone and myosin VI KD. This last result extends our immunoelectron microscopy results and suggests that myosin VI KD and dynasore treatment have a different effect on the mobility of microvillar NHE3.
DISCUSSION
Our study presents the strongest evidence to date that myosin VI moves cargo down the microvillus length of intestinal cells in addition to its role in subsequent steps of cargo endocytosis. By studying the Na+-absorptive Caco-2/Bbe cell, in which myosin VI was knocked down by short hairpin RNA (shRNA), we showed that NHE3 function and localization in the microvillus are dependent on myosin VI. Basal microvillus NHE3 was increased by myosin VI KD, whereas the second-messenger-induced (FSH and CCH) decrease in microvillus NHE3 activity and amount was blocked. Because these two agents have been shown to stimulate endocytosis without altering the rates of exocytosis (Gekle et al., 2002; Lee-Kwon et al., 2003; Musch et al., 2007; Zachos et al., 2013), we propose that both basal and stimulated endocytosis require myosin VI. Additionally, because EGF treatment of myosin VI KD cells still led to an increase in microvillar NHE3 activity, our results further indicate that myosin VI is not involved in the stimulated exocytosis of NHE3 in the Na+-absorptive cell. Importantly, our immunoelectron microscopy finding that microvillar NHE3 exhibited different microvillus distributions in control, myosin VI KD and dynasore-treated cells, convincingly demonstrates that myosin VI is also responsible for moving NHE3 down the microvillus length. Additionally, although the combination of myosin VI KD and dynasore treatment did not alter the microvillar distribution of NHE3 protein found in the myosin VI KD cells (our immunoelectron microscopy results), it significantly reduced the mobile fraction of microvillus NHE3 compared to that found in the myosin VI KD cells (our FRAP results).
Our results on NHE3 in this study complement and extend those reported for several small intestine apical transporters in the Snell's walzer mouse, which lacks myosin VI (Ameen and Apodaca, 2007; Hegan et al., 2012). For example, the cystic fibrosis transmembrane conductance regulator (CFTR)-related Cl− secretion and cell surface expression of CFTR are increased relative to that in wild-type mice, whereas its basal and stimulated internalization are reduced (Ameen and Apodaca, 2007). Similarly, increased apical expression of NHE3 and NaPi2b (also known as SLC34A2) in the jejunum of these mice and their reduced trafficking to subapical endosomes were observed (Hegan et al., 2012). Our immunoelectron microscopy analysis showing that KD of myosin VI in Caco-2/Bbe cells resulted in an increased proportion of NHE3 in the upper half of microvillus (>80%, Fig. 6; Fig. 7B) as compared to that in control (40%) or control plus dynasore-treated cells (60%), suggests very strongly that myosin VI regulates the movement of NHE3 down the microvillus.
Myosin 1A is a plus-directed motor that has been convincingly shown to move membrane cargo towards the microvillus tip, where the barbed ends of the actin filaments attach and actin monomers are added (Tyska and Nambiar, 2010). Studies of brush border transporters in the intestine of myosin-1A-null mice have shown that there is a virtually complete endocytic removal of transporters such as CFTR into the terminal web region and beyond. In contrast, although there is a significant increase of microvillar NHE3 in the myosin VI KD cells and Snell's walzer mouse intestine (T.C. and M.D., unpublished results), an intracellular pool of NHE3 remains that can be recruited upon treatment of cells with EGF (see Fig. 3E). This supports previous studies in which suggestions were made that there are multiple intracellular pools of the transporter: an NHE3 (basal) pool that is continually cycling to and from the microvillus; a reserve pool that requires a signal to move to the microvillus; and, perhaps, a non-cycling (biogenetic and/or degradative) pool (Alexander et al., 2005; Li et al., 2001; Murtazina et al., 2006).
Several outstanding questions remain. How does myosin VI physically associate with microvillus NHE3, an interaction needed to move NHE3 down the microvillus? NHE3 itself does not bind myosin VI (T.C. and M.D., unpublished results). This is not surprising given that many studies have shown that myosin VI binds to its cargos indirectly through adaptors (Tumbarello et al., 2013) and that the binding motifs in the adaptors vary considerably. For example, the adaptor Dab2 uses its C-terminal serine- and proline-rich regions (PTB domain and SYF residues) to bind to myosin VI (Morris et al., 2002a; Spudich et al., 2007), whereas GIPC uses a PDZ domain (Naccache et al., 2006), optineurin uses coiled-coil and ubiquitin-binding domains, T6BP and NDP52 bind to zinc-finger domains, and Tom1 uses an IEXWL amino acid motif (Tumbarello et al., 2012; Tumbarello et al., 2013; Morriswood et al., 2007; Sahlender et al., 2005). None of these motifs are present in the cytoplasmic C-terminus of NHE3. Thus, future studies should be directed towards identifying the adaptors that mediate the interaction between myosin VI and NHE3, and the mechanisms that regulate NHE3 movements along the microvillus.
MATERIALS AND METHODS
Materials
Reagents and antibodies were purchased from the following sources: EZ-Link Sulfo-NHS-SS-biotin (Pierce Chemical, Rockford, IL); restriction endonucleases (New England Biolabs, Ipswich, MA): 2′7′-Bis (2-carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester (BCECF-AM) (Invitrogen, Carlsbad, CA); mouse monoclonal anti-hemagglutinin (HA) antibodies (Covance Research Products, Princeton, NJ); mouse monoclonal anti-myosin VI antibodies (Sigma, St. Louis, MO); DNA primers (Operon Biotechnologies, Huntsville, AL); and donkey anti-mouse-IgG conjugated to 12-nm gold particles (Jackson ImmunoResearch Laboratories, Bar Harbor, Me). Rabbit polyclonal anti-myosin VI antibody was kindly provided by Folma Buss (Cambridge Institute for Medical Research, Cambridge, UK).
Cell culture and adenoviral infection
The Caco-2/Bbe cell line, originally derived from a human adenocarcinoma, was obtained from Mark Mooseker (Yale University, New Haven, CT) and Jerrold Turner (University of Chicago, Chicago, IL). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) plus 25 mM NaHCO3, 0.1 mM nonessential amino acids, 10% fetal bovine serum, 4 mM glutamine, 50 U/ml penicillin and 50 µg/ml streptomycin, pH 7.4, at 37°C in 5% CO2. To achieve polarity, cells were cultured on collagen-coated semi-permeable membranes (Transwells, polycarbonate or Anapore) for 12 days after becoming confluent. Cells infected with adenovirus containing HA–NHE3 (Ad-HA-NHE3) (Sarker et al., 2008) were first exposed to 6 mM EGTA in serum-free medium for 2 h at 37°C to increase virus access to the basolateral surface. Appropriate amounts of viral particles (109–1010 particles/ml) in serum-free medium were added to both chambers of EGTA-treated cells. After 6 or more hours, cells were rinsed and incubated further in normal growth medium. Cells used in transport assays were grown to confluence on small pieces of polycarbonate membranes glued to plastic coverslips (0.4 µm pore size, Corning; called ‘filter slips’). Adenoviral infection of cells on filter slips occurred 12 days after confluence and transport assays were performed 48 h later.
Lentivirus-shRNA KD of myosin VI
Sequence-verified shRNA lentiviral plasmids in the hairpin-pLKO14-puromycin vector were used for myosin VI gene silencing. Human myosin VI was targeted with three independent shRNAs obtained through the Johns Hopkins HiT center from Open Biosystems (Huntsville, AL) and were used to generate lentivirus particles (listed in supplementary material Table S1). pLKO.1 vector containing shRNA, a packaging vector pCMVdR8.91 containing gag, pol and rev genes, and envelope vector pCMV-VSV-G were co-transfected into human embryonic kidney 293T cells. Plasmids were prepared using an EndoFree Plasmid Maxi kit (Qiagen, Valencia, CA). 2×107 human embryonic kidney 293T cells were plated on a 10-cm Petri dish and 24 h later the medium was changed to Opti-MEM serum-free medium. Lipofectamine 2000 reagents were used for transfection according to the manufacturer's protocol (Invitrogen). The transfection conditions were: 10 µg of packaging plasmid plus 6 µg of envelope-encoding plasmid and 10 µg of shRNA-coding plasmid solution. Production of lentiviruses was enhanced by replacement of culture medium at 16-h post-transfection with 5 ml fresh medium containing 10 mM sodium butyrate for an additional 7–8 h (Gasmi et al., 1999; Sakoda et al., 1999), after which 5 ml of fresh medium was added for 16 h before virus harvesting. Lentivirus supernatants were passed through filters (0.45 µm pore PVDF Durapore, ThermoFisher Scientific, Bedford, MA) and used immediately or stored at −80°C. Negative controls were empty vector and Lenti-shGFP-puromycin (a lentiviral-shRNA construct specific for GFP, which is not endogenously expressed in mammalian cells).
For lentiviral transductions, Caco-2/Bbe cells were plated in six-well dishes (Transwell) and cultured for 24 h to achieve 40–50% confluence. Viral particles were mixed with an equal volume of complete medium, Polybrene was added (90 µg/ml, 30 min, 37°C) and the entire mixture was added to the well. After 24 h, the mixture was replaced with complete medium containing 20 µg/ml puromycin. Cells were analyzed after three or four passages.
The efficiency of myosin VI KD was assessed by western blot and immunofluorescence confocal microscopy using specific antibodies against myosin VI. All shRNAs gave similar levels of knockdown and similar phenotypes; construct no. 3 was subsequently used for these studies. Caco-2/Bbe cells transduced with control or myosin VI shRNA were studied 14 days after confluence.
Measurement of Na+/H+ exchange activity
Cellular Na+/H+ exchange activity in cells expressing HA–NHE3 was determined fluorometrically using the intracellular pH-sensitive dye BCECF-AM (Molecular Probes, Eugene, OR). Filter-grown cells (polycarbonate) were infected with Ad-HA-NHE3 at day 12 after reaching confluence, and were then serum-starved 48 h later for at least 4 h before Na+/H+ exchange activity (NHE3) was determined as described previously (Janecki et al., 1998; Li et al., 2004; Murtazina et al., 2007; Sarker et al., 2011). HOE-694 (50 µM) was included to inhibit the contributions of NHE1, NHE2 and NHE8. Cells were loaded for 1 h at 37°C with 10 µM BCECF-AM in Na+/NH4Cl solution (98 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgSO4, 1 mM NaH2PO4, 25 mM glucose, 20 mM HEPES, and 40 mM NH4Cl, pH 7.4). During dye loading and the NH4Cl prepulse, cells were treated with 25 µM FSK for 15 min or 80 µM dynasore for 30 min, or a volume control was added. For CCH treatment, 25 µM carbachol were added to basal side in TMA+ medium [130 mM tetramethylammonium (TMA) chloride, 5 mM KCl, 2 mM CaCl2, 1 mM MgSO4,1 mM NaH2PO4, 25 mM glucose, 20 mM HEPES pH 7.4] (∼6 min). The filters were mounted in a cuvette, placed in a fluorometer (Photon Technology international, Lawrenceville, NJ), and perfused from both sides with TMA+ medium to rapidly acidify the intracellular space. After 2–3 min, the apical TMA+ medium was replaced with Na+ medium containing 130 mM NaCl and no TMA. The basal side continued to be perfused in TMA+ medium. Initial rates of Na+-dependent intracellular alkalinization (efflux of H+, in micromolar per second) were calculated for a given pHi over the first 1 min and expressed as ΔpH/Δt. Mean±s.e.m. were determined from at least three experiments.
Quantification of surface NHE3 by a biotinylation assay
NHS-SS-biotin was used to determine the percentage of total NHE3 on the surface as described previously (Cha et al., 2006). Filter-grown cells were infected with Ad-HA-NHE3 at 12 days post-confluence and studied 48 h later. Cells were serum-starved for 4–6 h, rinsed three times in cold PBS (150 mM NaCl, 20 mM Na2HPO4 pH 7.4), then once in borate buffer (154 mM NaCl, 1.0 mM boric acid, 7.2 mM KCl and 1.8 mM CaCl2, pH 8.0). 1 mg/ml NHS-SS-biotin in borate buffer was added at 0 and 30 min. Cells were then washed three times with quenching buffer (120 mM NaCl, 20 mM Tris-HCl pH 7.4) to scavenge unreacted biotin, three times in PBS and solubilized with 1 ml N+ buffer (60 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM KCl, 5 mM Na3EDTA, 3 mM EGTA and 1% Triton X-100). The cell lysate was centrifuged at 2348 g for 10 min and the post-nuclear supernatant was collected, protein concentrations were measured by a Bradford assay [Sigma-Aldrich (St. Louis, MO)] and adjusted to 1 µg/µl. Of the 1 ml of cell lysate supernatant, 0.9 ml was incubated with streptavidin–Agarose beads (Pierce Chemical, Rockford, IL) for 3 h at 4°C. After sedimenting the beads, the supernatant was retained as the intracellular fraction and the avidin–agarose beads were washed five times in N− buffer (60 mM HEPES pH 7.4, 150 mM NaCl, 3 mM KCl, 5 mM Na3EDTA and 3 mM EGTA) with 0.1% Triton X-100 to remove nonspecifically bound proteins. The proteins bound to the avidin–agarose beads, which represent plasma membrane NHE3, were solubilized in 90 µl of loading buffer (5 mM Tris-HCl pH 6.8, 1% SDS, 10% glycerol and 1% 2-mercaptoethanol), boiled for 10 min. Two dilutions (30 µl and 60 µl) of total lysate, surface and intracellular proteins from each group were loaded, size-fractionated by SDS-PAGE (10% gel) and then electrophoretically transferred onto nitrocellulose. After blocking with 5% nonfat milk in PBS, the blots were probed with monoclonal anti-HA antibody, rinsed, incubated with anti-mouse-IgG conjugated to IRDye® 488 secondary antibodies (LI-COR) and visualized. Signals were quantified on an Odyssey Infrared Imaging System (Li-Cor, Lincoln, NE). The signal intensity derived by linear regression was used to obtain a single value for each sample. The percentage of surface NHE3 was calculated [(surface NHE3 signal/total NHE3 signal) × dilution factor of surface and total NHE3 samples] and expressed as percentage of total NHE3.
Immunocytochemistry, confocal microscopy and image quantification
Caco-2/Bbe cells were grown on Anapore filters (25 mm, 0.02 µm, Nunc) then infected at 12 days post-confluence with Ad-HA-NHE3 as described above. After 48 h, cells were washed with ice-cold PBS and fixed for 30 min at 4°C with 3% paraformaldehyde (PFA) in PBS. For dynasore treatment, cells were serum starved for 4 h and incubated with dynasore (80 µM for 30 min) or an equal volume of DMSO and then processed for fixation. Fixed cells were blocked and permeabilized with 1% BSA and 0.075% saponin in PBS for 1 h at 4°C. Cells were incubated 1 h at room temperature with primary antibodies in 1% BSA in PBS, rinsed in 0.1% BSA in PBS (three times for 5 min), then incubated with secondary antibodies in 1% BSA in PBS for 1 h, and rinsed again in 0.1% BSA in PBS (three times for 5 min). Cells were rinsed with 0.1% BSA and 0.075% saponin in PBS prior to mounting. Membrane inserts were detached from wells, placed on glass microscope slides, mounted with Fluorogel (Invitrogen), and examined with a Zeiss LSM510 confocal microscope. Images were acquired using a 63× (1.4 NA) water objective or 100× (1.6 NA) oil objective.
For quantitative determination of surface NHE3 using confocal microscopy, cells were grown on filters for 12 days, then infected with Ad-HA-NHE3 and processed for immunostaining 48 h later. Cells were fixed in 3% PFA for 30 min at 4°C as described above, except that saponin was omitted, and then incubated with WGA conjugated to Alexa Fluor 568 (Fl-WGA) to mark the apical surface. After rinsing, the cells were permeabilized with 0.075% saponin and labeled with Alexa-Fluor-488-conjugated mouse anti-HA. The total NHE3 fluorescence intensity and percentage of NHE3 on the surface were quantified using Volocity software as described previously (Zinchuk and Grossenbacher-Zinchuk, 2011). Signal intensity was represented as pixels. Overlap was determined with MetaMorph colocalization software.
Intact ileum from C57BL/6 wild-type and myosin VI-null mice were fixed in 3.5% PFA in PBS at 4°C and embedded in paraffin. Histologic sections (4 µm thick) were mounted onto Superfrost microscope slides (Fisher Scientific Co., Arlington, VA) and heat fixed. Slides were microwaved for antigen recovery in 10 mM sodium citrate buffer, pH 6 (Sigma Chemical Company, St. Louis, MO) at power level setting 9 (Panasonic Model# NN-C980B Conventional Microwave Oven, Secaucus, NJ), for 2–5 min. After cooling for 30 min, sections were washed in PBS and preblocked with 5% normal goat serum (NGS) in PBS for 30 min at room temperature. Sections were then incubated overnight (4°C) with mouse monoclonal Aquaporin7 antibody and rabbit polyclonal myosin VI antibody diluted 1∶100 in 5% NGS. Ileal sections were then washed twice in PBS for 10 min and incubated with anti-mouse-IgG conjugated to Alexa Fluor 488 (for aquaporin 7) and anti-rabbit-IgG conjugated to Alexa Fluor 568 (for myosin VI) secondary antibodies, each diluted 1∶100 for 1 h at room temperature. Nuclei were counterstained with Hoechst 33342. Sections were washed twice with PBS and mounted with Gel Mount. Ileal sections were imaged using a Zeiss LSM/510 confocal fluorescence microscope as above. Overlap of myosin VI with the microvillus marker aquaporin 7 was determined with MetaMorph software colocalization analysis.
Electron microscopy and immunoelectron microscopy
Electron microscopy of control and myosin VI KD cells
Polycarbonate-filter-grown cells were processed for conventional electron microscopy analysis at 14 days post-confluence. After several rinses in PBS, cells were fixed at room temperature for 1 h in 1.6% PFA, 2.5% glutaraldehyde, 3% sucrose and 0.1M sodium cacodylate pH 7.4. Cells were rinsed once in 0.1 M cacodylate plus 3% sucrose, and stored overnight at 4°C. After rinsing in the same solution, cells were incubated for 1 h on ice in reduced osmium (0.1 M cacodylate buffer with 0.4% postassium ferrocyanide and 1% OsO4), rinsed quickly at room temperature in double-distilled water, incubated for 30 min at room temperature in 2% uranyl acetate in double-distilled water, and dehydrated through a graded series of ethanol to 100%. Samples were incubated overnight in 1:1 EPON:ethanol at 4°C, warmed to room temperature, transferred for 1 h into 100% EPON plus DMP30, then left overnight in fresh EPON plus catalyst with gentle shaking. The next day, fresh EPON+ catalyst was added at 2-h intervals and incubated in a vacuum chamber for another 2 h. Filters were cut out of the holders, further cut into small pieces (∼2×2 mm), embedded in EPON and cured for 2–3 days at 65°C. Sections were cut on a Riechert Ultracut E with a Diatome Diamond knife, collected on formvar-coated 1×2 mm copper slot grids, and then stained with uranyl acetate followed by lead citrate. Grids were viewed on a Hitachi 7600 TEM operating at 80 kV and digital images of the apical regions of control and KD cells were captured with an AMT 1K×1K CCD camera.
Determination of microvillar lengths
The lengths of individual microvillus were measured from images of randomly chosen cells using MetaMorph software and a calibrated ruler set perpendicular to the baseline of the microvilli. At least two microvilli were measured per cell and a total of ∼300 microvilli were measured from each of the control and KD groups.
Immunoelectron microscopy
Ultrastructural localization studies were carried out on control and myosin VI KD Caco-2/Bbe cells grown on polycarbonate filters (Transwell, 0.4 µm, Corning Inc.) for 12 days then infected with Ad-HA-NHE3. After 48 h, cells were serum-starved for 5 h, treated with dynasore (80 µM for 30 min) or equal volume of DMSO and fixed in 2% periodate-lysine-paraformaldehyde for 30 min at 4°C. Fixed cells were brought to RT, rinsed in PBS (2×5 min), quenched in 50 mM NH4Cl (30 min), blocked in 1% BSA-PBS (30–60 min) and incubated for 1 h with anti-HA monoclonal antibody in blocking buffer. Controls consisted of infected cells fixed as above but incubated in blocking buffer without primary antibody. After one rinse in 0.2% BSA-PBS, cells were incubated for 2–3 h with anti-mouse IgG-12 nm gold in 0.2% BSA-PBS, rinsed twice in PBS, then fixed in GA/PFA, processed and embedded as described above. Oriented blocks were sectioned, so that each section contained ∼80–100 cells and each cell was visible from its apical to basolateral surface. Unstained sections were examined at 40,000–50,000×magnification and images of all gold particles associated with microvillus and the inter-microvillus region were acquired across each section. ∼25% of all cells were labeled with gold in a given section, requiring examination of multiple sections for each condition to accumulate sufficient numbers of gold particles for analysis. Three independent experiments were performed.
Analysis
The distribution of gold particles along microvillus was determined in micrographs using MetaMorph software. The distance of each gold particle from its microvillus base was recorded (0 was the base), the gold particles located 0–600 nm along the microvillus length and those present >600 nm from the microvillus base were grouped separately and each expressed as a percentage of the total gold particles along the microvillus. Comparison was performed with the two-sample Wilcox rank-sum (Mann–Whitney) test, as described previously (Fagerland and Sandvik, 2009).
Fluorescence recovery after photobleaching
To quantify the mobility of NHE3 at the apical domain of polarized Caco-2/Bbe cells, FRAP was used as previously reported (Cha et al., 2004). Cells were cultured on 12-well Transwell plates (#3401, Costar; Transwell Permeable Support, 0.4 mm polycarbonate membrane 12 mm insert, 12 well plate) for 12 days postconfluence. Caco-2/Bbe cells were then infected with Ad-HA-NHE3 (Sarker et al., 2008). Briefly, on the day of 12 days postconfluence, Caco-2/Bbe cells were treated with 6 mM EGTA in Caco-2/Bbe serum-free medium for 2 h before infection. Cells were then infected with Ad-HA-NHE3 and incubated for 6 or more hours. Then cells were washed with fresh serum-free Caco-2/Bbe medium and kept overnight before the FRAP experiment. To perform the FRAP study, surface HA–NHE3 was labeled with monoclonal anti-HA antibody (primary) in 1% BSA in PBS for 1 h and labeled with anti-mouse-IgG conjugated to Alexa Fluor 488 antibody in PBS plus 1% BSA solution for 1 h. Cells were then washed with PBS three times and kept in serum-free D-MEM/F-12 medium without Phenol Red (Life Technology) before FRAP measurement.
FRAP was performed on a stage heated to 37°C of a Zeiss LSM 510/Meta confocal microscope using the 488 nm line of a 400-milliwatt Kr/Ar laser in conjunction with a 100× Zeiss 1.4 NA Plan Apochromat oil immersion objective, with signal collected in the cell apical domain (0.3 µm optical sections starting at the outer limit of the microvillus), and mobile fractions and diffusion coefficients calculated as described previously (Cha et al., 2010). The Zeiss LSM software package allowed autofocusing on the coverslip surface in the reflection mode during the time lapse imaging. Fluorescence was determined within the bleach area (pre-bleach intensity) and then the area was photobleached with full laser power (100% power, 100% transmission). Recovery was followed with low laser power at 9-s intervals, which included up to 30 images, until the intensity had reach a new steady plateau. The mobile fraction was determined by comparing the fluorescence intensity in the bleached region after full recovery (F) with the fluorescence intensity before bleaching (Fi) and just after bleaching (Fo). Mobile and immobile fractions were calculated by comparing the intensity ratio in regions of interest (ROI) inside and outside the bleached area just before the bleach and after recovery as described previously (Cha et al., 2010). The postbleach intensities were normalized to correct for maximal loss of fluorescence due to the photobleach. Fluorescence intensity was normalized to the prebleach intensity (Fi), which was set to 100 in each experiment.
All data are shown as mean±s.e.m., which were obtained in at least three identical experiments, unless stated otherwise. Statistical comparison was performed by unpaired Student's t-test.
Supplementary Material
Acknowledgments
We acknowledge the generous contribution of anti-myosin-VI polyclonal antibody by Folma Buss (Cambridge Institute for Medical Research, Cambridge, UK).
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
T.C., A.H. and M.D. designed, performed and interpreted experiments and data, and wrote the paper. R.M. performed mouse experiments. J.P. analyzed immuno-EM data and performed statistical analysis. J.Y. performed image analysis. B.C. designed and performed FRAP experiments. R.S. made Adeno-HA-NHE3 virus.
Funding
This study was supported in part by the National Institute of Health, National Institute of Diabetes and Digestive and Kidney Diseases [grant numbers R01-DK26523, R01-DK61765, P01-DK072084 and P30-DK89502]; The Hopkins Digestive Diseases Basic and Translational Research Core Center; and the Hopkins Center for Epithelial Disorders. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.149930/-/DC1
References
- Alexander R. T., Furuya W., Szászi K., Orlowski J., Grinstein S. (2005). Rho GTPases dictate the mobility of the Na/H exchanger NHE3 in epithelia: role in apical retention and targeting. Proc. Natl. Acad. Sci. USA 102, 12253–12258 10.1073/pnas.0409197102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameen N., Apodaca G. (2007). Defective CFTR apical endocytosis and enterocyte brush border in myosin VI-deficient mice. Traffic 8, 998–1006 10.1111/j.1600-0854.2007.00587.x [DOI] [PubMed] [Google Scholar]
- Aschenbrenner L., Lee T., Hasson T. (2003). Myo6 facilitates the translocation of endocytic vesicles from cell peripheries. Mol. Biol. Cell 14, 2728–2743 10.1091/mbc.E02-11-0767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg J. S., Powell B. C., Cheney R. E. (2001). A millennial myosin census. Mol. Biol. Cell 12, 780–794 10.1091/mbc.12.4.780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biemesderfer D., Mentone S. A., Mooseker M., Hasson T. (2002). Expression of myosin VI within the early endocytic pathway in adult and developing proximal tubules. Am. J. Physiol. 282, F785–F794 [DOI] [PubMed] [Google Scholar]
- Birn H., Verroust P., Nexo E., Hager H., Jacobsen C., Christensen E. I., Moestrup S. K. (1997). Characterization of an epithelial ∼460kDa protein that facilitates endocytosis of intrinsic factor-vitamin B12 and binds receptor associated protein. J. Biol. Chem. 272, 26497–26504 [DOI] [PubMed] [Google Scholar]
- Bobulescu I. A., Dwarakanath V., Zou L., Zhang J., Baum M., Moe O. W. (2005). Glucocorticoids acutely increase cell surface Na+/H+ exchanger-3 (NHE3) by activation of NHE3 exocytosis. Am. J. Physiol. Renal Physiol. 289, F685–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher A. (1982). Characterization and ultrastructural role of the major components of the intestinal microvillus cytoskeleton. Cold Spring Harb. Symp. Quant. Biol. 46, Pt 2 871–879 [DOI] [PubMed] [Google Scholar]
- Buss F., Arden S. D., Lindsay M., Luzio J. P., Kendrick-Jones J. (2001). Myosin VI isoform localized to clathrin-coated vesicles with a role in clathrin-mediated endocytosis. EMBO J. 20, 3676–3684 10.1093/emboj/20.14.3676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha B., Kenworthy A., Murtazina R., Donowitz M. (2004). The lateral mobility of NHE3 on the apical membrane of renal epithelial OK cells is limited by the PDZ domain proteins NHERF1/2, but is dependent on an intact actin cytoskeleton as determined by FRAP. J. Cell Sci. 117, 3353–3365 10.1242/jcs.01180 [DOI] [PubMed] [Google Scholar]
- Cha B., Tse M., Yun C., Kovbasnjuk O., Mohan S., Hubbard A., Arpin M., Donowitz M. (2006). The NHE3 juxtamembrane cytoplasmic domain directly binds ezrin: dual role in NHE3 trafficking and mobility in the brush border. Mol. Biol. Cell. 17, 2661–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha B., Zhu X. C., Chen W., Jones M., Ryoo S., Zachos N. C., Chen T. E., Lin R., Sarker R., Kenworthy A. K. et al. (2010). NHE3 mobility in brush borders increases upon NHERF2-dependent stimulation by lyophosphatidic acid. J. Cell Sci. 123, 2434–2443 10.1242/jcs.056713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen E. I., Nielsen S. (1991). Structural and functional features of protein handling in the kidney proximal tubule. Semin. Nephrol. 11, 414–439 [PubMed] [Google Scholar]
- Collaco A., Jakab R., Hegan P., Mooseker M., Ameen N. (2010). Alpha-AP-2 directs myosin VI- dependent endocytosis of cystic fibrosis transmembrane conductance regulator chloride channels in the intestine. J. Biol. Chem. 285, 17177–17187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collazo R., Fan L., Hu M. C., Zhao H., Wiederkehr M. R., Moe O. W. (2000). Acute regulation of Na+/H+ exchanger NHE3 by parathyroid hormone via NHE3 phosphorylation and dynamin-dependent endocytosis. J. Biol. Chem. 275, 31601–31608 10.1074/jbc.M000600200 [DOI] [PubMed] [Google Scholar]
- Coudrier E., Reggio H., Louvard D. (1982). The cytoskeleton of intestinal microvilli contains two polypeptides immunologically related to proteins of striated muscle. Cold Spring Harb. Symp. Quant. Biol. 46, Pt 2, 881–892 10.1101/SQB.1982.046.01.082 [DOI] [PubMed] [Google Scholar]
- Donowitz M., Li X. (2007). Regulatory binding partners and complexes of NHE3. Physiol Rev. 87, 825–872 10.1152/physrev.00030.2006 [DOI] [PubMed] [Google Scholar]
- Fagerland M. W., Sandvik L. (2009). The Wilcoxon-Mann-Whitney test under scrutiny. Stat. Med. 28, 1487–1497 10.1002/sim.3561 [DOI] [PubMed] [Google Scholar]
- Gasmi M., Glynn J., Jin M. J., Jolly D. J., Yee J. K., Chen S. T. (1999). Requirements for efficient production and transduction of human immunodeficiency virus type 1-based vectors. J. Virol. 73, 1828–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gekle M., Serrano O. K., Drumm K., Mildenberger S., Freudinger R., Gassner B., Jansen H. W., Christensen E. I. (2002). NHE3 serves as a molecular tool for cAMP-mediated regulation of receptor-mediated endocytosis. Am. J. Physiol. Renal Physiol. 283, F549–F558 [DOI] [PubMed] [Google Scholar]
- Gottlieb T. A., Ivanov I. E., Adesnik M., Sabatini D. D. (1993). Actin microfilaments play a critical role in endocytosis at the apical but not the basolateral surface of polarized epithelial cell. J. Cell Biol. 120, 695–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasson T. (2003). Myosin VI: two distinct roles in endocytosis. J. Cell Sci. 116, 3453–3461 10.1242/jcs.00669 [DOI] [PubMed] [Google Scholar]
- Hegan P. S., Giral H., Levi M., Mooseker M. S. (2012). Myosin VI is required for maintenance of brush border structure, composition, and membrane trafficking functions in the intestinal epithelial cell. Cytoskeleton (Hoboken) 69, 235–251 10.1002/cm.21018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge T., Cope M. J. (2000). A myosin family tree. J. Cell Sci. 113, 3353–3354 [DOI] [PubMed] [Google Scholar]
- Janecki A. J., Montrose M. H., Zimniak P., Zweibaum A., Tse C. M., Khurana S., Donowitz M. (1998). Subcellular redistribution is involved in acute regulation of the brush border Na+/H+ exchanger isoform 3 in human colon adenocarcinoma cell line Caco-2. Protein kinase C-mediated inhibition of the exchanger. J. Biol. Chem. 273, 8790–8798 [DOI] [PubMed] [Google Scholar]
- Janecki A. J., Janecki M., Akhter S., Donowitz M. (2000). Basic fibroblast growth factor stimulates surface expression and activity of Na(+)/H(+) exchanger NHE3 via mechanism involving phosphatidylinositol 3-kinase. J. Biol. Chem. 275, 8133–8142 10.1074/jbc.275.11.8133 [DOI] [PubMed] [Google Scholar]
- Kravtsov D. V., Caputo C., Collaco A., Hoekstra N., Egan M. E., Mooseker M. S., Ameen N. A. (2012). Myosin Ia is required for CFTR brush border membrane trafficking and ion transport in the mouse small intestine. Traffic 13, 1072–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Kwon W., Kim J. H., Choi J. W., Kawano K., Cha B., Dartt D. A., Zoukhri D., Donowitz M. (2003). Ca2+-dependent inhibition of NHE3 requires PKC alpha which binds to E3KARP to decrease surface NHE3 containing plasma membrane complexes. Am. J. Physiol. 285, C1527–C1536 10.1152/ajpcell.00017.2003 [DOI] [PubMed] [Google Scholar]
- Levine S. A., Montrose M. H., Tse C. M., Donowitz M. (1993). Kinetics and regulation of three cloned mammalian Na+/H+ exchangers stably expressed in a fibroblast cell line. J. Biol. Chem. 268, 25527–25535 [PubMed] [Google Scholar]
- Li X., Galli T., Leu S., Wade J. B., Weinman E. J., Leung G., Cheong A., Louvard D., Donowitz M. (2001). Na+-H+ exchanger 3 (NHE3) is present in lipid rafts in the rabbit ileal brush border: a role for rafts in trafficking and rapid stimulation of NHE3. J. Physiol. 537, 537–552 10.1111/j.1469-7793.2001.00537.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Zhang H., Cheong A., Leu S., Chen Y., Elowsky C. G., Donowitz M. (2004). Carbachol regulation of rabbit ileal brush border Na+/H+ exchanger 3 (NHE3) occurs through changes in NHE3 trafficking and complex formation and is Src dependent. J. Physiol. 556, 791–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macia E., Ehrlich M., Massol R., Boucrot E., Brunner C., Kirchhausen T. (2006). Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 10, 839–850 10.1016/j.devcel.2006.04.002 [DOI] [PubMed] [Google Scholar]
- Marzesco A. M., Wilsch-Bräuninger M., Dubreuil V., Janich P., Langenfeld K., Thiele C., Huttner W. B., Corbeil D. (2009). Release of extracellular membrane vesicles from microvilli of epithelial cells is enhanced by depleting membrane cholesterol. FEBS Lett. 583, 897–902 [DOI] [PubMed] [Google Scholar]
- Matsudaira P. T. (1983). Structural and functional relationship between the membrane and the cytoskeleton in brush border microvilli. Ciba. Found. Symp. 95, 233–252 [DOI] [PubMed] [Google Scholar]
- McConnell R. E., Benesh A. E., Mao S., Tabb D. L., Tyska M. J. (2011). Proteomic analysis of the enterocyte BB. Am. J. Physiol. 300, G914–G926 10.1152/ajpgi.00005.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooseker M. S., Pollard T. D., Wharton K. A. (1982). Nucleated polymerization of actin from the membrane-associated ends of microvillar filaments in the intestinal brush border. J. Cell Biol. 95, 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooseker M. S. (1983). Actin binding proteins of the brush border. Cell 35, 11–13 [DOI] [PubMed] [Google Scholar]
- Mooseker M. S., Bonder E. M., Conzelman K. A., Fishkind D. J., Howe C. L., Keller T. C. (1984). The cytoskeletal apparatus of the intestinal brush border. Kroc. Found. Ser. 17, 287–307 [PubMed] [Google Scholar]
- Morris S. M., Arden S. D., Roberts R. C., Kendrick-Jones J., Cooper J. A., Luzio J. P., Buss F. (2002a). Myosin VI binds to and localises with Dab2, potentially linking receptor-mediated endocytosis and the actin cytoskeleton. Traffic 3, 331–341 10.1034/j.1600-0854.2002.30503.x [DOI] [PubMed] [Google Scholar]
- Morriswood B., Ryzhakov G., Puri C., Arden S. D., Roberts R., Dendrou C., Kendrick-Jones J., Buss F. (2007). T6BP and NDP52 are myosin VI binding partners with potential roles in cytokine signalling and cell adhesion. J. Cell Sci. 120, 2574–2585 10.1242/jcs.007005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtazina R., Kovbasnjuk O., Donowitz M., Li X. (2006). Na+/H+ exchanger NHE3 activity and trafficking are lipid Raft-dependent. J. Biol. Chem. 281, 17845–17855 10.1074/jbc.M601740200 [DOI] [PubMed] [Google Scholar]
- Murtazina R., Kovbasnjuk O., Zachos N. C., Li X., Chen Y., Hubbard A., Hogema B. M., Steplock D., Seidler U., Hoque K. M., Tse C. M., De Jonge H. R., Weinman E. J., Donowitz M. (2007). Tissue- specific regulation of sodium/proton exchanger isoform 3 activity in Na+/H+ exchanger regulatory factor 1 (NHERF1) null mice. cAMP inhibition is differentially dependent on NHERF1 and exchange protein directly activated by cAMP in ileum versus proximal tubule. J. Biol. Chem. 282, 25141–25151 [DOI] [PubMed] [Google Scholar]
- Musch M. W., Arvans D. L., Walsh-Reitz M. M., Uchiyama K., Fukuda M., Chang E. B. (2007). Synaptotagmin I binds intestinal epithelial NHE3 and mediates cAMP- and Ca2+-induced endocytosis by recruitment of AP2 and clathrin. Am. J. Physiol. 292, G1549–G1558 10.1152/ajpgi.00388.2006 [DOI] [PubMed] [Google Scholar]
- Naccache S. N., Hasson T., Horowitz A. (2006). Binding of internalized receptors to the PDZ domain of GIPC/synectin recruits myosin VI to endocytic vesicles. Proc. Natl. Acad. Sci. USA 103, 12735–12740 10.1073/pnas.0605317103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquier A. D., Lee D. H., McDonough A. A. (2009). Renal NHE3 and NaPi2 partition into distinct membrane domains. Am. J. Physiol. 296, C900–C910 10.1152/ajpcell.00526.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlender D. A., Roberts R. C., Arden S. D., Spudich G., Taylor M. J., Luzio J. P., Kendrick-Jones J., Buss F. (2005). Optineurin links myosin VI to the Golgi complex and is involved in Golgi organization and exocytosis. J. Cell Biol. 169, 285–295 10.1083/jcb.200501162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakoda T., Kasahara N., Hamamori Y., Kedes L. (1999). A high-titer lentiviral production system mediates efficient transduction of differentiated cells including beating cardiac myocytes. J. Mol. Cell. Cardiol. 31, 2037–2047 10.1006/jmcc.1999.1035 [DOI] [PubMed] [Google Scholar]
- Sarker R., Grønborg M., Cha B., Mohan S., Chen Y., Pandey A., Litchfield D., Donowitz M., Li X. (2008). Casein kinase 2 binds to the C terminus of Na+/H+ exchanger 3 (NHE3) and stimulates NHE3 basal activity by phosphorylating a separate site in NHE3. Mol. Biol. Cell 19, 3859–3870 10.1091/mbc.E08-01-0019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarker R., Valkhoff V. E., Zachos N. C., Lin R., Cha B., Chen T. E., Guggino S., Zizak M., de Jonge H., Hogema B. et al. (2011). NHERF1 and NHERF2 are necessary for multiple but usually separate aspects of basal and acute regulation of NHE3 activity. Am. J. Physiol. 300, C771–C782 10.1152/ajpcell.00119.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich G., Chibalina M. V., Au J. S., Arden S. D., Buss F., Kendrick-Jones J. (2007). Myosin VI targeting to clathrin-coated structures and dimerization is mediated by binding to Disabled-2 and PtdIns(4,5)P2. Nat. Cell Biol. 9, 176–183 10.1038/ncb1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbarello D. A., Waxse B. J., Arden S. D., Bright N. A., Kendrick-Jones J., Buss F. (2012). Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat. Cell Biol. 14, 1024–1035 10.1038/ncb2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbarello D. A., Kendrick-Jones J., Buss F. (2013). Myosin VI and its cargo adaptors – linking endocytosis and autophagy. J. Cell Sci. 126, 2561–2570 10.1242/jcs.095554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyska M. J., Nambiar R. (2010). Myosin-1a: A motor for microvillar membrane movement and mechanics. Commun. Integr. Biol. 3, 64–66 10.4161/cib.3.1.10141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells A. L., Lin A. W., Chen L-Q., Safer D., Cain S. M., Hasson T., Carragher B. O., Milligan R. A., Sweeney H. L. (1999). Myosin VI is an actin-based motor that moves backwards. Nature 401, 505–508 10.1038/46835 [DOI] [PubMed] [Google Scholar]
- Yang L. E., Maunsbach A. B., Leong P. K., McDonough A. A. (2005). Redistribution of myosin VI from top to base of proximal tubule microvilli during acute hypertension. J. Am. Soc. Nephrol. 16, 2890–2896 [DOI] [PubMed] [Google Scholar]
- Yang L. E., Sandberg M. B., Can A. D., Pihakaski-Maunsbach K., McDonough A. A. (2008). Effects of dietary salt on renal Na+ transporter subcellular distribution, abundance, and phosphorylation status. Am. J. Physiol. Renal Physiol. 295, F1003–F1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachos N. C., Lee L. J., Kovbasnjuk O., Li X., Donowitz M. (2013). PLC-γ directly binds activated c-Src, which is necessary for carbachol-mediated inhibition of NHE3 activity in Caco-2/BBe cells. Am. J. Physiol. 305, C266–C275 10.1152/ajpcell.00277.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinchuk V., Grossenbache-Zinchuk O. (2011). Quantitative co-localization analysis of confocal fluorescence microscopy images. Curr. Protoc. Cell Biol. Chapter 4, Unit 4.19 10.1002/0471143030.cb0419s52 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.