

ABSTRACT
Information from multiple signaling axes is integrated in the determination of cellular phenotypes. Here, we demonstrate this aspect of cellular decision making in glioblastoma multiforme (GBM) cells by investigating the multivariate signaling regulatory functions of the protein tyrosine phosphatase SHP2 (also known as PTPN11). Specifically, we demonstrate that the ability of SHP2 to simultaneously drive ERK1/2 and antagonize STAT3 pathway activities produces qualitatively different effects on the phenotypes of proliferation and resistance to EGFR and c-MET co-inhibition. Whereas the ERK1/2 and STAT3 pathways independently promote proliferation and resistance to EGFR and c-MET co-inhibition, SHP2-driven ERK1/2 activity is dominant in driving cellular proliferation and SHP2-mediated antagonism of STAT3 phosphorylation prevails in the promotion of GBM cell death in response to EGFR and c-MET co-inhibition. Interestingly, the extent of these SHP2 signaling regulatory functions is diminished in glioblastoma cells that express sufficiently high levels of the EGFR variant III (EGFRvIII) mutant, which is commonly expressed in GBM. In cells and tumors that express EGFRvIII, SHP2 also antagonizes the phosphorylation of EGFRvIII and c-MET and drives expression of HIF-1α and HIF-2α, adding complexity to the evolving understanding of the regulatory functions of SHP2 in GBM.
KEY WORDS: Signal transducer and activator of transcription-3 (STAT3), Extracellular signal-regulated kinase-1/2 (ERK1/2), Epidermal growth factor receptor (EGFR), HGF receptor (c-MET), Hypoxia inducible factors, Gefitinib
INTRODUCTION
Cells integrate information from multiple signaling pathways to execute decision-making processes (Janes et al., 2005; Wolf-Yadlin et al., 2006; Kumar et al., 2007; Lazzara and Lauffenburger, 2009). Whereas some signaling pathway intermediates act predominantly in one pathway, others exert substantial effects in multiple pathways, thus expanding their ability to control cell fate determination. One such protein is SH2 domain-containing phosphatase-2 (SHP2, also known as PTPN11), which plays key roles in development, homeostatic maintenance and disease. Here, we investigate the ability of SHP2 to simultaneously regulate the extracellular signal-regulated kinase-1/2 (ERK1/2, hereafter referred to as ERK) and signal transducer and activator of transcription-3 (STAT3) pathways, as well as other signaling events that we identify as SHP2-regulated for the first time, and the net effect of this regulation on cellular proliferation and response to co-inhibition of EGFR and the HGF receptor c-MET.
SHP2 was the first phosphatase to be identified as a proto-oncogene (Chan and Feng, 2007; Chan et al., 2008) and is primarily regarded as a mediator of proliferative and pro-survival signaling. Indeed, the best-studied signaling role of SHP2 is to promote ERK activity (Neel et al., 2003). The catalytic activity of SHP2, which is required for this function, is promoted through engagement of its N-terminal SH2 domains by phosphotyrosines on various receptor tyrosine kinases or adapter proteins, such as GRB2-associated binding protein-1 (GAB1) (Gu and Neel, 2003; Neel et al., 2003). SHP2 can also negatively regulate STAT3 activation downstream of the interleukin-6 receptor (Schmitz et al., 2000), and one recent study even described a ‘tumor-suppressor’ role for SHP2 in hepatocellular carcinoma through its regulation of STAT3 (Bard-Chapeau et al., 2011). SHP2 can also positively or negatively impact AKT pathway activity (Wu et al., 2001; Zhang et al., 2002). Through these signaling regulatory functions, the magnitude of which may depend on cell type or disease context, SHP2 is able to control cellular phenotypes including proliferation (Cai et al., 2002; Furcht et al., 2012), oncogenic transformation (Zhan et al., 2009), tumor progression (Aceto et al., 2012), response to therapeutics (Furcht et al., 2012) and senescence (Sturla et al., 2011).
A specific setting of interest where SHP2 influences multiple complex phenotypes is glioblastoma multiforme (GBM), the most common and lethal form of adult brain cancer (Zhang et al., 2012). One study described the ability of SHP2 to suppress cellular senescence in the GBM cell lines U87MG and A172, and reported simultaneous SHP2-mediated activation of ERK and inhibition of STAT3, although no causal relationships between ERK or STAT3 signaling and the senescence phenotype were established (Sturla et al., 2011). SHP2 function has also been linked to tumorigenicity of GBM cells that express the EGFR variant III (EGFRvIII), a mutant prevalent in GBM (Zhan et al., 2009). Of course, the role of ERK and STAT3 in promoting proliferation and survival across cancer types has been described in depth (Hodge et al., 2005; Roberts and Der, 2007). For example, in GBM cells, ERK activity promotes resistance to the chemotherapy drug cisplatin (Zhan and O'Rourke, 2004), and STAT3 is an important regulator of proliferation that has been recognized as a potential therapeutic target (Lo et al., 2008; de la Iglesia et al., 2009). Because SHP2 regulates the ERK and STAT3 pathways in qualitatively different ways, and the ERK and STAT3 signaling pathways promote qualitatively similar effects across multiple phenotypes, how the multivariate signaling roles of SHP2 are integrated to determine phenotype in GBM cells is unclear. Gaining a more complete understanding might help to address a number of outstanding issues, such as how GBM resistance to targeted inhibitors can be overcome (Sathornsumetee et al., 2007; Argyriou and Kalofonos, 2009; De Witt Hamer, 2010) and the potential efficacy of targeting SHP2 in glioblastoma.
Here, we evaluate the effects of the signaling roles of SHP2 on GBM cell proliferation and resistance to inhibitors of EGFR and c-MET, oncogenic receptors that drive GBM progression and chemoresistance. In a panel of GBM cell lines, SHP2 depletion reduced cellular proliferation but, surprisingly, also promoted resistance to EGFR and c-MET co-inhibition. These results appear to derive from the ability of SHP2 to drive ERK and antagonize STAT3 pathway activities in the panel of cell lines and the differential abilities of those pathways to control different phenotypes. That is, even though ERK and STAT3 both promote proliferation and survival, SHP2-mediated ERK activity is dominant in determining cellular proliferation rates, whereas suppression of STAT3 phosphorylation through SHP2 exerts the dominant effect in determining response to EGFR and c-MET co-inhibition. Interestingly, the ability of SHP2 to regulate these pathways was greatly diminished in cells that expressed sufficiently high levels of EGFRvIII, where SHP2 was basally sequestered with the receptor. We further found that SHP2 negatively regulates EGFRvIII and c-MET phosphorylation and drives expression of hypoxia-inducible factors 1 and 2 alpha (HIF-1α and HIF-2α, respectively; hereafter referred to as HIF-1/2α) in cultured cells and tumor xenografts. These results expand our understanding of SHP2 as a multivariate regulator of signaling and GBM cell phenotype and raise additional questions about how SHP2 function may be perturbed in different GBM contexts.
RESULTS
SHP2 depletion differentially impacts key GBM cell phenotypes and associated signaling pathways
Using the GBM cell lines U87MG, LN18, T98G and U118MG, we first evaluated the effect of small hairpin RNA (shRNA)-mediated SHP2 knockdown on cellular proliferation. As expected based on reports that used other glioblastoma cells (Zhan et al., 2009) and several other cell settings, SHP2 knockdown reduced cellular proliferation rates in all four cell lines (Fig. 1A). Interestingly, SHP2 knockdown also promoted cell survival in response to co-inhibition of EGFR and c-MET using the inhibitors gefitinib and PHA665752, respectively (Fig. 1B). Thus, in response to SHP2 knockdown, cells were generally less proliferative but significantly more resistant to EGFR and c-MET co-inhibition. The latter effect was unexpected, given previous findings that SHP2 knockdown enhances cell death in response to EGFR inhibition in non-small cell lung cancer (NSCLC) cells (Furcht et al., 2012) and that SHP2 antagonizes p73-dependent apoptosis (Amin et al., 2007). Western blot analysis revealed that SHP2 knockdown was accompanied by a concomitant decrease in ERK phosphorylation and increase in STAT3 phosphorylation (Fig. 1C), which could explain why proliferation was impeded while survival in response to EGFR and c-MET co-inhibition was enhanced. That is, ERK activity might contribute more substantially to determining proliferation rates, and STAT3 activity might contribute more substantially to survival in response to EGFR and c-MET co-inhibition.
Fig. 1.

SHP2 knockdown differentially impacts GBM cell proliferation and survival. (A) U87MG, LN18, T98G and U118MG cells that express control or SHP2-targeting shRNA were seeded at 100,000 cells/well, and cells were counted 72 h later. Counts are represented as the mean ± s.e.m. (n = 3); *P<0.05. (B) The indicated cell lines were co-treated with 20 µM gefitinib + 1 µM PHA665752 (G+P) or with DMSO as a control (DMSO). After 72 h, the percentage of TO-PRO-3-positive cells was measured by flow cytometry (n = 3); *P<0.05. (C) The indicated cell lines expressing control or SHP2-targeting shRNA were maintained in complete medium. Lysates were analyzed by western blotting using antibodies against the indicated proteins. Densitometry data are represented as the mean ± s.e.m. (n = 3); *P<0.05.
To explore this idea further, we used the data shown in Fig. 1A-C to assign quantitative values to the individual contributions of SHP2-controlled ERK and STAT3 pathway activities towards cellular proliferation and survival. We assumed that the quantitative measurement of a particular phenotype Xi under a particular cellular condition i (in this case, control or SHP2 knockdown) can be described as a linear combination of the phosphorylation levels of ERK and STAT3 (pE,i and pS,i, respectively), where the specific contribution of each pathway to Xi is determined by the product of a weighting coefficient for ERK or STAT3 (wE or wS, respectively) and the phosphorylation level of the protein. With these assumptions, Xi is defined as:
To evaluate pathway contributions to survival in response to therapeutics, the percentage of dead cells shown in Fig. 1B was subtracted from 100% to determine the percentage of surviving cells. Western blot signals of phosphorylated ERK and STAT3 were normalized to the corresponding signals of total protein, as shown in Fig. 1C. Finally, phosphorylation and phenotype data were normalized to values obtained from cells treated with control shRNA for each cell line, which led to wE and wS summing to one when the equation above was evaluated for the control condition. Performing the analysis for the proliferation phenotype for each cell line and averaging the data, we found average wE and wS values of 0.77 and 0.23, respectively. For cell survival in response to EGFR and c-MET co-inhibition, we found average wE and wS values of −0.14 and 1.14, respectively. These results suggest that ERK and STAT3 play dominant roles in proliferation and survival response, respectively. We note that a negative value for wE in the survival analysis might seem to suggest that ERK activity somehow negatively contributes to cell survival, but this is not the case. Rather, this result arises owing to the form of our model for Xi, which produces a wE <0 whenever the fold-increase in survival exceeds the fold-increase in STAT3 phosphorylation, and the fold-increase in ERK phosphorylation does not exceed that for STAT3 phosphorylation, which is the case for three of the four cell lines analyzed.
ERK and STAT3 inhibition further suggests differential pathway control of proliferation and survival in GBM cells
We next used the ERK and STAT3 inhibitors CI-1040 and Stattic, respectively, to confirm independently the relative contributions of ERK and STAT3 to cell phenotypes. Cellular proliferation was reduced with either ERK or STAT3 pathway inhibition (Fig. 2A,B; supplementary material Fig. S1A). Note that the incomplete inhibition of STAT3 phosphorylated at residue Y705 (37% reduction) observed in Fig. 2B resulted from our selection of a STAT3 inhibitor concentration that was low enough to produce relatively low levels of cell death as a single agent across the panel of cell lines. Using a lower concentration of gefitinib than the one used in the experiments shown in Fig. 1B to reduce baseline cell death, we also found that ERK or STAT3 inhibition promoted cell death in response to EGFR and c-MET co-inhibition (Fig. 2C). With the exception of U118MG cells where Stattic produced a substantial amount of cell death by itself, the effect of ERK inhibition on proliferation was generally greater than that of STAT3 inhibition. By contrast, the effect of STAT3 inhibition on cell death in response to gefitinib and PHA665752 was larger than that of ERK inhibition. Given that the same concentrations of CI-1040 and Stattic were used in the experiments shown in Fig. 2A,C, we interpret these data as indicating that both the ERK and STAT3 pathways participate in the regulation of cellular proliferation and survival, but confirming the weighting coefficient analysis conclusion that ERK is the stronger determinant of proliferation and STAT3 the stronger determinant of survival in response to EGFR and c-MET co-inhibition. This suggests that the elevated levels of phosphorylated STAT3 observed with SHP2 knockdown promoted resistance to EGFR and c-MET co-inhibition despite the impairment of ERK activity. To confirm this, we demonstrated that combining Stattic with the concentrations of gefitinib and PHA665752 used as shown in Fig. 1B increased the cell death response of cells in which SHP2 was knocked down (Fig. 2D).
Fig. 2.

ERK and STAT3 pathways both control proliferation and survival of GBM cells. (A) U87MG, LN18, T98G and U118MG cells were seeded at 100,000 cells/well and treated 24 h later with 6 µM CI-1040 (C), 4 µM Stattic (S) or DMSO as a control (DMSO) for 72 h prior to cell counting. Counts are represented as mean ± s.e.m. (n = 3); *P<0.05. (B) The indicated cell lines were treated with the same concentrations of inhibitor as shown in A for 30 min prior to lysis. Lysates were analyzed by western blotting with antibodies against the indicated proteins. Images are representative of three sets of biological replicates. (C) The indicated cell lines were treated with 6 µM CI-1040 (C) or 4 µM Stattic (S), as in A, or with 10 µM gefitinib + 1 µM PHA665752 (G+P), with 10 µM gefitinib + 1 µM PHA665752 + 6 µM CI-1040 (G+P+C), with 10 µM gefitinib + 1 µM PHA665752 + 4 µM Stattic (G+P+S), or with DMSO as a control (DMSO). The gefitinib concentration in this set of experiments was lower than that used in the experiments shown in Fig. 1B and lower than the concentration used in the experiments shown in panel D, in order to reduce cellular death in response to G+P treatment. After 96 h, the percentage of TO-PRO-3-positive cells was measured by flow cytometry (n = 3); *P<0.05. (D) The indicated cell lines expressing control or SHP2-targeting shRNA were treated with DMSO as a control (DMSO), with 4 µM Stattic (S), with 20 µM gefitinib + 1 µM PHA665752 (G+P), or with 20 µM gefitinib + 1 µM PHA665752 + 4 µM Stattic (G+P+S). After 72 h, the percentage of TO-PRO-3-positive cells was measured by flow cytometry (n = 3); *P<0.05.
We note as well that, in some cell lines, increases in STAT3 Y705 phosphorylation might involve a mechanism wherein ERK negatively regulates STAT3 Y705 phosphorylation by phosphorylating STAT3 S727 (Chung et al., 1997). Evidence for this potential connectivity between ERK and STAT3 is provided by our finding that MEK inhibition promoted STAT3 phosphorylation in some cell lines (supplementary material Fig. S1B).
The ability of SHP2 to regulate signaling and phenotypes is modulated by elevated expression of EGFRvIII
To evaluate the regulatory functions of SHP2 in the context of EGFRvIII expression, we stably depleted SHP2 in a panel of U87MG cell lines that expressed low, medium or high levels of EGFRvIII, or a high level of kinase-dead EGFRvIII (U87MG-L, U87MG-M, U87MG-H or U87MG-DK, respectively) (Huang et al., 2007). SHP2 depletion reduced proliferation in all four cell lines (Fig. 3A). Similar to effects observed in Fig. 1B, SHP2 knockdown also promoted survival in response to EGFR and c-MET co-inhibition in U87MG-DK, U87MG-L and U87MG-M cells, but there was no effect in U87MG-H cells (Fig. 3B). To confirm specificity of the effects of SHP2 knockdown, we used an additional, non-overlapping SHP2 shRNA to deplete SHP2 in U87MG-M cells, where cells with SHP2 knockdown were again more resistant to EGFR and c-MET co-inhibition (supplementary material Fig. S2A). To understand the reason for the lack of effect of SHP2 knockdown on survival response in U87MG-H cells, we first investigated signaling pathways in resting cells by using western blotting (Fig. 3C; supplementary material Fig. S2B). STAT3 phosphorylation was increased by SHP2 depletion in all cell lines including U87MG-H, which was surprising given our previous findings that STAT3 controlled survival response and that SHP2 knockdown did not rescue U87MG-H cells following treatment with gefitinib and PHA665752. We also noticed reduced ERK phosphorylation in all cell lines with SHP2 knockdown, although the reduction in ERK phosphorylation in U87MG-H cells was very modest – an effect which we explore further in experiments shown in Fig. 4. In addition, we noticed potential effects of SHP2 knockdown on the expression and phosphorylation of EGFRvIII, and on phosphorylation of c-MET – findings which we also revisit later.
Fig. 3.

Sufficiently high EGFRvIII expression diminishes the ability of SHP2 to promote ERK activity and to reduce STAT3 phosphorylation in the presence of EGFR and c-MET inhibitors. (A) U87MG-DK, U87MG-L, U87MG-M and U87MG-H cells expressing control or SHP2-targeting shRNA were seeded at 75,000 cells/well and were counted 72 h later. Counts are represented as mean ± s.e.m. (n = 3); *P<0.05. (B) The indicated cell lines expressing control or SHP2-targeting shRNA were treated with DMSO as a control (DMSO) or co-treated with 20 µM gefitinib + 1 µM PHA665752 (G+P). After 72 h, the percentage of TO-PRO-3-positive cells was measured by flow cytometry (n = 3); *P<0.05. (C) The indicated cell lines were grown in complete medium for 72 h, and lysates were analyzed by western blotting using antibodies against the indicated proteins. Images are representative of five sets of biological replicates. (D) U87MG-M and U87MG-H cells expressing control or SHP2-targeting shRNA were co-treated with 20 µM gefitinib + 1 µM PHA665752 (G+P) for 24 h and analyzed by western blotting using antibodies against the indicated proteins. Images are representative of three sets of biological replicates. (E) The indicated cell lines expressing control or SHP2-targeting shRNA were treated with DMSO as a control (DMSO) or with 20 µM gefitinib + 1 µM PHA665752 (G+P) for 24 h and lysed. STAT3 immunoprecipitates were analyzed by western blotting using antibodies against the indicated proteins. Images are representative of three sets of biological replicates. (F) U87MG-M cells were analyzed as shown in panel B with the addition of 4 µM Stattic (S) where indicated (n = 3); *P<0.05.
Fig. 4.

Sufficiently high EGFRvIII expression suppresses EGF-mediated ERK phosphorylation by SHP2 sequestration. (A) Serum-starved U87MG-DK, U87MG-L, U87MG-M and U87MG-H cells were treated with or without 10 ng/ml EGF for 5 min and lysed. SHP2 immunoprecipitates were analyzed by western blotting using antibodies against the indicated proteins. Images are representative of three sets of biological replicates. (B) Membrane and cytosolic fractions from serum-starved U87MG-DK and U87MG-H cells were analyzed by western blotting using antibodies against indicated proteins. Images are representative of four sets of biological replicates. (C) Serum-starved U87MG-L and U87MG-H cells were treated with or without 10 ng/ml EGF for 5 min, and slides were prepared for SHP2 immunofluorescence. Images are representative of multiple frames from three biological replicates. (D) EGF endocytosis rate constants (ke) were measured for the indicated cell lines by using 125I-EGF. Data are represented as mean ± s.e.m. (n = 6); *P<0.05. (E) The indicated serum-starved cell lines were treated with 10 ng/ml EGF for up to 15 min and lysates were analyzed by western blotting using antibodies against the indicated proteins. Images are representative of three sets of biological replicates.
To delve further into the lack of effect of SHP2 knockdown on cellular response to inhibitors in U87MG-H cells, we compared the effects of SHP2 knockdown in U87MG-M and U87MG-H cells co-treated with gefitinib and PHA665752 (Fig. 3D; supplementary material Fig. S2C). Interestingly, in the presence of the inhibitors, SHP2 depletion significantly increased STAT3 phosphorylation in U87MG-M cells but had essentially no effect in U87MG-H cells. Since STAT3 can be activated through direct binding to EGFR (Shao et al., 2003; de la Iglesia et al., 2009) or EGFRvIII (Fan et al., 2013), we hypothesized that elevated STAT3 Y705 phosphorylation is EGFRvIII-dependent in SHP2-depleted U87MG-H cells but EGFRvIII-independent in SHP2-depleted U87MG-M cells. This scenario would lead to a preferential reduction in STAT3 phosphorylation with EGFR inhibition in U87MG-H cells. Consistent with this model, association of STAT3 and pEGFRvIII was much higher in U87MG-H cells than in any other line (Fig. 3E; supplementary material Fig. S2D). In U87MG-H cells, association of STAT3 and pEGFRvIII was further promoted by SHP2 knockdown, presumably because of the concomitant increase in phosphorylated levels of EGFRvIII observed in this cell line. The unchanged association of STAT3 and pEGFRvIII for SHP2-depleted U87MG-M cells is consistent with the notion that EGFRvIII is not a primary driver of STAT3 phosphorylation below a threshold level of EGFRvIII expression. Moreover, co-treatment with gefitinib and PHA665752 eliminated association of STAT3 and pEGFRvIII in U87MG-H cells, consistent with reduced STAT3 phosphorylation in response to gefitinib and PHA665752 co-treatment (Fig. 3E). It should also be noted that EGFRvIII and c-MET phosphorylation were greatly reduced in cells co-treated with gefitinib and PHA665752 relative to DMSO-treated cells with or without SHP2 knockdown, which eliminates the possibility that a failure to reduce receptor phosphorylation was responsible for the resistance to EGFR and c-MET co-inhibition in cells with SHP2 knockdown (supplementary material Fig. S2E). Finally, as in parental U87MG cells, combining Stattic with gefitinib and PHA665752 enhanced cell death in SHP2-depleted U87MG-M cells (Fig. 3F).
At high expression levels EGFRvIII is able to sequester SHP2
In the same way that STAT3 can preferentially bind to EGFRvIII, other proteins may be sequestered by EGFRvIII when the receptor is expressed at high levels. We hypothesized that such a preferential binding effect for SHP2 could explain the modest effect that knockdown of SHP2 has on ERK phosphorylation in U87MG-H cells, as well as the gradual reduction in basal phosphorylation of ERK in control cells with increasing expression of EGFRvIII (Fig. 3C). Such an effect would be analogous to one we described in lung cancer cells, wherein kinase-activated and internalization-impaired EGFR mutants appear to sequester adapter-bound SHP2 in such a way that full ERK activation is prevented (Furcht et al., 2012). To explore this, we first investigated the basal and EGF-induced associations of SHP2 with GAB1 and with EGFRvIII. Increasing EGFRvIII expression clearly promoted association of GAB1 and SHP2 (Fig. 4A; supplementary material Fig. S3A), as well as the previously reported association of pEGFRvIII and SHP2 (Zhan et al., 2009). Although association of GAB1 and SHP2 was EGF-inducible in U87MG-DK, U87MG-L and U87MG-M cells, the high basal association observed in U87MG-H cells was so elevated that it was not augmented further by treatment with EGF. We further noted a larger fraction of SHP2 in the membrane compartment along with EGFRvIII in U87MG-H cells than in U87MG-DK cells (Fig. 4B; supplementary material Fig. S3B); an ∼1.5-fold increase of the SHP2 signal within the membrane compartment was observed in U87MG-H cells relative to U87MG-DK cells. Thus, the activity of EGFRvIII can promote sequestration of SHP2 in the membrane fraction. We also found that elevated EGFRvIII expression altered the basal intracellular distribution of SHP2, as observed by immunofluorescence. Whereas SHP2 moved from the cell interior towards the cell periphery in response to EGF in U87MG-L cells, basal SHP2 was already peripherally distributed in U87MG-H cells and did not redistribute in response to EGF (Fig. 4C; supplementary material Fig. S3C,D). Given that EGF-mediated endocytosis of wild-type EGFR was significantly reduced in U87MG-H cells (Fig. 4D) and that EGFRvIII itself is also endocytosis impaired (Huang et al., 1997; Grandal et al., 2007), our data are, indeed, consistent with the notion that active adapter- and EGFRvIII-bound SHP2 is sequestered at the plasma membrane in U87MG-H cells. The effect of this on ERK activation seems so pronounced that ERK phosphorylation cannot be induced in U87MG-H cells by exogenous EGF, whereas ERK induction does occur in U87MG-DK, U87MG-L and U87MG-M cells (Fig. 4E; supplementary material Fig. S3E). To explore whether altered expression of MKP3, the primary phosphatase for ERK, is responsible for the failure of EGF to induce ERK phosphorylation in U87MG-H cells, we investigated MKP3 expression across the panel of cell lines. However, we observed no trends in MKP3 expression that would explain our data (supplementary material Fig. S3F).
SHP2 negatively regulates phosphorylation of EGFRvIII and c-MET
As previously noted, the results shown in Fig. 3C suggest the ability for SHP2 to regulate EGFRvIII and c-MET phosphorylation. Specifically, the data show that total levels of phosphorylated EGFRvIII and c-MET were increased when SHP2 was knocked down in U87MG-H cells, but that EGFRvIII phosphorylation was reduced when SHP2 was knocked down in U87MG-L and U87MG-M cells. The apparent effect in U87MG-L and U87MG-M cells might arise because of the concomitant decrease in EGFRvIII expression when SHP2 is knocked down, which might result from impaired ERK activity (supplementary material Fig. S4A). To clarify this further, we ectopically expressed constitutively active SHP2 (SHP2E76A) in all four cell lines. This had a minimal effect on EGFRvIII expression but increased ERK phosphorylation and reduced EGFRvIII, c-MET and STAT3 phosphorylation (Fig. 5A; supplementary material Fig. S4B). SHP2E76A expression also promoted cell sensitivity to gefitinib and PHA665752 co-treatment in U87MG-M cells (Fig. 5B). To further investigate how SHP2 regulates EGFRvIII, c-MET and STAT3 phosphorylation, we transiently expressed SHP2WT or the substrate-trapping SHP2 double mutant SHP2D425A/C459S [SHP2DM; (Agazie and Hayman, 2003)] in all four U87MG cell lines. This double mutation abrogates the catalytic activity of SHP2 and causes irreversible binding of the catalytic domain to substrates. Phosphorylated EGFRvIII and c-MET co-immunoprecipitated with SHP2DM (Fig. 5C; supplementary material Fig. S4C), but STAT3 did not (supplementary material Fig. S4D), suggesting that EGFRvIII and c-MET are substrates of SHP2. These specific interactions have not been reported previously, but it has been reported that SHP2 can directly dephosphorylate other receptors, including HER2 (Zhou and Agazie, 2009), based on experiments using SHP2DM.
Fig. 5.

Expression of SHP2 mutants reveals negative regulation of EGFRvIII, c-MET and STAT3 phosphorylation. (A) Lysates of U87MG-DK, U87MG-L, U87MG-M and U87MG-H cells transduced with an empty pBabe.Puro vector (pBP) or SHP2E76A (E76A) and grown in full medium for 72 h were analyzed by western blotting using antibodies against the indicated proteins. Images are representative of three sets of biological replicates. (B) U87MG-M cells transduced with pBP or SHP2E76A were treated with DMSO as a control (DMSO) or with 20 µM gefitinib + 1 µM PHA665752 (G+P). After 72 h, the percentage of TO-PRO-3-positive cells was measured by flow cytometry (n = 3); *P<0.05. (C) Serum-starved cells of the indicated cell lines transiently transfected with SHP2WT or the double mutant SHP2D425A/C459S (SHP2DM) were lysed. SHP2 immunoprecipitates were analyzed by western blotting using antibodies against the indicated proteins. Images are representative of three sets of biological replicates.
SHP2 knockdown impedes tumor xenograft growth and expression of hypoxia-inducible factors
Female Nu/Nu immunodeficient nude mice were injected subcutaneously in both flanks with U87MG-M control or SHP2 knockdown cells. Tumors arising from control cells grew well and were highly responsive to gefitinib and PHA665752 co-treatment (Fig. 6A,B), suggesting that this co-treatment strategy can be effective in EGFRvIII-expressing GBM. We had hoped to be able to grow tumors arising from SHP2 knockdown cells to investigate the potential ability of SHP2 depletion to promote tumor resistance to gefitinib and PHA665752 co-treatment. However, after reaching an average maximum volume of 40 mm3, tumors arising from cells with SHP2 knockdown gradually shrank and never reached a sufficient size to begin treatment (Fig. 6B). Interestingly, HIF-1/2α expression was reduced in tumors arising from SHP2 knockdown cells compared with controls (Fig. 6C), which may explain their failure to form tumors. In vitro studies revealed a similar effect of SHP2 knockdown on HIF-2α expression under hypoxic and normoxic conditions, and on HIF-1α expression for normoxic culture (Fig. 6D). Control cells treated with the MEK inhibitor U0126 displayed diminished HIF-1α expression in normoxia and HIF-2α expression in both normoxia and hypoxia, suggesting that SHP2 regulates HIF-1/2α expression through controlling ERK activity.
Fig. 6.

Gefitinib and PHA665752 co-treatment or SHP2 knockdown impairs U87MG tumor xenograft growth. Mice were subcutaneously injected with control or SHP2-depleted U87MG-M cells. When tumors reached an average size of 50 mm3 (control shRNA only), mice were treated daily either with vehicle or 100 mg/kg gefitinib + 30 mg/kg PHA665752 (G+P) for 7 days (black arrow indicates the start of the treatment). (A) After treatment concluded, pictures were taken and tumors were harvested. (B) Tumor volumes were measured before and throughout treatment. Data are shown as mean ± s.e.m. (control shRNA: vehicle, SHP2 shRNA: no treatment and control shRNA: G + P; n = 12, 26 and 14 tumors, respectively). (C) Tumor lysates were analyzed by western blotting using antibodies against the indicated proteins. Densitometry data are shown as mean ± s.e.m. (n = 3). (D) U87MG-M cells expressing control or SHP2-targeting shRNA were pretreated with DMSO or 40 µM U0126 (control shRNA only) for 24 h prior to hypoxic culture for 24 h. Lysates were analyzed by western blotting using antibodies against the indicated proteins. Densitometry data are shown as mean ± s.e.m. (n = 3); *P<0.05.
DISCUSSION
Our results demonstrate for the first time that the ability of SHP2 to exert multivariate control over signaling in GBM cells enables it to regulate simultaneously and differentially the phenotypes of proliferation and resistance to therapy. This effect arises, at least in part, because ERK and STAT3, which are regulated in qualitatively different ways by SHP2, play dominant roles in the regulation of proliferation and therapeutic resistance, respectively. We uncovered a number of other previously undocumented SHP2 regulatory functions, including SHP2-mediated antagonism of EGFRvIII and c-MET phosphorylation, and regulation of HIF-1/2α expression, which may also play roles in determining GBM cell and tumor phenotypes. These integrated SHP2 signaling mechanisms and the ways they impact GBM cell phenotypes are summarized in Fig. 7.
Fig. 7.
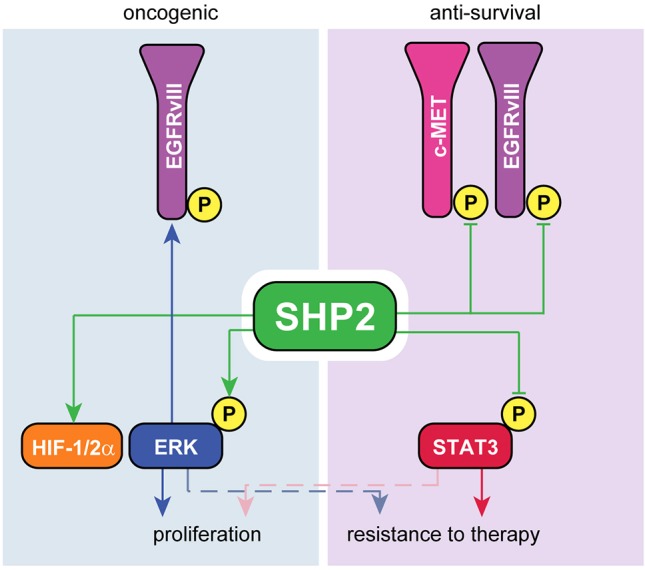
Summary of the oncogenic and anti-survival functions of SHP2 in GBM cells. Consistent with its best-described role, SHP2 possesses oncogenic functions by promoting the phosphorylation of ERK, which augments expression of EGFRvIII and HIF-1/2α. Conversely, SHP2 antagonizes survival signaling by apparent activity against EGFRvIII and c-MET as well as negative regulation of multiple modes of STAT3 phosphorylation.
A focus of our study is the impact of the ability of SHP2 to simultaneously promote ERK activity and suppress STAT3 phosphorylation. It has been shown in other contexts, and we show it explicitly for GBM cells, that the ERK and STAT3 pathways both promote proliferation and survival (here in response to co-inhibition of EGFR and c-MET). Overlap in the control of transcriptional events by ERK and STAT3 helps to explain their overlapping control of phenotypes. For example, ERK and STAT3 both drive expression of proteins that promote cell cycle progression and proliferation, promote expression of anti-apoptotic proteins and downregulate proteins in apoptotic pathways (Zhang and Liu, 2002; Yu and Jove, 2004; Dauer et al., 2005; Lu and Xu, 2006; Krens et al., 2008). This functional overlap in the regulation of broad classes of genes also contains overlap of specific gene products such as VEGF (Yu and Jove, 2004; Krens et al., 2008) and c-MYC (Zhang and Liu, 2002; Yu and Jove, 2004). Even with this partial overlap, in GBM cell lines the positive regulation of ERK by SHP2 is dominant in determining the effect of SHP2 expression on cellular proliferation, whereas SHP2-mediated suppression of STAT3 dominates in determining the effect of SHP2 expression on cellular sensitivity to co-inhibition of EGFR and c-MET. This updated view of the consequences of the multivariate control of SHP2 regarding signaling and phenotype fits within the general paradigm that cells integrate and interpret multivariate signaling information in different ways in the execution of cellular decisions (Janes et al., 2005; Kumar et al., 2007; Lazzara and Lauffenburger, 2009).
Other new aspects of our findings include the discovery that sufficiently high expression of EGFRvIII reduces the antagonism of STAT3 phosphorylation in the presence of kinase inhibitors, in addition to reducing the contribution of SHP2 to ERK activation, and the mechanisms underlying these effects. Others have noted a suppression of ERK activity when EGFRvIII is expressed (Moscatello et al., 1996), but the mechanistic basis for this had not previously been explored. Our data suggest that this effect is related to a mechanism we have elucidated previously for structurally distinct EGFR mutants in NSCLC cells, wherein the kinase-activated EGFR mutants, which display reduced ability to activate ERK and a reduced rate of ligand-mediated endocytosis, also promote basal sequestration of SHP2 with EGFR and GAB1 at the cell periphery (Lazzara et al., 2010; Furcht et al., 2012). It is also interesting to note that in NSCLC cells that express wild-type EGFR, SHP2 knockdown promotes an enhanced response to gefitinib (Furcht et al., 2012) rather than resistance. This presumably occurs because SHP2 knockdown produces generally small effects on STAT3 Y705 phosphorylation in NSCLC cells (Furcht et al., 2012) compared with what we observed here in GBM cell lines, further highlighting the contextual dependence of the functions of SHP2. Interestingly, the ability of EGFRvIII to sequester proteins also underlies loss of SHP2 control of STAT3 phosphorylation in the presence of EGFR and c-MET inhibitors at high EGFRvIII levels. As we demonstrate in Fig. 3E, this effect involves a shift in the ability of EGFRvIII to bind STAT3 when EGFRvIII is expressed at the highest levels. It is also worth noting that the ability of SHP2 to negatively regulate EGFR and c-MET phosphorylation was most apparent when EGFRvIII was expressed at high levels. This could also result from sequestration of active SHP2 at the plasma membrane where it has ready access to these receptors. We also note that the range of EGFRvIII expression explored here is consistent with that observed in tumors (Moscatello et al., 1995). Thus, the dependence of SHP2 functions on EGFRvIII expression might be clinically relevant.
There are, of course, additional signaling pathways regulated by EGFRvIII and SHP2 that have not been explored here but which could play roles in some apparent quantitative inconsistencies between the effects of altered SHP2 expression on signaling pathways and the phenotypic roles we have ascribed to those pathways. One example pertains to our observations in Fig. 3 in U87MG-H cells, where control cells were highly proliferative despite displaying relatively low basal phosphorylation of ERK, and SHP2 knockdown produced a modest effect on ERK phosphorylation but a large effect on proliferation. As just one possible explanation for this, the increased abundance of EGFRvIII in U87MG-H cells might promote the activity of other pathways that could compensate for ERK in promoting proliferation. If the activities of those other pathways are also regulated by SHP2 in a way that promotes proliferation, a large drop in proliferation could still accompany SHP2 knockdown with only a modest effect on ERK. In the future, it will be important to investigate these issues more broadly and quantitatively to fully understand the potential impact of targeting SHP2 or EGFRvIII in GBM.
Our data on the effects of SHP2 knockdown, expression of constitutively active SHP2 and expression of substrate-trapping SHP2 all suggest that SHP2 negatively regulates EGFRvIII phosphorylation, potentially through direct interaction. This contrasts with previous reports that SHP2-mediated ERK activity increases levels of phosphorylated EGFRvIII (Zhan and O'Rourke, 2004). This discrepancy might be explained by our additional finding that SHP2-mediated ERK activity enhances EGFRvIII expression, an effect that has also been noticed by others when knocking down SHP2 in certain GBM cell lines (Zhan et al., 2009). Thus, SHP2 appears to exert two countervailing effects, either of which might be dominant, in the determination of total cellular phosphorylated EGFRvIII levels. The notion that SHP2 can negatively regulate EGFRvIII phosphorylation might seem at odds with our finding that SHP2 knockdown impairs xenograft growth or the analogous findings of Zhan et al. (Zhan et al., 2009) where a catalytically inactive SHP2 was expressed in an EGFRvIII-positive tumor model. We interpret these aggregate results as indicating that any potential ability for SHP2 to impair tumorigenesis by negatively regulating EGFRvIII phosphorylation is trumped by the positive regulatory functions of SHP2 in tumorigenesis, including its apparent ability to control HIF-1/2α expression, at least in the cell line model used here.
Given ongoing efforts to develop specific SHP2 inhibitors for clinical use, it is worth noting that two distinct effects of SHP2 inhibition could arise in GBM cells and tumors. Based on our results, SHP2 inhibition would be expected to inhibit ERK activity but, simultaneously, to promote STAT3 phosphorylation. In cell culture, the integrated effect of these signaling perturbations was to slow cell growth while simultaneously promoting resistance to EGFR and c-MET co-inhibition. Based on this alone, it is unclear whether SHP2 inhibition is a useful therapeutic approach. Our finding that SHP2 controls expression of HIF-1/2α and growth of GBM tumor xenografts may obviate potential concerns about the ability of SHP2 inhibition to promote survival signaling through the STAT3 pathway; however, this remains to be demonstrated in more-detailed GBM tumor models that explore the potential ability of very high EGFRvIII expression to modulate SHP2 function. Assuming that SHP2 function is, indeed, controlled by EGFRvIII levels in vivo, recent advances in detecting EGFRvIII protein through magnetic resonance imaging (Tykocinski et al., 2012), as opposed to traditional tumor tissue biopsy approaches, may eventually advance our ability to predict the impact of SHP2 inhibition.
It should also be noted that our xenograft studies show promise for combining EGFR and c-MET inhibitors in EGFRvIII-positive GBM. This has not been previously demonstrated in vivo, although a previous study demonstrated the suitability of combining an HGF-targeted antibody with an EGFR inhibitor (Lal et al., 2009). Interestingly, the usefulness of certain irreversible EGFR inhibitors in EGFRvIII-positive GBM may obviate the need to combine c-MET and EGFR inhibitors (Vivanco et al., 2012). Whether or not c-MET inhibitors are needed moving forward, SHP2 inhibitors might eventually offer an attractive alternative in the treatment of GBM where resistance to other inhibitors arises. Of course, our data support the potential usefulness of STAT3 inhibitors in treating GBM. STAT3 has previously been identified as a key regulator of GBM cell survival (Lo et al., 2008; de la Iglesia et al., 2009) and at least one clinical trial (ClinicalTrials.gov NCT01904123) is scheduled to begin recruiting patients later this year to test the efficacy of STAT3 inhibition in cancers including GBM.
MATERIALS AND METHODS
Cell culture
LN18, T98G and U118MG cells were obtained from the American Type Culture Collection (Manassas, VA). U87MG parental cells and cells expressing low (1×106 receptors/cell), medium (1.5×106 receptors/cell) or high (2×106 receptors/cell) levels of EGFRvIII (U87MG-L, U87MG-M or U87MG-H, respectively), or a dead-kinase mutant of EGFRvIII (U87MG-DK, 2×106 receptors/cell) were a generous gift from Frank Furnari (University of California San Diego, San Diego, CA). All cells were maintained in DMEM supplemented with 10% FBS, 1 mM L-glutamine, 100 units/ml penicillin and 100 µg/ml streptomycin. U87MG cells expressing EGFRvIII were cultured with 400 µg/ml G418. Where indicated, cells were treated with EGF (Peprotech, Rocky Hill, NJ) following 8–16 h starvation in medium containing 0.1% FBS. All cell culture reagents were from Life Technologies (Carlsbad, CA). For experiments under hypoxic conditions, cells were cultured for 24 h in 0.5% O2 by using an Invivo2 400 hypoxia workstation (Ruskinn Technology, Grandview, MO) prior to lysis.
shRNA and stable expression constructs
DNA oligonucleotides encoding short hairpins targeting human SHP2 (Integrated DNA Technologies, San Jose, CA) were cloned into the pSicoR vector (Tyler Jacks, MIT, Cambridge, MA) (Ventura et al., 2004). The main shRNA targeted nucleotides 1780–1798 of SHP2 mRNA (5′-GGACGTTCATTGTGATTGA-3′). Control vectors were created by using shRNA sequences that do not target a known human mRNA. We also used an alternative, non-overlapping SHP2 shRNA targeting nucleotides 5890–5908 (5′-GTATTGTACCAGAGTATTA-3′) of human SHP2 in a cell proliferation experiment in the presence of drugs (supplementary material Fig. S2A). Combined with the data showing the effects of SHP2E76A expression, the data in supplementary material Fig. S2A help to demonstrate the SHP2 specificity of effects observed using the primary SHP2-targeting hairpin. To engineer cells that express shRNA, lentivirus was produced by using calcium-phosphate-mediated transfection of 293FT cells (Life Technologies) with pSicoR plasmids together with the packaging plasmids pCMV-VSV-G, pMDL-gp-RRE, and pRSV-Rev (Marilyn Farquhar, University of California San Diego, San Diego, CA). Virus was collected 48 and 72 h post-transfection and filtered using 0.45 µm syringe filters prior to infecting target cells.
SHP2 cDNA encoding SHP2E76A (Ben Neel, Ontario Cancer Institute, Toronto, ON, Canada) was inserted at the EcoRI site of the pBabe vector. Retrovirus was produced by calcium phosphate-mediated transfection of amphotropic Phoenix cells (Gary Nolan, Stanford University, Stanford, CA) with vector. Virus was harvested 24, 30 and 48 h post-transfection and used to infect target cells, which were selected in 2 µg/ml puromycin (Sigma-Aldrich, St Louis, MO). All expression and shRNA constructs were validated by sequencing, and protein knockdown was validated by western blot.
Transient expression of wild-type or substrate-trapping SHP2
Expression constructs for wild-type and substrate-trapping double mutant (DM; D425A/C459S) SHP2 in the pMT2 vector backbone were provided by Yehenew Agazie (West Virginia University, Morgantown, WV). U87MG cells were transfected with pMT2 plasmids using calcium phosphate. Cells were lysed in immunoprecipitation lysis buffer (Cell Signaling Technology, Danvers, MA; #9803) 48 h after transfection.
Inhibitors
Stocks of gefitinib (LC Laboratories, Woburn, MA), U0126 (LC Laboratories), CI-1040 (LC Laboratories), PHA665752 (Santa Cruz Biotechnologies, Dallas, TX) and Stattic (Sigma-Aldrich) were prepared in DMSO.
Cell death quantification
Cells were seeded at a density of 75,000–100,000 cells/well in six-well dishes, and treated 24 h later with different combinations of gefitinib, PHA665752, CI-1040 and Stattic, or with DMSO (control). After 72–96 h, floating and adherent cells were pooled and stained for permeability to TO-PRO-3 (Life Technologies). Cells were analyzed by flow cytometry within 1 h of resuspension. Flow cytometry was performed on a Becton Dickinson Biosciences FACSCalibur cytometer and data were analyzed using FlowJo.
Proliferation measurements
Cells were seeded at an initial density of 75,000 or 100,000 cells/well in six-well dishes. After growing for 72 h, cells were trypsinized, suspended in complete medium and counted by using a hemocytometer.
XTT viability assay
Cell proliferation in the presence of inhibitors was assessed using the XTT Cell Proliferation Assay (Roche, Indianapolis, IN). Cells were seeded in 96-well plates, grown for 16–24 h, and switched to complete medium containing up to 20 µM gefitinib and/or 1 µM PHA665752 for an additional 3 days. Subsequently, fresh medium and XTT reagent were added to wells, and plates were incubated for 2–4 h at 37°C to maximize signal-to-background. Absorbance was measured at 450 nm with a reference wavelength at 660 nm. The percentage of viable cells was determined by normalizing absorbance to that of cells treated with DMSO. Each experiment was performed on at least three separate days with each condition plated in three replicate wells on each day, except where noted.
Tumor xenografts
Female Nu/Nu immunodeficient nude mice (Charles River, Wilmington, MA) were subcutaneously injected in both flanks with control or SHP2-depleted U87MG-M cells (control shRNA: 750,000 cells; SHP2 shRNA: 2,500,000 cells). The difference in injected cell numbers was based upon observations of different rates of proliferation in vivo and in vitro with or without SHP2 knockdown. When tumors reached an average size of 50 mm3 (achieved by control tumors only), 7 day treatment with gefitinib and PHA665752 (Selleck Chemicals, Houston, TX) began. Gefitinib was resuspended in an aqueous solution containing 0.5% hydroxypropylmethylcellulose (Sigma-Aldrich) and 0.1% Tween-80 (Sigma-Aldrich) and was delivered at 100 mg/kg/day daily by oral gavage. PHA665752 was resuspended in an aqueous solution containing 1% dimethyl acetamide (Sigma-Aldrich), 10% propylene glycol (Sigma-Aldrich), and 1.05 moles L-lactic acid (Sigma-Aldrich) per mole of PHA665752, and was delivered at 30 mg/kg/day daily by intraperitoneal injection. Tumors were measured with a caliper before and throughout treatment, and tumor volume was calculated as π/6×A×B2, where A and B are the larger and smaller tumor diameters, respectively. Excised tumors were homogenized in immunoprecipitation lysis buffer before proceeding with western blotting. All experiments were approved by the University of Pennsylvania Institutional Animal Care and Use Committee and performed in accordance with NIH guidelines.
Subcellular fractionation
Serum-starved cells were washed with PBS and collected in hypotonic buffer (10 mM Tris-HCl pH 7.4, 1 mM MgCl2, 1 mM EDTA) supplemented with 1 mM PMSF, additional protease inhibitors, and phosphatase inhibitors. Crude lysates were generated with a Dounce homogenizer and centrifuged at 800 and 7900 g, for 5 min at each speed, to remove nuclei and mitochondria, respectively. Cleared lysates were centrifuged at 100,000 g for 60 min to separate membrane and cytosol fractions. Membrane pellets were washed with PBS, resuspended in hypotonic buffer and centrifuged again at 100,000 g. After additional washes, membrane pellets were resuspended in immunoprecipitation lysis buffer to solubilize proteins. To improve signals, membrane fractions were 10× more concentrated than cytosolic fractions in terms of the relative amount of total lysate loaded.
EGF internalization assay
EGF-mediated EGFR endocytosis rate constants (ke) were measured using 125I-EGF as described previously (Wiley and Cunningham, 1982; Lund et al., 1990).
Western blotting
Whole-cell lysates were prepared in a standard cell extraction buffer (Life Technologies; FNN0011) prepared with protease inhibitors and phosphatase inhibitors (Sigma-Aldrich). Lysates were cleared by centrifugation at 16,100 g for 10 min, and total protein concentrations were determined by micro-bicinchoninic acid (BCA) assay (Thermo Fisher Scientific, Waltham, MA). Proteins were resolved on 4–12% gradient polyacrylamide gels (Life Technologies) under denaturing and reducing conditions and transferred to 0.2 µm nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA). Membranes were imaged using a LI-COR Odyssey Imaging System. When needed, membranes were stripped with 0.2 M NaOH.
Immunofluorescence
Cells were seeded at 150,000 cells/well on 18 mm glass coverslips in six-well culture dishes. After serum starvation for 16 h, cells were treated with 10 ng/ml EGF for up to 30 min. Cells were then washed and fixed for 20 min in 4% paraformaldehyde and permeabilized with 0.25% Triton X-100 in PBS for 5 min. Coverslips were again washed and incubated with the SHP2 antibody in a humidified chamber for 3 h at 37°C. Washed coverslips were incubated with Alexa-Fluor-488-conjugated secondary antibody (Life Technologies) and Hoechst DNA stain (Sigma-Aldrich) for 1 h at 37°C. Coverslips were mounted on microscope slides using Prolong Gold Antifade mounting media (Life Technologies) and dried overnight. Fixed slides were imaged on a Zeiss Axiovert 40 CFL microscope using an A-Plan 100× oil objective and a SPOT Insight CCD camera. Specificity of the SHP2 antibody for immunofluorescence was verified using U87MG-M cells with or without SHP2 knockdown (supplementary material Fig. S3C).
Immunoprecipitation
Cells were lysed in immunoprecipitation lysis buffer, with 1 mM PMSF, protease inhibitors, and phosphatase inhibitors. After lysate centrifugation at 16,100 g and 4°C for 10 min and determination of protein concentrations by BCA assay, 400–500 µg of protein was incubated with agarose beads conjugated to SHP2 or STAT3 antibodies at 4°C overnight. Beads were washed three times with cold lysis buffer, re-suspended in LDS sample buffer (Life Technologies) and boiled before western blotting. Immunoprecipitation specificity was validated with comparisons to a rabbit control antibody (IgG; Santa Cruz Biotechnology) (supplementary material Figs S2D, S3A, S4C).
Antibodies
Antibodies against EGFR (catalog no. 2232), c-MET pY1234/1235 (catalog no. 3126), STAT3 pY705 (catalog no. 9145), ERK (catalog no. 4695) and ERK pT202/Y204 (catalog no. 4377) were from Cell Signaling Technology. SHP2 (sc-280) and STAT3 (sc-482) antibodies were from Santa Cruz Biotechnology. Antibodies against actin (MAB1501) and GAB1 (catalog no. 06-579) were from Millipore (Billerica, MA). Antibody against EGFR pY1068 (catalog no. 1727) was from Epitomics (Burlingame, CA). Antibodies against HIF-1α (catalog no. 10006421) and HIF-2α (NB100-122) were from Cayman Chemical (Ann Arbor, MI) and Novus Biologicals (Littleton, CO), respectively. Infrared dye-conjugated secondary antibodies were from Rockland Immunochemicals (Gilbertsville, PA). All antibodies were used according to manufacturers' recommendations.
Statistics
Statistical analyses were performed using a paired two-tailed Student's t-test.
Supplementary Material
Acknowledgments
We thank Donald M. O'Rourke for reviewing the manuscript and providing helpful feedback. We thank Ben Neel, Tyler Jacks, Marilyn Farquhar, Gary Nolan, Frank Furnari, and Yehenew Agazie for generously providing reagents.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
C.M.F., J.M.B. and A.M.R. performed cell culture experiments. N.S., L.K.M., C.M.F. and J.M.B. performed mouse studies. C.M.F., J.M.B., M.C.S. and M.J.L. designed experiments. C.M.F., J.M.B. and M.J.L. wrote the manuscript.
Funding
C.M.F. was supported in part by the University of Pennsylvania Cell and Molecular Biology Training Grant [grant number T32 GM-07229]; Training Program in Cancer Pharmacology [grant number R25 CA101871-07]; and a fellowship from the Ashton Foundation. J.M.B. was supported in part by the National Institutes of Health under Ruth L. Kirschstein National Research Service Award 2T32HL007954 from the NIH-NHLBI; and the National Science Foundation Graduate Research Fellowship [grant number DGE-08220]. A.M.R. received support from the University of Pennsylvania Institute for Regenerative Medicine. This project was funded, in part, under a grant with the Pennsylvania Department of Health. The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. This work was also supported in part by funds from the Howard Hughes Medical Institute and funds to M.J.L. from the University of Pennsylvania. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.150862/-/DC1
References
- Aceto N., Sausgruber N., Brinkhaus H., Gaidatzis D., Martiny-Baron G., Mazzarol G., Confalonieri S., Quarto M., Hu G., Balwierz P. J. et al. (2012). Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop. Nat. Med. 18, 529–537 10.1038/nm.2645 [DOI] [PubMed] [Google Scholar]
- Agazie Y. M., Hayman M. J. (2003). Development of an efficient “substrate-trapping” mutant of Src homology phosphotyrosine phosphatase 2 and identification of the epidermal growth factor receptor, Gab1, and three other proteins as target substrates. J. Biol. Chem. 278, 13952–13958 10.1074/jbc.M210670200 [DOI] [PubMed] [Google Scholar]
- Amin A. R., Thakur V. S., Paul R. K., Feng G. S., Qu C. K., Mukhtar H., Agarwal M. L. (2007). SHP-2 tyrosine phosphatase inhibits p73-dependent apoptosis and expression of a subset of p53 target genes induced by EGCG. Proc. Natl. Acad. Sci. USA 104, 5419–5424 10.1073/pnas.0700642104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argyriou A. A., Kalofonos H. P. (2009). Molecularly targeted therapies for malignant gliomas. Mol. Med. 15, 115–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard-Chapeau E. A., Li S., Ding J., Zhang S. S., Zhu H. H., Princen F., Fang D. D., Han T., Bailly-Maitre B., Poli V. et al. (2011). Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell 19, 629–639 10.1016/j.ccr.2011.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai T., Nishida K., Hirano T., Khavari P. A. (2002). Gab1 and SHP-2 promote Ras/MAPK regulation of epidermal growth and differentiation. J. Cell Biol. 159, 103–112 10.1083/jcb.200205017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan R. J., Feng G. S. (2007). PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood 109, 862–867 10.1182/blood-2006-07-028829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan G., Kalaitzidis D., Neel B. G. (2008). The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 27, 179–192 10.1007/s10555-008-9126-y [DOI] [PubMed] [Google Scholar]
- Chung J., Uchida E., Grammer T. C., Blenis J. (1997). STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol. Cell. Biol. 17, 6508–6516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer D. J., Ferraro B., Song L., Yu B., Mora L., Buettner R., Enkemann S., Jove R., Haura E. B. (2005). Stat3 regulates genes common to both wound healing and cancer. Oncogene 24, 3397–3408 10.1038/sj.onc.1208469 [DOI] [PubMed] [Google Scholar]
- de la Iglesia N., Puram S. V., Bonni A. (2009). STAT3 regulation of glioblastoma pathogenesis. Curr. Mol. Med. 9, 580–590 10.2174/156652409788488739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Witt Hamer P. C. (2010). Small molecule kinase inhibitors in glioblastoma: a systematic review of clinical studies. Neuro-oncol. 12, 304–316 10.1093/neuonc/nop068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Q. W., Cheng C. K., Gustafson W. C., Charron E., Zipper P., Wong R. A., Chen J., Lau J., Knobbe-Thomsen C., Weller M. et al. (2013). EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell 24, 438–449 10.1016/j.ccr.2013.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furcht C. M., Munoz Rojas A. R., Nihalani D., Lazzara M. J. (2012). Diminished functional role and altered localization of SHP2 in non-small cell lung cancer cells with EGFR-activating mutations. Oncogene 32, 2346–2355, e1-e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandal M. V., Zandi R., Pedersen M. W., Willumsen B. M., van Deurs B., Poulsen H. S. (2007). EGFRvIII escapes down-regulation due to impaired internalization and sorting to lysosomes. Carcinogenesis 28, 1408–1417 10.1093/carcin/bgm058 [DOI] [PubMed] [Google Scholar]
- Gu H., Neel B. G. (2003). The “Gab” in signal transduction. Trends Cell Biol. 13, 122–130 10.1016/S0962-8924(03)00002-3 [DOI] [PubMed] [Google Scholar]
- Hodge D. R., Hurt E. M., Farrar W. L. (2005). The role of IL-6 and STAT3 in inflammation and cancer. Eur. J. Cancer 41, 2502–2512 10.1016/j.ejca.2005.08.016 [DOI] [PubMed] [Google Scholar]
- Huang H. S., Nagane M., Klingbeil C. K., Lin H., Nishikawa R., Ji X. D., Huang C. M., Gill G. N., Wiley H. S., Cavenee W. K. (1997). The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J. Biol. Chem. 272, 2927–2935 10.1074/jbc.272.5.2927 [DOI] [PubMed] [Google Scholar]
- Huang P. H., Mukasa A., Bonavia R., Flynn R. A., Brewer Z. E., Cavenee W. K., Furnari F. B., White F. M. (2007). Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc. Natl. Acad. Sci. USA 104, 12867–12872 10.1073/pnas.0705158104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes K. A., Albeck J. G., Gaudet S., Sorger P. K., Lauffenburger D. A., Yaffe M. B. (2005). A systems model of signaling identifies a molecular basis set for cytokine-induced apoptosis. Science 310, 1646–1653 10.1126/science.1116598 [DOI] [PubMed] [Google Scholar]
- Krens S. F., Corredor-Adámez M., He S., Snaar-Jagalska B. E., Spaink H. P. (2008). ERK1 and ERK2 MAPK are key regulators of distinct gene sets in zebrafish embryogenesis. BMC Genomics 9, 196 10.1186/1471-2164-9-196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N., Wolf-Yadlin A., White F. M., Lauffenburger D. A. (2007). Modeling HER2 effects on cell behavior from mass spectrometry phosphotyrosine data. PLOS Comput. Biol. 3, e4 10.1371/journal.pcbi.0030004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal B., Goodwin C. R., Sang Y., Foss C. A., Cornet K., Muzamil S., Pomper M. G., Kim J., Laterra J. (2009). EGFRvIII and c-Met pathway inhibitors synergize against PTEN-null/EGFRvIII+ glioblastoma xenografts. Mol. Cancer Ther. 8, 1751–1760 10.1158/1535-7163.MCT-09-0188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzara M. J., Lauffenburger D. A. (2009). Quantitative modeling perspectives on the ErbB system of cell regulatory processes. Exp. Cell Res. 315, 717–725 10.1016/j.yexcr.2008.10.033 [DOI] [PubMed] [Google Scholar]
- Lazzara M. J., Lane K., Chan R., Jasper P. J., Yaffe M. B., Sorger P. K., Jacks T., Neel B. G., Lauffenburger D. A. (2010). Impaired SHP2-mediated extracellular signal-regulated kinase activation contributes to gefitinib sensitivity of lung cancer cells with epidermal growth factor receptor-activating mutations. Cancer Res. 70, 3843–3850 10.1158/0008-5472.CAN-09-3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo H. W., Cao X., Zhu H., Ali-Osman F. (2008). Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin. Cancer Res. 14, 6042–6054 10.1158/1078-0432.CCR-07-4923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z., Xu S. (2006). ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life 58, 621–631 10.1080/15216540600957438 [DOI] [PubMed] [Google Scholar]
- Lund K. A., Opresko L. K., Starbuck C., Walsh B. J., Wiley H. S. (1990). Quantitative analysis of the endocytic system involved in hormone-induced receptor internalization. J. Biol. Chem. 265, 15713–15723 [PubMed] [Google Scholar]
- Moscatello D. K., Holgado-Madruga M., Godwin A. K., Ramirez G., Gunn G., Zoltick P. W., Biegel J. A., Hayes R. L., Wong A. J. (1995). Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res. 55, 5536–5539 [PubMed] [Google Scholar]
- Moscatello D. K., Montgomery R. B., Sundareshan P., McDanel H., Wong M. Y., Wong A. J. (1996). Transformational and altered signal transduction by a naturally occurring mutant EGF receptor. Oncogene 13, 85–96 [PubMed] [Google Scholar]
- Neel B. G., Gu H., Pao L. (2003). The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 28, 284–293 10.1016/S0968-0004(03)00091-4 [DOI] [PubMed] [Google Scholar]
- Roberts P. J., Der C. J. (2007). Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26, 3291–3310 10.1038/sj.onc.1210422 [DOI] [PubMed] [Google Scholar]
- Sathornsumetee S., Reardon D. A., Desjardins A., Quinn J. A., Vredenburgh J. J., Rich J. N. (2007). Molecularly targeted therapy for malignant glioma. Cancer 110, 13–24 10.1002/cncr.22741 [DOI] [PubMed] [Google Scholar]
- Schmitz J., Weissenbach M., Haan S., Heinrich P. C., Schaper F. (2000). SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP2 recruitment site of gp130. J. Biol. Chem. 275, 12848–12856 10.1074/jbc.275.17.12848 [DOI] [PubMed] [Google Scholar]
- Shao H., Cheng H. Y., Cook R. G., Tweardy D. J. (2003). Identification and characterization of signal transducer and activator of transcription 3 recruitment sites within the epidermal growth factor receptor. Cancer Res. 63, 3923–3930 [PubMed] [Google Scholar]
- Sturla L. M., Zinn P. O., Ng K., Nitta M., Kozono D., Chen C. C., Kasper E. M. (2011). Src homology domain-containing phosphatase 2 suppresses cellular senescence in glioblastoma. Br. J. Cancer 105, 1235–1243 10.1038/bjc.2011.345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tykocinski E. S., Grant R. A., Kapoor G. S., Krejza J., Bohman L. E., Gocke T. A., Chawla S., Halpern C. H., Lopinto J., Melhem E. R. et al. (2012). Use of magnetic perfusion-weighted imaging to determine epidermal growth factor receptor variant III expression in glioblastoma. Neuro-oncol. 14, 613–623 10.1093/neuonc/nos073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A., Meissner A., Dillon C. P., McManus M., Sharp P. A., Van Parijs L., Jaenisch R., Jacks T. (2004). Cre-lox-regulated conditional RNA interference from transgenes. Proc. Natl. Acad. Sci. USA 101, 10380–10385 10.1073/pnas.0403954101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivanco I., Robins H. I., Rohle D., Campos C., Grommes C., Nghiemphu P. L., Kubek S., Oldrini B., Chheda M. G., Yannuzzi N. et al. (2012). Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2, 458–471 10.1158/2159-8290.CD-11-0284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley H. S., Cunningham D. D. (1982). The endocytotic rate constant. A cellular parameter for quantitating receptor-mediated endocytosis. J. Biol. Chem. 257, 4222–4229 [PubMed] [Google Scholar]
- Wolf-Yadlin A., Kumar N., Zhang Y., Hautaniemi S., Zaman M., Kim H. D., Grantcharova V., Lauffenburger D. A., White F. M. (2006). Effects of HER2 overexpression on cell signaling networks governing proliferation and migration. Mol. Syst. Biol. 2, 54 10.1038/msb4100094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. J., O'Rourke D. M., Feng G. S., Johnson G. R., Wang Q., Greene M. I. (2001). The tyrosine phosphatase SHP-2 is required for mediating phosphatidylinositol 3-kinase/Akt activation by growth factors. Oncogene 20, 6018–6025 10.1038/sj.onc.1204699 [DOI] [PubMed] [Google Scholar]
- Yu H., Jove R. (2004). The STATs of cancer—new molecular targets come of age. Nat. Rev. Cancer 4, 97–105 10.1038/nrc1275 [DOI] [PubMed] [Google Scholar]
- Zhan Y., O'Rourke D. M. (2004). SHP-2-dependent mitogen-activated protein kinase activation regulates EGFRvIII but not wild-type epidermal growth factor receptor phosphorylation and glioblastoma cell survival. Cancer Res. 64, 8292–8298 10.1158/0008-5472.CAN-03-3143 [DOI] [PubMed] [Google Scholar]
- Zhan Y., Counelis G. J., O'Rourke D. M. (2009). The protein tyrosine phosphatase SHP-2 is required for EGFRvIII oncogenic transformation in human glioblastoma cells. Exp. Cell Res. 315, 2343–2357 10.1016/j.yexcr.2009.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Liu H. T. (2002). MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 12, 9–18 10.1038/sj.cr.7290105 [DOI] [PubMed] [Google Scholar]
- Zhang S. Q., Tsiaras W. G., Araki T., Wen G., Minichiello L., Klein R., Neel B. G. (2002). Receptor-specific regulation of phosphatidylinositol 3′-kinase activation by the protein tyrosine phosphatase Shp2. Mol. Cell. Biol. 22, 4062–4072 10.1128/MCB.22.12.4062-4072.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Zhang W., Cao W. D., Cheng G., Zhang Y. Q. (2012). Glioblastoma multiforme: Molecular characterization and current treatment strategy. Exp. Ther Med. 3, 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Agazie Y. M. (2009). Molecular mechanism for SHP2 in promoting HER2-induced signaling and transformation. J. Biol. Chem. 284, 12226–12234 10.1074/jbc.M900020200 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.