

Abstract
With the emergence of high-throughput discovery platforms, robust preclinical small-animal models, and efficient clinical trial pipelines, it is becoming possible to envision a time when the treatment of human neurologic diseases will become personalized. The emergence of precision medicine will require the identification of subgroups of patients most likely to respond to specific biologically based therapies. This stratification only becomes possible when the determinants that contribute to disease heterogeneity become more fully elucidated. This review discusses the defining factors that underlie disease heterogeneity relevant to the potential for individualized brain tumor (optic pathway glioma) treatments arising in the common single-gene cancer predisposition syndrome, neurofibromatosis type 1 (NF1). In this regard, NF1 is posited as a model genetic condition to establish a workable paradigm for actualizing precision therapeutics for other neurologic disorders.
Neurofibromatosis type 1 (NF1) is one of the most common monogenic disorders in which affected individuals develop benign and malignant tumors.1 NF1 impacts 1:2,500 people worldwide, and individuals with NF1 are prone to the development of peripheral (neurofibromas, malignant peripheral nerve sheath tumors) and central (optic pathway glioma, malignant glioma) nervous system tumors.2,3 Similar to other autosomal dominant cancer predisposition syndromes,4,5 people with NF1 start life with a germline mutation in one copy of the NF1 tumor suppressor gene; however, tumors require somatic (acquired) inactivation of the remaining functional NF1 allele, leading to complete loss of NF1 expression in specific cell types.6,7 For example, complete Nf1 gene inactivation in neuroglial8,9 or Schwann cell10,11 progenitors is required for murine optic glioma or neurofibroma formation, respectively.
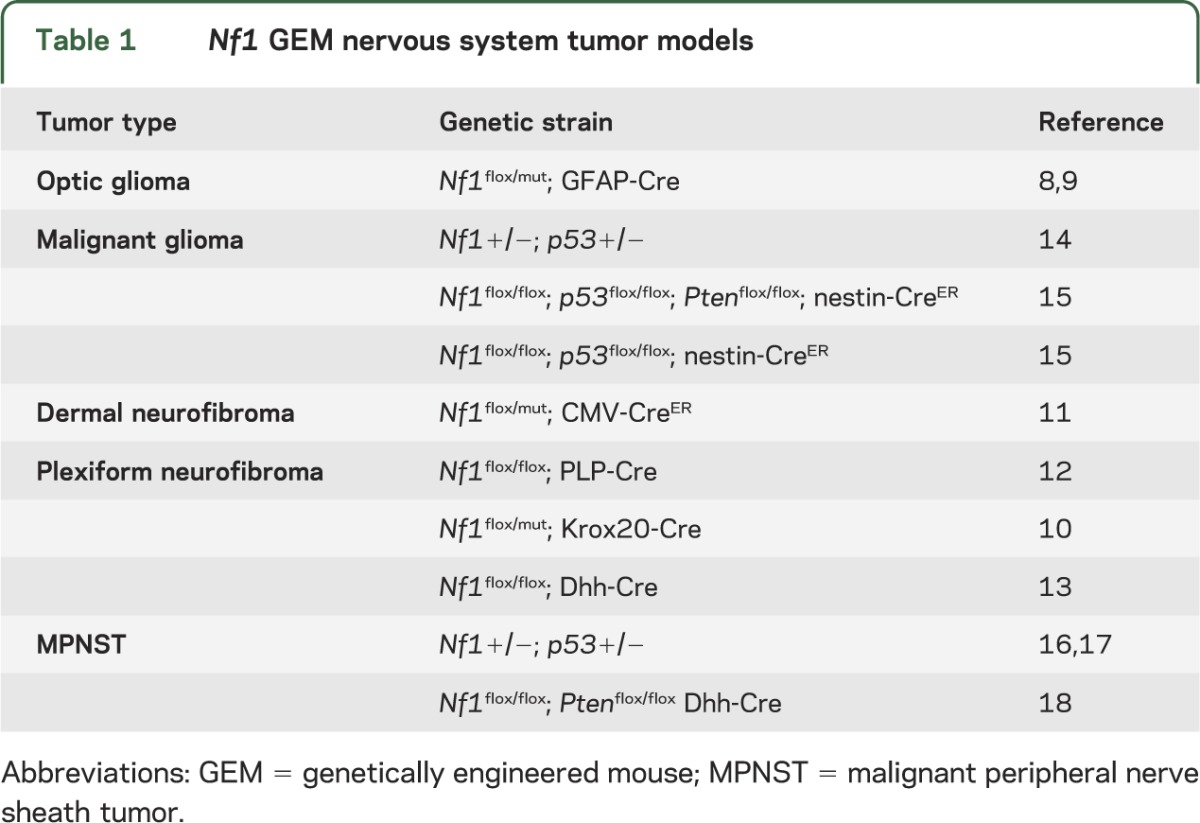
With the development of numerous accurate small-animal (genetically engineered mouse; GEM) models of NF1-associated nervous system tumors (table 1),8–18 the creation of the NF Clinical Trials Consortium,19 and the establishment of response criteria for NF1 clinical trials,20 the stage has been set for the discovery and validation of promising therapeutic strategies and their translation to people affected with NF1. However, despite these advances, there are currently no effective therapies, which likely reflects the striking biological and clinical heterogeneity inherent to this condition. This review uses NF1-associated brain tumors (optic glioma) as an illustrative platform to discuss the barriers and challenges to developing and implementing effective targeted therapies.
Table 1.
Nf1 GEM nervous system tumor models
NF1-ASSOCIATED OPTIC PATHWAY GLIOMA
NF1-associated optic pathway gliomas (NF1-OPGs) are largely pediatric tumors typically arising in children younger than 7 years of age.21,22 As such, 15%–20% of children with NF1 will develop World Health Organization grade I pilocytic astrocytomas (PAs) anywhere along the optic pathway, from the retro-orbital optic nerve to the postchiasmatic optic tracts (figure 1). In addition to neoplastic glial cells, 30%–50% of the cells in these tumors are non-neoplastic cells (microglia) harboring one functional copy of the NF1 gene and one nonfunctional NF1 allele (germline NF1 gene mutation).23
Figure 1. MRI reveals different types of optic gliomas arising in children with NF1.
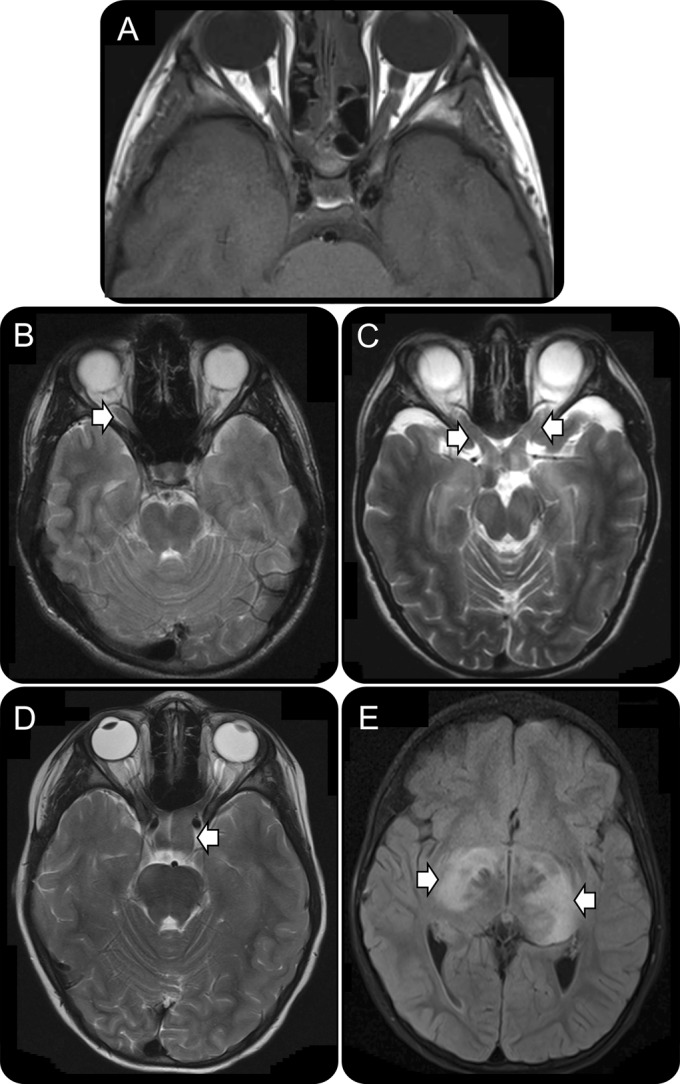
(A) Normal optic nerves and chiasm in a child with neurofibromatosis type 1 (NF1) but no optic pathway glioma. (B) Right optic glioma (arrow). (C) Bilateral optic nerve gliomas (arrows). (D) Optic chiasm glioma (arrow). (E) Postchiasmal optic tract glioma (arrows).
Since baseline MRI is not useful in predicting clinical outcome,24 it is currently not possible to identify children at greatest risk for developing NF1-OPG. In addition, following NF1-OPG identification, there are no reliable methods for determining which child is most likely to experience continued visual decline and require treatment, necessitating frequent MRI (often requiring sedation) and neuro-ophthalmologic evaluations, which can be unreliable in preverbal children.25,26 At present, affected children typically receive chemotherapy routinely used for sporadic PA (carboplatin/vincristine), with varying success. Radiation therapy is not used because of the elevated risk of secondary cancer development in this cancer predisposition syndrome.27 Despite radiographic evidence of tumor stabilization or response following chemotherapy, only a minority of children with NF1-OPG experience improvements in vision.28,29 In addition, there is increasing concern that chemotherapy in this young age group may result in cognitive decline as a late secondary sequela, negatively impacting long-term quality of life.30 Collectively, these issues highlight the pressing need to develop management strategies that reflect the unique biology of NF1-OPG.
USE OF NF1 GEM STRAINS TO UNDERSTAND NF1-OPG PATHOGENESIS
There are numerous important unanswered questions relevant to the care of children with NF1-OPG that may lead to the improved management of these brain tumors, ranging from explaining the unique spatial and temporal distribution of these tumors to understanding the cellular and molecular constraints that drive glioma formation and progression. Ideally, these answers would emanate from discovery efforts using human tissue specimens; however, in the case of NF1-OPG, few patients undergo tumor biopsy or removal prior to or following treatment. Moreover, many of the available NF1 brain tumor specimens represent tumors with unusual features, such as those arising in older children or in brain regions other than the optic pathway,31 and may not be representative of the more commonly encountered OPGs in children with NF1.
In light of the above challenges with human tissue specimens, Nf1 GEM strains have been developed that model the optic gliomas arising in children with NF1.8,9,32 While not perfect representations of the human condition, Nf1 optic glioma mice have yielded several unanticipated observations critical to the future design and execution of clinical trials for these tumors. It should be appreciated that these findings were only made possible through the use of Nf1 GEM strains.
Understanding the unique temporal and spatial pattern of NF1-OPG.
To address the question of why NF1 gliomas display a propensity for the optic pathway and brainstem of young children, researchers used a series of Nf1 mutant mice in which somatic Nf1 gene inactivation could be experimentally manipulated. Murine optic gliomas require complete Nf1 gene inactivation in specific neuroglial progenitors (neural stem cells; NSCs) during mid to late embryogenesis, thus creating a narrow developmental window in which tumor initiation must occur.32,33 The ability of these NSCs to generate gliomas following Nf1 loss during embryonic development, but not in more differentiated glial cell types after birth, likely accounts for the predilection for these tumors to arise in young children.
Second, NSCs from some brain regions, but not others, increase their growth following Nf1 gene inactivation. In these studies, Nf1 gene inactivation in NSCs residing in the third ventricle or brainstem results in stem cell expansion and increased glial differentiation, whereas no effect is observed following Nf1 loss in cortical or lateral ventricle NSCs.32,34 These findings partly explain the distinct brain region distribution of these tumors (optic pathway/brainstem but rarely in the cortex).
Identifying NF1-OPG targeted therapies.
Further analysis of the molecular basis underlying the above brain region– and cell type–specific effects revealed that the impact of Nf1 gene inactivation is dependent on how the downstream RAS signaling pathway functions in specific cell types. The NF1 gene codes for the protein neurofibromin, which contains a 300 amino acid domain similar to other proteins that function as negative regulators of the RAS proto-oncogene.35 While neurofibromin controls cell growth in numerous different cell types in a RAS-dependent manner,36,37 there are several distinct molecules that transmit the RAS growth signal, including MEK and AKT. Whereas RAS/MEK signaling is critical for murine Nf1 plexiform neurofibroma growth,38 Nf1 loss uniquely affects NSCs from the third ventricle and brainstem by activating AKT in a mammalian target of rapamycin (mTOR)–regulated manner,34 thus identifying the mTOR pathway as a key mediator of NF1-OPG growth. This cell type–specific growth dependence led to the evaluation of rapamycin, initially in preclinical mouse optic glioma models39 and then in a clinical trial for children with NF1-associated glioma (ClinicalTrials.gov identifier NCT01158651).
The fact that NF1-PAs are cellularly heterogeneous tumors composed of several distinct cell types raises the intriguing possibility that non-neoplastic stromal cells (e.g., microglia) may actively participate in NF1-OPG formation and growth. Consistent with this hypothesis, Nf1 optic glioma formation in mice requires the interplay between noncancerous cells (e.g., microglia) harboring one mutated Nf1 allele and precancerous cells (astroglial progenitors/astrocytes) with complete neurofibromin loss. As such, GEM strains only lacking Nf1 gene expression in neuroglial progenitors do not form optic gliomas40: optic gliomagenesis necessitates that Nf1 loss in neuroglial progenitors occur in mice with a germline inactivating Nf1 gene mutation in all cells in the brain.8,9 Additional support for a critical role for microglia in glioma development and growth derives from numerous complementary studies employing Nf1 GEM strains. In these experiments, silencing microglia function using either pharmacologic or genetic strategies reduces optic glioma tumor growth.41,42 Moreover, impairing microglia migration into the region of the developing mouse optic glioma delays tumor formation.43 The obligate role for noncancerous cells in the tumor microenvironment (stroma) offers new potential strategies for stroma-directed therapy. Studies are currently under way in several laboratories to identify these glioma-associated microglial factors.44,45
The key role of the microenvironment in tumor development and growth is further underscored by analogous observations involving mast cells and macrophages in Nf1 GEM plexiform neurofibromas.46 Similar to Nf1 murine optic gliomas, Nf1 plexiform neurofibroma formation requires that Nf1 loss in Schwann cell precursors occur in mice with a germline inactivating Nf1 gene mutation (Nf1+/−mice).10 Using bone marrow transplantation, Nf1+/−bone marrow–derived mast cells, but not those from normal mice, induce plexiform neurofibroma development in normal mice with Nf1 loss in Schwann cell precursors only.47 Moreover, the replacement of Nf1+/−bone marrow–derived mast cells with normal ones reduced plexiform neurofibroma growth in Nf1+/−mice with Nf1 loss in Schwann cell precursors. The dependence on mast cells led to the identification of the KIT receptor as a potential target for antitumoral therapy. In addition to promoting mast cell infiltration relevant to plexiform neurofibroma growth, KIT is also critical for tumor macrophage accumulation.48 As such, blocking KIT activation in Nf1 plexiform neurofibroma–bearing mice with imatinib resulted in attenuation of tumor growth, which is currently being explored further in human clinical trials.
Defining the cellular and molecular basis for NF1-OPG–associated visual loss.
One of the major disappointments has been the limited visual recovery in children following treatment with chemotherapy for clinically progressive NF1-OPG. Using Nf1 optic glioma mice, visual impairment was shown to result from tumor-induced retinal ganglion cell (RGC) axonal dysfunction, leading to RGC death by apoptosis.49 The mechanism underlying this neuronal death involves neurofibromin regulation of cyclic adenosine monophosphate (cAMP) production, such that restoring normal cAMP homeostasis in Nf1 optic glioma mice partially ameliorates the RGC apoptosis.50 This finding raises intriguing questions about the role of neuroprotective approaches in reversing visual loss in children with NF1-OPG.
Identifying genomic factors that influence NF1-OPG development and progression.
The low incidence of NF1-OPG could reflect the factors discussed above; however, it is also possible that genomic variation contributes to glioma development. As such, defining the genomic determinants that favor optic gliomagenesis has potential value for predictive risk assessment. In this regard, many Nf1 GEM studies employ mice maintained on a specific inbred genetic background, whereas others use mice on a mixed background. Humans by definition harbor a mixed genomic composition that reflects subtle chromosomal polymorphisms inherited from each of our parents. Studies have begun to unravel the genomic contributions to tumorigenesis in Nf1 GEM strains: whereas Nf1+/−;p53+/−mice develop high-grade astrocytomas (gliomas) when maintained on a C57BL/6 background, Nf1+/−;p53+/−mice maintained on a 129sv background exhibit a significantly reduced frequency of glioma formation.51,52 Similarly, >90% of Nf1flox/mut; GFAP-Cre mice maintained on a C57BL/6 background develop optic gliomas,8 compared to ∼20% maintained on a mixed genetic background.9 While astrocytoma predisposition genomic modifiers have been identified in mice, their application to the human condition has not been fully explored.
Another genomic factor that might influence NF1-OPG is the sex of the patient. As such, one exciting observation to emerge from Nf1 GEM studies is the impact of sex on NF1-OPG–induced vision loss. In Nf1 optic glioma mice, reduced cAMP levels and higher levels of RGC apoptosis are observed in females, leading to reduced visual acuity in female mice only.53 Moreover, while the frequency of OPG development is similar in girls and boys with NF1, 3 times more females with NF1-OPG require treatment for visual decline.53 These findings support a role for sex as a critical factor that underlies clinical outcomes in children with NF1-OPG.
TRANSLATING BASIC SCIENCE DISCOVERIES TO HUMAN THERAPEUTIC CLINICAL TRIALS
Despite these exciting advances in molecular/cellular biology and the ability to efficiently evaluate drugs in accurate preclinical models prior to human clinical trials, there are presently no effective treatments for most NF1-associated tumors. In part, this may reflect the manner in which Nf1 GEM preclinical drug study results are translated to the clinical workplace. As such, biologically based therapies frequently exhibit dramatic antitumoral responses in mice, but limited tumor shrinkage in human clinical trials. For example, imatinib was shown to be highly effective at attenuating Nf1 GEM plexiform neurofibroma growth,47 but it exhibited far less efficacy for treating human NF1-associated plexiform neurofibromas (<20% tumor response).54 Future preclinical studies will need to incorporate outcome expectations (e.g., number of mice exhibiting a response, percent of tumor reduction, and the durability of the effect) that parallel those employed in human clinical trials.
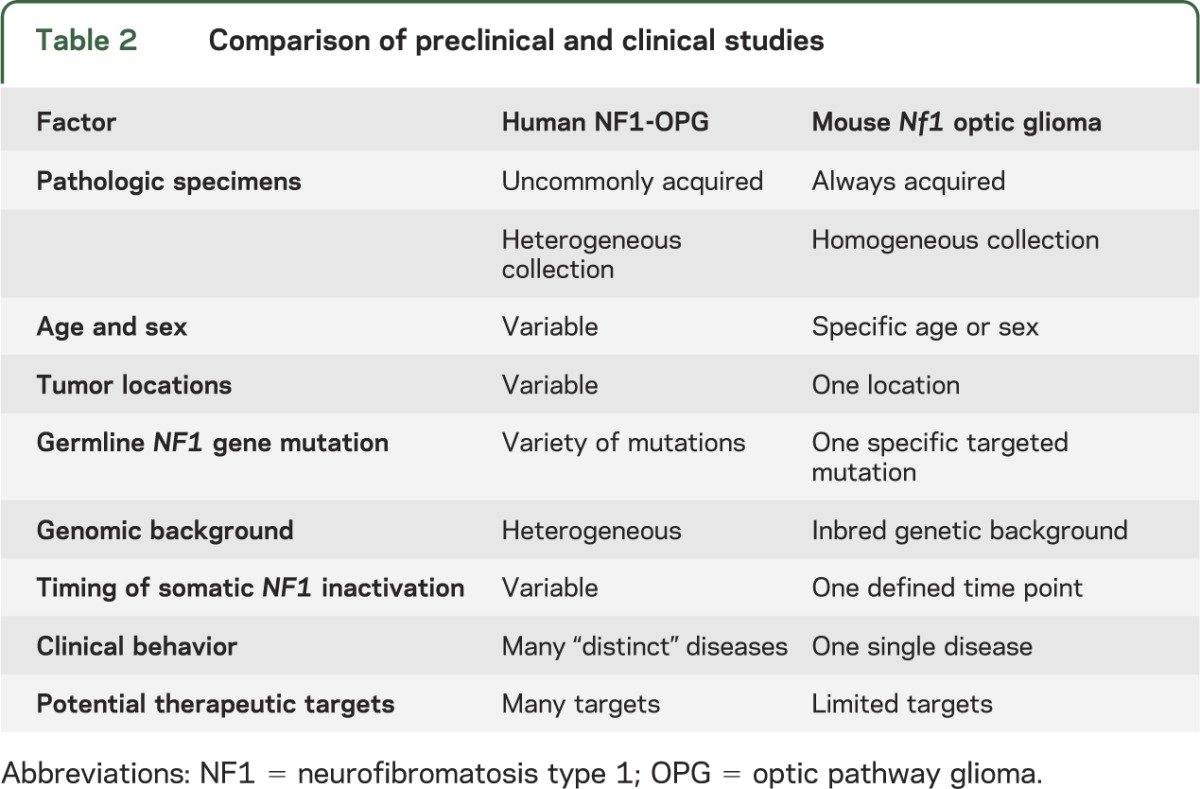
There are significant differences inherent in Nf1 GEM preclinical and NF1 patient clinical trials (table 2). Taking NF1-OPG as an illustrative example, human NF1 clinical trials do not involve the collection of pathologic specimens and they enroll patients of different ages, sexes, and tumor locations. As such, the genomic background, the timing of the somatic NF1 gene inactivation event, and the specific germline NF1 gene mutation vary from patient to patient. For this reason, the clinical outcomes reflect the fact that the patients enrolled in these trials constitute a diverse population of distinct disease subgroups, likely defined by the factors that influence NF1-OPG heterogeneity, with potentially different molecular drivers of tumor growth and druggable therapeutic targets. In addition, the lack of pathologic specimens prevents assessments of CNS drug penetration and target inhibition in the tumor, causing investigators to rely solely on changes in radiographic appearance (tumor volume) and visual improvement. Since tumor volume does not correlate with visual improvement and vision changes may require months (or even years) to fully manifest,28,29 the outcomes of current clinical trials may not be interpreted accurately.
Table 2.
Comparison of preclinical and clinical studies
In contrast, each Nf1 optic glioma GEM strain represents a homogeneous population of mice in which the genetic background, inherited Nf1 mutation, timing of somatic Nf1 loss, age, sex, and tumor location are identical. As such, these preclinical Nf1 GEM strains model only one disease subgroup with a more limited number of druggable targets. Moreover, optic glioma specimens are routinely acquired in these preclinical studies, enabling a demonstration of drug bioavailability (CNS penetration) and target inhibition. In this manner, while the use of a single homogeneous population of Nf1 optic glioma mice provides more interpretable experimental outcomes, the resulting findings may only be applicable to one subtype of NF1-OPG.
NF1-OPG IS A DISEASE OF HETEROGENEITY
As outlined above, it is highly likely that NF1-OPG comprises multiple distinct diseases defined by specific factors, including patient age, sex, tumor location, NF1 germline mutation, genomic background, and other genetic changes (figure 2). The identification of the responsible risk factors will likely yield clinically relevant subgroups of patients who could be stratified for tailored therapeutic approaches that best match their unique subtype.
Figure 2. Determinants of NF1-OPG disease heterogeneity.

Neurofibromatosis type 1–optic pathway glioma (NF1-OPG) heterogeneity is determined by a confluence of individual factors that individually affect cell biology and glioma risk. For example, the specific germline NF1 gene mutation creates differential effects on cyclic adenosine monophosphate (cAMP) levels and retinal ganglion cell (RGC) death in neurons (denoted R in the subgroup bar code), chemokine and growth factor production in microglia (denoted M in the subgroup bar code), and RAS pathway activation in neoplastic progenitors/glia (denoted S in the subgroup bar code). Similarly, other genetic alterations (KIAA1549:BRAF or PTEN mutation) alter the activity of the RAS pathway relevant to NF1-deficient tumor cell growth. In addition, patient sex leads to differences in cAMP levels (neurons) or RAS pathway activity (neoplastic progenitors/glia) to affect RGC survival or optic glioma growth. Likewise, the timing of and cell type with somatic NF1 gene inactivation influences NF1-OPG brain location (optic nerve vs postchiasmal tracts, denoted L in the subgroup bar code) or clinical features (clinical progression, denoted P in the subgroup bar code). Finally, the genomic background represents another strong determinant of tumor development and progression. Together, these factors (depicted as colored boxes to illustrate their relative effects) could be used to construct risk assessment algorithms that inform clinical practice.
Age.
In a large multicenter study, children younger than 2 years were more likely to experience visual decline secondary to NF1-OPG.55 While the etiology of this observation remains unknown, it may reflect the cell of origin and the developmental period during which NF1 inactivation occurs.
Sex.
As mentioned above, female mice as well as female humans with NF1-OPG are more likely to experience visual decline and require treatment than their male counterparts.53 These observations raise the possibility that females have different epigenetic programming due to X-chromosome influences, which change the intracellular context in which changes in neurofibromin expression affect biological outcomes. Identifying these epigenetic determinants may reveal new genes for risk stratification and therapeutic targeting.
It is also possible that sex influences optic glioma outcomes through differential hormonal production. In this scenario, female hormones (e.g., estrogen) may directly act on RGC neurons to influence intracellular cAMP levels and cell survival. As such, reduced neurofibromin function in RGC neurons may have different consequences in females, in whom heterotrimeric G protein–induced cAMP production is uniquely modulated, relative to their male counterparts. By leveraging this potential hormonal mechanism, it may be possible to envision therapies that target this receptor by repurposing drugs used to treat hormonally responsive male and female cancers.
Location.
Several studies have demonstrated that NF1-OPGs involving the postchiasmatic optic tracts are more likely to cause visual decline and require treatment.55 Moreover, this effect is independent of sex, suggesting that it reflects the primary biology of these tumors.53 As more diverse GEM models of NF1-OPG are developed, it might be possible to mechanistically determine why tumors in this location have such different outcomes relevant to the design of targeted therapies.
Germline NF1 gene mutation.
Emerging evidence from numerous laboratories has begun to reveal that the NF1 germline mutation in people with NF1 may have distinct consequences on the spectrum of the clinical features observed. As such, recognized genotype–phenotype correlations include individuals from different families harboring the c1756-1759_delACTA mutation, who, despite having the other features of NF1, do not develop dermal neurofibromas.56 Similarly, individuals with 5′ end NF1 gene frameshift and premature truncation mutations are prone to developing optic gliomas.57,58 In addition, patients with NF1 locus microdeletions that delete the entire NF1 gene are prone to the development of malignancies (malignant peripheral nerve sheath tumor, high-grade glioma), which may reflect the co-deletion of other tumor suppressor genes (e.g., SUZ12 gene) in the region.59,60 Together, these data suggest that not all germline NF1 gene mutations are equal in their biological effects.
In addition, it is important to recognize that the differential impact of these germline NF1 gene mutations on non-neoplastic cells (neurons and microglia) may be profound: mutations that mildly impair neurofibromin function in neurons would be predicted to have less deleterious effects on glioma-induced RGC survival and vision than mutations that completely abrogate neurofibromin expression from that allele. Similarly, microglia with NF1 germline mutations that result in significantly reduced neurofibromin expression may elaborate higher levels or a different spectrum of gliomagens (growth factors and chemokines) that promote glioma growth than those with relatively normal neurofibromin function. In this regard, correlating the germline NF1 mutation with tissue-specific neurofibromin expression levels may provide meaningful insights into NF1-OPG development and outcome as well as the design of future stroma-directed therapies and neuroprotective strategies. Studies are currently under way to generate Nf1 GEM strains harboring specific NF1 patient-derived germline Nf1 gene mutations for such mechanistic studies.
Genomic factors.
While genome-wide association studies (GWAS) have not yielded genomic predictors of glioma development in the general population owing to the diversity of initiating and cooperating genetic events required for malignant gliomagenesis, all patients with NF1-OPG share a similar genetic etiology (germline mutation in the NF1 tumor suppressor gene). Recent studies have begun to reveal single nucleotide polymorphisms that may predict NF1-OPG development (Dr. Joshua Rubin, written communication, 2014). While still early in their validation and application to predictive testing, they may permit early glioma risk assessment in a population with a known propensity for brain tumor formation. Similarly, the use of twin studies may also facilitate the identification of these genomic factors.61 The preselection of at-risk children for intense monitoring changes the current anticipatory management paradigm to one of more directed medical monitoring.
Additional genetic mutations.
While the vast majority of NF1-associated low-grade gliomas analyzed by next-generation sequencing harbor only NF1 gene inactivation,23 recent data suggest that some NF1-OPGs may have monoallelic loss of PTEN gene expression or concurrent KIAA1549:BRAF alterations.62 Recent Nf1 GEM studies in our laboratory in which these genetic changes were introduced revealed differential effects on tumor volume and proliferation. Moreover, the extent and diversity of growth control signaling pathway activation (e.g., MEK or AKT activation) is also different, raising the possibility that tailored treatment regimens will be required to suppress the growth of these NF1-OPGs compared to their counterparts harboring only neoplastic cell NF1 gene inactivation.
CHALLENGE FOR PRECISION MEDICINE
The implementation of individualized therapies will require information not currently available that can only be derived from large collaborative studies that leverage resources not widely available in the NF community. First, large data sets for epidemiologic investigations aimed at identifying clinical associations using population science approaches are required to provide hypotheses for future mechanistic laboratory investigation. For example, with the launch of the NF1 Patient Registry Initiative, a Web-based, patient-driven registry,63 relationships between specific medical conditions and NF1 brain tumor risk have begun to emerge (Dr. Kimberly Johnson, written communication, 2014). Second, the availability of genomic DNA and NF1 germline mutation data may enable GWAS that reveal new genomic predictors of NF1-OPG development as well as novel genotype–phenotype correlations. The resulting data, when confirmed, may allow clinicians to more accurately predict which children with NF1 are likely to develop OPG as well as which at-risk children are most likely to require treatment following NF1-OPG formation.
Third, there is a paucity of renewable human biospecimens for drug evaluation. The assembly of large induced pluripotent stem cell repositories for reprogramming into neurons, neuroglial progenitors, and microglia offers unprecedented opportunities for translational research. The use of human cells from patients with NF1 may set the stage for more personalized therapies. Fourth, future preclinical studies might consider the use of multiple GEM models of NF1-OPG that differ with respect to the germline Nf1 gene mutation, the timing of somatic Nf1 gene inactivation, the genetic background of the mouse, and the sex of the animal. The availability of a heterogeneous “clinic population” of NF1-OPG GEM strains may allow investigators to identify those distinct disease subgroups most likely to respond to specific targeted therapies.
With the successful deployment of the above strategies, it is possible to envision a future proactive model of NF1 clinical care in which young children with café-au-lait macules (initial presenting feature of NF1) are assessed for their risk of optic glioma development and then stratified into clinical subgroups based on predictive modeling for targeted therapies that reflect their individual underlying disease pathogenesis and biology. Of importance, the lessons learned from and paradigms created for NF1-OPG will likely apply to other neurologic disorders similarly characterized by clinical heterogeneity.
ACKNOWLEDGMENT
The author thanks Drs. Joshua Rubin and Kimberly Johnson for permission to cite their unpublished work and for their helpful comments during the preparation of this review.
GLOSSARY
- cAMP
cyclic adenosine monophosphate
- GEM
genetically engineered mouse
- GWAS
genome-wide association studies
- mTOR
mammalian target of rapamycin
- NF1
neurofibromatosis type 1
- NSC
neural stem cell
- OPG
optic pathway glioma
- PA
pilocytic astrocytoma
- RGC
retinal ganglion cell
STUDY FUNDING
This work was partly funded by a grant from the Department of Defense (W81XWH-12-0032).
DISCLOSURE
The author reports no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Upadhyaya M. Neurofibromatosis type 1: diagnosis and recent advances. Expert Opin Med Diagn 2010;4:307–322 [DOI] [PubMed] [Google Scholar]
- 2.Jett K, Friedman JM. Clinical and genetic aspects of neurofibromatosis 1. Genet Med 2010;12:1–11 [DOI] [PubMed] [Google Scholar]
- 3.Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol 2009;61:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990;250:1233–1238 [DOI] [PubMed] [Google Scholar]
- 5.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 1971;68:820–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jouhilahti EM, Peltonen S, Heape AM, Peltonen J. The pathoetiology of neurofibromatosis 1. Am J Pathol 2011;178:1932–1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laycock-van Spyk S, Thomas N, Cooper DN, Upadhyaya M. Neurofibromatosis type 1-associated tumours: their somatic mutational spectrum and pathogenesis. Hum Genomics 2011;5:623–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bajenaru ML, Hernandez MR, Perry A, et al. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res 2003;63:8573–8577 [PubMed] [Google Scholar]
- 9.Zhu Y, Harada T, Liu L, et al. Inactivation of NF1 in CNS causes increased glial progenitor proliferation and optic glioma formation. Development 2005;132:5577–5588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science 2002;296:920–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le LQ, Shipman T, Burns DK, Parada LF. Cell of origin and microenvironment contribution for NF1-associated dermal neurofibromas. Cell Stem Cell 2009;4:453–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayes DA, Rizvi TA, Cancelas JA, et al. Perinatal or adult Nf1 inactivation using tamoxifen-inducible PlpCre each cause neurofibroma formation. Cancer Res 2011;71:4675–4685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu J, Dombi E, Jousma E, et al. Preclinical testing of sorafenib and RAD001 in the Nf(flox/flox): DhhCre mouse model of plexiform neurofibroma using magnetic resonance imaging. Pediatr Blood Cancer 2012;58:173–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet 2000;26:109–113 [DOI] [PubMed] [Google Scholar]
- 15.Alcantara-Llaguno S, Chen J, Kwon CH, et al. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell 2009;15:45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cichowski K, Shih TS, Schmitt E, et al. Mouse models of tumor development in neurofibromatosis type 1. Science 1999;286:2172–2176 [DOI] [PubMed] [Google Scholar]
- 17.Vogel KS, Klesse LJ, Velasco-Miguel S, Meyers K, Rushing EJ, Parada LF. Mouse tumor model for neurofibromatosis type 1. Science 1999;286:2176–2179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keng VW, Rahrmann EP, Watson AL, et al. PTEN and NF1 inactivation in Schwann cells produce a severe phenotype in the peripheral nervous system that promotes the development and malignant progression of peripheral nerve sheath tumors. Cancer Res 2012;72:3405–3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gutmann DH, Blakeley JO, Korf BR, Packer RJ. Optimizing biologically targeted clinical trials for neurofibromatosis. Expert Opin Investig Drugs 2013;22:443–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plotkin SR, Blakeley JO, Dombi E, et al. Achieving consensus for clinical trials: the REiNS International Collaboration. Neurology 2013;81:S1–S5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr 1994;125:63–66 [DOI] [PubMed] [Google Scholar]
- 22.Listernick R, Darling C, Greenwald M, Strauss L, Charrow J. Optic pathway tumors in children: the effect of neurofibromatosis type 1 on clinical manifestations and natural history. J Pediatr 1995;127:718–722 [DOI] [PubMed] [Google Scholar]
- 23.Gutmann DH, McLellan MD, Hussain I, et al. Somatic neurofibromatosis type 1 (NF1) inactivation characterizes NF1-associated pilocytic astrocytoma. Genome Res 2013;23:431–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.King A, Listernick R, Charrow J, Piersall L, Gutmann DH. Optic pathway gliomas in neurofibromatosis type 1: the effect of presenting symptoms on outcome. Am J Med Genet 2003;122:95–99 [DOI] [PubMed] [Google Scholar]
- 25.Fisher MJ, Avery RA, Allen JC, et al. Functional outcome measures for NF1-associated optic pathway glioma clinical trials. Neurology 2013;81:S15–S24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Listernick R, Ferner RE, Liu GT, Gutmann DH. Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations. Ann Neurol 2007;61:189–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharif S, Ferner R, Birch JM, et al. Second primary tumors in neurofibromatosis 1 patients treated for optic glioma: substantial risks after radiotherapy. J Clin Oncol 2006;24:2570–2575 [DOI] [PubMed] [Google Scholar]
- 28.Dalla Via P, Opocher E, Pinello ML, et al. Visual outcome of a cohort of children with neurofibromatosis type 1 and optic pathway glioma followed by a pediatric neuro-oncology program. Neuro Oncol 2007;9:430–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalin-Hajdu E, Décarie JC, Marzouki M, Carret AS, Ospina LH. Visual acuity of children treated with chemotherapy for optic pathway gliomas. Pediatr Blood Cancer 2013. Epub Aug 19 [DOI] [PubMed] [Google Scholar]
- 30.Riva D, Massimino M, Giorgi C, et al. Cognition before and after chemotherapy alone in children with chiasmatic-hypothalamic tumors. J Neurooncol 2009;92:49–56 [DOI] [PubMed] [Google Scholar]
- 31.Leonard JR, Perry A, Rubin JB, King AA, Chicoine MR, Gutmann DH. The role of surgical biopsy in the diagnosis of glioma in individuals with neurofibromatosis-1. Neurology 2006;67:1509–1512 [DOI] [PubMed] [Google Scholar]
- 32.Lee DY, Gianino SM, Gutmann DH. Innate neural stem cell heterogeneity determines the patterning of glioma formation in children. Cancer Cell 2012;22:131–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Solga AC, Gianino SM, Gutmann DH. NG2 cells are not the cell of origin for murine neurofibromatosis-1 (Nf1) optic glioma. Oncogene 2013;33:289–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee DY, Yeh TH, Emnett RJ, White CR, Gutmann DH. Neurofibromatosis-1 regulates neuroglial progenitor proliferation and glial differentiation in a brain region-specific manner. Genes Dev 2010;24:2317–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu GF, O'Connell P, Viskochil D, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 1990;62:599–608 [DOI] [PubMed] [Google Scholar]
- 36.Bollag G, Clapp DW, Shih S, et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet 1996;12:144–148 [DOI] [PubMed] [Google Scholar]
- 37.DeClue JE, Papageorge AG, Fletcher JA, et al. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell 1992;69:265–273 [DOI] [PubMed] [Google Scholar]
- 38.Jessen WJ, Miller SJ, Jousma E, et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest 2013;123:340–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hegedus B, Banerjee D, Yeh TH, et al. Preclinical cancer therapy in a mouse model of neurofibromatosis-1 optic glioma. Cancer Res 2008;68:1520–1528 [DOI] [PubMed] [Google Scholar]
- 40.Bajenaru ML, Zhu Y, Hedrick NM, Donahoe J, Parada LF, Gutmann DH. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol 2002;22:5100–5113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Daginakatte GC, Gutmann DH. Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum Mol Genet 2007;16:1098–1112 [DOI] [PubMed] [Google Scholar]
- 42.Daginakatte GC, Gianino SM, Zhao NW, Parsadanian AS, Gutmann DH. Increased c-Jun-NH2-kinase signaling in neurofibromatosis-1 heterozygous microglia drives microglia activation and promotes optic glioma proliferation. Cancer Res 2008;68:10358–10366 [DOI] [PubMed] [Google Scholar]
- 43.Pong WW, Higer SB, Gianino SM, Emnett RJ, Gutmann DH. Reduced microglial CX3CR1 expression delays neurofibromatosis-1 glioma formation. Ann Neurol 2013;73:303–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warrington NM, Woerner BM, Daginakatte GC, et al. Spatiotemporal differences in CXCL12 expression and cyclic AMP underlie the unique pattern of optic glioma growth in neurofibromatosis type 1. Cancer Res 2007;67:8588–8595 [DOI] [PubMed] [Google Scholar]
- 45.Pyonteck SM, Akkari L, Schuhmacher AJ, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 2013;19:1264–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang FC, Staser K, Clapp DW. The plexiform neurofibroma microenvironment. Cancer Microenviron 2012;5:307–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang FC, Ingram DA, Chen S, et al. Nf1-dependent tumors require a microenvironment containing Nf1+/– and c-kit-dependent bone marrow. Cell 2008;135:437–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prada CE, Jousma E, Rizvi TA, et al. Neurofibroma-associated macrophages play roles in tumor growth and response to pharmacological inhibition. Acta Neuropathol 2013;125:159–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hegedus B, Hughes FW, Garbow JR, et al. Optic nerve dysfunction in a mouse model of neurofibromatosis-1 optic glioma. J Neuropathol Exp Neurol 2009;68:542–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown JA, Gianino SM, Gutmann DH. Defective cyclic AMP generation underlies the sensitivity of central nervous system neurons to neurofibromatosis-1 heterozygosity. J Neurosci 2010;30:5579–5589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reilly KM, Tuskan RG, Christy E, et al. Susceptibility to astrocytoma in mice mutant for Nf1 and Trp53 is linked to chromosome 11 and subject to epigenetic effects. Proc Natl Acad Sci USA 2004;101:13008–13013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amlin-Van Schaick JC, Kim S, DiFabio C, Lee MH, Broman KW, Reilly KM. Arlm1 is a male-specific modifier of astrocytoma resistance on mouse Chr 12. Neuro Oncol 2012;14:160–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diggs-Andrews KA, Brown JA, Gianino SM, Rubin JB, Wozniak DF, Gutmann DH. Sex is a major determinant of neuronal dysfunction in neurofibromatosis type 1. Ann Neurol 2013. Epub Dec 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robertson KA, Nalepa G, Yang FC, et al. Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: a phase 2 trial. Lancet Oncol 2012;13:1218–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fisher MJ, Loguidice M, Gutmann DH, et al. Visual outcomes in children with neurofibromatosis type 1-associated optic pathway glioma following chemotherapy: a multicenter retrospective analysis. Neuro Oncol 2012;14:790–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Upadhyaya M, Huson SM, Davies M, et al. An absence of cutaneous neurofibromas associated with a 3-bp in-frame deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am J Hum Genet 2007;80:140–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sharif S, Upadhyaya M, Ferner R, et al. A molecular analysis of individuals with neurofibromatosis type 1 (NF1) and optic pathway gliomas (OPGs), and an assessment of genotype-phenotype correlations. J Med Genet 2011;48:256–260 [DOI] [PubMed] [Google Scholar]
- 58.Bolcekova A, Nemethova M, Zatkova A, et al. Clustering of mutations in the 5' tertile of the NF1 gene in Slovakia patients with optic pathway glioma. Neoplasma 2013;60:655–665 [DOI] [PubMed] [Google Scholar]
- 59.Kehrer-Sawatzki H, Vogt J, Mußotter T, Kluwe L, Cooper DN, Mautner VF. Dissecting the clinical phenotype associated with mosaic type-2 NF1 microdeletions. Neurogenetics 2012;13:229–236 [DOI] [PubMed] [Google Scholar]
- 60.De Raedt T, Brems H, Wolkenstein P, et al. Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet 2003;72:1288–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rieley MB, Stevenson DA, Viskochil DH, Tinkle BT, Martin LJ, Schorry EK. Variable expression of the neurofibromatosis 1 in monozygotic twins. Am J Med Genet 2011;155A:478–485 [DOI] [PubMed] [Google Scholar]
- 62.Rodriguez FJ, Ligon AH, Horkayne-Szakaly I, et al. BRAF duplications and MAPK pathway activation are frequent in gliomas of the optic nerve proper. J Neuropathol Exp Neurol 2012;71:789–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Johnson KJ, Hussain I, Williams K, Santens R, Mueller NL, Gutmann DH. Development of an international internet-based neurofibromatosis Type 1 patient registry. Contemp Clin Trials 2013;34:305–311 [DOI] [PubMed] [Google Scholar]