

Abstract
Specific docking interactions between mitogen-activated protein kinases (MAPKs), their regulators, and their downstream substrates, are crucial for efficient and accurate signal transmission. To identify novel substrates of the c-Jun N-terminal kinase (JNK) family of MAPKs, we searched the human genome for proteins that contained (1), a predicted JNK-docking site (D-site); and (2), a cluster of putative JNK target phosphosites located close to the D-site. Here we describe a novel JNK substrate that emerged from this analysis, the functionally uncharacterized protein Smoothelin-like 2 (SMTNL2). SMTNL2 protein bound with high-affinity to multiple MAPKs including JNK1-3 and ERK2; furthermore, the identity of conserved amino acids in the predicted docking site (residues 180-193) was necessary for this high-affinity binding. In addition, purified full-length SMTNL2 protein was phosphorylated by JNK1-3 in vitro, and this required the integrity of the D-site. Using mass spectrometry and mutagenesis, we identified four D-site-dependent phosphoacceptor sites in close proximity to the docking site, at S217, S241, T236 and T239. A short peptide comprised of the SMTNL2 D-site inhibited JNK-mediated phosphorylation of the ATF2 transcription factor, showing that SMTNL2 can compete with other substrates for JNK binding. Moreover, when transfected into HEK293 cells, SMTNL2 was phosphorylated by endogenous JNK in a D-site dependent manner, on the same residues identified in vitro. SMTNL2 protein was expressed in many mammalian tissues, with notably high expression in skeletal muscle. Consistent with the hypothesis that SMTNL2 has a function in skeletal muscle, SMTNL2 protein expression was strongly induced during the transition from myoblasts to myotubes in differentiating C2C12 cells.
Keywords: MAP kinase, SMTNL2, JNK, D-Site, Docking, Phosphorylation
1. Introduction
Mitogen-activated protein kinase (MAPK) signaling cascades execute a number of critical functions in eukaryotic systems, including cell division, differentiation, and stress responses [1]. In mammalian cells, there are three major MAPK families, each containing multiple MAPK paralogs: the JNK (c-Jun N-terminal kinase) family, the ERK (extracellular-signal regulated kinase) family, and the p38 family [2]. JNK-family MAPKs are primarily activated by stress-associated signals, including UV radiation, ribotoxic stress, and cytokines, and are central to the regulation of stress responses and metabolic homeostasis [3]. Misregulated signal transduction involving the JNK cascade is linked to many human diseases including cancer, diabetes and muscular dystrophy, and to neurological disorders including stoke, Alzheimer's, Huntington's and Parkinson's diseases [4-7].
JNKs and other MAPKs phosphorylate their substrates on Serine or Threonine residues that are immediately followed by Proline residues. This S/T-P motif, however, is too degenerate to fully determine target specificity, as it is found in greater than 80% of all proteins. Thus, a critical feature of MAPK-substrate recognition is the binding of MAPKs to a short docking motif that resides on many substrates, distal to the target phosphorylation site(s) [8, 9]. For example, when JNK2 phosphorylates its substrate c-Jun, it first tethers itself to a docking site located within c-Jun residues 30-45, and then phosphorylates c-Jun on distal S-P sites such as Ser63 and Ser73. There are several classes of MAPK-docking sites, which differ in their sequence, and contact different regions of the MAPK. The abundant ‘D-site’ class of MAPK docking sites has a consensus sequence that consists of a cluster of basic residues, followed by a short spacer of 1-6 residues and a hydrophobic- X-hydrophobic submotif (K/R-X1–6-φ-X-φ). D-sites have been identified in a number of JNK substrates, including Heterogeneous Nuclear Ribonucleoprotein K (hnRNP-K), Insulin Receptor Substrate 1 (IRS-1), and transcription factors such as ATF2, c-Jun, Elk, Net, Gli1, Gli3, and NFAT4 [10]. D-sites are also found in the upstream MKKs that phosphorylate and activate JNK [11-13], in JNK scaffolding proteins such as JIP-1, and in MAPK phosphatases that dephosphorylate JNK [14, 15]. Docking sites facilitate the efficient and specific interaction of MAPKs with their substrates and binding partners [8]. Interfering with docking by mutagenesis, or by the use of blocking peptides, dramatically reduces the efficiency of substrate phosphorylation [16-18]. For these reasons, there is interest in targeting MAPK docking interactions as a therapeutic strategy. In this regard it is notable that a D-site peptide derived JIP-1 has been shown to have a protective effect in a rat model for stroke [19].
In order to better understand MAPK signaling pathways, and improve our ability to target these pathways to treat human disease, it is essential to identify additional MAPK-interacting proteins and substrates. One attractive strategy for the unbiased identification of novel substrates and regulators is to employ a computational bioinformatics approach to search protein sequence databases. Unlike phosphoproteomics and related biochemical methods, a computational approach is not biased against weakly-expressed proteins, or against proteins in little-studied cell types. While the MAPK target phosphorylation site S/T-P is too degenerate to be used as the basis of such a genome-wide search procedure, we have previously shown that D-sites can be used in this manner, provided that a sufficiently sophisticated search algorithm is employed. To this end, we developed D-finder, which uses a combination of expert knowledge and machine learning to predict D-sites in individual protein sequences or whole genomes [10]. D-finder identified previously unrecognized D-sites in known substrates (e.g. hnRNP-K) and it also uncovered new MAPK substrates of biomedical importance (e.g. Gli1 and Gli3), which we verified using rigorous and well-established biochemical assays [10].
Functionally verified D-sites in known substrates are typically located nearby (within 100 residues on the linear sequence) to the target phosphosites whose phosphorylation they promote [20]. Thus, while it is impractical to make potential phosphosites the focus of a genome-wide search, we hypothesized that the presence of nearby phosphosites might be used to augment and prioritize D-finder's predictions. Here we report an initial attempt to combine docking site and phosphosite prediction to identify new MAPK substrates. Furthermore, we provide a detailed investigation of SMTNL2, one of the high-priority hits obtained from this combined approach.
2. Materials and Methods
2.1 Construction of position weight matrixes for the identification of potential JNKphosphorylation sites
We compiled known in vitro and in vivo JNK1/2/3 phosphosites, found in the current database of PhosphoSitePlus [21] (Table S2; 89, 42, and 11 peptides respectively), and used these data to create position weight matrices (PWMs). PWMs were computed for JNK1, JNK2 and JNK3, for phosphoserine and phosphothreonine sites independently, thus resulting in a total of 6 PWMs. These PWMs take as input 15-residue peptide substrings centered on the SP or TP residue being evaluated. We calculated background frequencies for each amino acid from the set of every phosphoserine- and phosphothreonine-containing peptide within PhosphoSitePlus. This approach for determining background frequencies (as opposed to using global coding sequence frequencies, for example) should in principle reduce any bias towards over-scoring a peptide based on global properties (charge, surface accessibility, intrinsic disorder) that the set of all phosphoacceptor peptides may be enriched for.
These PWMs were then scanned over known JNK substrates, using LAsearch as implemented in PoSSuM 1.3 [22, 23], which calculates a p-value for each S/T site based on the expected number of matches for all permutations of the PWM for a background sequence of the same length as the input (E-value). The p-value threshold used when predicting JNK phosphosites was tuned so that 80% of the input JNK1 and JNK2 peptides would be predicted correctly as a JNK phosphosites. This threshold was chosen as it minimized the sum of false negatives plus false positives in known JNK substrates.
2.2 Human genes
The human MAPK genes used in this study were JNK1α 1 (MAPK8, NCBI Accession Number NM_002750), JNK2α2 (MAPK9, NM_002752), JNK3α1(MAPK10, NM_002753) and ERK2 (MAPK1, NM_002754). The accession number for SMTNL2 is NM_001114974.1.
2.3 Proteins & Peptides
Fusions of glutathione S-transferase (GST) to human ATF219-96 were purchased from Cell Signaling Technology. Activated human JNK1α1, JNK2α2, and JNK3α1 were purchased from Upstate Cell Signaling Solutions/Millipore. Activated mouse ERK2 was purchased from New England Biolabs.
The soluble peptides used in this study were synthesized by Mimotopes. Their sequences are as follows:
| SMTNL2: | FSAPDPPRPRPVSLSLRLP |
| MKK4: | MQGKRKALKLNFANPP |
| MKK4EAG: | MQGEAKALKGNFANPP |
MKK4EAG is a negative-control mutant version of MKK4 in which three consensus residues of the D-site (K, R, and L) have been mutated (to E, A and G, respectively).
2.4 Plasmids for in vitro transcription and translation
The SMTNL2 cDNA sequence (NM_001114974) was obtained from Open Biosystems (Huntsville, AL). Regions of interest were amplified by PCR using Pfu Ultra DNA polymerase (Stratagene). PCR products were purified (Qiagen) and digested with restriction enzymes designed into the primers, run on 1% agarose gels, and excised for gel extraction (Qiagen). Digested, gel purified products were inserted into pGEM-4Zstop [10]. Insert sequences were verified by DNA sequencing. To create D-site mutant (DSM) constructs, basic and hydrophobic residues in the predicted D-site were substituted with alanine residues using appropriate primers and the Quickchange site-directed mutagenesis kit (Stratagene). Mutations were verified by DNA sequencing.
2.5 Plasmids for the production of GST fusion proteins and cell culture
SMTNL2 coding sequences were excised from the pGEM4Z SMTNL2 constructs described above, and subcloned into pGEX-LB (a derivative of pGEX-4T-1) and pcDNA 3.1 (+) as described [24].
2.6 In vitro transcription and translation
Proteins labeled with [35S]-methionine were produced by coupled transcription and translation reactions (SP6, Promega). Translation products were partially purified by ammonium sulfate precipitation [9] and resuspended in binding buffer (20mM Tris-HCl (pH 7.5), 125mM KOAc, 0.5mM EDTA, 1mM DTT, 0.1% (v/v) Tween20, 12.5% (v/v) Glycerol). Comparable translation products were normalized for GST pull-down assays by SDS-PAGE and quantification using a Typhoon phosphorimager.
2.7 GST pull-down assays
Comparable amounts of [35S]-methionine labeled proteins (i.e. wild-type vs. D-site mutant) were pre-cleared with BSA-blocked glutathione Sepharose beads, then incubated with GST-MAPK fusion proteins (or GST alone as a control) that had been pre-bound to glutathione Sepharose beads. The incubation was carried out in binding buffer (see 2.6 above) for 15 min at 30°C, followed by and additional 30 min at room temperature with gentle rocking. Complexes bound to the beads were then pelleted by centrifugation, washed extensively with binding buffer, and heated in reducing SDS sample buffer. Samples were separated by SDS-PAGE, fixed in 40% Methanol/12% Acetic Acid, stained with GelCode Blue (Pierce) to visualize the GST-fusion proteins, then dried, and the radiolabelled bands were quantified using a phosphorimager. To generate values for percent binding, bands from lanes incubated with GST protein were normalized to the 5% input lane.
2.8 Purification of recombinant GST proteins
Expression of recombinant GST fusion proteins was induced in Escherichia coli BL21 competent cells (Stratagene) at 30°C for 2 h by addition of 1-thio-β-D-galactopyranoside [IPTG, 0.6 mM final]. Cell pellets were resuspended in lysis buffer (1× PBS, 1mM EDTA, 5mM DTT, 0.1% Triton, 1mM PMSF, 15% Glycerol), and the resulting extract was sonicated, clarified with 20% Triton X-100, and centrifuged at 12,000×g for 10 min to remove cell debris and nucleic acids. GST fusion proteins contained within the supernatants were purified by affinity chromatography using glutathione-Sepharose (Amersham Biosciences), eluted from beads using 10mM reduced glutathione, and dialyzed overnight against binding buffer. Eluted proteins were quantified against BSA standards.
2.9 Protein kinase assays
Kinase reactions (20 μl) contained 1× MAP Kinase Buffer (50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 1 mM EGTA, and 2 mM DTT), 50 mM ATP, 1 mCi of [γ-32P]-ATP, limiting amounts of enzyme, and substrate. Enzymes used included active human JNK1α1, JNK2α2, and JNK3α1 (Millipore): and active mouse ERK2 (New England Biosciences). The GST-SMTNL2 substrates were used at a concentration of 250 nM (366 ng/20 μl). Reactions were incubated at 30 °C for 30 min, then stopped with SDS sample buffer, separated by SDS-PAGE, fixed in 40% methanol/12% acetic acid, stained with GelCode Blue (Pierce) for visualization of the substrate band, dried, and quantified using a phosphorimager for measurment of 32P incorporation.
2.10 Peptide competition assays
Protein kinase reactions were performed as specified above. In addition, reactions contained the indicated concentration of the specified peptide.
2.11 Liquid chromatography-tandem mass spectrometry (LC MS/MS)
Protein kinase reactions were as described above except that ATP was increased to 200 mM, the kinase concentration was increased 10-fold, and the radioactive ATP tracer was omitted. Following real or mock kinase phosphorylation reactions, the products were separated by SDS-PAGE, and bands corresponding to the mass of the SMTNL2 fragments were excised from the gel and digested with trypsin or chymotrypsin. The resulting peptide digests were extracted and analyzed by LC-MS/MS using an LTQ-Orbitrap XL MS (Thermo Scientific, San Jose, CA) with on-line Eksigent NanoLC system (Eksigent, Dublin, CA) as described [25]. Briefly, peptides were separated using a capillary column (100 μm ID × 150 mm long) packed in house with C18 resins (GL Sciences) and eluted using a linear gradient of 2-35% B in 35 min (solvent A: 100% H2O/0.1% formic acid; solvent B: 100% acetonitrile/0.1% formic acid). A cycle of one full FT scan mass spectrum (350-1800 m/z, resolution of 60,000 at m/z 400) was followed by ten data-dependent MS/MS acquired in the linear ion trap with normalized collision energy (setting of 35%). Target ions selected for MS/MS were dynamically excluded for 30 sec. Protein identification and characterization was carried out by database searching using the Batch-Tag within the developmental version of Protein Prospector (v 5.8.0) [25] against Swissprot database as well as a targeted database consisting of a various JNK substrates including the engineered GST-tagged WT and DSM SMTNL2 sequences. The mass accuracy for parent ions and fragment ions were set as ± 20 ppm and 0.6 Da, respectively. A maximum of two missed cleavages were allowed. Protein N-terminal acetylation, methionine oxidation, N-terminal conversion of glutamine to pyroglutamic acid, and phosphorylation of serine and threonine were selected as the variable modifications. MS/MS spectra of phosphorylated peptides were inspected manually.
2.12 Tissue culture, Transfections and C2C12 differentiation
Human Embryonic Kidney (HEK) 293 cells were cultured using Dulbecco's modified Eagle's medium enriched with 10% heat-inactivated fetal bovine serum (Invitrogen), penicillin, streptomycin, and sodium bicarbonate. The culture was maintained in a humidified environment at 37°C and 5% CO2.
Transient transfections were performed with Lipofectamine (Invitrogen) following the manufacturer's recommended procedures. Cells were seeded at a density of 3×105 cells per well in a 6-well dish in antibiotic free media. HEK 293 cells were transfected with 1 μg of plasmid DNA encoding either V5-tagged SMTMNL WT, DSM, M4, or empty vector. After 24 h, the cells were treated with anisomycin (500 nM, 30 min) to activate the JNK pathway. In some cases, cells were treated with the JNK inhibitor SP600125 (Calbiochem, San Diego, CA) for 30 min prior to ansiomycin stimulation. Following treatment, cells were washed with ice cold PBS, harvested and lysed in 2 ml Triton Lysis Buffer (TLB, 20 mM Tris-HCl (pH 7.5), 137 mM NaCl, 2 mM EDTA, 10% glycerol (v/v), 0.1% Triton X-100 (v/v), 25 mM β-glycerophosphate, 1 mM sodium vanadate, 1:100 protease inhibitor cocktail (Sigma)) and centrifuged at 14,000 × g for 15 min at 4°C. The supernatant was collected and quantified by Bradford analysis (Bio-Rad).
C2C12 mouse myoblasts were grown in DMEM supplemented with 10% FCS. For differentiation, cells were grown on collagen-1 coated plates and allowed to come to confluence (Day 0). Differentiation media (DMEM with 2% Horse Serum) was then added to the cells and changed daily for 8 days.
2.13 Immunohistochemistry
Embryonic and adult muscle samples were fixed in 10% formalin for 48 hours at 4C, dehydrated in an ethanol gradient (30-80%), then embedded in paraffin. Tissues were sectioned at 8 M onto positively charged specimen slides and allowed to dry for 24-48 hours at 37C. Slides were exposed to 100 °C antigen retrieval in 10mM sodium citrate (pH 8) for 10 minutes, cooled to room temperature, peroxidase blocked for 10 minutes (Dako dual endogenous block), protein blocked for one hour (Dako serum free block), and incubated with primary Smtnl2 antibody (Sigma, HPA025706) at 1:500 overnight at 4 °C. Biotinylated anti-rabbit secondary at 1:500 was incubated for 1 hour at room temperature, followed by ABC reagent (Dako) for 1 hour and development using DAB chromagen (Fisher). Counterstaining was with 1:5 diluted Mayers Haemotxylin (Fisher) for 15-30 seconds; the slides were then mounted with permount.
3. Results
3.1 Ranking of putative phosphoproteins with MAPK docking-sites
We started with a list of 394 human proteins with putative docking sites of the D-site class, identified by D-finder and published previously [10]. D-finder was developed using a training set of literature-verified D-sites found in JNK substrates and binding partners, and thus D-finder's predictions should be enriched for JNK substrates relative to ERK/p38 substrates.
Each protein on the list of D-finder predictions was scanned for SP and TP sites, as these constitute potential MAPK phosphosites. Proteins were scored based on the number of S/T-P sites they contained within a 100 amino acid window on either side of their putative docking site (Table S1). In this manner, 308 proteins were identified as potential substrates, meaning that they contained at least one potential phosphosite within 100 residues of the predicted docking site. Of these 308, 232 contained a cluster of 2 or more S/T-P sites near the D-site.
To further analyze these 308 putative substrates for potential JNK phosphosites, we used a probability-weight matrix (PWM)-based approach to scan for S/T-P sites that were in a local sequence context that indicated that they might be efficient JNK phosphosites. PWMs for JNK1, JNK2 and JNK3 were constructed from the sequences of known JNK phosphosites obtained from the PhosphoSitePlus database [21]. A threshold p-value was chosen that was able to predict known JNK phosphosites with high accuracy (80%), without over-predicting (presumably) unphosphorylated S/T-P sites (see Materials and Methods for further details).
The output resulting from applying these PWMs to three known JNK targets (ATF2, JUNB, and JUN [20]) is shown in Fig. 1A. For ATF2, the JNK PWMs correctly identified the known JNK phosphosites T69, T71 and S112, while also correctly rejecting the many other ST/TP sites found throughout the protein. For JUNB, the JNK PWMs correctly identified the known JNK phosphosites T102 and T104, while also correctly rejecting the 3 other ST/TP sites in the polypeptide. Finally, for the canonical JNK substrate JUN, the JNK PWMs displayed a similar degree of accuracy, although it did falsely predict a single SP site (this lone false postive, however, was not within 100 amino acids of the D-site). From these examples it can be concluded that the JNK PWMs we constructed are both sensitive and specific.
Fig. 1.
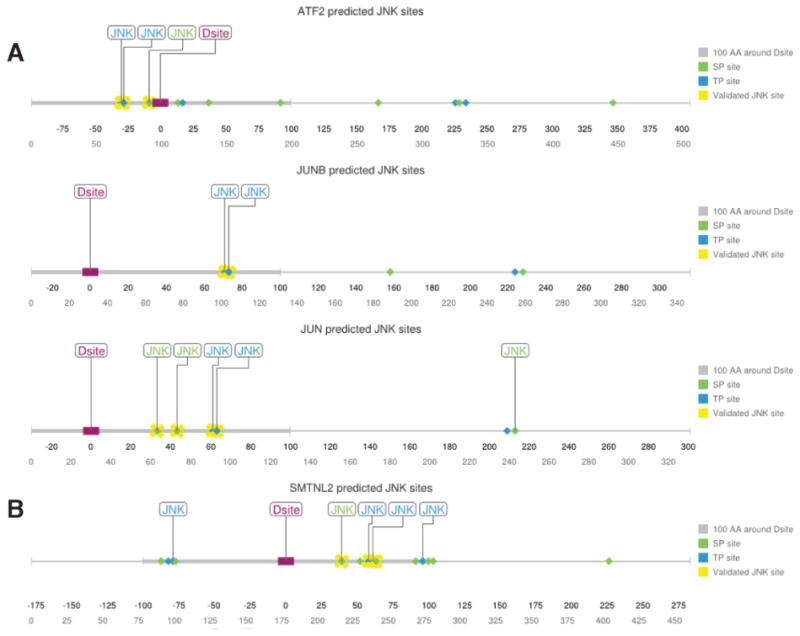
Prediction of JNK target phosphoacceptor sites close to a JNK-docking site.
A) Linear representation of known JNK targets ATF2, JUNB, and JUN with all potential ST/TPphosphosites shown. Those sites predicted by our PWM method to be likely JNK targets arelabeled as ‘JNK’. Literature-validated phosphosites are indicated with yellow crosses. The D-finder-predicted D-site is shown as a purple rectangle; the 100 amino acid window to either sideof the D-site is indicated by a thickened gray line. B) Linear representation of full-lengthSMTNL2 protein and all potential ST/TP phosphosites. Labeling as above. Phosphositesidentified in the mass spectrometry/mutagenesis experiments described further below areindicated with yellow crosses
When this PWM approach was used to scan the 308 putative substrates listed above, 61 were found to contain at least one putative JNK phosphosite proximal to the D-site, with 33 containing two or more such JNK sites (Table S1).
3.2 SMTNL2 is a predicted MAPK substrate
The human SMTNL2 protein was found to contain 11 minimal putative MAPK phosphoacceptor sites (S/T-P) within 100 amino acids of the D-site; furthermore, 5 of these were predicted to be likely JNK phosphosites by the JNK PWM approach (Fig. 1B).
SMTNL2 (Fig. 2) is a broadly conserved and poorly characterized 460 amino acid protein named for its similarity to the smooth muscle protein Smoothelin. Like Smoothelin, SMTNL2 contains a single calponin homology domain. The predicted D-site resides within amino acids 180-193, whereas the calponin homology domain resides near the C-terminus (Fig 2A). Moreover, the predicted D-site of SMTNL2 was high scoring, surface accessible and strongly conserved (Fig. 2B, Table S1, data not shown). Indeed, within the mammalian lineage, substitutions in residues known to be critical for D-site function have been largely conservative (e.g. K for R in the basic submotif, and F for L in the hydrophobic submotif; Fig. 2C); this suggests that there has been evolutionary pressure to preserve D-site functionality.
Fig. 2.

SMTNL2 protein and its predicted D-site. A) Diagram of human SMTNL2 protein showing the predicted D-site (amino acids 180-193) and the calponin homology (CH) domain (351-455). B) SMTNL2's D-site sequence aligned with known D-sites in selected JNK substrates (JUN, JUNB, ATF2, ELK1, ELK4) and in the upstream kinase MKK4. C) Conservation of the SMTNL2 JNK-docking site in other mammalian species. Residues different from the human sequence are italicized.
The human protein atlas and GEO database [26, 27] report that SMTNL2 is most abundantly expressed in skeletal muscle, but is also expressed in many other tissues including brain, liver, lung, and kidney. Recently, the first putative function for SMTNL2 emerged in a screen for regulators of epithelial cell architecture and morphogenesis in 3D culture [28]. In this same study, many of these genes, including SMTNL2, were shown to be downregulated in kidney cancer and breast tumors compared to the corresponding normal tissue [28].
Because of the evidence connecting both muscle physiology and cell polarity to MAPK signaling (see Discussion for further details), we pursued the further characterization of SMTNL2 as a putative MAPK-binding partner and substrate.
3.3 SMTNL2 protein binds specifically to multiple MAPKs
D-site-containing substrates bind to their cognate MAPKs with relatively high affinity, yielding dissociation constants (Kd's) typically in the range of 50-500 μM [9]. Interactions of this affinity can typically be detected in a Glutathione S-Transferase (GST) co-sedimentation (“pull-down”) assay. Therefore, we used this assay to determine if SMTNL2 could bind to any of the JNK-family MAP kinases (Fig. 3A).
Fig. 3.
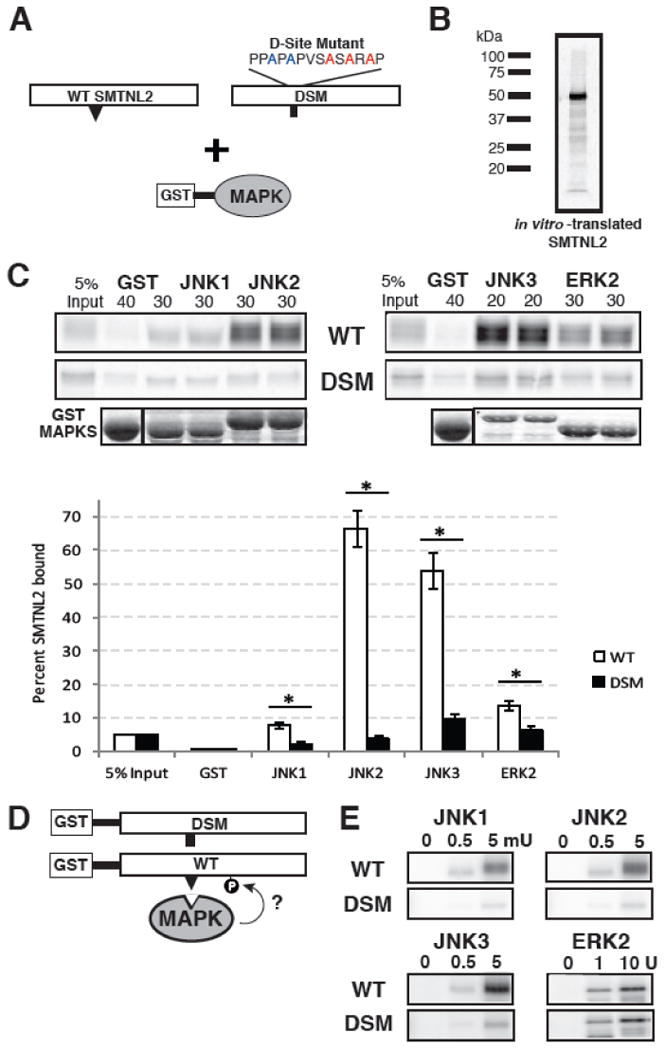
SMTNL2 is a novel substrate with a functional D-site. A) Full-length wild-type (WT) SMTNL2, or a D-site mutant (DSM) derivative, were tested for binding to GST-MAPK proteins. B) Representative in vitro transcription and translation product of full length SMTNL2. C) As shown in A, 35S-radiolabeled full-length SMTNL2 protein and a D-site mutant derivative were prepared by in vitro translation and partially purified by ammonium sulfate precipitation, and portions (5% of the amount added in the binding reactions) were resolved on a 10% SDS-polyacrylamide gel (lane 1 of each column of panels). Samples (∼1 pmol) of the same proteins were incubated with 40 μg of GST (lane 2 of each column) or with 30 μg of GST-JNK1, JNK2, JNK3 or ERK2 proteins (lanes 3-6 of each column, duplicate points shown), bound to glutathione-Sepharose beads, and the resulting bead-bound protein complexes were isolated by sedimentation, and resolved by 10% SDS-PAGE on the same gel. The gel was analyzed by staining with Coomassie Blue (CB) for visualization of the bound GST fusion protein (lowest row) and by Phosphorimager analysis for visualization of the bound radiolabeled protein (upper two rows). Graph of the results of multiple independent repetitions of the binding assay shown in A and B, with duplicate points in each repetition. Standard error bars are shown (n = 6 for WT, 3 for DSM). D) Wild-type (WT) and D-site-mutant (DSM) versions of SMTNL2 were tested as substrates for in vitro phosphorylation by active MAPKs. E) As shown in D, 0.25 μM of each GST-SMTNL2 protein was incubated with enzyme (0, 0.5 or 5 milliunits JNK1-3 or 0, 1, or 10 units ERK2) and [γ-32]ATP for 20 min. Samples were resolved on a 10% SDS-polyacrylamide gel and visualized by autoradiography.
First, to produce a radiolabeled version of SMTNL2 suitable for biochemical binding studies, full length SMTNL2 cDNA was obtained and subcloned into a vector (pGEM4Z-Stop) appropriate for generation of [35S]-labeled protein by in vitro translation. In vitro translation of full-length SMTNL2 resulted in the production of a polypeptide of apparent molecular weight ∼50 kDa (Fig. 3B) consistent with the predicted molecular weight of 50.2 kDa.
Next, Human JNK1, JNK2, JNK3 were fused at their N-termini to Schistosoma japonicum GST, and the resulting fusion proteins were expressed in bacteria and purified by adsorption to glutathione-sepharose beads. MAPKs prepared in this manner are obtained in their unphosphorylated, inactive states. The bead-bound GST-MAPKs (or GST-alone as a negative control) were then incubated with [35S]-labeled full-length human SMTNL2 protein. Bead-bound complexes were collected by sedimentation, washed extensively, and analyzed by SDS-PAGE and subsequent autoradiography. As shown in Fig. 3C (top panel labeled ‘WT’), full-length SMTNL2 bound efficiently to all three JNK proteins; moreover, this binding was specific, because only trace precipitation of SMTNL2 occurred when GST was used instead of the GST-JNK fusion proteins. Quantification of these data by phosphorimager analysis revealed that 8% of the SMTNL2 that was input into the assay bound to JNK1, whereas 65% and 50% of the input SMTNL2 protein bound JNK2 and JNK3, respectively (Fig. 3C). The GST moiety by itself pulled down less than 0.1% of the input SMTNL2.
As explained above, D-finder's predictions are expected to be enriched for JNK substrates relative to substrates for other MAPK families. Nevertheless, JNK, ERK and p38 D-sites all share a core consensus, and many D-sites have been shown to bind to members of more than one MAPK family. Moreover, ERK2 has been shown exhibit somewhat promiscuous D-site binding [17]. Thus, we also asked if SMTNL2 could bind to ERK2. Indeed, radiolabeled SMTNL2 protein did bind to purified GST-ERK2 (18% of input, data not shown).
Based on the quantitative binding data presented above, we calculated approximate dissociation constants (Kd's) of 50 μM, 2 μM, and 4 μM for the interaction of SMTNL2 with JNK1, JNK2 and JNK3, respectively. The SMTNL2-ERK2 interaction had a Kd of ∼20 μM. These Kd's are in a range that is typical of D-site promoted interactions [9].
3.4 The D-site in SMTNL2 facilitates MAPK binding
To determine if the predicted D-site contributed to SMTNL2's ability to bind with high affinity to multiple MAPK proteins, we mutated the consensus basic and hydrophobic residues in the predicted D-site of SMTNL2 to alanine via multistep site-directed mutagenesis (Fig. 3A). The resulting mutant protein was produced by in vitro transcription/translation and tested for binding to the panel of GST-MAPK proteins. Compared to wild-type SMTNL2 protein, D-site mutant (DSM) SMTNL2 exhibited substantially reduced binding to JNK1 (∼5-fold reduction), JNK2 (∼40-fold) and JNK3 (∼40-fold) (Fig. 3C). In contrast, binding of the SMTNL2 mutant to ERK2 was reduced by only 2-fold. These results demonstrate that the predicted D-site in SMTNL2 protein is indeed a fully functional, bone fide D-site, and that it is required for the high-affinity binding of SMTNL2 protein to JNK1, JNK2 and JNK3. The D-site also makes a modest contribution to the binding efficiency of the ERK-SMTNL2 interaction.
3.5 SMTNL2 is an in vitro, D-site-directed substrate of JNK
To facilitate further biochemical studies, we subcloned the full-length wild-type (WT) and D-site mutant (DSM) SMTNL2 coding sequences into a vector (pGEX-LB) suitable for generating soluble, GST-fused versions of the encoded proteins. Both the GST-SMTNL2 and the GST-SMTNL2-DSM fusion proteins were efficiently expressed and mostly soluble in E. coli, and could be produced at a reasonably high yield (∼50 μg obtained from a 100 ml culture, data not shown). The proteins were purified by glutathione affinity chromatography, eluted from the matrix with reduced glutathione, and then dialyzed to remove the glutathione, which can inhibit downstream assays.
In order to ascertain if SMTNL2 was an in vitro substrate of one or more MAPKs, the purified GST-SMTNL2 WT and DSM proteins were incubated with purified activated MAPK proteins and radiolabeled ATP in a standard in vitro kinase assay (Fig. 3D). In three independent experiments, each MAPK enzyme was tested at two different concentrations for its ability to phosphorylate wild-type or D-site-mutated SMTNL2; a no kinase control was also included. As shown in Fig. 3E, SMTNL2 was phosphorylated efficiently by JNK1, JNK2, JNK3, and ERK2. Furthermore, JNK-mediated phosphorylation of SMTNL2-DSM by all three JNK proteins was significantly reduced as compared to wild-type SMTNL2, thus indicating that the D-site in SMTNL2 is essential for efficient JNK-mediated phosphorylation.
Consistent with the minimal impact the D-site mutation had on ERK2 binding, the DSM protein was phosphorylated by ERK2 at roughly the same level as was the wild-type.
In summary, SMTNL2 is an in vitro substrate and binding partner of JNK-family and ERK-family MAPKs, and the identified D-site plays a major role in directing JNK-mediated binding and phosphorylation of SMTNL2.
3.6 The SMTNL2 D-site inhibits JNK-mediated phosphorylation of ATF2
The relative strength of a D-site-mediated interaction can be determined in a peptide competition assay. The peptide competition assay measures the specific binding of D-sites to their cognate MAPKs by their ability to inhibit phosphorylation of another D-site-containing substrate (Fig. 4A) [17]. Previous work determined that a D-site peptide derived from MKK4 strongly inhibited JNK phosphorylation of ATF2, while a mutant peptide (MKK4 EAG) containing substitutions at critical basic and hydrophobic residues of the D-site consensus did not [11, 12, 17]. These MKK4 peptides, shown in Fig. 4B, were used as positive and negative controls for the competition assay. As shown in Fig. 4C, the SMTNL2 D-site peptide was an effective inhibitor of JNK1 phosphorylation of ATF2. This inhibition was comparable to the MKK4 WT positive control peptide at four separate peptide concentrations, whereas the MKK4 EAG negative control peptide showed no inhibition of phosphorylation. This assay was repeated for JNK2 (Fig. 4D) and JNK3 (Fig. 4E) and gave similar results.
Fig. 4.
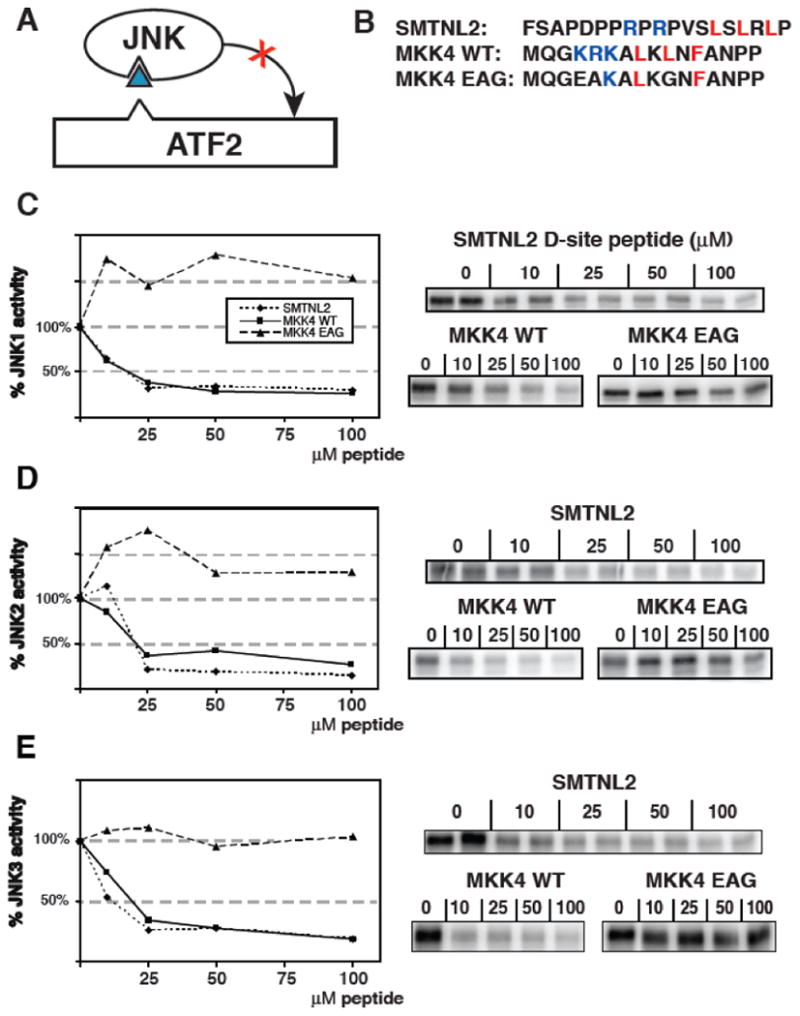
Inhibition of JNK phosphorylation of ATF2 by an SMTNL2 D-site peptide. A) D-site peptides (triangle) were used to inhibit JNK-mediated phosphorylation of the ATF transcription factor. B) Sequence of the D-site peptides used. C-E) Purified GST-ATF2 (0.5 μM) was incubated with 2.5 nM purified active JNK1 and [γ-32P] ATP for 30 min in the absence or presence of the specified concentrations of the indicated peptides. Phosphate incorporation into ATF2 was analyzed by SDS-PAGE and quantified on a phosphorimager. Results are plotted as a percent phosphorylation relative to that observed in the absence of any added peptide. Data are the average of three experiments, with duplicate points in each experiment. An autoradiogram of a representative experiment is shown on the right. D) As in C, except that the enzyme was purified active JNK2 (2.5 nM) (E) As in C, except that the enzyme was purified active JNK3 (2.5 nM).
Based on these data, we calculated the IC50 (the concentration of peptide that gives 50% inhibition) of the SMTNL2 D-site for JNK1, JNK2, and JNK3 as being in the range of 10-20 μM; this is broadly consistent with the dissociation constants (2-50 μM) calculated from the GST pull-down assays in Fig. 3.
In summary, the SMTNL2 D-site peptide binds to JNKs, and it competes with D-site-containing transcription factors such as ATF2 for JNK binding.
3.7 Identification of JNK phosphorylation sites on SMTNL2 by Mass Spectrometry
To identify the target phosphoacceptor sites in SMTNL2 that were phosphorylated by JNK, in vitro kinase assays were carried out to generate three different samples, consisting of wild-type SMTNL2 treated with and without active JNK2, and D-site mutant (DSM) SMTNL2 treated with active JNK2. Following these phosphorylation reactions, the samples were separated by SDS-PAGE, in-gel digested by either trypsin or chymotrypsin, and analyzed by mass spectrometry (LC MS/MS). Shown in Fig. 5A is a representative mass spectrum of one of the phosphopeptides identified by this analysis (m/z 830.86+2, SGETSAAALpSPMSAATL). This phosphopeptide correlates to phosphorylation of S217 on the full-length SMTNL2 protein. Specific phosphorylation of WT- versus DSM-SMTNL2 protein was confirmed by comparing the extracted ion chromatograms of the phosphorylated peptide (m/z 830.86) from the three different treatments (Fig. 5B). As indicated, the S217 phosphopeptide is only found in the WT plus active JNK2 sample, and not in the DSM plus JNK2 sample, demonstrating that a functional D-site is required for efficient phosphorylation of S217. Three additional D-site-dependent phosphosites were also identified in this manner: S230, T236, and T239 (Fig. 3E and Table 1)
Fig. 5.
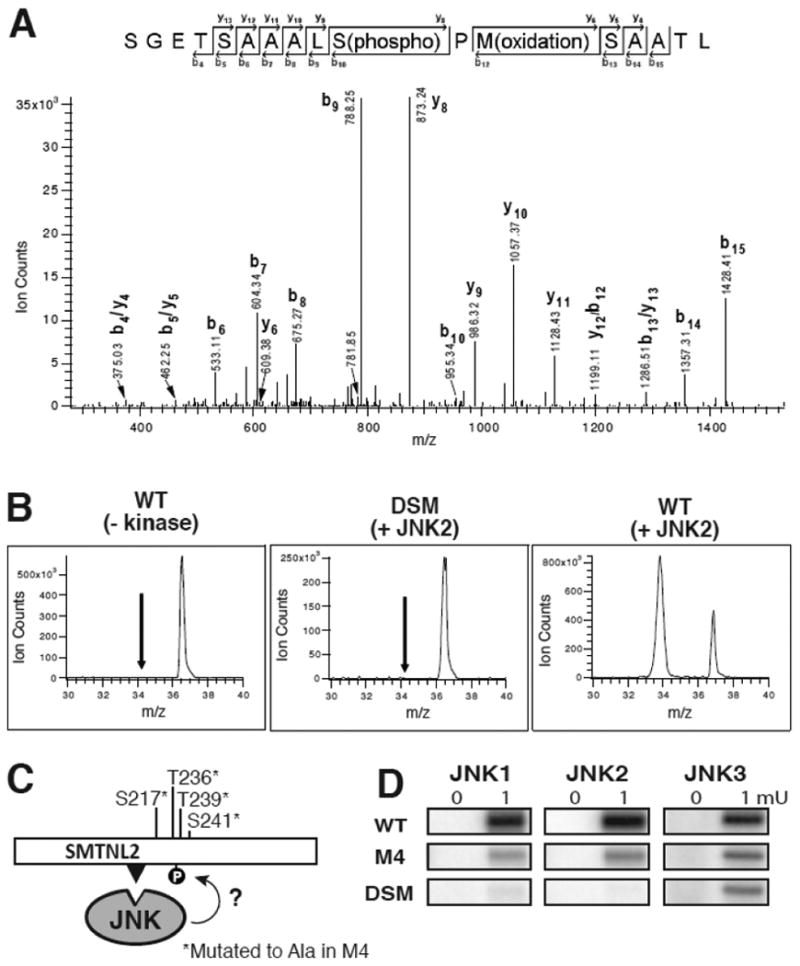
Tandem Mass spectrometry analysis of D-site-directed phosphorylation sites in SMTNL2. A and B) Representative MS analysis of an identified phosphorylated peptide (m/z 830.87, SGETSAAALS(phosphor)PM(oxidation)SAATL). Three samples were analyzed: WT with no kinase, WT with kinase, and DSM with kinase. A) MS/MS spectra of the identified peptide in the WT plus active JNK2 sample. B) Extracted ion chromatograms (XIC) of the parent ion in the three samples during LC MS runs. C) The 4 phosphoacceptor sites identified by mass spectrometry were changed to alanine using site-directed mutagenesis. This protein (termed M4) was fused to GST for use as a substrate in an in vitro kinase assay. The D-site is shown as a black triangle. D) Results of an in vitro kinase assay using 0.5 μM SMTNL2 WT, M4, and DSM as substrate and 0.1 or 1 mU JNK1-3 as the kinase.
Table 1. Jnk2 and Jnk3 in Vitro Phosphorylation sites identified by Tandem Mass Spectrometry in SMTNL2.
| m/z | Z | ppm | Peptide | Kinase |
|---|---|---|---|---|
| 767.3583 | 2 | 7.1 | GGLNPSPS(Phospho)EVITPW | Jnk3 |
| 899.0865 | 3 | 6 | GGLNPSPSEVIT(Phospho)PWTPSPSEKNSSF | Jnk2/Jnk3 |
| 1348.1265 | 2 | 6.3 | GGLNPSPSEVITPWT(Phospho)PSPSEKNSSF | Jnk2/Jnk3 |
| 899.0895 | 3 | 9.4 | GGLNPSPSEVITPWTPSPSEKNS(Phospho)SF | Jnk2/Jnk3 |
| 830.8671 | 2 | 8 | SGETSAAALS(Phospho)PM(Oxidation)SAATL | Jnk2/Jnk3 |
| 822.868 | 2 | 6.1 | SGETSAAALS(Phospho)PMSAATL | Jnk2 |
| 630.7652 | 2 | 6.9 | T(Phospho)PSPSEKNSSF | Jnk2/Jnk3 |
| 630.7664 | 2 | 8.8 | TPS(Phospho)PSEKNSSF | Jnk2 |
| 774.328 | 2 | 4.7 | TPS(Phsospho)PSEKNSSFTW | Jnk2/Jnk3 |
To confirm the phosphosites identified by mass spectrometry, we mutated all four of these phosphosites to alanine. This protein, designated SMTNL2-M4 (Fig. 5C), was compared with the WT and DSM alleles as a substrate in an in vitro kinase assay using JNK1, JNK2 or JNK3 as the enzyme (Fig. 5D). For all three JNK proteins, phosphorylation of the M4 mutant was significantly diminished compared to the wild type; indeed, phosphorylation of the M4 mutant was reduced down to the low levels seen with the D-site mutant. These results indicate that the four sites identified by mass spectrometry are targets of the majority of JNK-mediated phosphorylation observed.
As noted above, the JNK-specific position weight matrixes that we constructed predicted 5 of the 14 SP/ TP sites within 100 amino acids of the D-site as being putative high-efficiency JNK target sites. Of these 5 sites, 3 were validated by mass spectrometry (S217, T236 and T239), and two were not (T99 and T274). In addition, the mass spectrometry identified one site (S217) that was not predicted by the JNK PWMs. Thus, the PWM approach as applied to SMTNL2 identified 3 true positives, two false positives, 8 true negatives, and 1 false negative.
3.8 SMTNL2 is an in vivo JNK substrate
To determine if SMTNL2 was phosphorylated by JNK in mammalian cells, we constructed V5-epitope tagged versions of the WT, DSM, and M4 SMTNL2 proteins, and transiently transfected them into Human Embryonic Kidney (HEK) 293 cells, which are known to express JNK1 and JNK2. Twenty-four hours post transfection; the cells were stimulated for 30 minutes with the protein synthesis inhibitor anisomycin. Anisomycin treatment is known to trigger a ribotoxic stress signal that leads to activation of the JNK pathway [29]. Following treatment, the cells were harvested, lysed, and analyzed by immunoblotting, using an antibody specific to the V5 epitope to detect the SMTNL2 protein (Fig. 6A) and a phospho-JNK specific antibody to verify JNK activation by anisomycin (Fig. 6C).
Fig. 6.
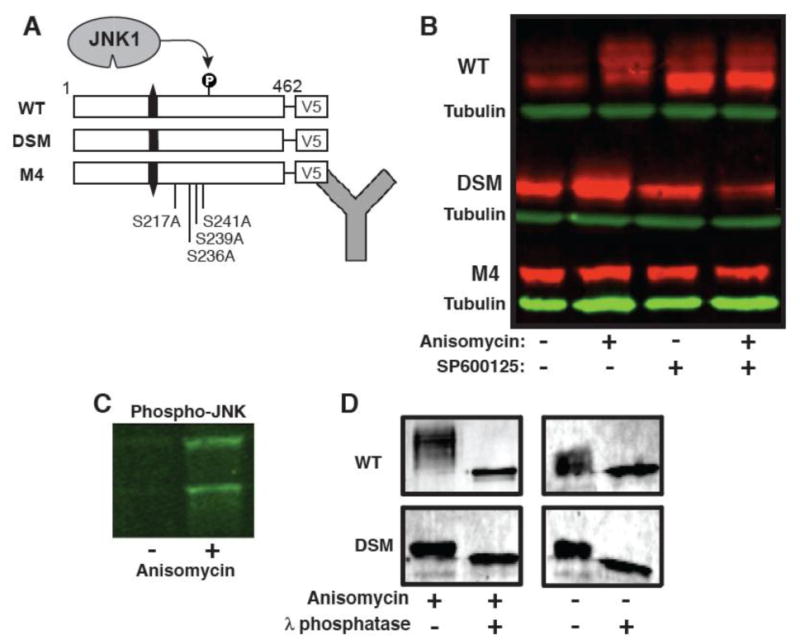
SMTNL2 is an in vivo JNK substrate. A) Wild-type and mutant versions of SMTNL2 were C-terminally-tagged with the V5 epitope and expressed in HEK-293 cells. Then, 24 hours post-transfection, the cells were treated with anisomycin to activate the JNK pathway and/or the JNK inhibitor SP600125. Following this treatment, whole cell protein extracts were prepared and SMTLN2 was visualized by Immunoblotting. B). Anti-V5 immunoblot of whole cell extracts prepared as described in A. C) Anti-phospho-JNK immunoblot of whole cell extracts prepared as described in A. D) WT and DSM-V5 SMTNL2 were immunoprecipitated from 293 cells with or without anisomycin, washed in 1× phosphatase buffer, and then treated lambda protein phosphatase (New England Biolabs) according to the supplied protocol.
As shown in Fig. 6B, in cells stimulated with anisomycin, there was a notable electrophoretic mobility shift of wild-type SMTNL2 protein, resulting in a series of bands of slower mobility (Fig. 6B, WT, compare lanes 1 and 2). Such a mobility shift is typical of phosphoproteins; indeed, as shown further below, treatment of this sample with lambda phosphatase confirmed that the shift was attributable to phosphorylation. This phosphorylation was dependent on activation of endogenous JNK, because pre-treatment of the cells with the JNK inhibitor SP600125 blocked the observed phospho-shift (compare lane 4 with lane 2). Although it is possible that the observed phosphorylation was catalyzed by a kinase downstream of JNK and not directly by JNK1/2, additional experiments with the DSM and M4 mutants described immediately below make this possibility very unlikely.
Further evidence that SMTNL2 is phosphorylated by JNK in vivo (and that this phosphorylation is responsible for the observed mobility shift) came from the behavior of the D-site mutant protein. In contrast to the wild type, the magnitude of the phospho-shift was considerably less with this mutant (Fig. 6B, DSM), strongly suggesting that there was reduced JNK-mediated phosphorylation of this mutant protein in vivo, just as was observed in vitro (Fig. 3). In addition, we assessed the anisomycin-stimulated phosphorylation of the SMTNL2 M4 mutant, which is lacking the 4 phosphosites (S217, S230, T236, and T239) that we showed are phosphorylated by JNK in vitro. Like the D-site mutant, the M4 mutant exhibited little or no detectable in vivo phospho-shift (Fig. 6B, M4), strongly suggesting that the phosphorylation that caused the phospho-shift in cells occurred on one or more of the residues identified by mass spectrometry of in vitro-phosphorylated SMTNL2.
To confirm that the observed electrophoretic mobility shift represented phosphorylated forms of SMTNL2, we immunoprecipitated SMTNL2 from stimulated HEK293 cells, split the immunoprecipitated pellets in two, and treated them either with or without lambda protein phosphatase for 30 minutes. Phosphatase treatment caused the shifted bands to collapse, confirming that phosphorylation was responsible for the observed mobility shift (Fig. 6D).
Taken together, the results obtained with wild-type SMTNL2 and with the D-site and M4 mutants strongly suggest that, in cells treated with the JNK-activating stimulus anisomycin, SMTNL2 protein became phosphorylated on S217, S230, T236, and/or T239 by endogenous JNK1 and JNK2. Thus we conclude that SMTNL2 is an in vivo JNK substrate.
3.9 SMTNL2 is highly expressed in skeletal muscle and is induced during differentiation of C2C12 myoblasts
In order to gain insight into possible functional roles of SMTNL2, we performed qRT-PCR to detect tissue expression in the mouse. We observed the highest expression in skeletal muscle, followed closely by heart (Fig. 7A). We also found significant mouse Smtnl2 mRNA expression in kidney, brain, skin, bladder, intestine, lung, stomach, and liver, along with low, but reproducibly detectable, expression in spleen (Fig. 7A). The mouse mRNA data agrees with human protein data obtained from the Human Protein Atlas [26], which shows strong expression of SMTNL2 in many human tissues, including muscle (Fig. 7A).
Fig. 7.
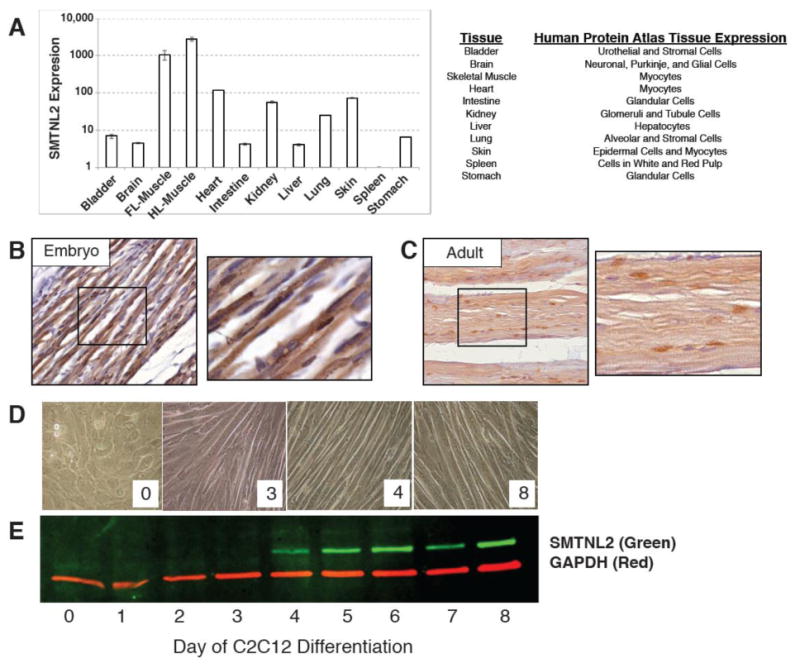
SMTNL2 is ubiquitously expressed in mouse and human tissue and is upregulated during myogenic differentiation. A) Smtnl2 mRNA expression from adult mouse tissues, as measured by qPCR, and matching human tissue expression from the Human Protein Atlas [26]. B) Immunohistochemistry of Smtnl2 protein in embryonic day 18.5 mouse skeletal muscle. C) Immunohistochemistry of Smtnl2 protein in adult 8-week mouse skeletal muscle. D) Images of C2C12 myogenic differentiation days –2 through 8, where day 0 corresponds to the initiation of differentiation by serum withdrawal E) Immunoblot of native Smtnl2 protein expression during myogenic differentiation in mouse C2C12 cells with representative images of the cells at Day 0, 3, 4, and 8 after initiation of differentiation.
In order to confirm the mRNA expression in mouse skeletal muscle, we performed immunohistochemistry on embryonic day 18.5, and also on eight week-old adult tissues, using a polyclonal anti-Smtnl2 antibody. As can be seen in Fig. 7B-C, Smtnl2 protein was localized to both the cytoplasm and nucleus at both ages. In order to gain a better understanding of SMTNL2 in muscle, we searched previously published microarray datasets utilizing the Gene Expression Omnibus (GEO) [27], and found that during differentiation of both primary hind limb skeletal muscle, and extra ocular muscle, SMTNL2 mRNA becomes highly upregulated [30]. Thus, SMTNL2 is a ubiquitously expressed protein in mammalian tissues, with very high expression in adult skeletal muscle.
To determine if SMTNL2 protein expression changes during myogenic differentiation, we used the well-characterized mouse myoblast cell line C2C12, which can be induced to differentiate from myoblasts to fused myotubes by changing the media to 2% horse serum [31]. As shown in Fig. 7E, SMTNL2 protein was not detectably expressed in growing C2C12 myoblasts, as determined by immunoblot analysis using an anti-SMTNL2 antibody. However, upon induction of differentiation with 2% horse serum, mouse SMTNL2 protein was induced, becoming highly expressed by day four, and remaining high throughout the remainder of the time course (Fig. 7E). Day 3 to 4 of C2C12 differentiation coincides with the fusion of myoblasts into myotubes and the start of myotube maturation (Fig. 7D); thus, increased Smtnl2 protein expression in C2C12 cells is associated with the stages of myotube formation and maturation.
4. Discussion
A common mechanism of interaction between MAP kinases and their substrates and binding partners involves the tethering of the MAPK to one or more docking sites on the binding partner [8]. In the case of MAPK substrates, after the MAPK docks to the substrate in question, it phosphorylates the substrate on one or more nearby serine or threonine residues. Here, with the goal of identifying new JNK substrates, we started with a list of human proteins that contained predicted JNK docking sites of the D-site class [10], and then used the presence of nearby consensus phosphosites as a second criterion to identify promising candidate proteins. We found a predicted D-site in the functionally uncharacterized protein Smoothelin-like 2 (SMTNL2), with multiple putative JNK phosphoacceptor sites nearby, and then verified SMTNL2 as JNK substrate both in vitro and in cells.
4.1. Combining docking site and phosphosite predictions to find new substrates
We have previously investigated the utility of identifying new MAP kinase substrates by searching protein sequence databases for the presence of MAPK-docking sites of the D-site class [10]. The goal of the present study was to investigate the utility of adding a second dimension to this analysis, the presence of likely MAPK phosphosites nearby the predicted D-site. Starting with a list of 394 human proteins predicted to have putative JNK D-sites (Table S1), we compared two approaches for predicting MAPK phosphosites. The first approach was to score any SP or TP dyad as a likely MAPK phosphosite. This approach appeared to be too broad, in that almost 80% of the proteins in the list had at least one S/T-P dyad within 100 residues of the predicted D-site, with almost 60% having two or more. Thus, we turned to an approach based on probability weight matrixes constructed from known JNK phosphorylation sites, with p-value cut offs carefully chosen to optimize the sensitivity and specificity of the predictions when applied to known JNK substrates. This approach prioritized a much smaller number of targets, with less than 10% of the list having two or more putative JNK phosphosites proximal to the D-site. We then showed that one of these candidates so prioritized, SMTNL2, was indeed a bone fide in vitro and in vivo substrate of JNK. Although our experimental verification of this approach is so far limited to a single candidate protein, our preliminary conclusion is that combining docking site and phosphosite predictions may facilitate the identification of novel substrates.
4.2. SMTNL2 is a JNK substrate in vitro and in cells
SMTNL2 protein bound to both ERK and JNK, and JNK binding was largely dependent on the integrity of the predicted D-site (Fig. 3). SMTNL2 protein was phosphorylated by ERK and JNK in vitro, (Fig. 3), and in the case of JNK, this phosphorylation was highly dependent upon the integrity of the predicted D-site. A peptide derived from the SMTNL2 D-site blocked JNK-mediated phosphorylation of the ATF2 transcription factor (Fig. 4), suggesting that SMTNL2 may compete with transcription factors and other substrates for binding to JNK (Fig. 4). We identified four JNK-dependent phosphoacceptor sites in SMTNL2 by mass spectrometry, and validated this result with site directed mutagenesis and in vitro kinase assays (Fig. 5). Finally, in cell-based assays, SMTNL2 was phosphorylated by JNK in vivo, and this phosphorylation required the D-site and occurred on the same residues that we identified in vitro (Fig. 6). We conclude that SMTNL2 is an authentic and novel JNK substrate that contains a functional D-site.
4.3. Functional characterization of SMTNL2
Little is known about SMTNL2 protein function, and there is no knockout mouse model of SMTNL2, so it is difficult at this time to explore the effect of JNK-mediated phosphorylation on SMTNL2 function. We were able to detect SMTNL2 mRNA and protein in multiple tissues including very high expression in mouse skeletal muscle by qPCR and immunohistochemistry (Fig. 7). To further explore the potential role of SMTNL2 in muscle, we examined the expression of SMTNL2 protein in differentiating C2C12 cells, which are a classic tissue culture model system to study myoblast differentiation. We found that SMTNL2 protein was strongly induced as C2C12 myocytes differentiated into myoblasts and formed myotubes (Fig. 7). Consistent with this, we found multiple conserved MyoD binding sites in the mouse and human SMTNL2 promoters (data not shown). Since SMTNL2 was not detectable in precursor myoblasts, its role in muscle is likely limited to differentiating or differentiated cells. We conclude that SMTNL2 protein may have an important function in skeletal muscle. That said, it is clear that SMTNL2 is expressed in other tissues and cells types was well, including cardiac muscle and non-muscle tissue (Fig. 7); furthermore, there is evidence that SMTNL2 has an important function in certain epithelial cells [28].
4.4. Additional clues about SMTNL2 function in muscle
The expression of SMTNL2 that we found in mouse tissues and in differentiating C2C12cells is consistent with the strong expression of human SMTNL2 protein observed in humanmuscle reported in the Human Protein Atlas [32]. Additionally, published high-throughput geneexpression studies, such as those found in the GEO database, can be mined for information onthe transcript's expression and change in experimental systems. In one study, SMTNL2 expression was shown to increase over time during differentiation of primary cell lines derived from extraocular muscle (EOM) and gastrocnemius skeletal muscle [30] consistent with our findings in C2C12 cells.
Additional exploration of GEO identified multiple studies with further information on SMTNL2 and its potential relationship to muscle. A dataset comparing normal human and Duchene muscular dystrophy skeletal muscle biopsies showed an average 50% reduction in SMTNL2 expression in the disease tissues [33]. Similarly, in a microarray study measuring gene expression of vastus lateralis (quadriceps) muscle 48 hours after knee immobilization, high initial expression of SMTNL2 was significantly reduced [34]. Furthermore, SMTNL2 expression in muscle was shown to be a predictive biomarker for response to aerobic exercise [35].
Interestingly, an extensive body of work implicates JNK signaling in muscle biology and pathology. JNK kinase activity is upregulated in skeletal muscle during contraction and in response to insulin and phorbol esters [36], during oxidative stress [37], and during C2C12 differentiation [38]. Increased activation of JNK1 has been found in mouse models of Duchenne Muscular Dystrophy (DMD) [39, 40]; Furthermore, when JNK1 was activated specifically in muscle, myotube viability and integrity became defective, similar to a dystrophic phenotype [41]. Finally, JNK1 in muscle has been shown to contribute to peripheral insulin resistance in response to diet-induced obesity [42]. The effect of JNK-mediated phosphorylation on SMTNL2 protein, and the consequences for the biology of muscle and other tissues in which SMTNL2 is expressed, is an intriguing question for future studies.
In conclusion, we used the combination of a predicted JNK-docking site and nearby predicted phosphosites to identify a new JNK substrate, SMTNL2, which we verified to be an authentic in vitro and in vivo substrate of the JNK family of MAPKs.
Supplementary Material
Highlights.
We searched the human genome for new MAP kinase substrates.
Our approach combined docking site and phosphosite predictions.
Smoothelin-like 2 (SMTNL2) is a novel JNK substrate.
SMTNL2 is phosphorylated by JNK in vitro and in cells.
SMTNL2 is highly expressed in skeletal muscle and several other tissues.
SMTNL2 expression is induced during myoblast differentiation.
Acknowledgments
This work was supported by NIH National Institute of General Medical Sciences research grants P50 GM76516 and R01 GM86883 (L.B.), as well as by R01 GM74830-06A1 (L.H.). E.A.G. was supported in part by the Center for Regenerative Medicine training grant TG2-01152. W.M.G. was supported in part by Systems Biology of Development training grant T32-HD60555. The work of E.A.G., R.M.K., V.P., T.C.W., M.Z. and P.B. was supported in part by grants National Science Foundation IIS-0513376, NIH LM010235, and NIH National Library of Medicine T15 LM07443 to P.B.. This work was also supported by National Cancer Institute Award Number P30CA062203. We thank Dr. Larry McReynolds and Dr. Bruce Gordon for commentary on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Avruch J. MAP kinase pathways: the first twenty years. Biochim Biophys Acta. 2007;1773:1150. doi: 10.1016/j.bbamcr.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 3.Bogoyevitch MA, Ngoei KR, Zhao TT, Yeap YY, Ng DC. c-Jun N-terminal kinase (JNK) signaling: Recent advances and challenges. Biochim Biophys Acta. 2010;1804:463. doi: 10.1016/j.bbapap.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Johnson GL, Nakamura K. The c-jun kinase/stress-activated pathway: regulation, function and role in human disease. Biochim Biophys Acta. 2007;1773:1341. doi: 10.1016/j.bbamcr.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antoniou X, Borsello T. The JNK signalling transduction pathway in the brain. Front Biosci (Elite Ed) 2012;4:2110. doi: 10.2741/e528. [DOI] [PubMed] [Google Scholar]
- 6.Sabio G, Davis RJ. cJun NH2-terminal kinase 1 (JNK1): roles in metabolic regulation of insulin resistance. Trends Biochem Sci. 2010;35:490. doi: 10.1016/j.tibs.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cellurale C, Sabio G, Kennedy NJ, Das M, Barlow M, Sandy P, Jacks T, Davis RJ. Requirement of c-Jun NH(2)-terminal kinase for Ras-initiated tumor formation. Mol Cell Biol. 2011;31:1565. doi: 10.1128/MCB.01122-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bardwell L. Mechanisms of MAPK signalling specificity. Biochem Soc Trans. 2006;34:837. doi: 10.1042/BST0340837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bardwell L, Shah K. Analysis of mitogen-activated protein kinase activation and interactions with regulators and substrates. Methods. 2006;40:213. doi: 10.1016/j.ymeth.2006.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whisenant TC, Ho DT, Benz RW, Rogers JS, Kaake RM, Gordon EA, Huang L, Baldi P, Bardwell L. Computational prediction and experimental verification of new MAP kinase docking sites and substrates including Gli transcription factors. PLoS Comput Biol. 2010;6 doi: 10.1371/journal.pcbi.1000908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ho DT, Bardwell AJ, Abdollahi M, Bardwell L. A docking site in MKK4 mediates high affinity binding to JNK MAPKs and competes with similar docking sites in JNK substrates. J Biol Chem. 2003;278:32662. doi: 10.1074/jbc.M304229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ho DT, Bardwell AJ, Grewal S, Iverson C, Bardwell L. Interacting JNK-docking sites in MKK7 promote binding and activation of JNK mitogen-activated protein kinases. J Biol Chem. 2006;281:13169. doi: 10.1074/jbc.M601010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grewal S, Molina DM, Bardwell L. Mitogen-activated protein kinase (MAPK)-docking sites in MAPK kinases function as tethers that are crucial for MAPK regulation in vivo. Cell Signal. 2006;18:123. doi: 10.1016/j.cellsig.2005.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Enslen H, Davis RJ. Regulation of MAP kinases by docking domains. Biol Cell. 2001;93:5. doi: 10.1016/s0248-4900(01)01156-x. [DOI] [PubMed] [Google Scholar]
- 15.Remenyi A, Good MC, Lim WA. Docking interactions in protein kinase and phosphatase networks. Curr Opin Struct Biol. 2006;16:676. doi: 10.1016/j.sbi.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 16.Bardwell AJ, Abdollahi M, Bardwell L. Docking sites on mitogen-activated protein kinase (MAPK) kinases, MAPK phosphatases and the Elk-1 transcription factor compete for MAPK binding and are crucial for enzymic activity. Biochem J. 2003;370:1077. doi: 10.1042/BJ20021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bardwell AJ, Frankson E, Bardwell L. Selectivity of docking sites in MAPK kinases. J Biol Chem. 2009;284:13165. doi: 10.1074/jbc.M900080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barr RK, Kendrick TS, Bogoyevitch MA. Identification of the critical features of a small peptide inhibitor of JNK activity. J Biol Chem. 2002;277:10987. doi: 10.1074/jbc.M107565200. [DOI] [PubMed] [Google Scholar]
- 19.Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, Bogousslavsky J, Bonny C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- 20.Bogoyevitch MA, Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol Mol Biol Rev. 2006;70:1061. doi: 10.1128/MMBR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beckstette M, Homann R, Giegerich R, Kurtz S. Fast index based algorithms and software for matching position specific scoring matrices. BMC Bioinformatics. 2006;7:389. doi: 10.1186/1471-2105-7-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Homann R, Beckstette M. PoSSuM software distribution. 2007 [Google Scholar]
- 24.Bardwell AJ, Flatauer LJ, Matsukuma K, Thorner J, Bardwell L. A conserved docking site in MEKs mediates high-affinity binding to MAP kinases and cooperates with a scaffold protein to enhance signal transmission. J Biol Chem. 2001;276:10374. doi: 10.1074/jbc.M010271200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaake RM, Milenkovic T, Przulj N, Kaiser P, Huang L. Characterization of cell cycle specific protein interaction networks of the yeast 26S proteasome complex by the QTAX strategy. J Proteome Res. 9:2016. doi: 10.1021/pr1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asplund A, Edqvist PH, Schwenk JM, Ponten F. Antibodies for profiling the human proteome-The Human Protein Atlas as a resource for cancer research. Proteomics. 2012;12:2067. doi: 10.1002/pmic.201100504. [DOI] [PubMed] [Google Scholar]
- 27.Barrett T, et al. NCBI GEO: archive for functional genomics data sets--10 years on. Nucleic Acids Res. 2011;39:D1005. doi: 10.1093/nar/gkq1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galvez-Santisteban M, et al. Synaptotagmin-like proteins control the formation of a single apical membrane domain in epithelial cells. Nat Cell Biol. 2012;14:838. doi: 10.1038/ncb2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fosbrink M, Aye-Han NN, Cheong R, Levchenko A, Zhang J. Visualization of JNK activity dynamics with a genetically encoded fluorescent biosensor. Proc Natl Acad Sci U S A. 2010;107:5459. doi: 10.1073/pnas.0909671107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Porter JD, Israel S, Gong B, Merriam AP, Feuerman J, Khanna S, Kaminski HJ. Distinctive morphological and gene/protein expression signatures during myogenesis in novel cell lines from extraocular and hindlimb muscle. Physiol Genomics. 2006;24:264. doi: 10.1152/physiolgenomics.00234.2004. [DOI] [PubMed] [Google Scholar]
- 31.Portier GL, Benders AG, Oosterhof A, Veerkamp JH, van Kuppevelt TH. Differentiation markers of mouse C2C12 and rat L6 myogenic cell lines and the effect of the differentiation medium. In Vitro Cell Dev Biol Anim. 1999;35:219. doi: 10.1007/s11626-999-0030-8. [DOI] [PubMed] [Google Scholar]
- 32.Ponten F, Schwenk JM, Asplund A, Edqvist PH. The Human Protein Atlas as a proteomic resource for biomarker discovery. J Intern Med. 2011;270:428. doi: 10.1111/j.1365-2796.2011.02427.x. [DOI] [PubMed] [Google Scholar]
- 33.Haslett JN, Sanoudou D, Kho AT, Han M, Bennett RR, Kohane IS, Beggs AH, Kunkel LM. Gene expression profiling of Duchenne muscular dystrophy skeletal muscle. Neurogenetics. 2003;4:163. doi: 10.1007/s10048-003-0148-x. [DOI] [PubMed] [Google Scholar]
- 34.Urso ML, Scrimgeour AG, Chen YW, Thompson PD, Clarkson PM. Analysis of human skeletal muscle after 48 h immobilization reveals alterations in mRNA and protein for extracellular matrix components. J Appl Physiol. 2006;101:1136. doi: 10.1152/japplphysiol.00180.2006. [DOI] [PubMed] [Google Scholar]
- 35.Timmons JA, et al. Using molecular classification to predict gains in maximal aerobic capacity following endurance exercise training in humans. J Appl Physiol. 2010;108:1487. doi: 10.1152/japplphysiol.01295.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leng Y, Steiler TL, Zierath JR. Effects of insulin, contraction, and phorbol esters on mitogen-activated protein kinase signaling in skeletal muscle from lean and ob/ob mice. Diabetes. 2004;53:1436. doi: 10.2337/diabetes.53.6.1436. [DOI] [PubMed] [Google Scholar]
- 37.Jing E, Emanuelli B, Hirschey MD, Boucher J, Lee KY, Lombard D, Verdin EM, Kahn CR. irtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc Natl Acad Sci U S A. 2011;108:14608. doi: 10.1073/pnas.1111308108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bennett AM, Tonks NK. Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science. 1997;278:1288. doi: 10.1126/science.278.5341.1288. [DOI] [PubMed] [Google Scholar]
- 39.Nakamura A, Yoshida K, Ueda H, Takeda S, Ikeda S. Up-regulation of mitogen activated protein kinases in mdx skeletal muscle following chronic treadmill exercise. Biochim Biophys Acta. 2005;1740:326. doi: 10.1016/j.bbadis.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 40.Smythe GM, Forwood JK. Altered mitogen-activated protein kinase signaling in dystrophic (mdx) muscle. Muscle Nerve. 2012;46:374. doi: 10.1002/mus.23312. [DOI] [PubMed] [Google Scholar]
- 41.Kolodziejczyk SM, Walsh GS, Balazsi K, Seale P, Sandoz J, Hierlihy AM, Rudnicki MA, Chamberlain JS, Miller FD, Megeney LA. Activation of JNK1 contributes to dystrophic muscle pathogenesis. Curr Biol. 2001;11:1278. doi: 10.1016/s0960-9822(01)00397-9. [DOI] [PubMed] [Google Scholar]
- 42.Sabio G, Kennedy NJ, Cavanagh-Kyros J, Jung DY, Ko HJ, Ong H, Barrett T, Kim JK, Davis RJ. Role of muscle c-Jun NH2-terminal kinase 1 in obesity-induced insulin resistance, Mol Cell Biol. 2010;30:106. doi: 10.1128/MCB.01162-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.