


Abstract
Toward achieving rapid and large scale genome modification directly in a target organism, we have developed a new genome engineering strategy that uses a combination of bioinformatics aided design, large synthetic DNA and site-specific recombinases. Using Cre recombinase we swapped a target 126-kb segment of the Escherichia coli genome with a 72-kb synthetic DNA cassette, thereby effectively eliminating over 54 kb of genomic DNA from three non-contiguous regions in a single recombination event. We observed complete replacement of the native sequence with the modified synthetic sequence through the action of the Cre recombinase and no competition from homologous recombination. Because of the versatility and high-efficiency of the Cre-lox system, this method can be used in any organism where this system is functional as well as adapted to use with other highly precise genome engineering systems. Compared to present-day iterative approaches in genome engineering, we anticipate this method will greatly speed up the creation of reduced, modularized and optimized genomes through the integration of deletion analyses data, transcriptomics, synthetic biology and site-specific recombination.
INTRODUCTION
With the advent of synthetic genomics comes the capacity to synthesize, reengineer and build whole new organisms with functionalities not present in nature. Large size synthetic DNA, a crucial component of this field is becoming more accessible due to the option of assembling up to genome size molecules from oligonucleotides or directly purchasing super-size synthetic DNA up to 2 Mb (http://sgidna.com/design.php). As our capacity to produce high-quality synthetic DNA molecules increases, so must our know-how to integrate functionality and expression of such designer DNA molecules across species barriers. At present, the challenge in grand scale genome engineering efforts such as whole genome modification, minimization, recoding or modularization is not re-synthesis of the genome, but instead the creation of a functional cell that can effectively accept the synthetic genome or genome segments of 100 kb or greater. There are few published examples of successful transfer and installation of a new genome—transplantation of a chemically synthesized synthetic genome into appropriate recipient cells to create the first synthetic bacterial cell (1–3), co-expression of a genome by megacloning to create hybrid cells with cross-species manifestations, raising the possibility of creating unique hybrid functional strains with cell-free DNA as the source (4), and more recently, iterative conjugation of large genome sections to create a full Escherichia coli chromosome with an altered genetic code (5,6). All of these approaches have limitations including cross species or kingdom barriers, issues with unwanted recombination, and/or an inability to transfer really large DNA regions.
Most genome manipulations for strain construction are still accomplished primarily by recombineering or with the help of phage derived site-specific recombinases such as the Cre-loxP or Flp-FRT or combination of both (7–10). The vast single gene knockout libraries are eventually combined to realize synergistic effects of multiple deletions on the phenotype of the organism. Single gene knock-outs made using λ Red recombination (a homologous recombination system provided by the γ, β, exo protein functions of the bacteriophage λ (7)), plus information gathered from random whole-genome transposon mutagenesis has been used toward stepwise genomic reduction that ultimately led to the stable reduced E. coli genome devoid of mobile elements and with desirable features like increased transformation (11,12).
Combining these individual deletions to develop the final strain is a time-consuming process and requires phage transduction or mating to pool the donor and recipient genotypes. The same methods can be used to make double deletions, but to greatly speed up whole genome modification what is truly needed is a method to integrate information from all available knockouts and reengineer large sections of the genome through simultaneous non-contiguous replacement. Although rational design toward a functional minimal genome is a goal still unrealized, it is not unrealistic to design multi-deletion genomes from metabolic modeling, comparative genomics and transcription analyses. As the first step toward achieving rapid whole genome modifications in the target organism without the use of genome transplantation or other genome transfer methods, we developed a process that uses synthetic biology methods, site-specific recombination, and recombineering to reengineer and minimize one segment of the E. coli genome. We used bioinformatics analyses of multiple E. coli genomes combined with data on essentiality of genes obtained from the Keio collection (13) to design a synthetic cassette carrying non-contiguous multiple deletions spanning 4–39 kb. This synthetic cassette was swapped with the targeted region using Cre-lox to create multiple non-contiguous modifications and minimizations over a 126-kb region of the E. coli genome. We anticipate this method can be adapted to a number of whole genome manipulation tasks—stabilization, alteration of the genetic code, integration of pathways, genome modularization and spatial characterization of pathways toward designer genomes with features that enable plug and play of parts and pathways as required for industrially relevant tasks.
MATERIALS AND METHODS
All manipulations were carried out in strains derived from E. coli BL21 (DE3), Accession NC_012971, Version NC_012971.2 gi:387825439, GenBank: CP001509.3. The strain of E. coli BL21 DE3 used in this study was devoid of the λ DE3 phage and some IS elements. These modifications were created during genome stabilization for an unrelated study and do not pertain to the results presented here.
DNA synthesis and assembly of the synthetic DNA construct
The synthetic DNA was provided by SGI-DNA (La Jolla, CA, USA). Two constructs were used in the Cre-lox-mediated synthetic replacement experiment. The synthetic cassette BAC-72 was designed to reconstruct 126.14 kb region on the E. coli BL21 DE3 genome by replacing it with 72.08 kb of minimized synthetic DNA, and BAC-52 replaces 103 kb of the genome with 52.96 kb. (The design of the synthetic cassettes is described in the ‘Results’ and ‘Supplementary Data’ sections.) The 78 339 bp DNA sequence was divided into 110 overlapping DNA sequences, which were individually assembled from overlapping oligonucleotides (Integrated DNA Technologies) and sequence verified (14). Using previously described in vitro (15,16) and in vivo DNA assembly methods (17,18), the 110 synthetic DNA cassettes were converted into a contiguous 78 kb DNA construct, following two assembly stages. The final constructs were assembled in pCC1BAC (Epicentre) and transformed into Epi300 cells (Epicentre®). A pure sequence-confirmed isolate was grown in LB (Luria-Bertani) medium containing 12.5 μg/ml chloramphenicol and 1× induction solution (provided by Epicentre) and the construct was prepared using a commercially available plasmid isolation kit (Qiagen).
λ Red and Cre recombinase-mediated engineering
The regions on the genome being reconstructed were floxed with two landing pads. The 126.14-kb region for BAC-72 (within coordinates 1 980 428…2 10 6568) and the 103-kb (within coordinates 1 98 0428…2 083 389) regions for BAC-52 were flanked by two landing pads—the loxm2/66-aac (aminoglycoside acetyl transferase for apramycin resistance) and the lox71-pac (puromycin acetyl transferase gene for puromycin resistance). The aac gene was derived from pIJ773 (19) and pac gene from Mini-Tn4001PsPuro (20) cloned into the pEL04 (21) backbone (pEL04-puromycin, this study). The landing pads were inserted using published Red recombination methods using plasmid pKD46 (7). Construction and insertion of the landing pad cassettes is described in detail in the Supplementary Data. The resulting strains were referred to as the swap recipient strains and given the following designations—CG349a2 swap recipient strain for BAC-72 and JZ794-7 swap recipient strain for BAC-52.
Induction of Cre recombinase, selection and screening of recombinants
The Cre recombinase was provided from plasmid 705-cre (Gene Bridges, Germany) driven by the temperature inducible λPR promoter (cI578) and maintained as low copy with Cre repression at 30°C. The recombinase induction at 37–42°C also results in loss of the plasmid. The swap recipient strains CG349a2 and JZ794-7 carrying the Cre plasmid were grown in LB with 34 μg/ml of chloramphenicol at 30°C, made electrocompetent using standard protocol and transformed with 500 ng of synthetic cassette DNA. Following electroporation, the cells were recovered in 1 ml LB for 2 h at 37°C to induce Cre recombinase, and selected for growth on LB containing antibiotics appropriate for selection of the synthetic cassette. BAC-72 transformations were selected on LB plus kanamycin (25 μg/ml) and tetracycline (10 μg/ml) and BAC-52 on LB kanamycin (25 μg/ml). The GFPuv ORF encodes a GFP variant optimized for maximal fluorescence when excited by standard UV light (360–400 nm) and was used as a method to screen for transfer of the full cassette whether it may be on the BAC or integrated into the genome. Ten to 20 colonies from each transformation plate were streaked on LB for single colonies and screened by Polymerase Chain Reaction (PCR).
RESULTS
Design of the synthetic cassette
The genomic region for replacement by a synthetic DNA construct was selected to have the following criteria: (i) at least one essential gene to retain, (ii) at least one conditionally essential gene and (iii) multiple large regions to eliminate from the replacement construct, including several that confer known phenotypes. Meeting these criteria, as well as watermarking individual replacement genes and adding selectable or screening markers, simplified proof that replacement was successful. These criteria were met by (a) keeping metG, (b) keeping the histidine biosynthesis operon and (c) removing 54 046 bp and 60 genes, including the colanic acid (CA) biosynthesis locus. We judged suitability for deletion according to availability and robustness of single gene deletion mutants in E. coli K-12, universality among 49 strains of E. coli (and two of Salmonella) as determined by PanOCT (Pan-genome ortholog clustering tool) (22) pan-genome analysis, functional annotation and comments in EcoGene 3.0 (23), and additional reports on essentiality, gene bloc deletion for genome minimization, and characterization of individual genes. Design of the synthetic cassette is described in detail in the Supplementary Data, Table S1 (in MS Excel) and illustrated in Figure 1.
Figure 1.
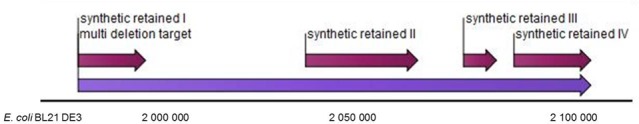
Design of the planned deletions and reconstruction of the 126,141 bp region of the E. coli BL21 DE3 genome (CLC genome analysis tool). Magenta arrows indicate the four regions retained in the synthetic construct BAC-72 and the three regions retained in BAC-52; the purple arrow indicates the 126-kb region of the genome being modified.
Two constructs were used in the Cre-loxP-mediated synthetic replacement experiment (Figure 2, top panel): (i) BAC-72, the full synthetic replacement cassette of 72 095 bp, carrying four non-contiguous essential/kept regions within genome coordinates 1 980 428…1 997 233, 2 036 265…2 064 080, 2 075 037…2 083 389 and 2 087 449…2 106 568) and (ii) BAC-52, the medium assembly of 52 979 bp carrying DNA sequence within coordinates 1 980 428…1 997 233, 2 036 265…2 064 080 and 2 075 037…2 083 389. The ‘kept’ regions were flanked by non-compatible lox sites loxm2/71 and lox66, each with a unique selectable marker that could only recombine with loxm2/66 and lox71 present on the genome landing pads (sequence of lox sites in Table S2 of Supplementary Data). The non-compatible lox sites on the genome landing pads and on the synthetic cassettes provide efficient unidirectional intermolecular exchange and stable integration following the Cre-mediated recombination event (24). The size of the full synthetic cassette BAC-72 with the loxm2/71-aph-sacB (aminoglycoside 3′-phosphotransferase for kanamycin resistance) and lox66-tetA-tetR (tetracycline resistance gene) is 78 339 bp, and the size of BAC-52 with loxm2/71-aph-sacB and lox66-GFP-uv is 57 309 bp. The sequences for the selectable markers on the synthetic cassettes were obtained from the following sources: the aph-sacB constructed from pKD4 (7) and pEL04 (21), the tetA-tetR from Tn10d, and GFPuv ORF sequence derived from pGFPuv vector (Clontech).
Figure 2.
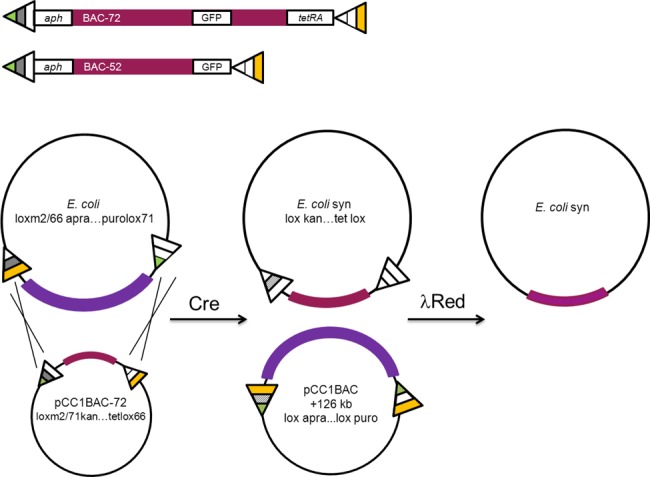
Top panel: Two synthetic cassettes were used in this study. The full synthetic cassette pCC1BAC-72 has 72 kb of synthetic redesigned genomic DNA flanked by lox sites and selection markers—loxm2/66 is linked with aph (sacB) and lox71 is linked with tetRA. This cassette also has the uv-GFP marker inserted after the first 52 kb. The BAC-52 is essentially the first part of the full synthetic cassette; it has 52 kb of synthetic reconstructed E. coli genomic DNA floxed by the loxm2/66-aph (sacB) and lox71-uvGFP. Bottom panel: Cre-mediated recombination occurs in the swap recipient strain between loxm2/66-aac and loxm2/71-aph-sacB at the 5′ or left end of the DNA segments and lox71-pac and lox66-tetA-tetR at the 3′ or right end. This results in exchange of the DNA flanked by the lox sites and the generation of lox sites not suitable for further recombination, thereby stably integrating the synthetic cassette into the E. coli genome. The swap is made seamless and markerless by removal of the landing pads using λ Red-mediated recombineering.
Modification of the E. coli genome through recombinase-mediated synthetic replacement
The overall design of the two synthetic cassettes and the strategy to create synthetically reduced E. coli genome is shown in Figure 2. Recipient strains carrying landing pads loxm2/66-aac (apramycin resistance) and the lox71-pac (puromycin resistance) were constructed using λ Red recombination (described in ‘Materials and Methods’ and ‘Supplementary Data’ sections). The resulting strains were referred to as CG349a2 swap recipient strain for BAC-72, and JZ794-7 swap recipient strain for BAC-52 and were transformed with pCC1BAC-72 and pCC1BAC-52 DNA, respectively. Following transformation the colonies were selected for resistance to the antibiotics present on the synthetic cassettes and screened for successful Cre-mediated recombination by PCR analysis from isolated genomic DNA. Successful Cre-lox-mediated exchange and minimization was achieved with both synthetic constructs at a rate of ∼30% in five independent experiments (see Supplementary Data for full explanation). PCR primers to screen the recombinants are described in Figure 3 where the primer numbers correspond to lane numbers in the agarose gels shown in Figure 4. The sequence of the primers and expected amplicon size is described in Supplementary Data, Table S4. Preliminary PCR analysis confirmed creation of new end junctions through exchange of aac and pac markers for the aph-sacB and tetRA for BAC-72, or aph-sacB and GFP for BAC-52 (Figure 3, primers 1 a, b and primers 8a, b and Figure 4, panels B and C) lanes 1 a, b and lanes 8 a, b). The recombinants containing the correct end junctions were sequenced (Sanger method) to verify correct lox recombination products. (The recombination between loxm2/66 and loxm2/71, and lox71 and lox66 created the expected non-compatible lox sequences on the genomic recombination site (24).) Primers were designed to amplify all the new junctions and to confirm loss of the non-contiguous regions from the swap strains (Figure 3). The strains containing the synthetic swaps BAC-72 and BAC-52 showed all the correct amplicons expected for the new junctions created in the genome through splicing of the non-contiguous ‘kept’ regions (Figure 3, primer sets 3–7 and Figure 4, panels B and C, lanes 3–7). The control E. coli BL21 DE3 wild-type showed amplification of products for some of the junction PCRs due to 10–17 bases of homology to the wild-type genome in the junction primers (Figure 4, panel A). The synthetic swap strains did not contain any DNA sequence from the non-contiguous regions targeted for deletion in the synthetic cassettes, as indicated by the absence of PCR product for primer sets 9–11. As expected, distinct amplicons were obtained when combinations of the screening primers were used in the wild-type and the swap strains (sets 12–14).
Figure 3.
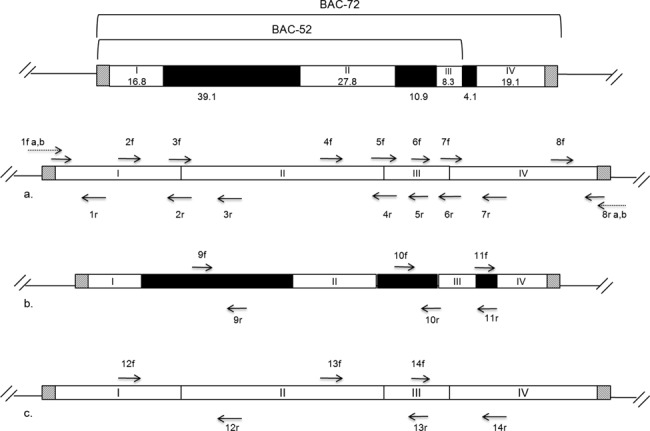
BAC-72 is the full synthetic cassette containing 78 kb of DNA of which 72 kb is minimized E. coli genomic sequence. The E. coli synthetic DNA is flanked by selectable markers and lox sites (gray boxes). The BAC-72 synthetic cassette was designed to replace 126 kb section of the E. coli chromosome. This event simultaneously deletes three non-contiguous regions (black boxes) and creates new junctions on the chromosome due to joining of the four regions in between (white boxes I–IV) that are retained in the synthetic DNA. BAC-52 is 57 kb with 52 kb of reduced E. coli DNA and replaces 103 kb of genomic DNA. This recombination event deletes two non-contiguous regions (39 and 27 kb) and creates new junction due to joining segments I, II and III. Primers to confirm the end junctions are set 1 and set 8. 1f(b) spans the new junction created through exchange of aac (apramycin) with the aph-sacB (kanamycin) and 8r (b) spans the new junction created through exchange of the pac (puromycin) landing pad with tetRA in BAC-72 or GFP-uv in BAC-52. Primers illustrated in dashed arrows (1f (a) and 8 r (a)) are ∼25 bp sequences of which 10–15 bases are inclusive of the precise start and end of the designed swap and thus have homology to both synthetic swap and wild-type. Similarly the junction primers 2r, 3f, 4r, 5f, 6r, 7f have 10–15 bases of homology with the kept regions and therefore depending on the PCR conditions are expected to give some amplicons in the wild-type background as well. Primers sets 9–11 are all internal to the regions deleted during construction of the synthetic cassette BAC-72 and are expected to amplify DNA only in the wild-type strain except for primer set 11 which will give a product in the BAC-52 swap strain (BAC-52 retains the 4.1 kb region). Sets 12, 13 and 14 are combinations of internal and junction primers designed to produce distinct amplicons in wild-type and the synthetic swap. The amplicon size for each PCR is described in Supplementary Data (Table S4).
Figure 4.
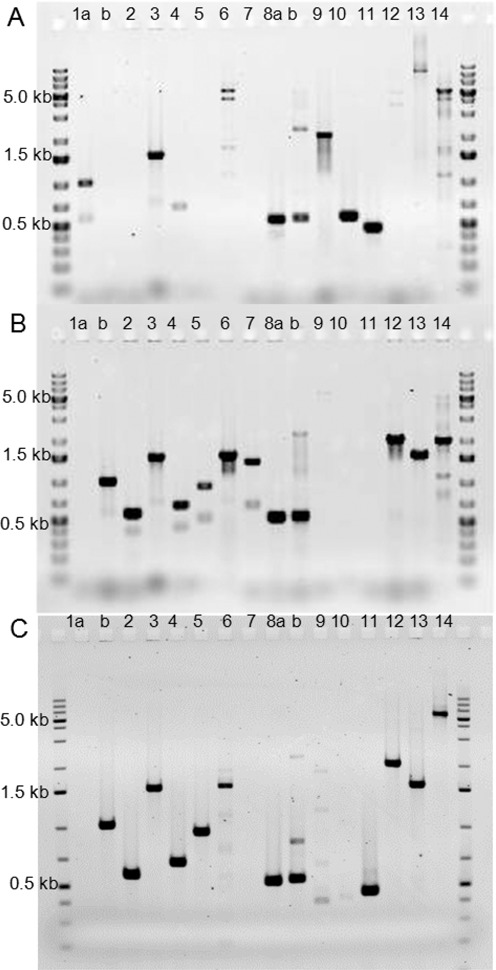
Agarose gels showing PCR analyses (with GeneRuler Ikb Plus DNA ladder, Thermo Scientific). Panel (A), control/wild-type; positive for amplicons 1(a), 8(a), 9, 10 and 11. Bands were observed in lanes 3, 6 and 8b due to some non-specific binding as well as the 10–17 bases of homology to the wild-type genome in the junction primers. Panel (B), synthetic swap with BAC-72; positive for amplicons 1b, junction primers 2–7 and 8(b). Primer set 1(b) and 8(b) confirms the insertion of the BAC-72. Primer sets 2–7 confirm presence of the three new internal junctions in the strain with the BAC-72 created through the joining of segments I-IV. PCR from primer sets 9–11 produce amplicons only in the negative control wild-type E. coli. Sets 12–14 produce different size amplicons for the synthetic swap strain and the control strain. Panel (C), synthetic swap with BAC-52; positive for amplicons 1b, junction primers 2–6, and 8(a) (b) and internal primer 11. Primer sets 1(b) and 8(b) confirm the insertion of the BAC-72. This strain gives a PCR product for 8(a) since the swap terminates prior to segment IV. Primer sets 2–6 confirm presence of the new internal junctions created through the joining of segments I, II and III. The internal primer set 11 gives an amplicon identical to the product seen in the wild-type panel (A) since the 4.11 kb region is retained after recombination with BAC-52. Sets 12 and 13 are identical to swap BAC-72, while set 14 gives a band similar to the wild-type strain but with a slightly higher molecular weight band due to presence of the selection marker in the BAC-52 swap.
Insertion of the full synthetic segment was confirmed for the BAC-72 synthetic swap by sequencing through the entire 78 kb region including the selectable markers, lox sites and at least 0.5 kb outside of the lox sites by Sanger method primer walks. All of the designed synonymous codon changes within the cassette were located, and the insertion of the full cassette was confirmed to be in the expected location. In addition, the BAC-72 synthetic swap strain and E. coli BL21 DE3 wild-type were sequenced using PacBio sequencing system (Pacific Biosciences). Reads from PacBio whole genome sequencing of the BAC-72 synthetic swap strain were mapped to the synthetic fragment and gave one contig with 100% perfect match. The E. coli BL21 DE3 wild-type reads were mapped to the synthetic fragment and gave four contigs, each with <100% perfect match (due to single nucleotide changes made in the design). To ensure the loss of the BAC and other episomal constructs, the confirmed synthetic swap strains were passaged 3–6 times in LB without antibiotics and screened for loss of apramycin and puromycin resistance (present on genomic landing pads and transferred to BACs after Cre-mediated recombination) and chloramphenicol (on pCC1BAC). No episomal constructs could be isolated from these strains using DNA isolation procedures specific for the enrichment and isolation of BACs.
The goal was to create genome modifications seamless and scar-free, so the modifications can progress as a continuous cycle either through the regeneration or introduction of new recombinase recognition sites and selectable markers. Toward creating such a strain, the selectable markers and associated lox sites were removed using λ Red-mediated recombination following published methods (25,26). The markers on the full cassette BAC-72 aph-sacB and tetR-tetA inserted into the genome after exchange are designed for positive and negative selection. The aph-sacB cassette and the associated lox sites were removed using PCR amplified cassettes with 300 bases of homology to the region immediately flanking the resistance markers to create a seamless reengineered section on the genome. The negative selection for removal of the tetR-tetA marker was unsuccessful in this strain of E. coli BL21 (27). Rather than screening a large number of colonies for removal of tetR-tetA by sensitivity to tetracycline, this marker was replaced with thyA, which was subsequently removed using the trimethoprim-based selection to create a seamless exchange in the genome (28).
DISCUSSION
Here we report a novel method to redesign and/or minimize E. coli using a combination of synthetic genomics and site-specific recombination. In this study, we have used Cre recombinase and large synthetic DNA cassettes to simultaneously delete multiple non-contiguous regions on the chromosome. We replaced a 126-kb region of the E. coli genome with a 72-kb synthetic cassette consisting of genes deemed to be favorable for robustness, while removing regions ranging from 4 to 39 kb that were interspaced between the kept regions. Our 72 kb synthetic swap could be the largest Cre-mediated insertion reported yet where the Cre recombinase successfully outcompeted homologous recombination (29,30). At this point, we have yet to determine the size limit of synthetic cassettes that can be integrated into bacterial and possibly even cyanobacterial and mammalian cell line genomes. Because of the versatility and high-efficiency of the Cre-lox system, this method can be adapted to any organism where this recombinase is functional (31). New high efficiency Cre variants and mutant recognition sites have been developed enabling reiterative precise scar-free genome engineering (32–35). Our targeted synthetic replacement method could also be modified to use with any highly precise genome engineering method for large-scale modifications in bacterial or eukaryotic systems. Ultimately the goal is to accomplish whole genome modifications in a reiterative and seamless fashion through (re)generation and/or removal of the selectable markers and to ‘walk’ around the genome in segments of several hundred kilo bases.
One issue we encountered was differentiating BAC transformations from the correct recombinants. We noticed a significant number of colonies that had undergone correct Cre-mediated recombination but continued to retain the landing pad associated selection markers after several passages in medium lacking any selective agent. Furthermore, some colonies displayed correct recombination at the lox sites but retained some wild-type regions (see Supplementary Data for explanation). Some of these issues could be averted by using a non-replicative BAC with ‘self-destruct’ mechanism such as CcdB toxin–antitoxin system (36), modulating increased expression of Cre and using a recombination deficient strain. But it is also conceivable that some recombinants retain a reduced version of the recombination-derived BAC. This in spite of the lack of the selective pressure of antibiotics, because some of the deleted regions although not deemed essential for life provided some evolutionary advantage to the population that retained it.
Although we used synthetic DNA, the recombinase-mediated synthetic replacements can be achieved with PCR amplified DNA, or with fragments obtained from restriction enzyme digested genomic DNA. Replacement cassettes of minimized genomic DNA, metabolic pathways from diverse organisms, modularized segments of the genome can be constructed by PCR, Gibson Assembly® (16) or yeast recombination (37–40). As we move toward reengineering larger sections of the genome, there may be issues of synthetic DNA toxicity or instability such as from assembling E. coli DNA in an E. coli host. We believe these problems can be addressed by using recoded DNA carrying synonymous changes, DNA from closely related organisms, or by moving the entire assembly process to yeast. For this study, the final assembly of the synthetic cassettes was done in Saccharomyces cerevisiae. Yeast is often used to bring together large pieces of prokaryotic and eukaryotic DNA using its recombination-based assembly methods such as TAR (transformation-associated recombination) (41), TREC (tandem repeat coupled with endonuclease cleavage) (37) etc., and to stably maintain the cloned DNA in linear or circular YACs. Recently, we published methods to assemble and stably maintain up to 200 kb of prokaryotic DNA from restriction enzyme digested genomic DNA. Through the addition of more yeast origins of replication, the size of the cloned DNA was increased to over 450 kb (42). Prokaryotic or eukaryotic DNA up to 2.4 Mb can be maintained as yeast artificial chromosomes (YAC) (43), and the method of transfer of the assembled cassette into the recipient cell can be chosen depending on the most effective and routinely used technique available for the specific system, be it plant cells, human cell lines or cyanobacteria. Once the cassette is generated, the selection for its functionality and toleration of deletions is always viability and growth rate of the recipient cell.
In this study, our design was based solely on comparative analysis of available E. coli genomes and compilation of deletion data, but in the future we anticipate incorporating in silico modeling and transcriptome data from the organism grown in a specific environment. Such analyses can reveal the essentiality and interactions of gene components specific to growth conditions, allowing us to tailor the genome design to meet exact requirements of an engineered microbial cell factory and move forward in genome minimization and reorganization.
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online.
Acknowledgments
We thank Ariel Schwartz, John Gill, Billyana Tsvetanova and Lixia Fu for technical assistance and data analysis.
Footnotes
Present address: Carissa Grose, Frederick National Laboratory for Cancer Research, P.O.Box B Frederick, MD 21702, Email: carissa.grose@nih.gov
FUNDING
Novartis Foundation and Synthetic Genomics Vaccines, Inc. (SGVI). Funding for open access charge: Novartis Foundation and Synthetic Genomics Vaccines, Inc. (SGVI).
Conflict of interest. Daniel G. Gibson is Vice President and holds employee stock options at Synthetic Genomics, Inc.
REFERENCES
- 1.Gibson D.G., Glass J.I., Lartigue C., Noskov V.N., Chuang R.Y., Algire M.A., Benders G.A., Montague M.G., Ma L., Moodie M.M., et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science. 2010;329:52–56. doi: 10.1126/science.1190719. [DOI] [PubMed] [Google Scholar]
- 2.Karas B.J., Jablanovic J., Sun L., Ma L., Goldgof G.M., Stam J., Ramon A., Manary M.J., Winzeler E.A., Venter J.C., et al. Direct transfer of whole genomes from bacteria to yeast. Nat. Methods. 2013;10:410–412. doi: 10.1038/nmeth.2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lartigue C., Vashee S., Algire M.A., Chuang R.Y., Benders G.A., Ma L., Noskov V.N., Denisova E.A., Gibson D.G., Assad-Garcia N., et al. Creating bacterial strains from genomes that have been cloned and engineered in yeast. Science. 2009;325:1693–1696. doi: 10.1126/science.1173759. [DOI] [PubMed] [Google Scholar]
- 4.Itaya M., Tsuge K., Koizumi M., Fujita K. Combining two genomes in one cell: stable cloning of the Synechocystis PCC6803 genome in the Bacillus subtilis 168 genome. Proc. Natl. Acad. Sci. U.S.A. 2005;102:15971–15976. doi: 10.1073/pnas.0503868102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isaacs F.J., Carr P.A., Wang H.H., Lajoie M.J., Sterling B., Kraal L., Tolonen A.C., Gianoulis T.A., Goodman D.B., Reppas N.B., et al. Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science. 2011;333:348–353. doi: 10.1126/science.1205822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lajoie M.J., Rovner A.J., Goodman D.B., Aerni H.R., Haimovich A.D., Kuznetsov G., Mercer J.A., Wang H.H., Carr P.A., Mosberg J.A., et al. Genomically recoded organisms expand biological functions. Science. 2013;342:357–360. doi: 10.1126/science.1241459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Datsenko K.A., Wanner B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun W., Wang S., Curtiss R., 3rd Highly efficient method for introducing successive multiple scarless gene deletions and markerless gene insertions into the Yersinia pestis chromosome. Appl. Environ. Microbiol. 2008;74:4241–4245. doi: 10.1128/AEM.00940-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H.H., Isaacs F.J., Carr P.A., Sun Z.Z., Xu G., Forest C.R., Church G.M. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460:894–898. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fukiya S., Mizoguchi H., Mori H. An improved method for deleting large regions of Escherichia coli K-12 chromosome using a combination of Cre/loxP and lambda Red. FEMS Microbiol. Lett. 2004;234:325–331. doi: 10.1016/j.femsle.2004.03.042. [DOI] [PubMed] [Google Scholar]
- 11.Hirokawa Y., Kawano H., Tanaka-Masuda K., Nakamura N., Nakagawa A., Ito M., Mori H., Oshima T., Ogasawara N. Genetic manipulations restored the growth fitness of reduced-genome Escherichia coli. J. Biosci. Bioeng. 2013;116:52–58. doi: 10.1016/j.jbiosc.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 12.Csorgo B., Feher T., Timar E., Blattner F.R., Posfai G. Low-mutation-rate, reduced-genome Escherichia coli: an improved host for faithful maintenance of engineered genetic constructs. Microb. Cell Fact. 2012;11:11. doi: 10.1186/1475-2859-11-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K.A., Tomita M., Wanner B.L., Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006;2:2006 0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dormitzer P.R., Suphaphiphat P., Gibson D.G., Wentworth D.E., Stockwell T.B., Algire M.A., Alperovich N., Barro M., Brown D.M., Craig S., et al. Synthetic generation of influenza vaccine viruses for rapid response to pandemics. Sci. Transl. Med. 2013;5:185ra168. doi: 10.1126/scitranslmed.3006368. [DOI] [PubMed] [Google Scholar]
- 15.Gibson D.G., Smith H.O., Hutchison C.A., 3rd, Venter J.C., Merryman C. Chemical synthesis of the mouse mitochondrial genome. Nat. Methods. 2010;7:901–903. doi: 10.1038/nmeth.1515. [DOI] [PubMed] [Google Scholar]
- 16.Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A., 3rd, Smith H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 17.Gibson D.G., Benders G.A., Andrews-Pfannkoch C., Denisova E.A., Baden-Tillson H., Zaveri J., Stockwell T.B., Brownley A., Thomas D.W., Algire M.A., et al. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science. 2008;319:1215–1220. doi: 10.1126/science.1151721. [DOI] [PubMed] [Google Scholar]
- 18.Gibson D.G., Benders G.A., Axelrod K.C., Zaveri J., Algire M.A., Moodie M., Montague M.G., Venter J.C., Smith H.O., Hutchison C.A., 3rd One-step assembly in yeast of 25 overlapping DNA fragments to form a complete synthetic Mycoplasma genitalium genome. Proc. Natl. Acad. Sci. U.S.A. 2008;105:20404–20409. doi: 10.1073/pnas.0811011106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gust B., Challis G.L., Fowler K., Kieser T., Chater K.F. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. U.S.A. 2003;100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Algire M.A., Lartigue C., Thomas D.W., Assad-Garcia N., Glass J.I., Merryman C. New selectable marker for manipulating the simple genomes of Mycoplasma species. Antimicrob. Agents Chemother. 2009;53:4429–4432. doi: 10.1128/AAC.00388-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee E.C., Yu D., Martinez de Velasco J., Tessarollo L., Swing D.A., Court D.L., Jenkins N.A., Copeland N.G. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics. 2001;73:56–65. doi: 10.1006/geno.2000.6451. [DOI] [PubMed] [Google Scholar]
- 22.Fouts D.E., Brinkac L., Beck E., Inman J., Sutton G. PanOCT: automated clustering of orthologs using conserved gene neighborhood for pan-genomic analysis of bacterial strains and closely related species. Nucleic Acids Res. 2012;40:e172. doi: 10.1093/nar/gks757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou J., Rudd K.E. EcoGene 3.0. Nucleic Acids Res. 2013;41:D613–D624. doi: 10.1093/nar/gks1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langer S.J., Ghafoori A.P., Byrd M., Leinwand L. A genetic screen identifies novel non-compatible loxP sites. Nucleic Acids Res. 2002;30:3067–3077. doi: 10.1093/nar/gkf421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hashimoto M., Ichimura T., Mizoguchi H., Tanaka K., Fujimitsu K., Keyamura K., Ote T., Yamakawa T., Yamazaki Y., Mori H., et al. Cell size and nucleoid organization of engineered Escherichia coli cells with a reduced genome. Mol. Microbiol. 2005;55:137–149. doi: 10.1111/j.1365-2958.2004.04386.x. [DOI] [PubMed] [Google Scholar]
- 26.Ellis H.M., Yu D., DiTizio T., Court D.L. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. U.S.A. 2001;98:6742–6746. doi: 10.1073/pnas.121164898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maloy S.R., Nunn W.D. Selection for loss of tetracycline resistance by Escherichia coli. J. Bacteriol. 1981;145:1110–1111. doi: 10.1128/jb.145.2.1110-1111.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong Q.N., Ng V.C., Lin M.C., Kung H.F., Chan D., Huang J.D. Efficient and seamless DNA recombineering using a thymidylate synthase A selection system in Escherichia coli. Nucleic Acids Res. 2005;33:e59. doi: 10.1093/nar/gni059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santos C.N., Regitsky D.D., Yoshikuni Y. Implementation of stable and complex biological systems through recombinase-assisted genome engineering. Nat. Commun. 2013;4:2503. doi: 10.1038/ncomms3503. [DOI] [PubMed] [Google Scholar]
- 30.Kuhlman T.E., Cox E.C. Site-specific chromosomal integration of large synthetic constructs. Nucleic Acids Res. 2010;38:e92. doi: 10.1093/nar/gkp1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y., Yau Y.Y., Perkins-Balding D., Thomson J.G. Recombinase technology: applications and possibilities. Plant Cell Rep. 2011;30:267–285. doi: 10.1007/s00299-010-0938-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee G., Saito I. Role of nucleotide sequences of loxP spacer region in Cre-mediated recombination. Gene. 1998;216:55–65. doi: 10.1016/s0378-1119(98)00325-4. [DOI] [PubMed] [Google Scholar]
- 33.Albert H., Dale E.C., Lee E., Ow D.W. Site-specific integration of DNA into wild-type and mutant lox sites placed in the plant genome. Plant J. 1995;7:649–659. doi: 10.1046/j.1365-313x.1995.7040649.x. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki E., Nakayama M. VCre/VloxP and SCre/SloxP: new site-specific recombination systems for genome engineering. Nucleic Acids Res. 2011;39:e49. doi: 10.1093/nar/gkq1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eroshenko N., Church G.M. Mutants of Cre recombinase with improved accuracy. Nat. Commun. 2013;4:2509. doi: 10.1038/ncomms3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bernard P., Gabant P., Bahassi E.M., Couturier M. Positive-selection vectors using the F plasmid ccdB killer gene. Gene. 1994;148:71–74. doi: 10.1016/0378-1119(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 37.Noskov V.N., Segall-Shapiro T.H., Chuang R.Y. Tandem repeat coupled with endonuclease cleavage (TREC): a seamless modification tool for genome engineering in yeast. Nucleic Acids Res. 2010;38:2570–2576. doi: 10.1093/nar/gkq099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noskov V.N., Chuang R.Y., Gibson D.G., Leem S.H., Larionov V., Kouprina N. Isolation of circular yeast artificial chromosomes for synthetic biology and functional genomics studies. Nat. Protoc. 2011;6:89–96. doi: 10.1038/nprot.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muller H., Annaluru N., Schwerzmann J.W., Richardson S.M., Dymond J.S., Cooper E.M., Bader J.S., Boeke J.D., Chandrasegaran S. Assembling large DNA segments in yeast. Methods Mol. Biol. 2012;852:133–150. doi: 10.1007/978-1-61779-564-0_11. [DOI] [PubMed] [Google Scholar]
- 40.Shao Z., Zhao H. DNA assembler, an in vivo genetic method for rapid construction of biochemical pathways. Nucleic Acids Res. 2009;37:e16. doi: 10.1093/nar/gkn991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larionov V., Kouprina N., Solomon G., Barrett J.C., Resnick M.A. Direct isolation of human BRCA2 gene by transformation-associated recombination in yeast. Proc. Natl. Acad. Sci. U.S.A. 1997;94:7384–7387. doi: 10.1073/pnas.94.14.7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Noskov V.N., Karas B.J., Young L., Chuang R.Y., Gibson D.G., Lin Y.C., Stam J., Yonemoto I.T., Suzuki Y., Andrews-Pfannkoch C., et al. Assembly of large, high G+C bacterial DNA fragments in yeast. ACS Synth. Biol. 2012;1:267–273. doi: 10.1021/sb3000194. [DOI] [PubMed] [Google Scholar]
- 43.Den Dunnen J.T., Grootscholten P.M., Dauwerse J.G., Walker A.P., Monaco A.P., Butler R., Anand R., Coffey A.J., Bentley D.R., Steensma H.Y., et al. Reconstruction of the 2.4 Mb human DMD-gene by homologous YAC recombination. Hum. Mol. Genet. 1992;1:19–28. doi: 10.1093/hmg/1.1.19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.