

Abstract
Next-generation sequencing (NGS) technologies enable new insights into the diversity of virus populations within their hosts. Diversity estimation is currently restricted to single-nucleotide variants or to local fragments of no more than a few hundred nucleotides defined by the length of sequence reads. To study complex heterogeneous virus populations comprehensively, novel methods are required that allow for complete reconstruction of the individual viral haplotypes. Here, we show that assembly of whole viral genomes of ∼8600 nucleotides length is feasible from mixtures of heterogeneous HIV-1 strains derived from defined combinations of cloned virus strains and from clinical samples of an HIV-1 superinfected individual. Haplotype reconstruction was achieved using optimized experimental protocols and computational methods for amplification, sequencing and assembly. We comparatively assessed the performance of the three NGS platforms 454 Life Sciences/Roche, Illumina and Pacific Biosciences for this task. Our results prove and delineate the feasibility of NGS-based full-length viral haplotype reconstruction and provide new tools for studying evolution and pathogenesis of viruses.
INTRODUCTION
Assessing the genetic diversity of intra-host pathogen populations has gained increasing interest in recent years. Especially viruses with high mutation rates and short replication cycles, such as human immunodeficiency virus type 1 (HIV-1), rapidly develop highly diverse intra-host populations, called viral quasispecies (1,2). To estimate the diversity of a viral quasispecies, next-generation sequencing (NGS) technologies can be applied, which produce thousands to millions of sequence reads in one run from a mixed sample.
Different NGS technologies are available, among them pyrosequencing by 454 Life Sciences/Roche (454/Roche), reversible terminator sequencing-by-synthesis by Illumina and single-molecule sequencing by Pacific Biosciences (PacBio) (3). They differ in sequencing chemistry, speed, throughput, read length, error rates and error patterns. Each of the three technologies can be used to analyze viral genomes, but none enables sequencing regions larger than a few hundred bases with sufficiently high accuracy. Consequently, virus diversity is typically assessed only by studying single nucleotide positions or mutations within a read length (4–10). Genome-wide reconstruction was only achieved with simulated data in silico, so far (11–14). Therefore, experimental amplification and sequencing strategies, combined with appropriate bioinformatics methods for haplotype inference, are needed to phase multiple overlapping sequence reads, and to allow reconstruction of whole genomes. HIV-1 is an attractive model system for developing such a global haplotype inference methodology due to its small genome, high intra-patient genetic diversity and clinical relevance.
Virus populations do not qualify for recent single-cell genomics approaches that have been applied to cancer cells and to some microbes (15), because these techniques require single-cell separation prior to genome amplification. Up to now, this segregation is not feasible for viruses, because they are too small. Thus, characterizing viral quasispecies has to rely on the analysis of assorted samples. The challenge is then to reliably solve a jigsaw puzzle of short error-prone sequence reads and to reconstruct the different constituent haplotypes over long genomic regions (16). Errors can occur at each of the required steps of viral genome amplification and during sequencing. Furthermore, estimating the relative frequencies of the viral haplotypes in the population is affected by amplification biases due to, for instance, primer mismatches and in vitro recombination (17). Specialized bioinformatics tools have been developed to cope with these error-prone NGS data and to reconstruct, from a multiple sequence alignment of reads, the haplotypes that constitute the viral population within a single host (16). Multiple read alignment can introduce additional uncertainty by placing insertions and deletions ambiguously.
To test genome-wide haplotype reconstruction on real data, we first combined five HIV-1 strains in a large batch to obtain similar aliquots of one mixture of heterogeneous viral haplotypes. In a second step to evaluate our analytical approach in a relevant clinical setting, we longitudinally studied a well-characterized HIV-1 infected individual who during the course of infection acquired a new viral strain by superinfection as documented by clonal sequencing of the partial env gene which encodes the viral envelope. The three NGS technologies 454/Roche, Illumina and PacBio were applied to sequence the full-length genomes of the five distinct HIV-1 strains, each requiring different protocols for sample preparation and sequencing. We used QuasiRecomb (18) and PredictHaplo (19) to correct sequencing errors, reconstruct the viral variants locally (within one read length) and globally (connecting overlapping reads).
MATERIALS AND METHODS
Viruses
293T cells were transfected separately with five different HIV-1 full-length plasmids using LipofectamineTM2000 (Invitrogen, Zug) according to the manufacturer's protocol. The HIV-1 full-length plasmids were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: pYK-JRCSF from Irvin S.Y. Chen and Yoshio Koyanagi; pNL4–3 from Malcolm Martin; pYU2 from Beatrice Hahn and George M. Shaw; p89.6 from Ronald G. Collman and pHXB2 was kindly provided by Marek Fischer. Cell supernatants were collected 48 h post-transfection after centrifugation for 5 min at 3000 revolutions per minute and then filtered through 0.22 μm Sterilflip® (Millipore, Zug) to obtain cell-free virus stocks. The virus stocks were quantified by real-time polymerase chain reaction (PCR) and pooled in approximate same amounts to obtain the 5-virus-mix, whose virus titer was 6.67 × 106 HIV-1 RNA copies/ml cell culture supernatant, as previously described (20). Numerous aliquots of this mix of intact virus particles were stored at −80°C.
Patient's samples were processed accordingly. This patient participates in the Zurich Primary HIV Infection (ZPHI) study, which is an observational, open label, non-randomized, single center study (www.clinicaltrials.gov; ID NCT00537966) (21,22). Written informed consent was obtained from the patient prior to inclusion.
HIV-1 full-length genome sequencing using the 454 Life Sciences/Roche platform
Approximately 106 HIV-1 RNA copies were isolated from the virus particles in the 5-virus-mix using the NucleoSpin® RNA Virus Kit (Macherey and Nagel, Oensingen) according to the manufacturer's protocol and eluted in 60 μl water. RNA was treated with 5 U DNase (DNase I recombinant, RNase-free; Roche, Mannheim) at 25°C for 30 min followed by 70°C for 15 min and then separated into three reverse transcription reactions, each containing 9.2 μl DNase treated RNA and a mix of 12 or 11 oligonucleotides for cDNA syntheses (0.2 μM end concentration for each oligonucleotide) (Supplementary Table S1). RNA plus oligonucleotide-mix was incubated at 65°C for 10 min followed by cooling at 4°C for 2 min. cDNA synthesis was performed with Transcriptor High Fidelity cDNA System (Roche) according to the manufacturer's protocol. The cDNA was treated with RNase H (New England Biolabs, Bioconcept, Allschwil) following the manufacturer's instructions. PCRs were performed using 5 μl of cDNA in a total volume of 30 μl: 94°C-5′, 30–35 x (94°C-15′, 57°C-45′, 72°C-60′) containing 0.4 mM dNTPs (Fermentas, Mont-sur-Lausanne), 3 U FastStart High Fidelity Polymerase (Roche) and 0.32 μM of each forward oligonucleotide primer A (5′–CGTATCGCCTCCCTCGCGCCA-TCAG-HIV-1–3′) and reverse oligonucleotide primer B (5′–CTATGCGCCTTGCCAGCCCGC-TCAG-HIV-1–3′) including A and B sequences at the 5′-ends necessary for the 454/Roche FLX/Titanium pyrosequencing method; HIV-1 specific oligonucleotide sequences are listed in Supplementary Table S1. All amplicons were gel purified and quantified with the Quant-iTTM PicoGreen® dsDNA Assay (Invitrogen). Equimolar amounts of gel-purified amplicons were pooled, clonally amplified on capture beads and sequenced using two large regions of of a 70 × 75 mm PicoTiterPlate. The standard bidirectional Amplicon protocol (Lib A) was followed according to manufacturer's guidelines (454 Life Sciences/Roche, Branford).
HIV-1 full-length genome sequencing using the Illumina platform
Approximately 105 HIV-1 RNA copies were isolated as described above except that the DNase treatment was done on the column after the first washing step and RNA was eluted in 25 μl water. Two multiplexed cDNA synthesis reactions were done with 10 μl DNase treated RNA containing two and three specific oligonucleotides (1 μM) and 0.5 mM dNTPs (Supplementary Table S1) were added and the mix was incubated at 65°C for 5 min followed by cooling at 4°C for 2 min. Reverse transcription was performed using the following conditions: 5x reaction buffer (Clontech, Saint-Germain-en-Laye), 20 U Protector RNase Inhibitor (Roche), 200 U PrimeScript Reverse Transcriptase (Clontech) and water to a total volume of 20 μl. The reaction mix was incubated at 42°C for 60 min followed by 70°C for 15 min. The cDNA was treated with RNase H (New England Biolabs) following the manufacturer's instructions. Five PCRs were performed with 2 μl of cDNA using 1 U of Platinum® Taq DNA Polymerase High Fidelity (Invitrogen), 0.4 mM of each dNTP and 0.5 μM oligonucleotides (Supplementary Table S1). The PCR cycling conditions were 94°C-2′, 35 x (94°C-30′, 55°C-30′, 68°C-2′40′). PCR products were purified with the Agencourt AMPure XP PCR Purification (Beckman Coulter, Krefeld) following the manufacturer's instructions and quantified as described above. The sequencing library was prepared with the Nextera XT DNA Sample Preparation Kit (Illumina, San Diego) according to the manufacturer's description and sequenced using a MiSeq Benchtop Sequencer generating paired-end reads of 2×250 bp length (v2 kit).
HIV-1 full-length genome sequencing using the Pacific Biosciences platform
Approximately 105 HIV-1 RNA copies were isolated as described above (Illumina). Five separate cDNA synthesis reactions were performed with 10 μl of DNase treated RNA, 1.2 mM of each dNTP and 0.6 μM oligonucleotide (Supplementary Table S1). The mix was incubated at 65°C for 5 min and 4°C for 2 min. cDNA synthesis was performed with 200 U of PrimeScript Reverse Transcriptase (Clontech) and 20 U Protector RNase Inhibitor (Roche) in a total volume of 20 μl at 42°C for 60 min and 70°C for 15 min. The cDNA was treated with RNase H (New England Biolabs) following the manufacturer's instructions. Eleven PCRs were performed with 2 μl cDNA using 0.4 U Platinum® Taq DNA Polymerase High Fidelity, 10x PCR reaction buffer, 2 mM MgSO4 (Invitrogen), 0.4 mM of each dNTP and 0.5 μM oligonucleotides (Supplementary Table S1). The PCR cycling conditions were 94°C-2′, 25–28 x (94°C-30′, 58°C-30′, 68°C-110′). PCR products were purified using NucleoSpin® Gel and PCR Clean-up (Machery and Nagel, Oensingen) following the manufacturer's instructions and purity was verified with the Bioanalyzer DNA 12000 Kit (Agilent Technologies, Basel). DNA was quantified as described above and amplicons were pooled to a total quantity of 2 μg. The pool was treated with PreCR® Repair Mix (New England Biolabs) to repair any DNA damage and the sequencing library was prepared using the DNA Template Prep Kit 2.0 (250 bp ≤ 3 kb) (Pacific Biosciences, Menlo Park), following the manufacturer's instructions. The Pacific Biosciences RS instrument was programmed to load the libraries on PacBio SMRT cells using the standard diffusion method. The circular consensus sequencing method has been used to ensure lower error rates. In order to achieve long read length, long movies of 120 min were taken.
HIV-1 full-length genome sequencing of clinical samples
Two clinical samples from an HIV-1 infected patient were used for HIV-1 full-length genome sequencing using both the Illumina (2×300 bp, v3 kit) and the Pacific Biosciences platforms. Sample preparation was performed as for the 5-virus-mix, except that 1 ml of plasma was centrifuged for 1 h at 50 000 g prior to RNA isolation. The cDNA synthesis was performed in two parallel reactions, each with three RT oligonucleotides (Supplementary Table S2), followed by nested or semi-nested PCR for the amplification of 5 and 11 amplicons, respectively (Supplementary Table S2).
Clonal env sequencing
Full length envelope single genome amplicons were derived from plasma virus by RNA extraction followed by DNase treatment (RNeasy, Qiagen, Hombrechtikon), reverse transcription using RT Superscript III (Invitrogen AG, Basel) and nested PCR amplification of diluted cDNA using Platinum Taq high fidelity polymerase (Invitrogen). For each sample 36 appropriate end-point dilutions containing ∼0.5 cDNA templates as determined by quantitative real-time PCR of the tat-rev-vpu region were processed. Bidirectional sequencing of 10 overlapping regions by dye terminator cycle sequencing (ABI Prism BigDye, Applied Biosystems, Rotkreuz) was accomplished as previously described (23). Sequences were assembled with SeqMan (DNASTAR Inc., Madison WI), checked for ambiguities, premature stop codons and hypermutations (www.hiv.lanl.gov/content/sequence/HYPERMUT) and analyzed by using BioEdit 7, Phylip 3.68 and MEGA 5.
Alignment
The read data sets from the three NGS platforms 454/Roche, Illumina and PacBio, were converted to fastq files. For 454/Roche and PacBio, the InDelFixer software (http://www.cbg.ethz.ch/software/InDelFixer/) was used to first extract reads and to perform quality clipping from both ends of the reads with a PHRED threshold of 30. Second, adaptors were removed and approximate matching regions were found in the reference genome using k-mer matching. Subsequently, each read was pairwise aligned to the HIV-1HXB2 reference sequence using a sensitive Smith–Waterman algorithm to create a multiple sequence alignment discarding insertions. Exchanging the HIV-1HXB2 reference sequence has no influence on the alignment result as long as the same HIV-1 subtype is chosen. For the Illumina data set, quality clipping was performed with sickle (https://github.com/najoshi/sickle) and alignments were produced with the Burrows–Wheeler Aligner BWA mem algorithm. Reads were discarded if three or more consecutive N base calls were present or if the read length was less than 150 nt, 300 nt and 1000 nt for Illumina, 454/Roche and PacBio, respectively.
Global haplotype reconstruction
Viral haplotype reconstruction of the 5-virus-mix was performed with the quasispecies assembly tool QuasiRecomb 1.2 (www.cbg.ethz.ch/software/quasirecomb) (18) employing the flags ‘-conservative’, because our focus was on dominant haplotypes, ‘-noRecomb’, as the underlying population structure was not subjected to recombination and the runtime reduced, and ‘-t 500’, specifying the number of restarts of the inference algorithm. In regions that are not known to harbor deletions ‘-noGaps’ was employed to weight down gaps and to increase convergence. For the clinical sample, QuasiRecomb was executed with default parameters. QuasiRecomb implements a hidden Markov model to infer a viral quasispecies from deep-coverage NGS data using an expectation maximization (EM) algorithm for maximum a posteriori (MAP) parameter estimation. It takes paired-end information explicitly into account. The model assumes that a complex quasispecies emerges from a few dominating haplotypes, called generators and represented as position-wise base probability distributions. Model selection, i.e. determining the number of generators, was performed gene-wise and five generators were always selected by the model without prior knowledge for the 5-virus-mix, unless haplotypes were missing. These generators, i.e. their mode, represented the haplotypes in the underlying quasispecies. The efficiency of global haplotype reconstruction is improved by informed generator initialization in the EM algorithm. We constructed initial generators hierarchically from previously reconstructed subregions by generating all combinations of the subregion haplotypes. For each combination, 10 random restarts were considered with initial generator probability tables drawn from Dirichlet distributions centered around each combined haplotype sequence, and the best combination was selected. Another 50 random restarts with initial generators randomized from the optimal combination were then used to find the final MAP estimates. All combinations of haplotypes were generated using ‘-mix -i region1.fasta -h region2.fasta’ and a specific initialization can be used with ‘-initMu haplotypes.fasta’. This approach is used for regions >1.5 kb, >2 kb and >5 kb, for Illumina, 454/Roche and PacBio, respectively.
Global haplotype reconstruction over long genomic regions, including genome-wide quasispecies assembly, was also performed with the PredictHaplo software, version 1.0 (http://bmda.cs.unibas.ch/HivHaploTyper/). PredictHaplo implements a fully probabilistic approach to quasispecies reconstruction. Given a set of aligned reads, its built-in Bayesian mixture model with a Dirichlet process prior allows for adaptively estimating the unknown number of underlying haplotypes. Inference is carried out via a Markov chain Monte Carlo sampling scheme that was optimized to scale with real-world NGS data sets. The reliability of a reconstructed haplotype sequence is estimated via an internal quality score which measures the amount of overlap of the subset of reads assigned to this sequence. The reconstruction process starts by locally clustering the aligned reads in order to identify the region with the highest number of high-quality clusters. This region then serves as a seed for globally reconstructing the quasispecies by propagating the mixture model. Once this process is completed, the internal quality scores are used for identifying reliable variant sequences. Prior parameters and threshold values have been set based on simulated data produced by the MetaSim program (24).
RESULTS
Experimental design
We combined five HIV-1 strains in a large batch to obtain similar aliquots of one mixture of heterogeneous viral haplotypes (Figure 1A). Three NGS technologies, namely, 454/Roche, Illumina and PacBio, were applied to sequence the full-length genomes of the mixture of HIV-1 strains, each requiring different protocols for sample preparation and sequencing. For 454/Roche, 35 ∼500 bp long amplicons were generated by reverse transcriptase-PCR (RT-PCR) overlapping in ∼250 bp (all oligonucleotides are given in Supplementary Table S1). For Illumina, five ∼2500 bp long amplicons were obtained by RT-PCR overlapping in ∼500 bp. Amplicons were enzymatically fragmented (Nextera tagmentation reaction) prior to sequencing, i.e. in contrast to the other two NGS technologies, we applied a shot-gun approach for Illumina. For PacBio, eleven ∼1500 bp long amplicons with ∼750 bp overlaps were amplified by RT-PCR (Figure 1A).
Figure 1.
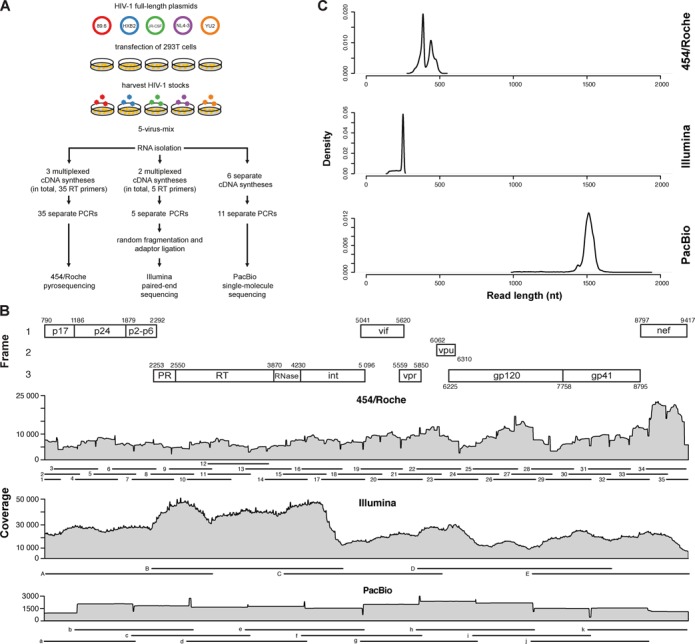
HIV-1 full-length genome sequencing using three different NGS technologies. (A) Experimental protocol. Five HIV-1 full-length plasmids were transfected into 293T cells to generate five different virus stocks. These clones were mixed in a large batch and aliquoted. RNA was isolated and amplified with three different protocols. DNA libraries were sequenced with either 454/Roche, Illumina or PacBio. (B) Coverage in overlapping reads per base pair. The map of the HIV-1 genome is shown on the top, with each subsequently analyzed gene indicated. The position numbering refers to the HIV-1HXB2 genome (GenBank accession number K03455). Amplicon layout is visualized for each NGS platform with individual numbering (Supplementary Table S1). (C) Read length distribution of each NGS technology, after preprocessing and alignment.
Sequencing of HIV-1 full-length genomes was successful for all amplicons, except amplicon 28 in the 454/Roche setup. The coverage had an average (range) of 7712 (2237–22 661), 23 010 (5084–47 232) and 1741 (450–3147), reads per nucleotide, for 454/Roche, Illumina and PacBio, respectively (Figure 1B). Average read lengths after pre-processing and alignment were (mean ± sd) 410 ± 41 bp, 237 ± 26 bp and 1499 ± 77 bp for 454/Roche, Illumina and PacBio, respectively (Figure 1C). To obtain the underlying ground truth, each of the five virus stocks was sequenced separately with Illumina prior to mixing them. Reads were aligned against the respective GenBank reference genome and a consensus sequence was called for each viral strain. As our goal was to define a consensus sequence for each strain, the coverage of high-quality reads was not sufficient to perform single-nucleotide variant (SNV) calling. Compared to the published GenBank sequences for those virus strains, we detected 2–36 mismatches per HIV-1 strain (Supplementary Table S3). The pair-wise relative Hamming distances of the 5-virus-mix were between 2.61% and 8.45% (Supplementary Table S4).
Gene-wise haplotype reconstruction reduces false SNV calls
We assessed the quality of gene-wise haplotype reconstruction by comparing SNV calls and haplotype predictions to the underlying ground truth for each of the three NGS platforms. The accuracy of SNV calls derived from gene-wise haplotype reconstructions was compared to the standard method based simply on alignment mismatches (Figure 2A). SNVs were called if their abundance was higher than a given threshold. For standard SNV calling, this threshold was the assumed technical error rate of 1%, except for deletions in the PacBio data set, where the cutoff was 5%, as the majority of PacBio errors are due to deletions: 32.2% and 7.3% of the positions harbor deletions over 1% and 5%, respectively. For haplotype-based SNV calling, all SNVs found on at least one reconstructed haplotype were called. Without haplotype reconstruction, SNV calls that appeared in the reads but are missing in the five ground truth sequences (false positives) were numerous and distributed all over the sequenced region, for PacBio and 454/Roche, but considerably less frequent for Illumina. The false positive calls in the 454/Roche and PacBio data sets were dominated by deletions, which are common errors for these two technologies. Illumina sequence reads showed a 9- and 17-fold lower false positive rate than 454/Roche and PacBio, respectively. SNVs that are present in the ground truth reference sequences but were missing in the reads (false negatives) were only found in regions with missing amplicons (amplicon 28 for 454/Roche and variant HIV-1YU2 in amplicon i for PacBio). Gene-wise haplotype reconstruction, including both local and global reconstruction depending on read and gene lengths, reduced the rates of false positive calls substantially to 0.04%, 0.08% and 0.0%, for 454/Roche, Illumina and PacBio, respectively (Figure 2B). Haplotype inference did not only eliminate insertion and deletion errors, but also almost all substitution errors. Comparing the false positive and false negative calls prior and after haplotype reconstruction between the three NGS technologies showed that the number of joint errors is low, except for deletions, where a substantial number of gaps were present in 454/Roche and PacBio sequence reads (Figure 2B). However, these errors were successfully removed during haplotype reconstruction.
Figure 2.
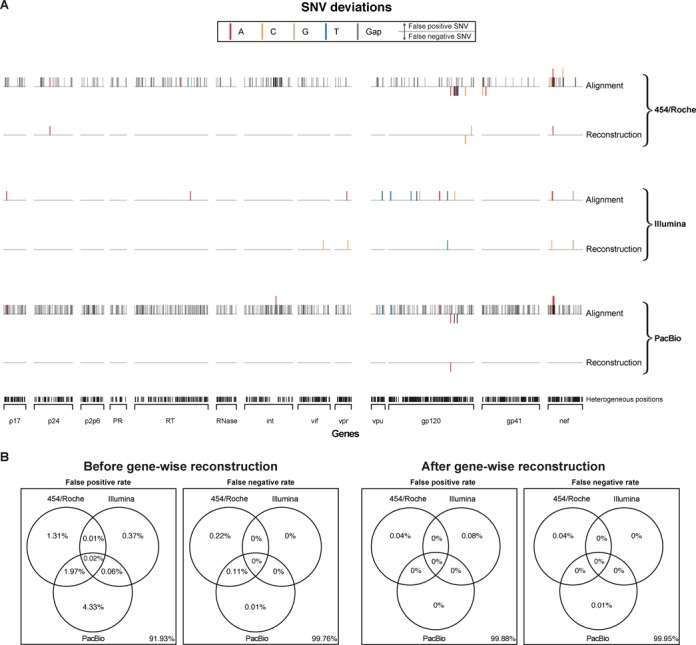
SNV calling based only on the alignment (naïve) versus haplotype reconstruction. (A) Distribution of SNVs across the full-length HIV-1 coding region compared to the ground truths for each NGS platform, on top shown as directly obtained from the alignment and on the bottom after gene-wise haplotype reconstruction. A SNV is called from the alignment, when its relative occurrence is higher than 1%, except for the deletions within the PacBio data, where a threshold of 5% was applied. SNVs from inferred haplotypes are called if found in at least one reconstructed haplotype. False positive SNVs and false negative SNVs are represented as upward and downward pointing bars, respectively. Bars are color coded for the four different nucleotides and the gap-symbol, bars may stack for multiple false calls at a single position, and false positives and false negatives may occur at the same position. (B) Co-occurrence of false positive and false negative SNVs among NGS platforms shown as Venn diagrams, with and without gene-wise haplotype reconstruction.
Gene-wise haplotype reconstruction and frequency estimation of HIV-1 variants
For each data set obtained from one of the three NGS platforms, we reconstructed haplotypes and estimated their frequencies on different spatial scales of the viral genome. Haplotypes were inferred for each viral gene separately using QuasiRecomb (18). The five haplotypes were correctly identified for all 13 HIV-1 genes using sequence reads obtained from any of the three NGS platforms (Figure 3), with the single exception of gp120 in the 454/Roche data, where one amplicon was missing (amplicon 28, Figure 1B). Successful amplification of all fragments is necessary, because missing overlaps generally disrupt haplotype reconstruction. Out of 38 gene-wise reconstructions in total, all five virus variants were predicted in 36 genes without any mismatch relative to the ground truth. In one case, from the Illumina sequence reads, only four gp41 haplotypes were perfectly reconstructed, because the relative Hamming distance between the two closest haplotypes is below 1%. Here, the short sequence reads as obtained by Illumina were disadvantageous. In the other case, the reconstruction of HIV-1YU2 gp120 had one mismatch relative to the ground truth using the PacBio data set, because this base was only covered by the missing variant HIV-1YU2 in amplicon i (Figure 1B).
Figure 3.
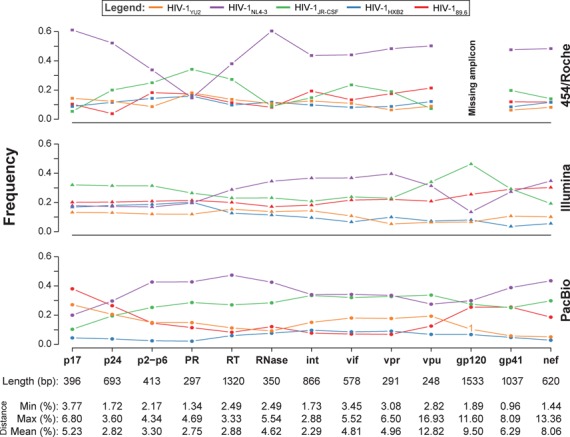
Gene-wise haplotype reconstruction. Haplotypes were reconstructed for the genes p17, p24, p2-p6 (functional regions in gag), PR, RT, RNase, Int (pol, polymerase), gp120, gp41 (env, envelope) and the accessory genes vif, vpr, vpu and nef using QuasiRecomb. The five distinct HIV-1 strains are color coded and their frequencies in each region are shown for each NGS platform. The length of each region is denoted in base pairs at the bottom of each column together with the genetic distances of the five HIV-1 variants against each other in the corresponding regions. A number instead of a symbol indicates the Hamming distance of the reconstructed haplotype to its closest match in the ground truth.
We previously estimated the frequencies of the virus strains in the 5-virus-mix by amplifying the protease gene using single-genome amplification (SGA) (20), the current gold standard for studying diversity of virus populations (25). Here, we used those data for comparison with the NGS-based frequency estimates (Figure 3). For each NGS platform, the gene-wise median haplotype frequencies did not differ significantly from those obtained by SGA (Table 1, p = 0.9776, Kruskal–Wallis test (26)). However, frequency estimates did fluctuate locally between genes.
Table 1. Estimated frequencies of reconstructed haplotypes across genes.
| Estimated frequencies% (mean ± sd)a | |||||
|---|---|---|---|---|---|
| Sequencing technology | HIV-1 89.6 | HIV-1 HXB2 | HIV-1 JR-CSF | HIV-1 NL4–3 | HIV-1 YU2 |
| 454/Roche | 13.7 ± 5.1 | 11.0 ± 2.4 | 18.2 ± 8.6 | 46.2 ± 12.9 | 10.9 ± 3.5 |
| Illumina | 22.1 ± 3.9 | 11.2 ± 5.9 | 28.0 ± 7.3 | 27.3 ± 9.2 | 11.1 ± 3.2 |
| PacBio | 16.5 ± 9.7 | 5.8 ± 2.6 | 27.2 ± 6.4 | 35.9 ± 7.9 | 14.6 ± 6.2 |
| SGA | 12.3 ± 2.3 | 9.2 ± 3.3 | 27.9 ± 4.6 | 38.4 ± 3.2 | 12.2 ± 0.7 |
NGS, next-generation sequencing.
aFor the NGS platforms (columns 1–3), the mean and standard deviation of each frequency across all 13 gene-wise predictions are reported. For single genome analysis (SGA), the estimates are based on three replicates with 168, 156 and 148 protease sequences.
Global haplotype reconstruction of HIV-1 full-length genomes
5-virus-mix
Next, we extended global haplotype reconstruction first to the HIV-1 gag-pol region spanning 4036 bp and subsequently to the entire coding region of HIV-1 spanning a total of 8627 bp (referred to as full-length, despite the omitted non-coding regions of the long terminal repeats). In the gag-pol region, all five virus strains were perfectly reconstructed using either QuasiRecomb or PredictHaplo (Figure 4). Sequence reads from all three NGS platforms were suitable for global reconstruction of the entire gag-pol region. For full-length haplotype reconstruction, we assembled reads over a genomic region six times longer than the ∼1.5 kb long amplicons used for PacBio single-molecule sequencing. With PacBio data, haplotype reconstruction using QuasiRecomb was almost error-free with only a single mismatch in HIV-1YU2 relative to the closest ground truth (Figure 4). Using PredictHaplo, we also reconstructed all five haplotypes correctly, but only three haplotypes passed PredictHaplo's internal quality control (Figure 4); exclusion of two haplotypes was due to insufficient overlap of reads. For the short Illumina reads, no haplotype passed PredictHaplo's conservative quality control, but full-length reconstruction was successful with QuasiRecomb, allowing only nine mismatches for HIV-1HXB2 in gp41 (Figure 4). We perfectly recovered the first 5272 bp of the HIV-1 coding region when using the 454/Roche data, but the assembly was discontinued due to the missing amplicon 28. When we merged the 454/Roche and PacBio data sets, all positions were covered by amplicons with sufficient overlap, and perfect full-length haplotype reconstruction was achieved using either PredictHaplo or QuasiRecomb (Figure 4). The frequency estimates were almost identical between the different NGS technologies and were also in agreement with the SGA results. Fluctuations in variant frequencies were substantially reduced for the frequency estimates based on the full-length reconstructed haplotypes with standard deviations around one order of magnitude smaller than for the gene-wise reconstruction (Tables 1 and 2).
Figure 4.
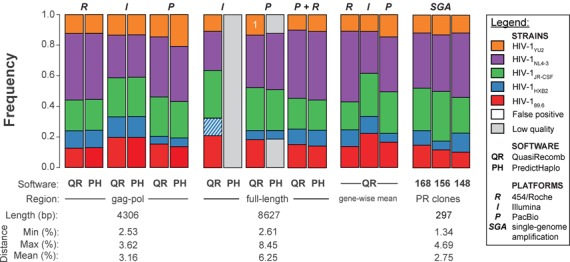
Global haplotype reconstruction of the HIV-1 gag-pol genomic region and of full-length genomes. Estimated frequencies are shown for the HIV-1 gag-pol region and the full-length coding region for each data set and each computational method. Gray shaded areas represent haplotypes that did not pass quality control; white striped area shows reconstructed HIV-1HXB2 with a Hamming distance of nine. The white number in the QuasiRecomb result indicates the Hamming distance to the closest ground truth. For comparison, the mean gene-wise frequencies for each data set using QuasiRecomb and the frequencies of each virus strain detected by SGA of the protease gene in three independent experiments (20). Numbers of analyzed clones are given below each bar.
Table 2. Estimated frequencies of reconstructed haplotypes over the full-length coding region.
| Estimated frequencies% (mean ± sd)a | |||||
|---|---|---|---|---|---|
| NGS platform | HIV-1 89.6 | HIV-1 HXB2 | HIV-1 JR-CSF | HIV-1 NL4–3 | HIV-1 YU2 |
| 454/Roche | 14.4 ± 0.4 | 11.0 ± 0.4 | 19.9 ± 0.5 | 43.5 ± 1.0 | 11.1 ± 0.4 |
| Illumina | 22.6 ± 0.3 | 10.0 ± 0.4 | 29.6 ± 0.4 | 26.9 ± 0.4 | 10.9 ± 0.3 |
| PacBio | 17.6 ± 0.3 | 5.9 ± 0.5 | 27.1 ± 0.5 | 36.2 ± 0.5 | 13.1 ± 0.4 |
NGS, next-generation sequencing.
aFor each platform, we computed mean and standard deviation of full-length reconstructed haplotype frequencies of 100 bootstrap samples from 179 521; 1 365 693 and 10 651 reads for 454/Roche, Illumina and PacBio, respectively.
Patient's samples
The first sample of this patient (sample A) was obtained ∼7 weeks after estimated day of primary HIV-1 infection (Figure 5A). Antiretroviral therapy was started at this time point and interrupted at week 91 according to the study protocol. The second sample (sample B) was obtained at week 99. Superinfection with a second HIV-1 strain presumably occurred within the weeks following week 91 as primarily revealed by extensive clonal env sequencing in two independent laboratories. At week 7, 9/9 env clones contained one haplotype and at week 107, 4/8 env clones contained another, distinct haplotype and the remaining 4 clones were recombinants between the first and the superinfecting HIV-1 strains.
Figure 5.
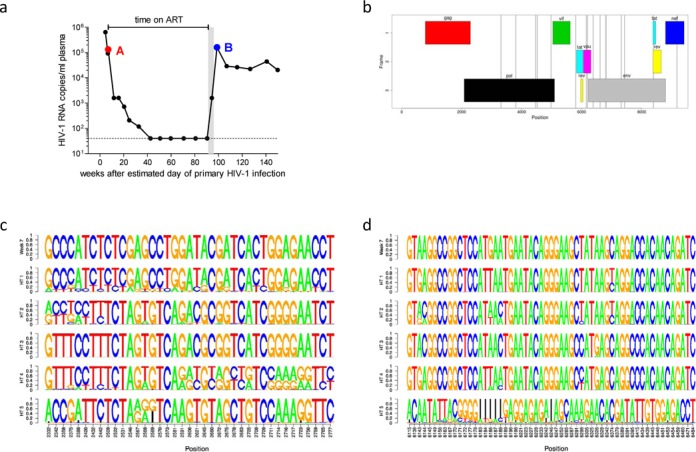
Global haplotype reconstruction of HIV-1 in an HIV-1 superinfected patient. (a) HIV-1 RNA load (black circles) and estimated time of superinfection (gray shaded area). Depicted are time points A (∼7 weeks after primary HIV-1 infection) and B (∼99 weeks after primary HIV-1 infection) that were chosen for HIV-1 full-length sequencing using the Illumina platform. Global haplotype reconstruction was performed using both computational methods, data obtained with the PredictHaplo model are depicted. The dotted line shows the detection limit of the viral load assay (40 HIV-1 RNA copies/ml plasma). (b) Mismatches between haplotype 1 (HT 1) to the haplotype from week 7 are shown as gray vertical lines within the HIV-1 gene map. (c and d) Sequence logo representation of all five reconstructed haplotypes (HT1 – HT 5) and the respective haplotype from week 7 in a window between position 2300 and 2800 (c) and between 6100 and 6500 (d). The frequency information in the sequence logos reflects the statistical properties of the PredictHaplo model, where haplotypes are represented as chains of probability tables over the four nucleotides plus gaps. Shown are only positions where at least one haplotype differs from the others.
Samples A and B were sequenced using the Illumina and Pacific Biosciences platforms. The coverage had an average (range) of 168 401 (30 650–255 500) and 3203 (1940–5524) reads per nucleotide, respectively. At week 7, we reconstructed a single full-length haplotype in the virus population. After superinfection at week 99, two haplotypes with frequencies of 97.2% and 2.8% were reconstructed by QuasiRecomb from the Illumina reads in a window that covered gp41 of the env region. The env clones were used to validate the reconstructed haplotypes and indeed, the haplotypes with frequencies of 97.2% and 2.8% matched with zero and two base pair mismatches to env clones of weeks 99 and 7, respectively. Sequence dissimilarity between the two haplotypes was 10.5%.
For global, i.e. genome-wide, haplotype reconstruction, we changed the computational setup in two important details: first, we used the PredictHaplo software which is specifically tailored for this purpose and contrary to QuasiRecomb which is more suitable for gene-wise reconstructions, and second, the Illumina reads were combined with the much longer PacBio reads to ensure sufficient overlaps around all amplicon borders. Global reconstruction with PredictHaplo based on these combined Illumina/PacBio reads yielded five global haplotypes at week 99. One of these haplotypes closely resembled the single full-length haplotype from week 7. On the whole genome these two sequences differ only at 16 positions (Figure 5B). Probably due to primer bias, the frequency estimate for this haplotype varied with the position in the genome, but the small median frequency over all positions of only 1.9% clearly indicates that this was a minor variant at week 99. The composition of all five reconstructed haplotypes varied over the whole genome. While in some local windows, four of the haplotypes (HT 1–HT 4) were composed of local variations of the sequence from week 7 and only one haplotype (HT 5) was substantially different (Figure 5C and D). In other local windows we found more complicated patterns that indicate the presence of at least three major variants and recombination events. Recombinant haplotypes of week 7 and week 99 have been reconstructed by PredictHaplo and QuasiRecomb. The number of recombination events varied spatially, from no recombination in gp41 to up to 19 recombination sites in the first 500 bp of the pol-region. We focused on gp41 of the env region in which two variants were detected with QuasiRecomb. A comparison with the five global haplotypes identified by PredictHaplo corroborates these previous findings: In this region, three of the five global haplotypes are identical and match perfectly to the dominating sequence found by QuasiRecomb, i.e. the sequence of the env-clones of week 99. These three haplotypes together have an estimated frequency of 98.1%, which is also very close to the QuasiRecomb frequency estimate. From the remaining 1.9% of the virus population, one global haplotype matches perfectly to the minor env variant found by QuasiRecomb, i.e. the env-clones of week 7. The last of the five global haplotypes might be a recombination of these two sequences, but given the tiny fraction of this haplotype and the relatively high diversity of the associated reads, this interpretation is not entirely clear. Further, such a recombination could not be verified by QuasiRecomb.
DISCUSSION
We have shown that global haplotype reconstruction of distinct viral genomes within a heterogeneous virus population is feasible using short sequence reads of a mixture of HIV-1 haplotypes in defined combinations of HIV-1 strains as well as in a real patient's sample. Perfect global reconstruction and correct frequency estimation were accomplished applying three different NGS platforms, namely, 454/Roche, Illumina and PacBio, despite their differences in read length, coverage and error patterns (3). Each of these technologies has specific advantages and disadvantages when used for viral haplotype reconstruction. Besides NGS technology, viral haplotype assembly also depends critically on the genetic heterogeneity of the underlying population and the diversity distribution along the genome.
454/Roche offers intermediate read length and coverage, and the errors occur mainly in homopolymeric regions (27). However, this technology is very labor-intensive and requires a high number of amplicons to cover the complete HIV-1 genome with sufficient overlap of neighboring amplicons, which increases the risk of interruptions in global haplotype reconstruction. Indeed, in our data set, the missing amplicon 28 (out of 35) in the 454/Roche data prevented full-length haplotype reconstruction. Illumina generates short reads at very high coverage and a low error rate (28). Full-length genomes of four out of five HIV-1 haplotypes were perfectly reconstructed, but the fifth haplotype could not be reconstructed correctly. The viral gene gp41 in the variants HIV-1HXB2 and HIV-1NL4–3 shows a distance of less than 1%, which is below the assumed technical error rate. The short read length did not allow to bridge this region of low diversity, thus, full-length haplotype reconstruction was only partly successful based on the Illumina data set. This finding shows that high coverage and low error rates cannot compensate for short read length. Pacific Biosciences enables long read length, but the coverage per run is low and the error rate high (29). Nevertheless, using this data set alone, perfect full-length haplotype reconstruction of all five HIV-1 viruses was successful. One mismatch was observed in the reconstructed HIV-1YU2 strain compared to the ground truth. This virus was not successfully amplified in one of 11 amplicons for the PacBio procedure. For gene-wise reconstruction, we have shown that a minimal relative Hamming distance of 1% is sufficient for successful reconstruction.
All three NGS technologies enabled perfect reconstruction of the entire gag-pol region of over 4000 nucleotides thereby exceeding the current maximal read length by several fold. We observed fluctuations in haplotype frequencies performing gene-wise reconstruction that most probably were due to PCR artifacts especially in the 454/Roche or PacBio data sets. Amplification biases can lead to the loss of a haplotype during PCR, either by primer mismatches or the potential PCR artifact of differential amplification or the combination of both (17,30,31), and therefore influence the frequency estimations of viral variants. We ruled out that coverage, genetic distance or read length caused these fluctuations (data not shown). However, we observed that these fluctuations were substantially reduced when we expanded the global haplotype reconstruction to the entire gag-pol or even the full-length genome. This shows that amplification biases can be averaged out by including several amplicons in the global haplotype reconstruction.
A recent study by Prosperi et al. (32) measured the performance of different assembly methods, including PredictHaplo but not QuasiRecomb. They used a mixed population with 20 genetically different HIV-1 variants of which they tried to reconstruct a 1.4 kb long region based on 350 bp long reads. Despite the six times shorter region than the full-length HIV-1 genome, none of the tested programs was able to reconstruct more than six correct haplotypes and all of them produced false positive haplotypes. We have improved and extended existing haplotype reconstruction methods considerably to accomplish HIV-1 full-length assembly. Specifically, we have developed a hierarchical assembly strategy based on QuasiRecomb's inference model, and we have introduced quality scoring as well as paired-end support to PredictHaplo. Both extensions are essential and the resulting methods are the first probabilistic models capable of HIV-1 full-length haplotype assembly. The more conservative approach of PredictHaplo avoids false positive calls, but may miss haplotypes if they cannot be explained by sufficiently overlapping reads. On the other hand, QuasiRecomb is less strict leading to a higher quote of reconstructed haplotypes, but it requires several restarts of the optimization procedure to obtain the correct haplotypes. Both methods could be further improved in several ways. For example, read alignment could be integrated into the reconstruction process to better handle insertions/deletions. In QuasiRecomb, deletions may be modeled explicitly as long jumps between generators to account for the alignment uncertainty in deletion-prone regions. When it comes to comparing the two software tools, we suggest as a general rule to use QuasiRecomb for gene-wise diversity and recombination studies, and the more conservative but computationally more efficient PredictHaplo software for genome-wide reconstructions.
In conclusion, we have shown that reconstruction of whole genomes from a sufficiently heterogeneous virus population is feasible using a variety of different NGS technologies. The most apparent difficulty in global reconstruction of haplotypes is the reconstruction of regions with low diversity. The sequence read overlaps need to be long enough to bridge such conserved region. The data generated in this study is based on the same mixture of HIV-1 strains using three of the most common NGS platforms. It can be used to benchmark current and upcoming tools for global haplotype reconstruction. Furthermore, as a proof of concept for future in vivo studies we successfully applied our approach to a well characterized intraclade superinfection case as defined by conventional clonal env sequencing. We were able to reconstruct the initial founder haplotype. After superinfection, we could reconstruct the haplotype of the superinfecting strain which was present at a frequency of 99%. In addition, we were able to find the initial founder haplotype again at a very low frequency of ∼2% after superinfection has occurred and the new superinfecting haplotypes in addition to recombinants were responsible for ∼98% of the virus population at week 99. Of note, by conventional clonal sequencing the initial founder strain could not be detected at this time point. Since the first case of superinfection was described (33), it is worried that superinfection could have detrimental implications on HIV therapy and future vaccines, thus, more detailed investigations of virus populations in HIV-1 superinfected patients are needed. Taken together our single-haplotype genomics approach provides a powerful instrument to investigate the evolution of HIV-1 and other viruses also in vivo.
ACCESSION NUMBER
The three data sets (454/Roche, Illumina and PacBio) of the 5-virus-mix have been deposited in the Sequence Read Archive under accession number SRP029432. The links to the data sets and the consensus sequences of the individual strains are available at https://github.com/armintoepfer/5-virus-mix.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We are grateful to our patients for their commitment and participation in the ZPHI study. We thank Ch. Grube, B. Hasse, U. Karrer, R. Oberholzer, L. Aceto, R. Laffer, U. von Both, M. Huber, K. Thierfelder, E. Presterl, Y. Flammer, S. Kuster, J. Nemeth, A. von Braun, D. Braun,, T. Frey and M. Flepp for excellent patient care, H. Kuster, F. Burgener and D. Klimpel for technical help and I. Nievergelt and C. Vögtli for administrative assistance. Illumina sequencing of the 5-virus-mix was performed in the Quantitative Genomics Facility at D-BSSE, ETH Zurich.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
Swiss National Science Foundation [CR32I2_127017 and 146331 to N.B., K.J.M., V.R. and H.F.G.] and [130865 to H.F.G.]; University of Zurich's Clinical Research Priority Program Viral Infectious Diseases: ZPHI study [to A.T. and H.F.G.].
Conflict of interest statement. M.D. has received travel grants from Abbott and MSD Sharp & Dohme, has received a research grant from MSD Sharp & Dohme and has been an adviser for MSD Sharp & Dohme, Gilead Sciences and Janssen & Cilag. H.F.G. has been an adviser and/or consultant for the following companies: GlaxoSmithKline, Abbott, Gilead, Novartis, Boehringer Ingelheim, Roche, Tibotec, Pfizer and Bristol-Myers Squibb, and has received unrestricted research and educational grants from Roche, Abbott, Bristol-Myers Squibb, Gilead, Astra-Zeneca, GlaxoSmithKline and Merck Sharp & Dohme (all money went to institution). K.J.M. received travel grants and honoraria from Gilead Sciences, Roche Diagnostics, Tibotec, Bristol-Myers Squibb and Abbott; the University of Zurich has received research grants from Gilead, Roche and Merck Sharp & Dohme for studies that K.J.M. serves as principal investigator and advisory board honoraria from Gilead Sciences.
REFERENCES
- 1.Nowak M.A. What is a quasispecies? Trends Ecol. Evol. 1992;7:118–121. doi: 10.1016/0169-5347(92)90145-2. [DOI] [PubMed] [Google Scholar]
- 2.Domingo E., Sheldon J., Perales C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012;76:159–216. doi: 10.1128/MMBR.05023-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Metzker M.L. Sequencing technologies—the next generation. Nat. Rev. Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 4.Macalalad A.R., Zody M.C., Charlebois P., Lennon N.J., Newman R.M., Malboeuf C.M., Ryan E.M., Boutwell C.L., Power K.A., Brackney D.E., et al. Highly sensitive and specific detection of rare variants in mixed viral populations from massively parallel sequence data. PLoS Comput. Biol. 2012;8:e1002417. doi: 10.1371/journal.pcbi.1002417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henn M.R., Boutwell C.L., Charlebois P., Lennon N.J., Power K.A., Macalalad A.R., Berlin A.M., Malboeuf C.M., Ryan E.M., Gnerre S., et al. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 2012;8:e1002529. doi: 10.1371/journal.ppat.1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bimber B.N., Dudley D.M., Lauck M., Becker E.A., Chin E.N., Lank S.M., Grunenwald H.L., Caruccio N.C., Maffitt M., Wilson N.A., et al. Whole-genome characterization of human and simian immunodeficiency virus intrahost diversity by ultradeep pyrosequencing. J. Virol. 2010;84:12087–12092. doi: 10.1128/JVI.01378-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poon A.F., Swenson L.C., Bunnik E.M., Edo-Matas D., Schuitemaker H., van ‘t Wout A.B., Harrigan P.R. Reconstructing the dynamics of HIV evolution within hosts from serial deep sequence data. PLoS Comput. Biol. 2012;8:e1002753. doi: 10.1371/journal.pcbi.1002753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tilton J.C., Wilen C.B., Didigu C.A., Sinha R., Harrison J.E., Agrawal-Gamse C., Henning E.A., Bushman F.D., Martin J.N., Deeks S.G., et al. A maraviroc-resistant HIV-1 with narrow cross-resistance to other CCR5 antagonists depends on both N-terminal and extracellular loop domains of drug-bound CCR5. J. Virol. 2010;84:10863–10876. doi: 10.1128/JVI.01109-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsibris A.M., Korber B., Arnaout R., Russ C., Lo C.C., Leitner T., Gaschen B., Theiler J., Paredes R., Su Z., et al. Quantitative deep sequencing reveals dynamic HIV-1 escape and large population shifts during CCR5 antagonist therapy in vivo. PLoS One. 2009;4:e5683. doi: 10.1371/journal.pone.0005683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zagordi O., Klein R., Daumer M., Beerenwinkel N. Error correction of next-generation sequencing data and reliable estimation of HIV quasispecies. Nucleic Acids Res. 2010;38:7400–7409. doi: 10.1093/nar/gkq655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schirmer M., Sloan W.T., Quince C. Benchmarking of viral haplotype reconstruction programmes: an overview of the capacities and limitations of currently available programmes. Briefings Bioinformat. 2012;15 doi: 10.1093/bib/bbs081. [DOI] [PubMed] [Google Scholar]
- 12.Prosperi M.C., Prosperi L., Bruselles A., Abbate I., Rozera G., Vincenti D., Solmone M.C., Capobianchi M.R., Ulivi G. Combinatorial analysis and algorithms for quasispecies reconstruction using next-generation sequencing. BMC Bioinformat. 2011;12:5. doi: 10.1186/1471-2105-12-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skums P., Mancuso N., Artyomenko A., Tork B., Mandoiu I., Khudyakov Y., Zelikovsky A. Reconstruction of viral population structure from next-generation sequencing data using multicommodity flows. BMC Bioinformat. 2013;14:S2. doi: 10.1186/1471-2105-14-S9-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zagordi O., Daumer M., Beisel C., Beerenwinkel N. Read length versus depth of coverage for viral quasispecies reconstruction. PLoS ONE. 2012;7:e47046. doi: 10.1371/journal.pone.0047046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stepanauskas R. Single cell genomics: an individual look at microbes. Curr. Opin. Microbiol. 2012;15:613–620. doi: 10.1016/j.mib.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 16.Beerenwinkel N., Gunthard H.F., Roth V., Metzner K.J. Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data. Front Microbiol. 2012;3:329. doi: 10.3389/fmicb.2012.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanagawa T. Bias and artifacts in multitemplate polymerase chain reactions (PCR) J. Biosci. Bioeng. 2003;96:317–323. doi: 10.1016/S1389-1723(03)90130-7. [DOI] [PubMed] [Google Scholar]
- 18.Töpfer A., Zagordi O., Prabhakaran S., Roth V., Halperin E., Beerenwinkel N. Probabilistic inference of viral quasispecies subject to recombination. J. Comput. Biol. 2013;20:113–123. doi: 10.1089/cmb.2012.0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prabhakaran S., Rey M., Zagordi O., Beerenwinkel N., Roth V. HIV haplotype inference using a propagating Dirichlet process mixture model. IEEE/ACM Trans. Comput. Biol. Bioinform. 2013;11 doi: 10.1109/TCBB.2013.145. [DOI] [PubMed] [Google Scholar]
- 20.Di Giallonardo F., Zagordi O., Duport Y., Leemann C., Joos B., Kunzli-Gontarczyk M., Bruggmann R., Beerenwinkel N., Gunthard H.F., Metzner K.J. Next-generation sequencing of HIV-1 RNA genomes: determination of error rates and minimizing artificial recombination. PLoS ONE. 2013;8:e74249. doi: 10.1371/journal.pone.0074249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Metzner K.J., Scherrer A.U., Preiswerk B., Joos B., von Wyl V., Leemann C., Rieder P., Braun D., Grube C., Kuster H., et al. Origin of minority drug-resistant HIV-1 variants in primary HIV-1 infection. J. Infect. Dis. 2013;208:1102–1112. doi: 10.1093/infdis/jit310. [DOI] [PubMed] [Google Scholar]
- 22.Rieder P., Joos B., Scherrer A.U., Kuster H., Braun D., Grube C., Niederost B., Leemann C., Gianella S., Metzner K.J., et al. Characterization of human immunodeficiency virus type 1 (HIV-1) diversity and tropism in 145 patients with primary HIV-1 infection. Clin. Infect. Dis. 2011;53:1271–1279. doi: 10.1093/cid/cir725. [DOI] [PubMed] [Google Scholar]
- 23.Manrique A., Rusert P., Joos B., Fischer M., Kuster H., Leemann C., Niederost B., Weber R., Stiegler G., Katinger H., et al. In vivo and in vitro escape from neutralizing antibodies 2G12, 2F5, and 4E10. J. Virol. 2007;81:8793–8808. doi: 10.1128/JVI.00598-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richter D.C., Ott F., Auch A.F., Schmid R., Huson D.H. MetaSim: a sequencing simulator for genomics and metagenomics. PLoS One. 2008;3:e3373. doi: 10.1371/journal.pone.0003373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salazar-Gonzalez J.F., Bailes E., Pham K.T., Salazar M.G., Guffey M.B., Keele B.F., Derdeyn C.A., Farmer P., Hunter E., Allen S., et al. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J. Virol. 2008;82:3952–3970. doi: 10.1128/JVI.02660-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kruskal W.H., Wallsi W.A. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 1952;47:583–621. [Google Scholar]
- 27.Margulies M., Egholm M., Altman W.E., Attiya S., Bader J.S., Bemben L.A., Berka J., Braverman M.S., Chen Y.J., Chen Z., et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bentley D.R., Balasubramanian S., Swerdlow H.P., Smith G.P., Milton J., Brown C.G., Hall K.P., Evers D.J., Barnes C.L., Bignell H.R., et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eid J., Fehr A., Gray J., Luong K., Lyle J., Otto G., Peluso P., Rank D., Baybayan P., Bettman B., et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 30.Jabara C.B., Jones C.D., Roach J., Anderson J.A., Swanstrom R. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc. Natl. Acad. Sci. U.S.A. 2011;108:20166–20171. doi: 10.1073/pnas.1110064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mild M., Hedskog C., Jernberg J., Albert J. Performance of ultra-deep pyrosequencing in analysis of HIV-1 pol gene variation. PLoS ONE. 2011;6:e22741. doi: 10.1371/journal.pone.0022741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prosperi M.C., Yin L., Nolan D.J., Lowe A.D., Goodenow M.M., Salemi M. Empirical validation of viral quasispecies assembly algorithms: state-of-the-art and challenges. Sci. Rep. 2013;3:2837. doi: 10.1038/srep02837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jost S., Bernard M.C., Kaiser L., Yerly S., Hirschel B., Samri A., Autran B., Goh L.E., Perrin L. A patient with HIV-1 superinfection. N. Engl. J. Med. 2002;347:731–736. doi: 10.1056/NEJMoa020263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.