


Abstract
The findings that microRNAs (miRNAs) are essential for early development in many species and that embryonic miRNAs can reprogram somatic cells into induced pluripotent stem cells suggest that these miRNAs act directly on transcriptional and chromatin regulators of pluripotency. To elucidate the transcription regulatory networks immediately downstream of embryonic miRNAs, we extended the motif activity response analysis approach that infers the regulatory impact of both transcription factors (TFs) and miRNAs from genome-wide expression states. Applying this approach to multiple experimental data sets generated from mouse embryonic stem cells (ESCs) that did or did not express miRNAs of the ESC-specific miR-290-295 cluster, we identified multiple TFs that are direct miRNA targets, some of which are known to be active during cell differentiation. Our results provide new insights into the transcription regulatory network downstream of ESC-specific miRNAs, indicating that these miRNAs act on cell cycle and chromatin regulators at several levels and downregulate TFs that are involved in the innate immune response.
INTRODUCTION
Embryonic stem cells (ESCs) originate from the inner cell mass of mammalian blastocysts. Due to their ability to self-renew as well as differentiate into various specialized cell types, they hold the promise of medical applications, such as stem cell therapy and tissue engineering. Therefore, the regulatory mechanisms behind pluripotency, stem cell fate and renewal are of great interest.
MiRNAs are short (∼22 nt long), single-stranded RNAs that post-transcriptionally regulate the expression of target genes (1). Computational and high-throughput studies suggest that a single miRNA can regulate hundreds of target genes (2,3) and that the majority of human mRNAs are regulated by miRNAs (4). Several studies found that the expression of ESC-specific miRNAs is required for initiation of stem cell differentiation and normal embryonic development (5–7). The ESC-specific miR-290-295 cluster accounts for ∼50% of the miRNA population of mouse ESCs (8–11) and its expression is downregulated relatively rapidly during differentiation (9,12). Interestingly, three of the seven miRNAs that are co-expressed from the miR-290-295 cluster, namely, miR-291a-3p, miR-294 and miR-295, are sufficient to force a G1→S transition (13) and promote induced pluripotency (14). All of these miRNAs, as well as those of another ESC-specific miRNA cluster, miR-302-367 (12,15), have the same sequence ‘AAGUGCU’ at positions 2-8 (also called the ‘seed’) which defines a family of miRNAs with related targets (4).
In contrast to the miR-290-295 cluster, miR-302-367 is also present in human and has been used to reprogram fibroblasts into induced pluripotent stem cells (iPSCs) (16). The reprogramming of differentiated cells into pluripotent stem cells by the ESC-specific miRNAs entails large gene expression and phenotypic changes that are likely to be due to regulatory cascades that involve several regulators. To identify transcriptional regulators that are immediate targets of the AAGUGCU seed family miRNAs, we analyzed data obtained in several previous studies that aimed to uncover the function of the miR-290-295 cluster.
These data consist of microarray-based measurements of mRNA expression in ESCs that were either deficient in miRNAs or expressed subsets of ESC-specific miRNAs (Supplementary Table S1). Sinkkonen et al. (17) analyzed mRNA expression of ESCs that express miRNAs (Dicer+/ −), ESCs that do not express miRNAs (Dicer−/ −) as well as Dicer−/ − ESCs transfected with the miR-290-295 cluster miRNAs (miR-290, miR-291a-3p, miR-292-3p, miR-293, miR-294 and miR-295 mimics). The study showed that the expression profile of ESCs can be restored to a large extent in Dicer−/ − ESCs through transfection of miR-290-295 cluster miRNAs, and that these miRNAs are important for appropriate de novo DNA methylation in differentiating ESCs. Hanina et al. (18) profiled mRNA expression in Dicer−/ − ESCs as well as in Dicer−/ − ESCs transfected with miR-294. Combining these expression data with a biochemical approach to isolate Argonaute 2 (Ago2)-bound mRNAs, the study identified miR-294 targets in ESCs. It further concluded that miR-294 regulates a subset of genes that are also targeted by the Myc transcriptional regulator and that some of the effects of miR-294 expression may be due to the indirect upregulation of pluripotency factors, such as Lin28. Employing mRNA expression profiling of Dgcr8−/ − ESCs, as well as miR-294-transfected Dgcr8−/ − ESCs, Melton et al. (19) showed that self-renewal and differentiation of ESCs is regulated in an antagonistic manner by miR-294 and let-7. Finally, Zheng et al. (11) profiled mRNA expression of miRNA expressing ESCs and Dicer−/ − ESCs and uncovered a pro-survival, anti-apoptotic function of the miR-290-295 cluster of miRNAs.
Altogether, these studies provide five separate experimental data sets that can be used to investigate the function of AAGUGCU seed family miRNAs in ESCs. They all determined mRNA expression profiles of ESCs with impaired miRNA expression (due to knockout of either Dgcr8 or Dicer components of the miRNA biogenesis pathway), as well as of ESCs that expressed miRNAs of the AAGUGCU seed family. The latter were either ES cells which expressed the full complement of miRNAs, or miRNA-deficient ESCs that were transfected with either miRNAs of the miR-290-295 cluster, or only miR-294. Although it has been observed that these studies resulted in sets of miRNA targets that are only partially overlapping (10), a meta-analysis that combines these data sets to identify the pathways that are most reproducibly targeted by the AAGUGCU miRNAs has not been performed.
In our study, we aimed to infer transcriptional regulators that are directly and consistently targeted by the AAGUGCU family of miRNAs, the pathways that these regulators control and the interactions that they have with each other. Toward this end, we modeled genome-wide mRNA expression in terms of computationally predicted target sites of both transcription factors (TFs) and miRNAs. This approach allowed us to identify a number of transcriptional regulators whose activity is consistently altered by miRNAs of the AAGUGCU seed family and that could contribute to the maintenance of pluripotency. Through reporter assays we validated these regulators as targets of AAGUGCU seed family miRNAs. Employing Dicer−/ − mouse ES cells we showed that the expression of the IRF2 TF is strongly upregulated in the absence of miRNAs and that the nuclear concentration of the RelA component of the nuclear factor kappa-B (NF-κB) pathway upon stimulation with tumor necrosis factor α (TNF-α) is also increased. Our results give new insights into the functions of miRNAs in the regulatory circuitry of ESCs.
MATERIALS AND METHODS
Experimental data sets
Supplementary Table S1 summarizes the data sets that we obtained from the Gene Expression Omnibus (GEO) database of the National Center for Biotechnology Information (NCBI) and that we have used in our study. Each data set covers at least two distinct experimental conditions, with three replicates per condition. The first condition of every data set corresponds to an ESC line deficient in mature miRNAs due to Dicer- or Dgcr8-knockout. The second condition corresponds to either an ESC line expressing the entire complement of embryonically expressed miRNAs or the knockout cell line transfected with miR-294 or with mimics of the miR-290 cluster miRNAs (mir-290, mir-291a-3p, mir-292-3p, mir-293, mir-294 and mir-295).
Microarray analysis
Computational analysis of Illumina MouseWG-6 v2.0 Expression BeadChips from Hanina et al. (2010)
We downloaded the processed data from the GEO database of NCBI (accession no. GSE20048). Probe-to-gene associations were made by mapping the probe sequences (provided by the authors) to the set of mouse transcript sequences (downloaded 2011-02-19 from the UCSC Genome Bioinformatics web site).
We computed average gene expression levels as weighted averages of the signals of all probes that perfectly matched to at least one transcript of the gene. Whenever a probe mapped to multiple genes, a weight of 1/n was assigned to each of the n genes that the probe matched. For a given replicate experiment, the log2 expression fold change of each gene was then determined by subtracting the log2-average expression of the gene in the first condition (control) from the log2-average expression in the second condition (treatment).
Computational analysis of Affymetrix Mouse Genome 430 2.0 chips from Sinkkonen et al. (2008) and Zheng et al. (2011)
We downloaded the data from the GEO database (accessions GSE8503, GSE7141 and GSE30012) and analyzed the CEL files with the R software (http://www.R-project.org) using the BioConductor affy package (20). We used the GCRMA algorithm (21) for background correction and the MClust R package (22) to fit a two-component Gaussian mixture model to the log2-probe intensities and classify probes as expressed or not expressed. A probe was considered for further analysis if it was consistently classified as expressed in all three replicates of at least one of the two experimental conditions. The remaining probes were quantile normalized across all conditions and replicates of a particular experiment. Probe-to-gene associations were made by mapping probe sequences (provided on the Affymetrix web site, http://www.affymetrix.com) to mouse transcript sequences (as used by motif activity response analysis (MARA), downloaded from UCSC Genome Bioinformatics web site as described above). We then computed log2-gene expression fold changes as described for Illumina Expression BeadChips (see above).
Computational analysis of Affymetrix Mouse Gene 1.0 ST chips from Melton et al. (2010)
We downloaded the data from the GEO database (accession no. GSE18840) and analyzed the CEL files with the R Bioconductor oligo package (23). We used the Robust Multi-array Average (RMA) algorithm (24) for background adjustment. The rest of the analysis, including the classification of probes into expressed/not expressed, the quantile normalization, and the calculation of log2 gene expression fold changes, was carried out as described above.
Proportions of AAGUGCU miRNA seed family targets among genes that are consistently downregulated in multiple experiments
For each gene and each experiment, we calculated the standard error in its log2 fold change across the replicates. A gene was considered significantly downregulated when it was down-regulated more than 1.96 standard-errors. We then determined the intersection set of significantly downregulated genes for every possible subset of the experiments S={MeltonDGCR8KOVs294, SinkkonenDicerKOVs290, SinkkonenDicerKOVsWT}. Subsequently, for every obtained intersection set, the proportion of AAGUGCU miRNA seed family targets (TargetScan aggregate PCT score predictions (4)) was determined and plotted against the size of the corresponding intersection set.
Combined MARA of TFs and miRNAs
We carried out the MARA (25) separately for each experimental data set. MARA relates the expression level E driven by individual promoters (measured by microarrays) to the number of binding sites N that various regulators have in the promoters using a simple linear model
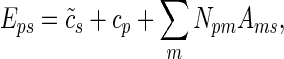 |
(1) |
where cp is a term reflecting the basal expression of promoter p, 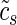 reflects the mean expression in sample s, and Ams is the (unknown) activity of binding motif m in sample s (where with ‘sample’ we refer to any individual replicate of any condition of a data set, see section ‘Experimental data sets’ above). That is, using the predicted site-counts Npm and the measured expression levels Eps we used an approximation (1) to infer the activities Ams of all motifs across all samples by ridge regression. In our analyses, we considered a curated set of 189 TF binding motifs (for detailed information about the motifs and the corresponding TFs see Supplementary Table S7). Furthermore, we included the binding sites in the 3’UTRs of mRNAs of 85 miRNA families by incorporating aggregate PCT scores as provided by TargetScan (4) (predictions downloaded on the 27th of March 2012 from the TargetScan web site, http://www.targetscan.org). miRNAs are grouped into families by their seed sequences and in particular the AAGUGCU seed family corresponds to the following miRNAs: mmu-miR-291a-3p, mmu-miR-294, mmu-miR-295, mmu-miR-302a, mmu-miR-302b and mmu-miR-302d. An aggregate PCT score was assigned to a promoter by averaging the aggregate PCT scores of transcripts associated with this promoter.
reflects the mean expression in sample s, and Ams is the (unknown) activity of binding motif m in sample s (where with ‘sample’ we refer to any individual replicate of any condition of a data set, see section ‘Experimental data sets’ above). That is, using the predicted site-counts Npm and the measured expression levels Eps we used an approximation (1) to infer the activities Ams of all motifs across all samples by ridge regression. In our analyses, we considered a curated set of 189 TF binding motifs (for detailed information about the motifs and the corresponding TFs see Supplementary Table S7). Furthermore, we included the binding sites in the 3’UTRs of mRNAs of 85 miRNA families by incorporating aggregate PCT scores as provided by TargetScan (4) (predictions downloaded on the 27th of March 2012 from the TargetScan web site, http://www.targetscan.org). miRNAs are grouped into families by their seed sequences and in particular the AAGUGCU seed family corresponds to the following miRNAs: mmu-miR-291a-3p, mmu-miR-294, mmu-miR-295, mmu-miR-302a, mmu-miR-302b and mmu-miR-302d. An aggregate PCT score was assigned to a promoter by averaging the aggregate PCT scores of transcripts associated with this promoter.
For a given motif m, MARA provides for each sample s motif activities 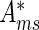 and associated errors σms. More specifically, marginalizing over all other motifs, the likelihood P(D|Ams) of the expression data D given the activity of a given motif is proportional to a Gaussian
and associated errors σms. More specifically, marginalizing over all other motifs, the likelihood P(D|Ams) of the expression data D given the activity of a given motif is proportional to a Gaussian
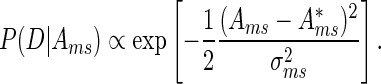 |
(2) |
Given that all analysed experiments were performed in multiple replicates we were interested in averaging motif activities across replicates and we used the following Bayesian approach. For each motif m separately, we assumed that the activities across a group g of replicates belonging to a specific condition of an experiment (see section ‘Experimental data sets’ above) are normally distributed around some (unknown) mean 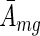 with (unknown) variance
with (unknown) variance 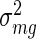
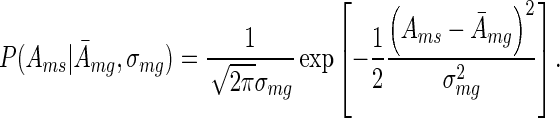 |
(3) |
By combining the prior from Equation (3) with the likelihood from Equation (2) for each replicate sample s ∈ g and integrating out the (unobserved) true activities Ams in each of the replicates, we obtained the probability of the form
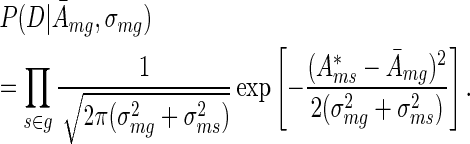 |
(4) |
Formally, we would next integrate out the unknown standard deviation of activities in the group σmg of this likelihood. Unfortunately, this integral cannot be performed analytically. We thus approximated the integral by the value of the integrand at its maximum, i.e. we numerically found the value of σmg that maximizes expression (4). Assuming an uniform prior over mean activity 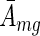 , we find that
, we find that 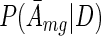 is again a Gaussian with mean
is again a Gaussian with mean
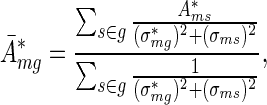 |
(5) |
and error
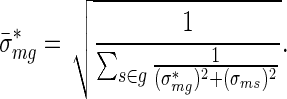 |
(6) |
where 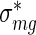 is the maximum likelihood estimate of Expression (4). We call the quantities defined in (5) and (6) averaged activities and averaged errors, respectively.
is the maximum likelihood estimate of Expression (4). We call the quantities defined in (5) and (6) averaged activities and averaged errors, respectively.
To identify motifs that consistently change in their activities across experiments, we wanted to further average motif activities across these experiments. However, because of the inherent differences in the scale of expression variation in the different experiments, the motif activities also varied in scale across the experiments. Thus, before averaging we first standardized the motif activities across the two conditions a and b. That is, for a given experiment we defined a scale L
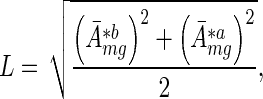 |
(7) |
and rescaled the activities
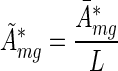 |
(8) |
and their errors
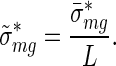 |
(9) |
These condition-specific, averaged and rescaled activities (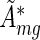 ) and errors (
) and errors (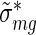 ) from the different experiments were then combined into two groups, i.e. the group of a conditions and the group of b conditions, and for each group we again averaged the activities exactly as described above for the replicates.
) from the different experiments were then combined into two groups, i.e. the group of a conditions and the group of b conditions, and for each group we again averaged the activities exactly as described above for the replicates.
To rank the activity changes between two different experimental conditions (presence/absence of miRNAs) we determined a 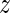 -value for every motif m by dividing the change in averaged activities between the two different conditions a and b by the averaged errors as follows
-value for every motif m by dividing the change in averaged activities between the two different conditions a and b by the averaged errors as follows
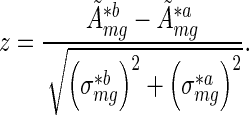 |
(10) |
Consequently, from the results of Equation (10) we obtained a global 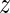 -value-based ranking of the motifs.
-value-based ranking of the motifs.
Motif–motif interaction network
To uncover which TFs were targeted by a particular motif m, we focused only on those TF genes, whose promoters were consistently (in all experiments) predicted by MARA to be targets of motif m. MARA computes a target score S for each potential target promoter of motif m. S corresponds to the log-likelihood ratio of the data D assuming the promoter is indeed a target, and assuming the promoter is independent of the regulator, i.e.
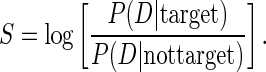 |
(11) |
Assuming a uniform prior of 1/2 that the promoter is indeed a target, the posterior probability p that the promoter is a target given the data is
 |
(12) |
To obtain a combined probability pc that a gene is a target of a particular motif across N different experiments the probability product was calculated by multiplying the probabilities pn obtained in individual experiments n, i.e.
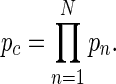 |
(13) |
Evaluating miR-294 targets with luciferase assays
Cloning, cell culture and luciferase assay
We polymerase chain reaction (PCR)-amplified 3′UTR fragments of the putative target genes from Normal Murine Mammary Gland (NMuMG) genomic DNA and cloned them into pGEM-T Easy vector (Promega; A1360). We used site-directed mutagenesis and the QuickChange II kit (Stratagene; 200524-5) to generate deletion mutant constructs that differed in a few nucleotides in the miR-294 seed-matching region from the wild-type construct. All constructs, wild-type and mutated, were verified by sequencing and then subcloned into the empty psiCHECK-2 vector (Promega; C8021) at XhoI - NotI restriction sites. The sequences of the primers used for cloning and mutagenesis can be found in Supplementary Tables S9 and S10, respectively. NMuMG cells were reverse-transfected with Lipofectamine2000 reagent (Invitrogen; 11668019), and the corresponding psiCHECK-2 constructs in the presence of 50nM Syn-mmu-miR-294-3p mimic (QIAGEN; MSY0000372), or 50 nM of non-targeting negative control siRNA (Microsynth). Between 36 and 48 h post-transfection cells were collected and both Renilla and firefly luciferase activities were measured using Dual Glo Luciferase Assay System (Promega; E2940).
For each gene, expression was measured for both constructs in 3 separate experiments, and each experiment contained 3 technical replicates.
Analysis of the luciferase data
We denote by 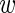 ir the logarithm (base 2) of the expression level of the luciferase construct containing the wild-type 3′UTR in experiment i replicate r and by mir the analogous expression for the mutant construct. For each gene the data thus consist of 9 values
ir the logarithm (base 2) of the expression level of the luciferase construct containing the wild-type 3′UTR in experiment i replicate r and by mir the analogous expression for the mutant construct. For each gene the data thus consist of 9 values 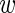 and 9 values m. We took into account two sources of variability, namely, true expression variability across experiments and ‘measurement noise’ between replicates. We first describe the measurement noise. Assuming the true expression of the wild type was
and 9 values m. We took into account two sources of variability, namely, true expression variability across experiments and ‘measurement noise’ between replicates. We first describe the measurement noise. Assuming the true expression of the wild type was 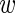 i, we assumed that the probability to measure expression level
i, we assumed that the probability to measure expression level 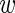 ir (in a given replicate r) follows a Gaussian distribution with a certain variance τi
ir (in a given replicate r) follows a Gaussian distribution with a certain variance τi
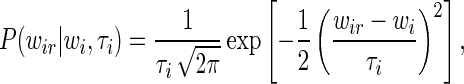 |
(14) |
thus allowing for the possibility that each experiment i has a different level of noise τi between replicates. The probability of the wild-type data of experiment i, assuming that τi is given, is simply the product of expressions P(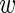 ir|
ir|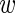 i, τi) over the three replicates r = 1 through 3. Using 〈
i, τi) over the three replicates r = 1 through 3. Using 〈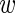 i〉 and var(
i〉 and var(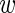 i) to denote the mean and variance of the measurement across the replicates, we can rewrite this as
i) to denote the mean and variance of the measurement across the replicates, we can rewrite this as
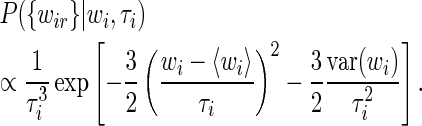 |
(15) |
Integrating over the unknown variable τi from 0 to infinity with a scale prior P(τi)∝1/τi we obtain
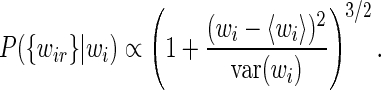 |
(16) |
Approximating this Student's t distribution by a Gaussian, that is, approximating the probability of the data in experiment i by a Gaussian with mean 〈 i〉 and variance var(
i〉 and variance var( i), we have
i), we have
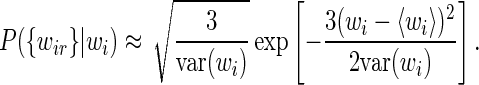 |
(17) |
Since the variability between replicates is much smaller than the variability across experiments, this approximation will have a negligible effect on the final outcome.
For the true variability between experiments, we denote by 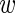 the ‘true’ average expression of the wild-type construct. We assume that the deviation of the level
the ‘true’ average expression of the wild-type construct. We assume that the deviation of the level 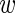 i in experiment i from the mean
i in experiment i from the mean 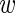 follows a Gaussian distribution with variance σ. We thus have
follows a Gaussian distribution with variance σ. We thus have
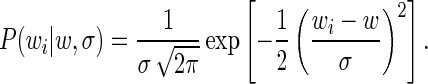 |
(18) |
To obtain the probability of the data given 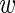 we multiply P({
we multiply P({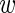 ir}|
ir}|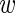 i) by P(
i) by P(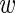 i|
i|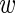 , σ) and integrate over the unknown expression level
, σ) and integrate over the unknown expression level 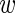 i. We then obtain
i. We then obtain
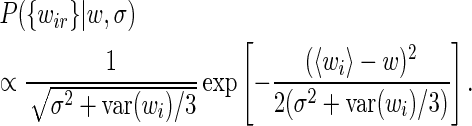 |
(19) |
The interpretation of this formula is straightforward. The deviation between the mean 〈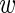 i〉 of the observations in experiment i, and the average level
i〉 of the observations in experiment i, and the average level 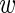 is Gaussian-distributed with a variance that is the sum of the variability σ2 across experiments, and the variability var(
is Gaussian-distributed with a variance that is the sum of the variability σ2 across experiments, and the variability var(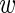 i)/3 associated with estimating
i)/3 associated with estimating 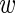 i from the 3 replicate measurements due to measurement noise.
i from the 3 replicate measurements due to measurement noise.
For the measurements of the mutant construct in experiment i we obtain an analogous equation
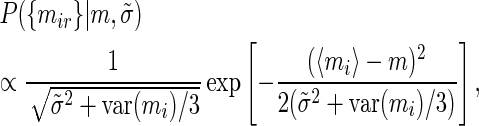 |
(20) |
where we have introduced the variability 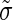 of the true expression of the mutant construct across replicates. What we are interested in is the difference
of the true expression of the mutant construct across replicates. What we are interested in is the difference
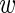 − m in log-expression of the wild-type and mutant construct. To this end, we define μ =
− m in log-expression of the wild-type and mutant construct. To this end, we define μ = 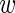 − m and y = (m +
− m and y = (m + 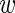 )/2 and integrate over y. We then obtain
)/2 and integrate over y. We then obtain
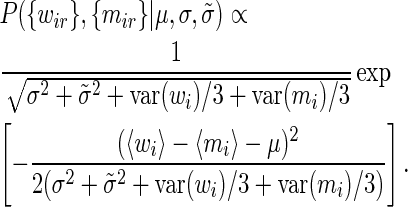 |
(21) |
This is again a Gaussian with mean 〈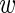 i〉 − 〈mi〉 and a variance that is the sum of all variances σ2,
i〉 − 〈mi〉 and a variance that is the sum of all variances σ2, 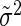 , var(
, var(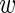 i)/3 and var(mi)/3.
i)/3 and var(mi)/3.
Clearly, although both σ2 and 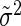 are unknown, the only variable that enters in our equations is their sum. We thus simplify the notation by defining this sum as
are unknown, the only variable that enters in our equations is their sum. We thus simplify the notation by defining this sum as
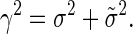 |
(22) |
Similarly, we redefine the variance associated with measurement noise as
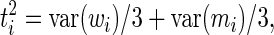 |
(23) |
which leads to
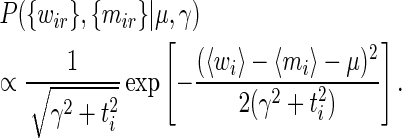 |
(24) |
We now combine the data from the different experiments and remove the final unknown variable γ. The probability of all data given the variable of interest μ and unknown variability parameter γ is simply the product
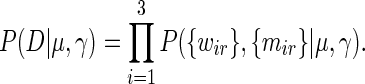 |
(25) |
To obtain the probability of the data D given μ we multiply this expression with a scale prior for γ, i.e. P(γ) = 1/γ, and integrate over γ
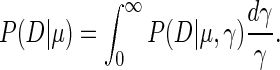 |
(26) |
We performed the integration numerically with Mathematica to obtain P(D|μ), and used Bayes’ theorem to compute the posterior distribution of the parameter μ, P(μ|D) as 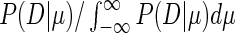 . Finally, we determined the 5 percentile, the 25 percentile, the median, the 75 percentile and the 95 percentile of this distribution again with the Mathematica software.
. Finally, we determined the 5 percentile, the 25 percentile, the median, the 75 percentile and the 95 percentile of this distribution again with the Mathematica software.
Mouse ESC (mESC) culture
The generation of Dicer(DCR)flox/flox and DCR−/ − mouse ES cell lines has been described elsewhere (26). The cells were routinely screened for both pluripotency and differentiation markers (see Supplementary Figure S4). Both mES cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco; 41966-029) supplemented with 15% of a special batch of fetal bovine serum tested for optimal growth of mESCs. In addition, the DMEM contained 1000 U/ml of a homegrown recombinant LIF (a kind gift of Thomas Grentzinger), 0.1mM 2β-mercaptoethanol (Millipore; ES-007-E), 1x L-Glutamine (Gibco; 25030-024), 1x Sodium Pyruvate (Gibco; 11360) and 1x Minimum Essential Medium, Non-Essential Amino Acids (MEM, NEAA) (Gibco; 11140-35). The cells were grown on gelatin-coated (Sigma; G1393) dishes. The medium was changed daily, and the cells were subcultured every 2–3 days. To induce NF-κB signaling, mESCs were treated with 20 ng/ml TNF-α (Cell Signaling Technology; 5178) for 24 h.
Quantitative reverse transcriptase-PCR (qRT-PCR)
Total RNA was extracted from mESCs using Tri Reagent (Sigma; T9424) following the supplier's protocol. Contaminating DNA was removed using the RQ1 RNase-Free DNase kit (Promega; M6101). The resulting DNA-free RNA was then purified using the RNeasy MinElute Cleanup kit (Qiagen; 74204) and quantified using Nanodrop. Superscript III (Invitrogen; 18080) was then used to create cDNA following the manufacturer's recommendations. The cDNA was finally purified using QIAquick PCR Purification kit (Qiagen; 74204), quantified using Nanodrop and diluted to 8 ng/μl. Each qRT-PCR reaction was run using 2 μl of the purified cDNA in triplicate (n = 3) using Power SYBR Green PCR Master Mix (Applied Biosystems; 4367659) on a StepOne Plus RT-PCR System (Applied Biosystems). The following primer pairs were used in this study:
Mouse IRF2 Fwd: 5′-CTG GGC GAT CCA TAC AGG AAA-3′
Mouse IRF2 Rev: 5′-CTC AAT GTC GGG CAG GGA AT-3′
Mouse E2F5 Fwd: 5′-GTT GTG GCT ACA GCA AAG CA-3′
Mouse E2F5 Rev: 5′-GGC CAA CAG TGT ATC ACC ATG A-3′
Mouse c-Myc Fwd: 5′-GTT GGA AAC CCC GCA GAC AG-3′
Mouse c-Myc Rev: 5′-ATA GGG CTG TAC GGA GTC GT-3′
Mouse GAPDH Fwd: 5′-CAT CAC TGC CAC CCA GAA GAC TG-3′
Mouse GAPDH Rev: 5′-ATG CCA GTG AGC TTC CCG TTC AG-3′
qRT-PCR data were normalized using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression and evaluated using the 2−ΔΔCt method (27). Significant changes in gene expression were identified based on Student's t-test.
Western blots
To extract total proteins from mESCs, radioimmunoprecipitation assay buffer supplemented with 1x Complete, ethylenediaminetetraacetic acid (EDTA)-free protease inhibitor cocktail (Roche; 11873580001) was used to lyze cell pellets. Cytosolic and nuclear protein fractions were enriched using a series of lysis buffers as follows:
Lysis Buffer 1 (LB1): 50 mM Hepes-KOH, pH 7.5; 140 mM NaCl; 1 mM EDTA, pH 8.0; 10% v/v Glycerol; 0.5% v/v NP-40; 0.25% v/v Triton X-100.
Lysis Buffer 2 (LB2): 10 mM Tris-HCl, pH 8.0; 200 mM NaCl; 1mM EDTA, pH 8.0; 0.5 mM EGTA, pH 8.0.
Lysis Buffer 3 (LB3): 10mM Tris-HCl, pH 8.0; 100 mM NaCl; 1 mM EDTA, pH 8.0; 0.5 mM EGTA, pH 8.0; 0.1% v/v Na-Deoxycholate; 30% v/v N-Lauroylsarcosine.
All lysis buffers were supplemented with the protease inhibitor cocktail immediately before use. The cytosolic fraction was extracted by lyzing the cell pellets in LB1 that leaves the nuclear membrane intact. The nuclei were then pelleted (1,350 x g; 4°C; 5 min), washed with LB2, pelleted once more and finally lyzed with LB3 to release the nuclear contents. All protein lysates were quantified using the BCA Protein Assay kit (Pierce; 23227). The following antibodies (dilution 1:1000) were used in this study:
Anti-IRF2 (Center) rabbit IgG (Abgent; AP11225c)
Anti-NF-κB p65 (D14E12) XP rabbit IgG (Cell Signaling Technology; 8242)
Anti-GAPDH (6C5) mouse IgG (Santa Cruz Biotechnology; sc-32233)
Anti-Histone H3 (C-16) goat IgG (Santa Cruz Biotechnology; sc-8654)
HRP-conjugated Polyclonal swine Anti-Rabbit (Dako; P0217)
HRP-conjugated Polyclonal rabbit Anti-Mouse (Dako; P0260)
HRP-conjugated Polyclonal rabbit Anti-Goat (Dako; P0449)
Western blot signals were visualized with the enhanced chemiluminescence blotting detection reagents (GE Healthcare; RPN2106). Cytosolic enrichment was confirmed via a postive GAPDH signal, while nuclear enrichment was confirmed by Histone H3. Western blot quantifications were performed using the ImageJ software by quantifying the pixels of each band and normalizing against a housekeeper, such as Histone H3.
RESULTS
General relationship between data sets
A common, though perhaps naive expectation is that combining data from experiments that have been independently performed in different labs, with different experimental procedures, allows one to identify essential properties of the system that are invariant with respect to details of the experimental approach. In our case, in any given experiment, confounding effects may have led to some genes being spuriously identified as targets of AAGUGCU miRNAs (false positives), and true targets of AAGUGCU miRNAs being missed (false negatives). For example, because it is unclear whether the miRNA processing enzymes solely function in this pathway, it is important to analyze data from ESCs in which the miRNA biogenesis has been impaired at different levels (Dicer in the studies of Sinkkonen et al. (17) and Hanina et al. (18) and Dgcr8 in the study of Melton et al. (19)). Furthermore, although ESCs expressing the full complement of miRNAs provide the most physiological reference point for the function of the miR-290-295 cluster miRNAs in normal, unstressed cells, the effect of these miRNAs in these cells is confounded by the effects of other co-expressed miRNAs. Similarly, if the profiled cell population was heterogeneous with respect to the pluripotency/differentiation status, the let-7 miRNAs may have masked the effect of miR-294, because these miRNAs have antagonistic effects (19).
Requiring targets to show consistent downregulation across multiple data sets can reduce the number of false positive miR-294 targets. On the other hand, requiring perfect consistency across a large number of experiments is likely to lead to too many false negatives, simply because different experiments have different levels of accuracy or confounding effects. Thus, we first investigated the relationship of gene-level expression changes between ESCs that did or did not express embryonic miRNAs in all pairs of experiments. Although pairwise Pearson's correlation coefficients were as low as 0.11 (Supplementary Figure S1), three of the five experimental data sets (Figure 1a), covering all described conditions (expression of miR-294, miR-290-295 cluster miRNAs or the entire complement of embryonically expressed miRNAs in a miRNA-deficient background) gave reasonably high pairwise correlation coefficients (Figure 1b). We therefore focused our discussion on these data sets, and for completeness, we present the results of a similar analysis of all five data sets in the Supplementary material (Supplementary Figure S2 and Tables S5 and S6). Of the ∼4000–5000 genes that were downregulated in a single experiment, a little less than 2000 genes were downregulated in all three experiments. Importantly, the proportion of predicted AAGUGCU seed family targets among downregulated genes increased when intersecting an increasing number of data sets (Figure 1c), indicating that the approach of a combined analysis of these data sets does have the potential to reveal important regulators that are immediately downstream of the AAGUGCU family of miRNAs. 252 of the genes downregulated in all three experiments were predicted AAGUGCU seed family targets (4) (Supplementary Table S2).
Figure 1.
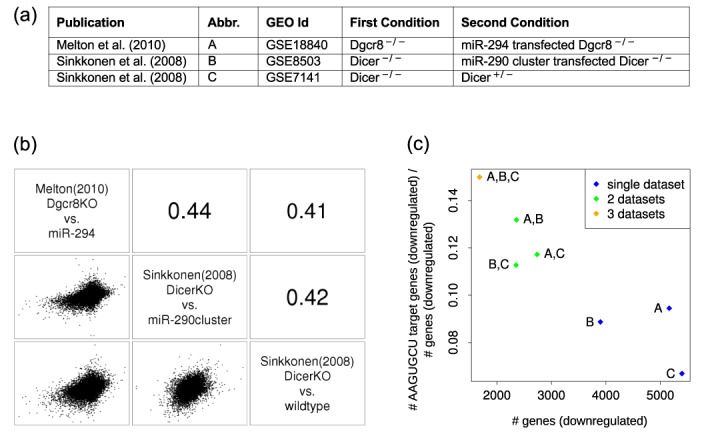
Overview of the mRNA expression data sets—(a) Data sources. (b) Matrix of scatter plots (below diagonal) and Pearson's correlation coefficients (above diagonal) of per-gene log2 fold changes in pairs of experiments. The names of the individual data sets are shown on the diagonal. (c) Proportion of predicted targets of the AAGUGCU seed family of miRNAs (TargetScan aggregate PCT score based predictions (4)) among genes that are consistently downregulated in all three (orange), pairs (green) or individual data sets (blue) (indicated by the labels, key given in the ‘Abbr.’ column of the table in panel (a)), plotted against the number of genes that are consistently downregulated in all of the considered data sets.
The transcriptional network regulated by the miRNAs of the AAGUGCU seed family in ESCs
As mentioned in the Introduction, the main aim of our study was to identify transcriptional regulators that are targeted by the AAGUGCU seed family and at the same time can account for the largest fraction of gene expression changes that are observed in cells that do or do not express the miRNAs. We therefore built on the MARA approach (28) that we recently made available in the form of an easy-to-use web application (25). In contrast to standard transcriptome analyses that strive to find genes (including transcription regulators) whose expression changes significantly between conditions, MARA aims to infer changes of the regulatory impact (also referred to as ‘activity’) of binding motifs. This is achieved by modeling gene expression as a linear function of the number of regulatory motif binding sites occurring in the promoter (for TFs) and 3′UTR (for miRNAs) of the gene and the unknown activity of each motif. The change in activity of a specific binding motif (e.g. of the Irf2 TF) in a specific condition (e.g. transfection of miR-294) is inferred from the expression changes of all (predicted) targets of this motif (determined by transcriptome profiling), taking into account the occurrences of sites for other regulators in these targets. For example, a decrease in Irf2 activity is inferred when the predicted Irf2 targets consistently show a decrease in expression that cannot be explained by the occurrence of binding sites for other regulatory motifs in the promoters or 3′UTRs of these targets. This means that MARA can uncover gene expression changes that are due not only to changes in the mRNA expression level of a regulator, but also to changes in the active form (e.g. for TFs through post-translational modifications, such as phosphorylation) of the regulator. MARA was initially developed for the characterization of transcription regulatory networks (28), and we have recently extended it to also model miRNA-dependent changes in mRNA stability (25). For this study we further extended the MARA approach to identify regulators whose activity not only changes most significantly between samples but also reproducibly across multiple data sets. Our approach is described in detail in the Materials and Methods section.
To verify that MARA can indeed uncover the key regulator in these experiments, namely, the miRNAs of the AAGUGCU seed family, we first applied MARA taking into account all TFs and miRNA seed families (see Supplementary Table S4). In subsequent analyses, however, we performed the MARA analysis with only the AAGUGCU seed family motif added to the full complement of TF motifs. This was because when all miRNAs are included in the analysis, MARA will also infer non-zero activities for other miRNAs, e.g. those with significantly overlapping sets of targets (29).
MARA quantifies the extent to which the activity of each motif varies across conditions by a  -statistic, that roughly corresponds to the ratio between the average deviation of the motif activity from zero and the standard deviation of the motif activity (see Materials and Methods). Supplementary Table S3 shows all motifs ranked by their absolute
-statistic, that roughly corresponds to the ratio between the average deviation of the motif activity from zero and the standard deviation of the motif activity (see Materials and Methods). Supplementary Table S3 shows all motifs ranked by their absolute  -values.
-values.
MARA also predicts which promoters or 3′UTRs are targeted by each motif, quantifying the confidence in each predicted motif-target interaction by a posterior probability (see Materials and Methods). We used these probabilities to construct a regulatory network of motif–motif interactions (Figure 2) that provides a synthetic view of the regulatory impact of the AAGUGCU seed family of miRNAs on the transcriptional network of pluripotent stem cells. An arrow was drawn from motif A to motif B whenever motif A was predicted by MARA to regulate a TF b whose binding specificity is represented by motif B. Only motif-TF interactions that were predicted in all data sets and that involved motifs with high significance (|z-value| > 5) are shown.
Figure 2.
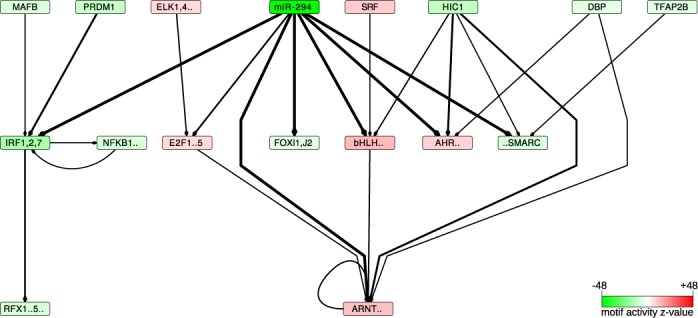
The transcriptional network inferred to be affected by the miRNAs of the AAGUGCU seed family (represented by miR-294)—A directed edge was drawn from a motif A to a motif B if A was consistently (across data sets) predicted to regulate a TF b whose sequence specificity is represented by motif B. The thickness of the edge is proportional to the product of the probabilities that A targets b. For the clarity of the figure, only motifs with absolute z-values >5 and only edges with a target probability product >0.3 are shown. The intensity of the color of a box representing a motif is proportional to the significance of the motif (the corresponding  -values can be found in Supplementary Table S3). Red indicates an increase and green a decrease in activity, corresponding to increased and decreased expression, respectively, of the tagets of the motif when the miRNAs are expressed. The full motif names as well as the corresponding TFs are listed in Supplementary Table S7.
-values can be found in Supplementary Table S3). Red indicates an increase and green a decrease in activity, corresponding to increased and decreased expression, respectively, of the tagets of the motif when the miRNAs are expressed. The full motif names as well as the corresponding TFs are listed in Supplementary Table S7.
The motif corresponding to the AAGUGCU seed family (represented by the dark green ‘miR-294’ motif in Figure 2) is by far the most significantly changing motif (see also Supplementary Table S3). Its negative change in activity upon miRNA expression is consistent with the destabilizing effect of the miRNA on its targets.
The motif with the second most significant change in activity, ‘IRF1,2,7’, is bound by the interferon regulatory factors. MARA predicts that this motif is directly targeted by miR-294, in line with previous suggestions that the interferon regulatory factors are targets of the miR-290 cluster miRNAs (18). We present a more detailed analysis of this motif in the next section.
A second motif whose activity decreases significantly upon miRNA expression is ‘FOX{I1,J2}’ (Figure 3a). Of the TFs associated with this motif, Foxj2 is predicted within all data sets to be directly regulated by miR-294 (Figure 2). Consistently, Foxj2 is downregulated upon miRNA expression on the mRNA level (Figure 3b). In order to validate that Foxj2 is a direct target of the miRNAs, as predicted by both ElMMo (30) and TargetScan (Figure 3b), we cloned the 3′UTR of Foxj2 downstream of a luciferase reporter and co-transfected this construct together with miR-294 in the murine mammary gland cell line NMuMG. For comparison, we generated a construct in which the presumed miRNA-294 target site was mutated and we performed similar co-transfection experiments. The results of this experiment clearly show that Foxj2 is indeed a functional target of miR-294 (Figure 3c). We carried out similar transfection experiments with control siRNAs, that do not target the reporter, and a standard analysis of these data is presented in Supplementary Figure S3. Little is known about the function of Foxj2 in cell fate. It appears to be expressed very early in development (31), but its overexpression has a negative effect on embryogenesis (32). Our results suggest that the AAGUGCU seed family of miRNAs contributes to the maintenance of an adequate expression of Foxj2 in pluripotent stem cells. The third most significant changing motif, basic-helix-loop-helix (referred to as ‘bHLH..’ in Figure 2), can be bound by many TFs (reviewed in (33)), some of which are predicted direct targets of miR-294.
Figure 3.
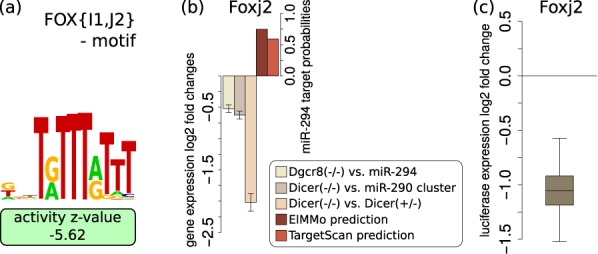
Foxj2 is a direct target of miR-294—(a) The ‘FOX{I1,J2}’ motif shows a negative change in activity in the presence of miR-294. (b) Foxj2 mRNA log2 fold changes (±1.96*SEM; n = 3) in the Melton et al. Dgcr8−/ − versus miR-294 transfection (yellow), Sinkkonen et al. Dicer−/ − versus miR-290-295 cluster transfection (dark brown) and Dicer−/ − versus Dicer+/ − (light brown) data sets, as well as the prediction scores for these genes as targets of miR-294 as given by ElMMo (30) (dark red) and TargetScan (aggregate PCT) (4) (light red). (c) A luciferase reporter construct carrying the 3′UTR of Foxj2 is downregulated upon co-transfection with miR-294 relative to a construct carrying the Foxj2 3′UTR but with a mutated miR-294 target site (n = 9).
To further elucidate the transcription regulatory network downstream of the AAGUGCU seed family of miRNAs, we analyzed in-depth the TFs whose associated motif had the most significant activity change (|z-value| > 5) and that were consistently predicted by MARA to be direct targets of the miR-294 seed family miRNAs across the multiple data sets (Table 1).
Table 1. TFs consistently predicted by MARA to be direct targets of miR-294 and whose absolute motif activity z-value is >5.
| Name | Motif | Motif Abbreviation | Activity z-value |
|---|---|---|---|
| Irf2 | IRF1,2,7.p3 | IRF1,2,7 | -16.29 |
| Mxd3 | bHLH_family.p2 | bHLH.. | 13.00 |
| Clock | bHLH_family.p2 | bHLH.. | 13.00 |
| Arnt2 | ARNT_ARNT2_BHLHB2_MAX_MYC_USF1.p2 | ARNT.. | 11.60 |
| Arnt2 | AHR_ARNT_ARNT2.p2 | AHR.. | 8.39 |
| BAF170 | DMAP1_NCOR{1,2}_SMARC.p2 | ..SMARC | -6.98 |
| E2f5 | E2F1..5.p2 | E2F1..5 | 6.62 |
| Foxj2 | FOX{I1,J2}.p2 | FOXI1,J2 | -5.62 |
We found that the majority of these direct target TFs fall into three categories that have previously been associated with pluripotency: NF-κB-related interferon response factors that control NF-κB signalling, cell cycle regulators and epigenetic regulators.
AAGUGCU seed family miRNAs modulate Irf2-dependent transcription
The ‘IRF1,2,7’ motif shows the second strongest activity change upon changes in miR-294 expression (Figure 4a and Supplementary Table S3). Of the individual factors associated with this motif, Irf2 is the one that was consistently predicted by our analysis to be a direct target of the AAGUGCU seed family miRNAs across data sets (Table 1), consistent with the predictions of both ElMMo and TargetScan (Figure 4b). Irf2 was downregulated at the mRNA level across all analyzed data sets (Figure 4b). Consistently, we found that Irf2 is strongly downregulated in DCRflox/flox compared to DCR−/ − ESCs, both at the mRNA level (Figure 4c) as well as at the protein level (Figure 4d). To validate Irf2 as a direct target of miR-294, we conducted luciferase assays as described above for Foxj2. Our results demonstrate that Irf2 is indeed targeted by miR-294 (Figure 4e). Although relatively little is known about the function of this factor in ESCs, a recent study showed that Irf2 overexpression causes differentiation of ESCs (34). The strong impact of AAGUGCU miRNAs on Irf2 levels and the relatively large impact of the ‘IRF1,2,7’ motif on gene expression suggest that this regulatory connection plays an important role in maintaining ESC pluripotency.
Figure 4.
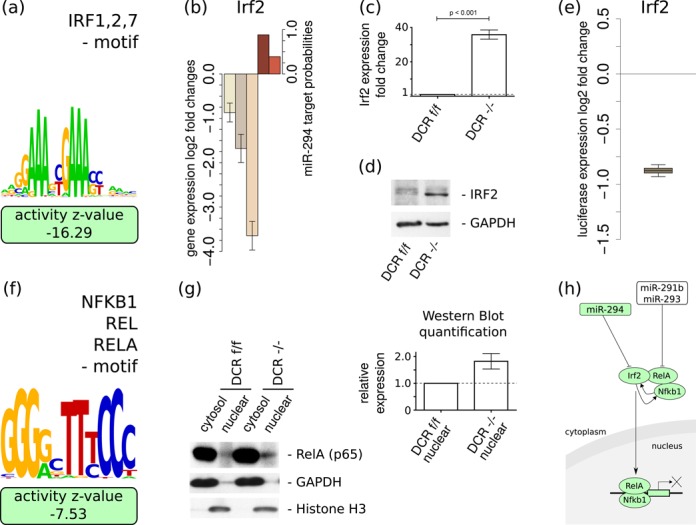
miR-294 targets the Irf2 TF and modulates ‘IRF1,2,7’ and ‘NFKB1_REL_RELA’ activities—(a) The activity of the ‘IRF1,2,7’ motif is strongly decreased in the presence of miR-294. (b) The expression of Irf2 is downregulated within all analysed data sets (±1.96*SEM; n = 3) and Irf2 is predicted by ElMMo and TargetScan to be a direct target of miR-294 (color scheme as in Figure 3). Low levels of Irf2 mRNA (c) and protein (d) in DCRflox/flox ES cells compared to miRNA deficient DCR−/ − ESCs are observed with qRT-PCR and western blot, respectively. qRT-PCR experiments were run in triplicate (± SEM; n = 3). (e) The luciferase reporter construct carrying the Irf2 3′UTR shows a strong response to miR-294 co-transfection compared to a similar construct but with a mutated Irf2 target site (n = 9). (f) Sequence logo of the ‘NFKB1_REL_RELA’ motif that is associated with the canonical NF-κB pathway and that exhibits a significant decrease in activity in the presence of miR-294. (g) Western blots of RelA, GAPDH and Histone H3 in nuclear and cytoplasmic fractions in ESCs that do and do not express miRNAs. The densitometric quantification indicates an increased level of nuclear RelA in the DCR−/ − ESCs compared to DCRflox/flox ESCs (± SEM; n = 3). (h) Proposed model of the inhibitory effect of miR-290-295 cluster miRNAs on the canonical NF-κB pathway in pluripotent stem cells. Regulatory motifs are denoted by colored rectangles and individual genes by ovals. See text for the evidence of individual interactions.
Like the ‘IRF1,2,7’ motif, the ‘NFKB1_REL_RELA’ motif also exhibits a significantly lower activity when the embryonic miRNAs are expressed (Figure 4f). Western blot confirms that after stimulation with TNF-α, DCRflox/flox ESCs have lower levels of nuclear NF-κB pathway-associated marker RelA compared with miRNA-deficient DCR−/ − ES cells (Figure 4g). This observation is consistent with a decreased activity of the canonical NF-κB signalling pathway in the presence of the miRNAs, which has been shown to be important for maintaining ESCs in a pluripotent state yet poised to undergo differentiation (35,36). Indeed, the Nanog pluripotency factor directly interacts with components of the NF-κB complex, inhibiting its transcriptional activity (35). Combining our results with recent reports that link the expression of the miR-290-295 cluster to signalling through the canonical NF-κB pathway and the latter to Irf2, the following model of the involvement of the miR-290-295 cluster in the regulation of NF-κB signalling emerges. Expression of the RelA component of the NF-κB complex is repressed post-transcriptionally by the miR-290-295 cluster members miR-291b-5p and miR-293 both of which do not belong to the AAGUGCU seed family of miRNAs (36). In humans, RelA recruitment to the nucleus, which is a pre-requisite for NF-κB complex-dependent transcription, appears to depend on IRF2 (37), whose knockdown interferes with transcriptional activation via NF-κB (37). Here we found that in mouse, IRF2 expression is also repressed by other members of the miR-290-295 cluster, namely, the AAGUGCU family of miRNAs. Thus, the miRNAs of the miR-290-295 cluster may act in concert to inhibit the canonical NF-κB signalling in ESCs (Figure 4h).
miRNAs of the AAGUGCU seed family impact the cell cycle at multiple levels
AAGUGCU seed family members of the miR-290-295 cluster were previously shown to accelerate the G1→S transition and promote proliferation of ESCs by targeting the cyclin E-Cdk2 regulatory pathway (13). Consistently, we found that these miRNAs increase the activity of transcription regulatory motifs associated with activation of the cell cycle (Figure 5a), in particular, the ‘ARNT_ARNT2_BHLHB2_MAX_MYC_USF1’ motif that is bound by Myc. This TF was previously found to increase upon miR-294 transfection (19). How the miRNAs, with intrinsically repressive function, increase the Myc activity on its targets is unknown. Our analysis suggests a few hypotheses.
Figure 5.
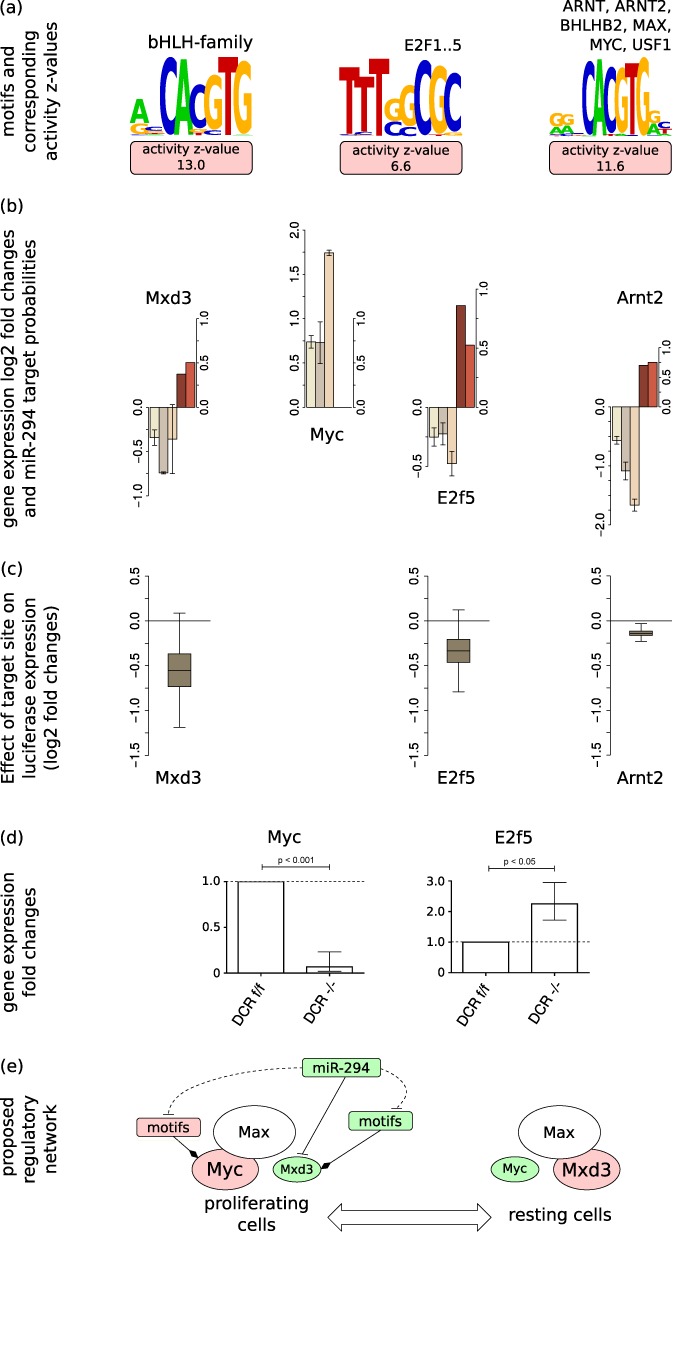
miR-294 impacts cell cycle regulation at multiple levels—(a) MARA analysis reveals that miR-294 induces positive activity changes of multiple motifs involved in cell cycle regulation. Shown are the sequence logos of these motifs: the Myc- and Arnt2-associated motif ‘ARNT_ARNT2_BHLHB2_MAX_MYC_USF1’, the putative Myc-regulating ‘E2F1..5′ motif and the Mxd3-associated ‘bHLH-family’ motif. (b) log2 mRNA fold changes (±1.96*SEM; n = 3) of Myc, Arnt2, E2f5 and Mxd3 (color scheme as in Figure 3) in the analyzed data sets. (c) Luciferase constructs carrying the 3′UTR of Arnt2, E2f5 or Mxd3, respectively, are downregulated upon co-transfection with miR-294 relative to constructs carrying the same 3′UTRs but with mutated miR-294 binding sites (n = 9). (d) qRT-PCR shows decreased expression of Myc and increased expression of E2f5 in DCR−/ − ESCs relative to DCRflox/flox ESCs. qRT-PCR experiments were run in triplicate (±SEM; n = 3). (e) Proposed model of miR-294-dependent regulation of the Myc-Max/Mxd-Max network. Shapes scheme is as in Figure 4. Green or red shapes represent negative or positive changes (in motif activities or gene expression fold changes), respectively. Dashed lines indicate indirect and solid lines direct regulatory links between motifs/genes.
Specifically, luciferase assays show that three cell cycle-associated TFs, namely, Mxd3 (also known as Mad3), E2f5 and Arnt2 are not only predicted but also experimentally confirmed direct targets of the AAGUGCU seed family miRNAs (Figure 5b and c and Table 1). Mxd3 is one of the so-called ‘Mad’ partners of the Max protein (reviewed in (38)). In contrast to Myc, which forms a heterodimeric complex with Max in proliferating cells (39), the Mad factors Mad1, Mad3 (i.e. Mxd3) and Mad4 are primarily expressed and form complexes with Max in differentiating, growth-arrested cells (40). Mxd3 was further shown to specifically regulate the S-phase (41).
Second, we found that E2f5, one of the TFs associated with the ‘E2F1..5′ motif, was consistently downregulated at the mRNA level in all analyzed data sets (Figure 5b) and luciferase assays further confirm that E2f5 is a target of miR-294 (Figure 5c), albeit with a small response to the miRNA. Consistently, E2f5 expression is increased in DCR−/ − ESCs compared to DCRflox/flox ESCs (Figure 5d). The positive activity change of the E2F1..5 motif in the presence of the miRNAs (Figure 5a) suggests that this TF acts predominantly as repressor (as proposed before, reviewed in (42)). Notably, Myc is among the predicted targets of E2F1..5, providing an indirect path to the upregulation of Myc upon the presence of the miRNAs (Figure 5b and d).
Finally, Arnt2, a TF associated with the ‘ARNT_ARNT2_BHLHB2_MAX_MYC_USF1’ motif, but also with the ‘AHR_ARNT_ARNT2’ motif that corresponds to the complex of Arnt2 and Ahr, is also a predicted direct target of the AAGUGCU seed family which we validated in a luciferase assay (Figure 5c). This TF forms heterodimers with the aryl-hydrocarbon receptor (AHR) (43) and appears to be involved in the differentiation of ESCs into endothelial cells under hypoxic conditions (44), but otherwise little is known about its function. Given that Arnt2 and Myc (45) share the same binding motif, an interesting hypothesis is that Arnt2 competes with Myc for binding to targets and that its downregulation by AAGUGCU miRNAs allows Myc to act at promoters which would otherwise be bound by Arnt2. This hypothesis is again consistent with a positive Myc activity in ESCs, in which these miRNAs are expressed.
The model that we propose based on these results is that miRNAs of the AAGUGCU family regulate the cell cycle and the G→S transition through multiple pathways that come together in the increased expression of the crucial Myc regulator (Figure 5e). The miRNAs are able to downregulate the Mxd3 antagonist of Myc, the E2f5 repressor which would in turn result in the increased expression of E2f5 targets including Myc, and can downregulate Arnt2 which may compete with Myc for binding to regulatory sites.
miRNAs of the AAGUGCU seed family control multiple epigenetic regulators
As TFs, epigenetic regulators are also enriched among the targets of miRNAs (46). A role for the miR-290-295 cluster in epigenetic regulation was already proposed by Sinkkonen et al. (17), who found that expression of retinoblastoma-like 2 (Rbl-2) protein, a known repressor of the de novo methyltransferases, is controlled by these miRNAs. Through our analysis we found that the AAGUGCU miRNAs directly target the epigenetic regulator BAF170 (Smarcc2), a component of ATP-dependent, BAF (BRG1-associated factor) complexes (also known as SWI/SNF complexes) that remodel the nucleosome structure and thereby regulate gene expression (reviewed in (47)). The activity of the BAF170 motif changed significantly upon AAGUGCU miRNA expression in miRNA-deficient ESCs (Figure 6a, Table 1), accompanied by consistent downregulation of BAF170 mRNA (Figure 6b). Comparing constructs with and without the putative miR-294 binding site in the BAF170 3′UTR in a luciferase assay we found that BAF170 is significantly downregulated by miR-294 (Figure 6c), indicating that BAF170 is indeed a direct target of miR-294.
Figure 6.
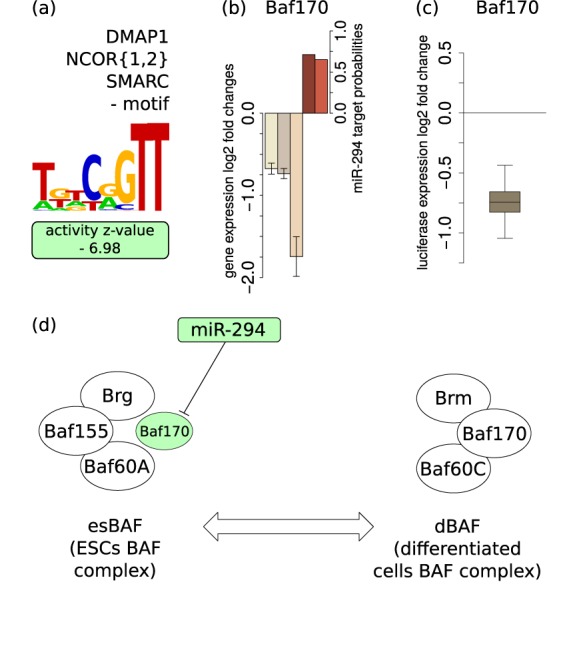
The BAF170 (Smarcc2) component of the dBAF chromatin remodeling complex is a direct target of miR-294—(a) MARA analysis reveals a negative activity change of the ‘DMAP1_NCOR{1,2}_SMARC’ motif in the presence of miR-294. (b) Expression of BAF170 (Smarcc2) is consistently downregulated in the presence of miR-294 in all considered experimental data sets (±1.96*SEM; n = 3; color scheme as in Figure 3). (c) A luciferase construct carrying the BAF170 3′UTR is downregulated upon co-transfection with miR-294 relative to a construct carrying a mutated 3′UTR (n = 9). (d) Model of the possible involvement of miR-294 in the maintenance of the ESC-specific chromatin remodeling complex esBAF. The miRNA-induced reduction in BAF170 levels may contribute to the maintenance of appropriate levels of esBAF complexes in ESCs thereby maintaining self-renewal and proliferation (48). Color, shapes and lines scheme is as in Figure 5.
Recently, it was shown that BAF170 is downregulated during miR-302-367-based reprogramming and that BAF170 knockdown increases the number of iPSC colonies in somatic cell reprogramming (49). As miRNAs of the miR-302-367 cluster share the seed sequence with miR-294, it is likely that miR-294 has similar effects on BAF170 expression and pluripotency.
The model that emerges from these studies is that the AAGUGCU family of miRNAs may play a role in the remodeling of BAF complexes. In ESCs, the BAF complex (esBAF), which contains a BAF155 subunit, shares a large proportion of target genes with the pluripotency-associated TFs Oct4, Sox2 and Nanog (50) and is required for the self-renewal and maintenance of pluripotency in mESCs (48). Consistently, overexpression of esBAF components was found to promote reprogramming (51). In differentiated cells, however, the so-called differentiated cell BAF complex (dBAF) (52), contains the BAF170 and not the BAF155 subunit (48). The fact that induced BAF170 expression in ESCs decreases the level of BAF155 protein suggested that BAF170 can displace BAF155 from esBAF, thereby increasing its degradation rate (48). By preventing expression of BAF components that are specific to differentiated cells and that antagonize embryonic state-specific BAF (Figure 6d), the AAGUGCU family of miRNAs may promote an ESC-specific epigenetic state.
DISCUSSION
It has been established that ESC-specific miRNAs that share an AAGUGCU seed region are among the regulatory factors that are necessary to maintain a pluripotent ESC state. Strikingly, overexpression of a cluster of ESC-specific miRNAs was found sufficient for inducing reprogramming of differentiated cells into iPSCs. This suggests that the miRNAs can set into motion an entire regulatory cascade that leads to cell reprogramming. Several studies determined the gene expression profiles of ESCs that did and did not express AAGUGCU family miRNAs. An insight emerging from these studies was that miR-290-295 miRNAs regulate the cell cycle and apoptosis, either directly or indirectly.
To better understand how the direct regulatory factor targets of these miRNAs contribute to pluripotency, we made use of a recently developed method, called MARA, that models gene expression in terms of computationally predicted regulatory sites. The approach originates in regression models that were first proposed by Bussemaker et al. (53) for inferring regulatory elements from gene expression data. However, MARA's goal is different. It uses predicted regulatory sites in combination with a linear model to infer from gene expression data the activities of transcriptional regulators. The first application of MARA (28) to the reconstruction of the core transcriptional regulatory network of a differentiating human cell line, demonstrated that the method can successfully infer key regulatory interactions ab initio. Notably, it was found that MARA accurately infers the activities of the key regulatory motifs, in spite of computational predictions of regulatory sites being error-prone, and of gene expression likely being a much more complex function of the regulatory sites. The power of the method stems from the fact that motif activities are inferred from the statistics of expression of hundreds to thousands of putative target genes of each regulatory motif. Here we have used an extended version of the MARA model, which also includes predicted miRNA binding sites, to infer both transcriptional and post-transcriptional regulators of mRNA expression levels. A similar approach was recently applied by Setty et al. (54) to reconstruct the regulatory networks in glioblastoma.
The TF targets of the AAGUGCU miRNAs that we identified with the extended MARA model had the following properties:
The activity of their corresponding motif changed significantly upon expression of the AAGUGCU miRNAs, meaning that the predicted targets of these regulators showed, on average, consistent expression changes.
Their expression was consistently downregulated at the mRNA level upon expression of the AAGUGCU miRNAs.
They were predicted as direct targets of the AAGUGCU family of miRNAs by miRNA target prediction programs.
They were consistently (i.e. within every analyzed data set) predicted by MARA to be directly regulated by the AAGUGCU seed family of miRNAs on the basis of the dependence of their expression changes on the presence of the miRNA binding sites in their 3′UTRs.
They could be confirmed as AAGUGCU miRNA targets with luciferase assays.
Altogether, these lines of evidence firmly establish these transcriptional regulators as direct targets of the AAGUGCU seed family miRNAs, forming the first layer downstream of this miRNAs in the regulatory network of pluripotency.
First, our analysis suggests that AAGUGCU miRNAs target the cell cycle, and in particular the G1→S transition, through multiple pathways. By targeting the repressive cell cycle regulator E2f5, the miRNAs might directly promote the G1→S transition. In addition, the miRNAs seem to increase the activity of the proliferation-associated TF Myc through multiple indirect routes, including shifting the balance between Myc and its antagonist Mxd3 within transcription regulatory complexes that act on Myc target genes. Second, we found that the AAGUGCU miRNAs may affect the balance between chromatin remodeling complexes that are active in ESCs and in differentiated cells, a function probably important for keeping specific genomic regions from being silenced through heterochromatin formation. Third, we found that the AAGUGCU miRNAs directly target the interferon regulatory factor Irf2, whose expression is strongly increased in DCR−/ − cells, consistent with a significant change in the regulatory impact that we inferred for this factor. Finally, our analysis uncovers a few transriptional regulators that have previously not been connected to the transcriptional network of pluripotent stem cells, including Foxj2, whose expression is strongly affected by the miRNAs and the Clock (circadian locomotor output cycles kaput) TF. Interestingly, circadian oscillations are not present in mouse ES cells, but are switched on during differentiation, and then disappear again upon reprogramming of differentiated cells into iPSCs (55). It is thus tempting to speculate that circadian oscillations in ESCs may be actively suppressed by the AAGUGCU miRNAs and that downregulation of these miRNAs during development may be necessary for the establishment of circadian rhythms. However, the response of the 3′UTR of Clock in luciferase assays was very variable in our hands, and we were not able to unambiguously validate it as a direct target of miR-294.
As mentioned before, the AAGUGCU seed motif is not unique to miRNAs of the mouse-specific miR-290-295 cluster. It also occurs in the miR-302 family of miRNAs that is present in human and in a shifted version (at positions 3–9 instead of 2–8) it occurs in the miR-17/20a miRNAs of the oncogenic miR-17-92 cluster. Although miR-19 has been reported to be the key oncogenic component of this cluster (56), the strong effects that AAGUGCU miRNAs exert on the cell cycle raise the question of whether miR-17 and miR-20a may not play a role similar to miR-294 in malignant cells.
In summary, our analysis demonstrates that combining accurate predictions of regulatory elements with analysis of transcriptome-wide mRNA expression changes in response to specific manipulations is a general and powerful approach to uncovering key regulators within gene expression networks. In the future, incorporation of measurements of miRNA expression as well as of predictions of TF binding sites in miRNA genes will enable identification of feedback loops between miRNAs and TFs that are known to operate in many systems.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank the members of the Zavolan group for feedback on the manuscript.
FUNDING
Swiss National Science [#31003A_127307]; European Research Council Starting Grant [to M.Z.]. Werner Siemens fellowship at the Biozentrum [to A.J.G.]. Source of open access funding: Biozentrum, University of Basel.
Conflict of interest statement. None declared.
REFERENCES
- 1.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Lim L.P., Lau N.C., Garrett-Engele P., Grimson A., Schelter J.M., Castle J., Bartel D.P., Linsley P.S., Johnson J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 3.Selbach M., Schwanhäusser B., Thierfelder N., Fang Z., Khanin R., Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 4.Friedman R.C., Farh K.K.-H., Burge C.B., Bartel D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanellopoulou C., Muljo S.A., Kung A.L., Ganesan S., Drapkin R., Jenuwein T., Livingston D.M., Rajewsky K.C.P. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 2005;19:489–501. doi: 10.1101/gad.1248505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murchison E.P., Partridge J.F., Tam O.H., Cheloufi S., Hannon G.J. Characterization of Dicer-deficient murine embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 2005;102:12135–12140. doi: 10.1073/pnas.0505479102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y., Medvid R., Melton C., Jaenisch R., Blelloch R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat. Genet. 2007;39:380–385. doi: 10.1038/ng1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Babiarz J.E., Ruby J.G., Wang Y., Bartel D.P., Blelloch R. Mouse ES cells express endogenous shRNAs, siRNAs, and other microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008;22:2773–2785. doi: 10.1101/gad.1705308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ciaudo C., Servant N., Cognat V., Sarazin A., Kieffer E., Viville S., Colot V., Barillot E., Heard E., Voinnet O. Highly dynamic and sex-specific expression of microRNAs during early ES cell differentiation. PLoS Genet. 2009;5:e1000620. doi: 10.1371/journal.pgen.1000620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leung A.K., Young A.G., Bhutkar A., Zheng G.X., Bosson A.D., Nielsen C.B., Sharp P.A.C.P. Genome-wide identification of Ago2 binding sites from mouse embryonic stem cells with and without mature microRNAs. Nat. Struct. Mol. Biol. 2011;18:237–244. doi: 10.1038/nsmb.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng G.X., Ravi A., Calabrese J.M., Medeiros L.A., Kirak O., Dennis L.M., Jaenisch R., Burge C.B., Sharp P.A.C.P. A latent pro-survival function for the mir-290-295 cluster in mouse embryonic stem cells. PLoS Genet. 2011;7:e1002054. doi: 10.1371/journal.pgen.1002054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Houbaviy H.B., Murray M.F., Sharp P.A. Embryonic stem cell-specific MicroRNAs. Dev. Cell. 2003;5:351–358. doi: 10.1016/s1534-5807(03)00227-2. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y., Baskerville S., Shenoy A., Babiarz J.E., Baehner L., Blelloch R. Embryonic stem cell-specific microRNAs regulate the G1-S transition and promote rapid proliferation. Nat. Genet. 2008;40:1478–1483. doi: 10.1038/ng.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Judson R.L., Babiarz J.E., Venere M., Blelloch R. Embryonic stem cell-specific microRNAs promote induced pluripotency. Nat. Biotechnol. 2009;27:459–461. doi: 10.1038/nbt.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuo C.-H., Deng J.H., Deng Q., Ying S.-Y. A novel role of miR-302/367 in reprogramming. Biochem. Biophys. Res. Commun. 2012;417:11–16. doi: 10.1016/j.bbrc.2011.11.058. [DOI] [PubMed] [Google Scholar]
- 16.Anokye-Danso F., Trivedi C.M., Juhr D., Gupta M., Cui Z., Tian Y., Zhang Y., Yang W., Gruber P.J., Epstein J.A., Morrisey E.E. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell. 2011;8:376–388. doi: 10.1016/j.stem.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sinkkonen L., Hugenschmidt T., Berninger P., Gaidatzis D., Mohn F., Artus-Revel C.G., Zavolan M., Svoboda P., Filipowicz W. MicroRNAs control de novo DNA methylation through regulation of transcriptional repressors in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 2008;15:259–267. doi: 10.1038/nsmb.1391. [DOI] [PubMed] [Google Scholar]
- 18.Hanina S.A., Mifsud W., Down T.A., Hayashi K., O'Carroll D., Lao K., Miska E.A., Surani M.A. Genome-wide identification of targets and function of individual MicroRNAs in mouse embryonic stem cells. PLoS Genet. 2010;6:e1001163. doi: 10.1371/journal.pgen.1001163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melton C., Judson R.L., Blelloch R. Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. Nature. 2010;463:621–626. doi: 10.1038/nature08725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gautier L., Cope L., Bolstad B.M., Irizarry R.A. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics (Oxford, England) 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 21.Wu Z., Irizarry R.A., Gentleman R., Martinez-Murillo F., Spencer F. A model-based background adjustment for oligonucleotide expression arrays. J. Am. Stat. Assoc. 2004;99:909–917. [Google Scholar]
- 22.Fraley C., Raftery A.E. mclust: Model-based clustering/normal mixture modeling. R package version 3. 2009 [Google Scholar]
- 23.Carvalho B.S., Irizarry R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics (Oxford, England) 2010;26:2363–2367. doi: 10.1093/bioinformatics/btq431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Irizarry R., Hobbs B., Collin F., D Beazer-Barclay Y., J Antonellis K., Scherf U., P Speed T. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics Oxford England. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 25.Balwierz P.J., Pachkov M., Arnold P., Gruber A.J., Zavolan M., van Nimwegen E. ISMARA: automated modeling of genomic signals as a democracy of regulatory motifs. Genome Res. 2014;24:869–884. doi: 10.1101/gr.169508.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ciaudo C., Jay F., Okamoto I., Chen C.-J., Sarazin A., Servant N., Barillot E., Heard E., Voinnet O. RNAi-dependent and independent control of LINE1 accumulation and mobility in mouse embryonic stem cells. PLoS Genet. 2013;9:e1003791. doi: 10.1371/journal.pgen.1003791. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif.) 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki H., Forrest A.R.R., van Nimwegen E., Daub C.O., Balwierz P.J., Irvine K.M., Lassmann T., Ravasi T., Hasegawa Y., de Hoon M.J.L., et al. The transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line. Nat. Genet. 2009;41:553–562. doi: 10.1038/ng.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsang J.S., Ebert M.S., van Oudenaarden A. Genome-wide dissection of microRNA functions and cotargeting networks using gene set signatures. Mol. Cell. 2010;38:140–153. doi: 10.1016/j.molcel.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaidatzis D., van Nimwegen E., Hausser J., Zavolan M. Inference of miRNA targets using evolutionary conservation and pathway analysis. BMC Bioinformat. 2007;8:69–92. doi: 10.1186/1471-2105-8-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Granadino B., Arias-de-la Fuente C., Pérez-Sánchez C., Párraga M., López-Fernández L.A., del Mazo J., Rey-Campos J. Fhx (Foxj2) expression is activated during spermatogenesis and very early in embryonic development. Mech. Dev. 2000;97:157–160. doi: 10.1016/s0925-4773(00)00410-x. [DOI] [PubMed] [Google Scholar]
- 32.Martín-de Lara F., Sánchez-Aparicio P., Arias de la Fuente C., Rey-Campos J. Biological effects of FoxJ2 over-expression. Transgenic Res. 2008;17:1131–1141. doi: 10.1007/s11248-008-9214-3. [DOI] [PubMed] [Google Scholar]
- 33.Skinner M.K., Rawls A., Wilson-Rawls J., Roalson E.H. Basic helix-loop-helix transcription factor gene family phylogenetics and nomenclature. Differentiation; Res. Biol. Diversity. 2010;80:1–8. doi: 10.1016/j.diff.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamizu K., Piao Y., Sharov A.A., Zsiros V., Yu H., Nakazawa K., Schlessinger D., Ko M. S.H. Identification of transcription factors for lineage-specific ESC differentiation. Stem Cell Rep. 2013;1:545–559. doi: 10.1016/j.stemcr.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Torres J., Watt F.M. Nanog maintains pluripotency of mouse embryonic stem cells by inhibiting NFkappaB and cooperating with Stat3. Nat. Cell Biol. 2008;10:194–201. doi: 10.1038/ncb1680. [DOI] [PubMed] [Google Scholar]
- 36.Lüningschrör P., Stöcker B., Kaltschmidt B., Kaltschmidt C. miR-290 cluster modulates pluripotency by repressing canonical NF-κB signaling. Stem Cells (Dayton, Ohio) 2012;30:655–664. doi: 10.1002/stem.1033. [DOI] [PubMed] [Google Scholar]
- 37.Chae M., Kim K., Park S.-M., Jang I.-S., Seo T., Kim D.-M., Kim I.-C., Lee J.-H., Park J. IRF-2 regulates NF-kappaB activity by modulating the subcellular localization of NF-kappaB. Biochem. Biophys. Res. Commun. 2008;370:519–524. doi: 10.1016/j.bbrc.2008.03.136. [DOI] [PubMed] [Google Scholar]
- 38.Hurlin P.J., Huang J. The MAX-interacting transcription factor network. Seminars Cancer Biol. 2006;16:265–274. doi: 10.1016/j.semcancer.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 39.Ayer D.E., Eisenman R.N. A switch from Myc:Max to Mad:Max heterocomplexes accompanies monocyte/macrophage differentiation. Genes Dev. 1993;7:2110–2119. doi: 10.1101/gad.7.11.2110. [DOI] [PubMed] [Google Scholar]
- 40.Hurlin P.J., Quéva C., Koskinen P.J., Steingrímsson E., Ayer D.E., Copeland N.G., Jenkins N.A., Eisenman R.N. Mad3 and Mad4: novel Max-interacting transcriptional repressors that suppress c-myc dependent transformation and are expressed during neural and epidermal differentiation. EMBO J. 1995;14:5646–5659. doi: 10.1002/j.1460-2075.1995.tb00252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quéva C., McArthur G.A., Iritani B.M., Eisenman R.N. Targeted deletion of the S-phase-specific Myc antagonist Mad3 sensitizes neuronal and lymphoid cells to radiation-induced apoptosis. Mol. Cell. Biol. 2001;21:703–712. doi: 10.1128/MCB.21.3.703-712.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cam H., Dynlacht B.D. Emerging roles for E2F: beyond the G1/S transition and DNA replication. Cancer Cell. 2003;3:311–316. doi: 10.1016/s1535-6108(03)00080-1. [DOI] [PubMed] [Google Scholar]
- 43.Sekine H., Mimura J., Yamamoto M., Fujii-Kuriyama Y. Unique and overlapping transcriptional roles of arylhydrocarbon receptor nuclear translocator (Arnt) and Arnt2 in xenobiotic and hypoxic responses. J. Biol. Chem. 2006;281:37507–37516. doi: 10.1074/jbc.M606910200. [DOI] [PubMed] [Google Scholar]
- 44.Han Y., Kuang S.-Z., Gomer A., Ramirez-Bergeron D.L. Hypoxia influences the vascular expansion and differentiation of embryonic stem cell cultures through the temporal expression of vascular endothelial growth factor receptors in an ARNT-dependent manner. Stem Cells (Dayton, Ohio) 2010;28:799–809. doi: 10.1002/stem.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith K., Dalton S. Myc transcription factors: key regulators behind establishment and maintenance of pluripotency. Regenerative Med. 2010;5:947–959. doi: 10.2217/rme.10.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gruber A.J., Zavolan M. Modulation of epigenetic regulators and cell fate decisions by miRNAs. Epigenomics. 2013;5:671–683. doi: 10.2217/epi.13.65. [DOI] [PubMed] [Google Scholar]
- 47.Wilson B.G., Roberts C.W.M. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer. 2011;11:481–492. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- 48.Ho L., Ronan J.L., Wu J., Staahl B.T., Chen L., Kuo A., Lessard J., Nesvizhskii A.I., Ranish J., Crabtree G.R. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc. Natl. Acad. Sci. U. S. A. 2009;106:5181–5186. doi: 10.1073/pnas.0812889106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Subramanyam D., Lamouille S., Judson R.L., Liu J.Y., Bucay N., Derynck R., Blelloch R. Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat. Biotechnol. 2011;29:443–448. doi: 10.1038/nbt.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ho L., Jothi R., Ronan J.L., Cui K., Zhao K., Crabtree G.R. An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proc. Natl. Acad. Sci. U. S. A. 2009;106:5187–5191. doi: 10.1073/pnas.0812888106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singhal N., Graumann J., Wu G., Araúzo-Bravo M.J., Han D.W., Greber B., Gentile L., Mann M., Schöler H.R. Chromatin-remodeling components of the BAF complex facilitate reprogramming. Cell. 2010;141:943–955. doi: 10.1016/j.cell.2010.04.037. [DOI] [PubMed] [Google Scholar]
- 52.He L., Liu H., Tang L. SWI/SNF chromatin remodeling complex: a new cofactor in reprogramming. Stem Cell Rev. 2012;8:128–136. doi: 10.1007/s12015-011-9285-z. [DOI] [PubMed] [Google Scholar]
- 53.Bussemaker H.J., Li H., Siggia E.D. Regulatory element detection using correlation with expression. Nat. Genet. 2001;27:167–171. doi: 10.1038/84792. [DOI] [PubMed] [Google Scholar]
- 54.Setty M., Helmy K., Khan A.A., Silber J., Arvey A., Neezen F., Agius P., Huse J.T., Holland E.C., Leslie C.S. Inferring transcriptional and microRNA-mediated regulatory programs in glioblastoma. Mol. Syst. Biol. 2012;8:605–620. doi: 10.1038/msb.2012.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yagita K., Horie K., Koinuma S., Nakamura W., Yamanaka I., Urasaki A., Shigeyoshi Y., Kawakami K., Shimada S., Takeda J., et al. Development of the circadian oscillator during differentiation of mouse embryonic stem cells in vitro. Proc. Natl. Acad. Sci. U. S. A. 2010;107:3846–3851. doi: 10.1073/pnas.0913256107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mu P., Han Y.-C., Betel D., Yao E., Squatrito M., Ogrodowski P., de Stanchina E., D'Andrea A., Sander C., Ventura A. Genetic dissection of the miR-17∼92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev. 2009;23:2806–2811. doi: 10.1101/gad.1872909. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.