


Abstract
Single-nucleotide polymorphisms, either inherited or due to spontaneous DNA damage, are associated with numerous diseases. Developing tools for site-specific nucleotide modification may one day provide a way to alter disease polymorphisms. Here, we describe the in vitro selection and characterization of a new deoxyribozyme called F-8, which catalyzes nucleotide excision specifically at thymidine. Cleavage by F-8 generates 3′- and 5′-phosphate ends recognized by DNA modifying enzymes, which repair the targeted deoxyribonucleotide while maintaining the integrity of the rest of the sequence. These results illustrate the potential of DNAzymes as tools for DNA manipulation.
INTRODUCTION
The integrity of DNA, the carrier of genetic information (1), is crucial for all living organisms, yet the genome is under constant threat from environmental insults such as UV radiation, intracellular attacks due to reactive oxygen species and intrinsic problems such as replication errors (2–4). In mammalian cells, these forces are estimated to give rise to as many as 104 damage events per cell (5). Many of these events may lead to single-nucleotide mutations in particular genes, affecting the expression and activity of the encoded protein. Many of these so-called single-nucleotide polymorphisms (SNPs) have been linked to human diseases, including phenylketonuria, hemophilia and certain cancers (6). Significant advances have been made in the past several years to develop accurate, rapid and cost-effective technologies for SNP detection, such as denaturing gradient gel electrophoresis (7), microfluidic devices (8), technologies based upon chip (9), allele-specific polymerase chain reaction (PCR) (10), strand displacement amplification (11), rolling circle amplification (12) and ligase chain reaction (13).
Because of the strong association between some SNPs and certain human diseases, replacement the SNP to the wild-type (non-disease) genotype may provide a therapeutic option. Accomplishing this requires developing methods to remove a target nucleotide in a specific DNA sequence and replace it with a desired one. Site specificity is key: this DNA manipulation should not alter sequences at other positions of the genome. In cells, protein enzymes such as RNase H and FEN-1 can efficiently excise ribonucleotides from oligonucleotides (14,15), but few tools are available to delete specific deoxyribonucleotides from oligonucleotides (5,16). Nevertheless, the feasibility of this approach was recently demonstrated when a series of deoxyribozymes (DNAzymes) were shown to be capable of precise, single-nucleotide excision repair (16).
The first step in single-nucleotide excision repair is site-specific cleavage at the target nucleotide, and many research groups have explored different cleavage approaches (17). Cleavage most frequently involves either (i) hydrolysis of phosphodiester linkages, called hydrolytic cleavage (Figure 1A); or (ii) oxidative cleavage of deoxyribose residues (17). Such oxidative cleavage occurs in vivo when DNA is exposed to oxidative stress or other insults that lead to formation of DNA adducts and lesions; DNA repair pathways may also use oxidative cleavage to cross-link damaged residues (18). One particularly well-studied type of oxidative cleavage is nucleotide excision, in which the glycosidic bond is cleaved, releasing a nucleobase and breaking the DNA strands (Figure 1B, inset) (19). This process occurs in cells when DNA glycosylases recognize damaged bases in the DNA sequence and initiate DNA repair (20–23). Nucleotide excision is an attractive approach for achieving DNA repair because it is simple and the resulting DNA termini can be manipulated enzymatically in a variety of ways (5,19). However, new tools must be developed, since glycosylases recognize only damaged bases and many clinically important DNA damage events involve base substitutions (24); in addition, glycosylases generate ‘unconventional’ DNA ends such as apurinic/apyrimidinic (AP) sites, which can lead to further DNA damage (5).
Figure 1.
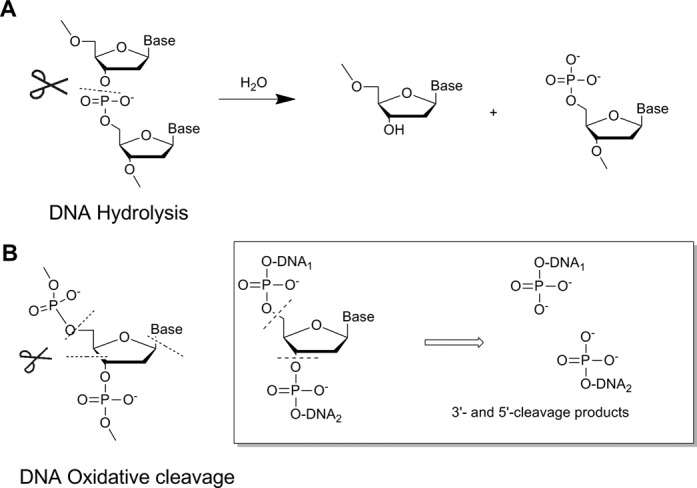
Major types of DNA cleavage. (A) Hydrolytic cleavage of DNA (P–O bond scission is shown). (B) Oxidative cleavage of DNA (possible cleavage sites at the sugar or base are shown). The inset shows the products of oxidative cleavage along the backbone (nucleotide excision).
As an alternative to enzymes, artificial DNA cutters have been constructed by combining, covalently or non-covalently, a DNA-cutting molecule and a sequence-recognizing molecule, such as an oligonucleotide or pyrrole-imidazole polyamide. Many of these cutters have been shown to cleave DNA at specific target sequences, though they show relatively poor efficiency (17). Few DNA cutters are capable of site-specific nucleotide excision; most that have this activity show little or no specificity, cleaving the target DNA at multiple sites (18,25).
Potentially much more efficient are catalytic DNAzymes, and several groups have used in vitro selection to obtain DNAzymes that can modify DNA site-specifically (26–32). Unfortunately only a few DNAzymes capable of promoting site-specific DNA nucleotide excision have been reported, and most of them work only at purine nucleotides (33–36). This reflects the fact that depurination is generally easier than depyrimidination at physiological pH because pyrimidine nucleobases lack the purine N7 atom, which can be activated (e.g. via protonation) to facilitate deglycosylation (35,37). An additional challenge with depyrimidination is that it occurs approximately 54-fold more slowly with thymine than with cytosine at pH 7.4. This is because thymine is released as an anion and cytosine as a neutral species; anion formation is less favorable in the negatively charged microenvironment of the phosphodiester backbone (37). The few DNAzymes that do promote depyrimidination through deglycosylation catalyze the reaction at multiple sites on the DNA substrate and they exhibit a strong preference for A or C over T (35).
In order to generate a DNAzyme that efficiently catalyzes site-specific excision at thymine nucleotide, we used in vitro selection to obtain F-8, which we characterize here structurally, kinetically and mechanistically. This new DNAzyme may become a useful tool for in vitro DNA manipulation.
MATERIALS AND METHODS
Materials
T4 polynucleotide kinase (PNK), T4 ligase and FastAPTM were purchased from MBI Fermentas. 9°N DNA ligase was purchased from New England Biolabs, Klentaq DNA polymerase from Huagene Biosciences (Fujian, China), and [γ-32P]ATP from Furui Biological Engineering (Beijing, China). ATP, dCTP and all oligonucleotides were PAGE-purified by Sangon Biotech (Shanghai, China). All reagents and chemicals, including H2O18, NaN3, DABCO, DMSO, H2O2, DTT and t-BuOH were analytical grade and used without further purification.
Labeling reaction
Oligonucleotides were phosphorylated using 10 units of PNK at 37°C for 1 h in 50 mM Tris-HCl (pH 7.8 at 23°C), 40 mM NaCl, 10 mM MgCl2, 1 mg/ml BSA and 10 μCi [γ-32P]ATP. The labeled product was purified by 10% denaturing PAGE.
Deoxyribozyme cleavage assays
Trans cleavage reactions were performed at 37°C using about 1 μM deoxyribozyme and 10 nM substrate in reaction buffer [50 mM HEPES (pH 7.4), 400 mM NaCl, 100 mM KCl, 10 mM MgCl2, 7.5 mM MnCl2, 50 μM CuCl2]. Before the reaction, deoxyribozyme-substrate was heated in H2O at 90°C for 30 s, then cooled at room temperature for 5 min. The cooled mixture was added to an equal volume of 2× reaction buffer and incubated at 37°C. Reactions were halted at predetermined times by adding EDTA (pH 8.0) to a final concentration of 30 mM. Trans cleavage products were separated on 10 or 20% denaturing PAGE and quantitated by Phosphorimager analysis.
Kinetic analyses
Time courses for each reaction condition were performed at least twice for eight time points. Data were fit to the exponential Y = Ymax [1 − e(−kobst)] using non-linear regression in GraphPad Prism 4. The observed rate constant (kobs) and maximum cleavage yield (Ymax) were determined from the regression curve.
RESULTS AND DISCUSSION
We used a library of randomized 40-nt oligonucleotides of randomized sequences flanked by upstream and downstream ‘binding arms’ that hybridize perfectly with the DNA substrate (Figure 2A). Like the well-known RNA/DNA-cleaving DNAzymes (35,38) that have common structure: (i) Highly conserved catalytic domain is flanked by two single-strand binding arms which will recognize substrate by forming duplex. (ii) The cleaving site is located in the middle of two binding arms and is in the opposition to the catalytic core. The binding arms are used as primers for PCR amplification during the selection cycles in order to avoid mutations in these regions. This strategy has worked well for in vitro selection of various DNAzymes (29–31). Our substrate contained a 6-nt open region intended to serve as the cleavage site, where the potential catalytic cores of DNAzymes would seek the favorite dinucleotide junction to cleave by themselves. Cleavage reactions were performed at 37°C in 50 mM HEPES (pH 7.4) containing 400 mM NaCl, 100 mM KCl, 10 mM MgCl2, 7.5 mM MnCl2 and 50 μM CuCl2. Here, we would like to identify new deoxyribozymes by in vitro selection in presence of Cu2+ and Mn2+ because both of them have redox activity which could promote the cleavage of DNA through oxidative pathway. Self-cleaved oligonucleotides were separated from intact ones by polyacrylamide gel electrophoresis (Supplementary Figure S2.1). Cleavage product was detected in the seventh round of selection, and the library pool was sequenced after 10 rounds of selection (Supplementary Figure S2.2).
Figure 2.
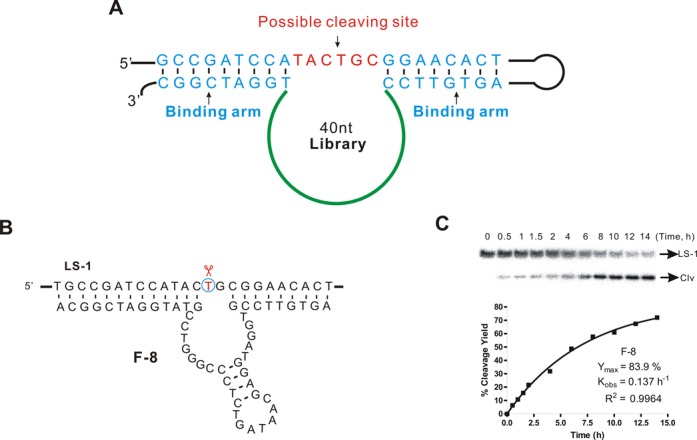
In vitro selection of DNA-cleaving DNAzymes. (A) Structure of the randomized library used for in vitro selection. A 40-nt randomized region was flanked by two binding arms that hybridized perfectly to the substrate strand containing the single-stranded target sequence TACTGC. (B) Structure of the in vitro-selected DNAzyme F-8. The thymidine at the cleavage site is circled. (C) PAGE analysis of an F-8 trans cleavage assay under single-turnover conditions (about 1 μM deoxyribozyme combined with 10 nM substrate) at 37°C in 50 mM HEPES (pH 7.4) containing 400 mM NaCl, 100 mM KCl, 10 mM MgCl2, 7.5 mM MnCl2 and 50 μM CuCl2. Representative results are shown for a 14-h time course; the observed rate constant (kobs ∼ 0.14 h−1, R2 = 0.9964) and maximum cleavage yield (Ymax) were derived using non-linear regression.
From the in vitro selection we obtained a new DNAzyme F-8, which trans-cleaved the target strand specifically at the desired thymidine with a kobs as high as 0.14 h−1 (Figure 2B and C and Supplementary Figure S3). We verified the precise location of DNA cleavage in systematic experiments in which we performed the cleavage reactions using DNA substrates radiolabeled with 32P on the 5′-, 3′-end and internally, and then analyzed the reaction products using high-resolution PAGE (Supplementary Figures S4, S5.1, S5.3 and S5.4). The cleavage generated the expected 3′- and 5′-phosphate DNA termini was confirmed by treating the cleavage products with phosphatase and DNA ligase and observing recovery of the original DNA construct (Supplementary Figures S5.2, S5.5 and S5.6). The results of these PAGE studies were further confirmed by analyzing the cleavage products using mass spectrometry (Figure 3B and C). The results of all these experiments lead us to conclude that F-8 catalyzes excision specifically at a thymidine but not purine nucleoside, and that it generates cleavage products with 3′- and 5′-phosphate termini. These termini can be further manipulated by natural enzymes.
Figure 3.
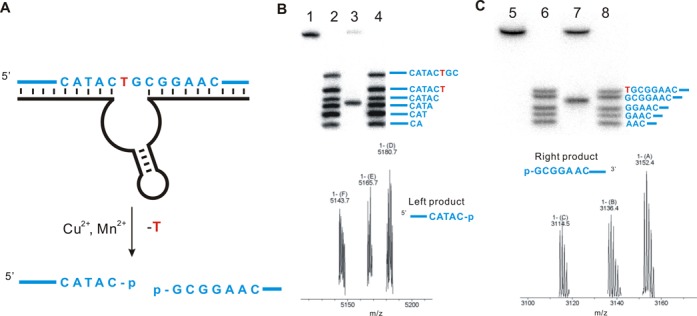
Nucleotide excision of DNA by the DNAzyme F-8. (A) The site of cleavage based on PAGE analysis and mass spectrometry is shown. (B) High-resolution 20% PAGE analysis (upper panel) and mass spectrometry (lower panel) to assign the upstream cleavage fragment generated by F-8. Lane 1, 5′-32P-radiolabeled substrate LS-1; lanes 2 and 4, 5’-32P-radiolabeled oligonucleotide standards based on the substrate LS-1; lane 3, DNA substrate cleaved by F-8 under standard conditions (calcd. 5142.8, found. 5143.7). (C) High-resolution 20% PAGE analysis (upper panel) and mass spectrometry (lower panel) to assign the downstream cleavage fragment generated by F-8. Lane 5, 3’-32P-radiolabeled substrate LS-1+8A (Figure S5.3); lanes 6 and 8, 3’-32P-radiolabeled oligonucleotide standards based on the substrate LS-1+8A; lane 7, DNA substrate cleaved by F-8 under standard conditions (calcd. 3114.5, found 3114.5). The assays indicate that F-8 generates products bearing 3′- and 5′-phosphate termini. Cleavage assays were under standard single-turnover conditions (about 1 μM deoxyribozyme combined with 10 nM substrate).
Having characterized the catalytic activity of F-8, we next analyzed the chemical requirements for its activity. F-8 requires both Mn2+ and Cu2+ as cofactors for full catalytic ability. In fact, F-8 activity strongly depends on the concentration of Cu2+, with maximal activity occurring near 12 μM (Figure 4C and Supplementary Figure S6.4). Above this concentration, however, F-8 gradually loses activity, perhaps because the high concentration of Cu2+ disrupts double-stranded structures (28,39). In contrast, F-8 is tolerant to quite high Mn2+ concentrations (Figure 4D and Supplementary Figure S6.5). Mn2+ and Cu2+ concentrations for optimal cleavage by F-8 were found to be, respectively, 40 mM and 12 μM. Cleavage efficiency did not vary with different concentrations of Na+, K+ or Mg2+ (data not shown). F-8 worked well over a wide range of pH (6.7–8.5) and temperature (15–50°C; Figure 4A and B, and Supplementary Figure S6.2 and S6.3). This robustness may make F-8 even more useful for future applications.
Figure 4.
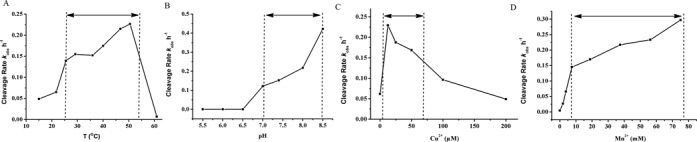
Cleavage of DNA substrate by F-8 under different reaction conditions. All reactions were conducted using trace amounts of 5’-32P-labeled substrates (about 10 nM) with 1 μM deoxyribozyme F-8 in 50 mM HEPES or other appropriate buffer containing 400 mM NaCl, 100 mM KCl and 10 mM MgCl2. (A) Reactions were performed under standard conditions (pH 7.4) at different temperatures. (B) Reactions were performed at 37°C at different pHs. (C) Reactions were performed at 37°C and at pH 7.4 in the presence of different Cu2+ concentrations. (D) Reactions were performed at 37°C and at pH 7.4 in the presence of different Mn2+ concentrations.
We examined the structure of F-8 in detail (Figure 5A). The secondary structure is predicted to consist of a stem–loop that ends in two single-stranded domains. Structure–function studies in which we systematically changed the nucleotide sequence of F-8 and monitored the cleavage activity of the variants showed that the binding arm oligonucleotides could be changed without loss of activity, as long as the complementarity of the double-stranded regions was preserved (Supplementary Figure S7.1 and S7.2). The optimal binding arm length was 11–12 nt (Supplementary Figure S7.3). The recognition site for F-8 is the tetranucleotide YTGC (Y = T/C), which has a frequency of one repeat every 128 bp in the human genome (Figure 5A). Deletion or substitution in this motif significantly reduced or abolished cleavage (Supplementary Figures S7.2 and S7.3). Replacing the T in the cleavage motif with A led to < 5% cleavage; replacing the T with G or C led to no detectable cleavage. These results indicate that F-8 cleaves at thymidine with high specificity.
Figure 5.
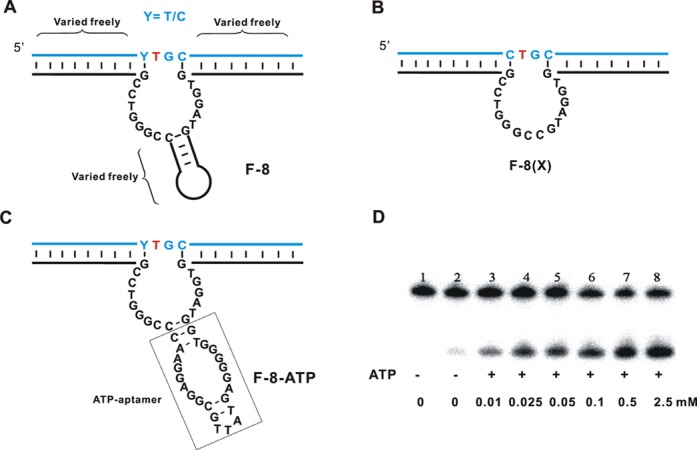
Structure-function studies of F-8. (A) Sequence in the structure of F-8 that were extensively varied. (B) Structure of F-8(X), a variant of F-8 lacking the hairpin but retaining cleavage activity. The catalytic core comprises 16 nt. (C) Sequence and structure of F-8-ATP, a variant of F-8 carrying an ATP-binding aptamer in the hairpin region intended to place the cleavage activity under allosteric regulation by ATP. (D) PAGE analysis of DNA substrate cleavage by F-8-ATP. Reactions were carried out for 4 h under standard conditions in the presence of 50 μM H2O2 and the indicated concentrations of ATP. Lane 1, 5′-32P-labeled DNA substrate. Lane 2, cleavage of F-8-ATP in the absence of additional ATP; lanes 3–8, cleavage reactions in the presence different concentrations of additional ATP. Cleavage assays were under standard single-turnover conditions (about 1 μM deoxyribozyme combined with 10 nM substrate).
We were able to freely vary the sequence of the hairpin in the catalytic core as well as the sequences of the binding arms (maintaining complementarity) without significantly affecting cleavage ability (Figure 5A). Similarly, lengthening or truncating the stem or altering its sequence did not affect cleavage (Supplementary Figure S7.3). In fact, substituting the entire hairpin with a GC dinucleotide junction to give the variant F-8(X) led to similar cleavage activity as observed with native F-8 (Figure 5B). To the best of our knowledge, F-8(X) is one of the shortest DNAzymes with DNA-cleaving catalytic ability (29,32). Shorter catalytic core regions allow more complete coverage of sequence space, and this could be achieved not only by in vitro selection but also by rational design.
These experiments clearly show that the hairpin of F-8 is not crucial for catalytic activity (Supplementary Figure S7.4). This further increases the potential usefulness of F-8 for future applications, since it may be possible to introduce functional oligonucleotides into the hairpin to achieve specific types of DNA manipulation. To test this possibility, we inserted an aptamer sequence into the hairpin to place F-8 under the allosteric regulation of ATP (Figure 5C and D) (40). As expected, this modification rendered F-8 dependent on ATP for efficient cleavage. These results suggest the possibility of ‘fine-tuning’ the activity of F-8 by insertion of other functional oligonucleotides.
DNAzymes that oxidatively cleave DNA react with molecular oxygen or hydrogen peroxide to generate a variety of reactive oxygen species or metal-oxo species that act as oxidative intermediates (18). In order to identify the active chemical species responsible for nucleotide excision by F-8, we performed cleavage reactions in the presence of radical scavengers (DMSO, t-BuOH), singlet oxygen quenchers [NaN3, 1,4-diazabicyclo[2,2,2]octane (DABCO)], a superoxide scavenger (superoxide dismutase, SOD), or a hydrogen peroxide scavenger (catalase). Cleavage activity was unaffected by addition of DMSO, t-BuOH or SOD (Supplementary Figure S8.1), allowing us to rule out the possibility that hydroxyl radical and superoxide were involved in cleavage reaction. The singlet oxygen scavenger DABCO did not significantly affect cleavage activity, while NaN3 significantly reduced cleavage (Supplementary Figure S8.2). We interpret these results to mean that singlet oxygen species are not involved; we attribute the inhibition in the presence of NaN3 to its ability to chelate metal ions (41). Addition of the H2O2 scavenger catalase completely inhibited the catalytic activity of F-8, suggesting that F-8 cleaved DNA through a mechanism involving H2O2. We have determined whether the DNA substrate could be cleaved only by an oxidant or reductant agent. As seen in Supplementary Figure S8.3, no evidence indicated that DNA cleavage was occurred in the presence of H2O2/DTT alone. We have also conducted a more detailed characterization of the DNA cleavage products in the presence of H2O2/DTT in order to assess the results of cleavage process. The cleavage generated the expected 3′- and 5′-phosphate DNA termini was consistent with that under standard cleavage conditions (Supplementary Figures S5.5, S5.6, S8.4 and S8.5). These results are consistent with an oxidative cleavage mechanism that makes use of hydrogen peroxide for the cleavage of DNA.
Consistent with this idea, we found that adding 50 μM H2O2 to the cleavage reaction significantly accelerated the reaction rate of F-8 (kobs ∼ 0.25 min−1) and of F-8(X) (kobs ∼ 0.084 min−1; Supplementary Figure S8.6 and S8.7). In addition, adding H2O2 as an oxidant or DTT as a reductant to the reaction, either together with Mn2+ and Cu2+ or in place of one or the other, greatly accelerated the reaction (Supplementary Figure S8.8 and S8.9).
Oxidative DNA cleavage involves a redox metal center, and we investigated the type and oxidation state of metal involved in F-8. Adding the Cu(I) chelator neocuproine (42) to the cleavage reaction inhibited F-8 activity (Supplementary Figure S8.10), suggesting that Cu(II) is reduced to Cu(I) during the reaction, and that Cu(I) participates in the cleavage reaction. Consistent with this idea, adding 50 μM Cu(I) to the reaction enhanced the catalytic activity of F-8 approximately 63-fold (Supplementary Figure S8.11). In contrast, adding the oxidant Mn3+ did not affect the cleavage reaction (Supplementary Figure S8.12), suggesting that DNA cleavage by F-8 is more likely to involve copper ion than manganese ion.
As further confirmation that F-8 does not cleave DNA by a hydrolytic mechanism, we performed the reaction in 18O-water as solvent. Mass spectrometry revealed no 18O in the phosphate groups of the cleavage products (Supplementary Figure S8.13). Taken together, our mechanistic studies suggest that F-8 cleaves DNA by the same mechanism by which DNA cutters oxidize 2-deoxyribose (18).
We examined whether we could combine DNAzyme F-8 with natural enzymes to achieve site-specific single-nucleotide excision repair while maintaining the integrity of the original DNA sequence in vitro. We focused on G·T mismatches, which are common errors during replication; these usually trigger base excision repair, in which the T is removed and the C is restored (43–45). To realize our strategy, Klentaq DNA polymerase, which owns higher fidelity, more heatstable activity and lacks the 5′-3′ exonuclease activity and thermal stable 9°N with DNA ligation activity were chosen. First we used F-8 to catalyze site-specific excision at the designated pyrimidine (T) to generate both 3′- and 5′-phosphate termini (Figure 6). Next we added T4 polynucleotide kinase (PNK) to remove only the 3′-phosphate group (46). Recover the processed DNA fragments from PAGE gel. Introducing a right DNA splint and dCTP allowed us to extend the free 3′-end of one DNA fragment by one deoxynucleotide (C), with sequential extension and ligation reactions in a one pot, the product DNA strand has a sequence identical to the starting DNA substrate, with the T replaced with C. The reaction conditions consist of a 3 min denaturation at 94°C followed by 23 cycles of 94°C (30 s) and 50°C (3 min) in the PCR thermal cycler. Each step of the process was monitored using radio-labeled oligonucleotides and PAGE analysis. The product in lane 5 (Figure 6) has sequence identical to the synthetic marker, while it was not visible on the gel when in the absence of dCTP (lane 5, Supplementary Figure S8.14). The product was also confirmed by the result from resistance to cleavage with F-8 (Supplementary Figure S8.15). In this way, the sequence of depyrimidination, extension and ligation allowed us to achieve in vitro single-deoxyribonucleotide repair in which T was replaced with C.
Figure 6.
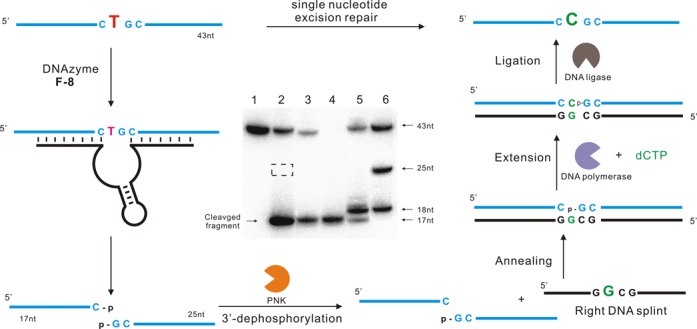
Conversion of a single deoxybonucleotide T (red, left) in a DNA strand to a deoxyribonucleotide C (green, right) using a combination of DNAzyme F-8 and natural enzymes. DNA substrate was cleaved by F-8 to generate both 3′- and 5′-phosphate termini. Then the 3′-phosphate group was removed by T4 polynucleotide kinase (PNK), and a right DNA splint was introduced. Subsequent extension and ligation restored the original DNA except for a substitution of T with C. Inset: PAGE analysis of the various stages of the single-nucleotide excision repair process. Lane 1, the original 43-nt 5′-32P-labeled DNA substrate. Lane 2, the upstream cleavage product generated by F-8. The cleaved fragment migrates slightly faster than the corresponding 18-nt marker because of its 3′-phosphate group. The dotted box indicates the expected position of the downstream cleavage product, which was invisible on the autoradiogram since it contained no 32P. Lane 3, the cleavage product after dephosphorylation with PNK. Lane 4, 5′-32P-labeled marker that comigrates with the 17-nt upstream cleavage fragment. Lane 5, the reaction products obtained after purifying the cleavage fragments, annealing them with right DNA splint, extending them by a C (18 nt) using DNA polymerase, and finally ligating to generate full-length substrate (43 nt). Lane 6, synthetic DNA markers. Cleavage assays were under standard single-turnover conditions (about 1 μM deoxyribozyme combined with 10 nM substrate).
CONCLUSION
We have generated, by in vitro selection, a new DNA-cleaving DNAzyme, called F-8, that catalyzes the site-specific cleavage of single-stranded DNA at thymidine but not purine nucleoside in the presence of Cu2+ and Mn2+ as cofactors. Adding H2O2 significantly accelerates the cleavage rate. This oxidative cleavage generates both 3′- and 5′-phosphate termini that we were able to modify using natural enzymes to achieve the site-specific single-nucleotide excision repair of a T for a C. Structure-function studies show that the catalytic activity of F-8 can tolerate extensive substitutions in the hairpin domain, and as an example, we added an aptamer to the hairpin and made cleavage activity ATP-dependent. This suggests that F-8 holds excellent potential as a tool for specific DNA manipulations. Our laboratory continues to use in vitro selection to identify novel DNAzyme tools.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Chinese Academy of Science [Hundred Talents Program]; the National Natural Science Foundation of China [21172215, 21102139, 21232005, 21321061 and 21322208]; National Program on Key Basic Research Projects of China (973 Program) [2012CB720603]. Funding for open access charge: National Natural Science Foundation of China [21322208].
Conflict of interest statement. None declared.
REFERENCES
- 1.Breaker R.R., Joyce G.F. A DNA enzyme with Mg(2+)-dependent RNA phosphoesterase activity. Chem. Biol. 1995;2:655–660. doi: 10.1016/1074-5521(95)90028-4. [DOI] [PubMed] [Google Scholar]
- 2.Swarts S.G., Gilbert D.C., Sharma K.K., Razskazovskiy Y., Purkayastha S., Naumenko K.A., Bernhard W.A. Mechanisms of direct radiation damage in DNA, based on a study of the yields of base damage, deoxyribose damage, and trapped radicals in d(GCACGCGTGC)(2) Radiat. Res. 2007;168:367–381. doi: 10.1667/RR1058.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kincaid K., Beckman J., Zivkovic A., Halcomb R.L., Engels J.W., Kuchta R.D. Exploration of factors driving incorporation of unnatural dNTPS into DNA by Klenow fragment (DNA polymerase I) and DNA polymerase alpha. Nucleic Acids Res. 2005;33:2620–2628. doi: 10.1093/nar/gki563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Halliwell B. Oxidative stress and neurodegeneration: where are we now? Neurochemistry. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 5.Baute J., Depicker A. Base excision repair and its role in maintaining genome stability. Crit. Rev. Biochem. Mol. Biol. 2008;43:239–276. doi: 10.1080/10409230802309905. [DOI] [PubMed] [Google Scholar]
- 6.Wabuyele M.B., Farquar H., Stryjewski W., Hammer R.P., Soper S.A., Cheng Y.W., Barany F. Approaching real-time molecular diagnostics: single-pair fluorescence resonance energy transfer (spFRET) detection for the analysis of low abundant point mutations in K-ras oncogenes. J. Am. Chem. Soc. 2003;125:6937–6945. doi: 10.1021/ja034716g. [DOI] [PubMed] [Google Scholar]
- 7.Myers R.M., Lumelsky N., Lerman L.S., Maniatis T. Detection of single base substitutions in total genomic DNA. Nature. 1985;313:495–498. doi: 10.1038/313495a0. [DOI] [PubMed] [Google Scholar]
- 8.Ng J.K.K., Feng H.H., Liu W.T. Rapid discrimination of single-nucleotide mismatches using a microfluidic device with monolayered beads. Anal. Chim. Acta. 2007;582:295–303. doi: 10.1016/j.aca.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 9.Van Bers N.E.M., Santure A.W., Van Oers K., De Cauwer I., Dibbits B.W., Materman C., Crooijmans R.P.M.A., Sheldon B.C., Visser M.E., Groenen M.A.M., et al. The design and cross-population application of a genome-wide SNP chip for the great tit Parus major. Mol. Ecol. Resour. 2012;12:753–770. doi: 10.1111/j.1755-0998.2012.03141.x. [DOI] [PubMed] [Google Scholar]
- 10.Duan X., Liu L., Wang S. Homogeneous and one-step fluorescent allele-specific PCR for SNP genotyping assays using conjugated polyelectrolytes. Biosens. Bioelectron. 2009;24:2095–2099. doi: 10.1016/j.bios.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 11.Shi C., Ge Y., Gu H., Ma C. Highly sensitive chemiluminescent point mutation detection by circular strand-displacement amplification reaction. Biosens. Bioelectron. 2011;26:4697–4701. doi: 10.1016/j.bios.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 12.Bi S., Li L., Zhang S. Triggered polycatenated DNA scaffolds for DNA sensors and aptasensors by a combination of rolling circle amplification and DNAzyme amplification. Anal. Chem. 2010;82:9447–9454. doi: 10.1021/ac1021198. [DOI] [PubMed] [Google Scholar]
- 13.Zhou L., Du F., Zhao Y., Yameen A., Chen H., Tang Z. DNAzyme based gap-LCR detection of single-nucleotide polymorphism. Biosens. Bioelectron. 2013;45:141–147. doi: 10.1016/j.bios.2013.01.061. [DOI] [PubMed] [Google Scholar]
- 14.Rydberg B., Game J. Excision of misincorporated ribonucleotides in DNA by RNase H (type 2) and FEN-1 in cell-free extracts. Proc. Natl. Acad. Sci. U.S.A. 2002;99:16654–16659. doi: 10.1073/pnas.262591699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim N., Huang S.N., Williams J.S., Li Y.C., Clark A.B., Cho J.E., Kunkel T.A., Pommier Y., Jinks-Robertson S. Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science. 2011;332:1561–1564. doi: 10.1126/science.1205016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiang Y., Wang Z., Xing H., Lu Y. Expanding DNAzyme functionality through enzyme cascades with applications in single nucleotide repair and tunable DNA-directed assembly of nanomaterials. Chem. Sci. 2013;4:398–404. doi: 10.1039/C2SC20763J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aiba Y., Sumaoka J., Komiyama M. Artificial DNA cutters for DNA manipulation and genome engineering. Chem. Soc. Rev. 2011;40:5657–5668. doi: 10.1039/c1cs15039a. [DOI] [PubMed] [Google Scholar]
- 18.Dedon P.C. The chemical toxicology of 2-deoxyribose oxidation in DNA. Chem. Res. Toxicol. 2008;21:206–219. doi: 10.1021/tx700283c. [DOI] [PubMed] [Google Scholar]
- 19.David S.S., Williams S.D. Chemistry of glycosylases and endonucleases involved in base-excision repair. Chem. Rev. 1998;98:1221–1262. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- 20.Schroeder G.K., Wolfenden R. Rates of spontaneous disintegration of DNA and the rate enhancements produced by DNA glycosylases and deaminases. Biochemistry. 2007;46:13638–13647. doi: 10.1021/bi701480f. [DOI] [PubMed] [Google Scholar]
- 21.Loverix S., Geerlings P., McNaughton M., Augustyns K., Vandemeulebroucke A., Steyaert J., Versees W. Substrate-assisted leaving group activation in enzyme-catalyzed N-glycosidic bond cleavage. J. Biol. Chem. 2005;280:14799–14802. doi: 10.1074/jbc.M413231200. [DOI] [PubMed] [Google Scholar]
- 22.Birck M.R., Schramm V.L. Nucleophilic participation in the transition state for human thymidine phosphorylase. J. Am. Chem. Soc. 2004;126:2447–2453. doi: 10.1021/ja039260h. [DOI] [PubMed] [Google Scholar]
- 23.Murray P.E., McNally V.A., Lockyer S.D., Williams K.J., Stratford I.J., Jaffar M., Freeman S. Synthesis and enzymatic evaluation of pyridinium-substituted uracil derivatives as novel inhibitors of thymidine phosphorylase. Bioorg. Med. Chem. 2002;10:525–530. doi: 10.1016/s0968-0896(01)00309-1. [DOI] [PubMed] [Google Scholar]
- 24.Fromme J.C., Banerjee A., Verdine G.L. DNA glycosylase recognition and catalysis. Curr. Opin. Struct. Biol. 2004;14:43–49. doi: 10.1016/j.sbi.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 25.Pogozelski W.K., Tullius T.D. Oxidative strand scission of nucleic acids: routes initiated by hydrogen abstraction from the sugar moiety. Chem. Rev. 1998;98:1089–1108. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- 26.Cuenoud B., Szostak J.W. A DNA metalloenzyme with DNA ligase activity. Nature. 1995;375:611–614. doi: 10.1038/375611a0. [DOI] [PubMed] [Google Scholar]
- 27.Carmi N., Shultz L.A., Breaker R.R. In vitro selection of self-cleaving DNAs. Chem. Biol. 1996;3:1039–1046. doi: 10.1016/s1074-5521(96)90170-2. [DOI] [PubMed] [Google Scholar]
- 28.Carmi N., Breaker R.R. Characterization of a DNA-cleaving deoxyribozyme. Bioorg. Med. Chem. 2001;9:2589–2600. doi: 10.1016/s0968-0896(01)00035-9. [DOI] [PubMed] [Google Scholar]
- 29.Chandra M., Sachdeva A., Silverman S.K. DNA-catalyzed sequence-specific hydrolysis of DNA. Nat. Chem. Biol. 2009;5:718–720. doi: 10.1038/nchembio.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao Y., Allen E.C., Silverman S.K. Merely two mutations switch a DNA-hydrolyzing deoxyribozyme from heterobimetallic (Zn2+/Mn2+) to monometallic (Zn2+-only) behavior. Chem. Commun. 2011;47:1749–1751. doi: 10.1039/c0cc04575f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao Y., Wehrmann R.J., Ibrahim N.A., Silverman S.K. Establishing broad generality of DNA catalysts for site-specific hydrolysis of single-stranded DNA. Nucleic Acids Res. 2012;40:1778–1786. doi: 10.1093/nar/gkr860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu H., Furukawa K., Weinberg Z., Berenson D.F., Breaker R.R. Small, highly active DNAs that hydrolyze DNA. J. Am. Chem. Soc. 2013;135:9121–9129. doi: 10.1021/ja403585e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheppard T.L., Ordoukhanian P., Joyce G.F. A DNA enzyme with N-glycosylase activity. Proc. Natl. Acad. Sci. U.S.A. 2000;97:7802–7807. doi: 10.1073/pnas.97.14.7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hobartner C., Pradeepkumar P.I., Silverman S.K. Site-selective depurination by a periodate-dependent deoxyribozyme. Chem. Commun. 2007:2255–2257. doi: 10.1039/b704507g. [DOI] [PubMed] [Google Scholar]
- 35.Dokukin V., Silverman S.K. Lanthanide ions as required cofactors for DNA catalysts. Chem. Sci. 2012;3:1707–1714. doi: 10.1039/C2SC01067D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Velez T.E., Singh J., Xiao Y., Allen E.C., Wong O.Y., Chandra M., Kwon S.C., Silverman S.K. Systematic evaluation of the dependence of deoxyribozyme catalysis on random region length. ACS Comb. Sci. 2012;14:680–687. doi: 10.1021/co300111f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berti P.J., McCann J.A. Toward a detailed understanding of base excision repair enzymes: transition state and mechanistic analyses of N-glycoside hydrolysis and N-glycoside transfer. Chem. Rev. 2006;106:506–555. doi: 10.1021/cr040461t. [DOI] [PubMed] [Google Scholar]
- 38.Santoro S.W., Joyce G.F. A general purpose RNA-cleaving DNA enzyme. Proc. Natl. Acad. Sci. U.S.A. 1997;94:4262–4266. doi: 10.1073/pnas.94.9.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rifkind J.M., Shin Y.A., Heim J.M., Eichhorn G.L. Cooperative disordering of single-stranded polynucleotides through copper crosslinking. Biopolymers. 1976;15:1879–1902. doi: 10.1002/bip.1976.360151002. [DOI] [PubMed] [Google Scholar]
- 40.Breaker R.R. Engineered allosteric ribozymes as biosensor components. Curr. Opin. Biotechnol. 2002;13:31–39. doi: 10.1016/s0958-1669(02)00281-1. [DOI] [PubMed] [Google Scholar]
- 41.El Amrani F.B., Perello L., Real J.A., Gonzalez-Alvarez M., Alzuet G., Borras J., Garcia-Granda S., Montejo-Bernardo J. Oxidative DNA cleavage induced by an iron(III) flavonoid complex: synthesis, crystal structure and characterization of chlorobis (flavonolato)(methanol) iron(III) complex. J. Inorg. Biochem. 2006;100:1208–1218. doi: 10.1016/j.jinorgbio.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 42.Oikawa S., Kawanishi S. Site-specific DNA damage induced by NADH in the presence of copper(II): role of active oxygen species. Biochemistry. 1996;35:4584–4590. doi: 10.1021/bi9527000. [DOI] [PubMed] [Google Scholar]
- 43.Wiebauer K., Jiricny J. In vitro correction of G.T mispairs to G.C pairs in nuclear extracts from human cells. Nature. 1989;339:234–236. doi: 10.1038/339234a0. [DOI] [PubMed] [Google Scholar]
- 44.Neddermann P., Jiricny J. The purification of a mismatch-specific thymine-DNA glycosylase from HeLa cells. J. Biol. Chem. 1993;268:21218–21224. [PubMed] [Google Scholar]
- 45.Maiti A., Drohat A.C. Dependence of substrate binding and catalysis on pH, ionic strength, and temperature for thymine DNA glycosylase: insights into recognition and processing of G.T mispairs. DNA Repair (Amst) 2011;10:545–553. doi: 10.1016/j.dnarep.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schurer H., Lang K., Schuster J., Morl M. A universal method to produce in vitro transcripts with homogeneous 3’ ends. Nucleic Acids Res. 2002;30:e56. doi: 10.1093/nar/gnf055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.