

Abstract
We previously identified the heterogeneous ribonucleoprotein SAF-A/hnRNP U as a substrate for DNA-PK, a protein kinase involved in DNA damage response (DDR). Using laser micro-irradiation in human cells, we report here that SAF-A exhibits a two-phase dynamics at sites of DNA damage, with a rapid and transient recruitment followed by a prolonged exclusion. SAF-A recruitment corresponds to its binding to Poly(ADP-ribose) while its exclusion is dependent on the activity of ATM, ATR and DNA-PK and reflects the dissociation from chromatin of SAF-A associated with ongoing transcription. Having established that SAF-A RNA-binding domain recapitulates SAF-A dynamics, we show that this domain is part of a complex comprising several mRNA biogenesis proteins of which at least two, FUS/TLS and TAFII68/TAF15, exhibit similar biphasic dynamics at sites of damage. Using an original reporter for live imaging of DNA:RNA hybrids (R-loops), we show a transient transcription-dependent accumulation of R-loops at sites of DNA damage that is prolonged upon inhibition of RNA biogenesis factors exclusion. We propose that a new component of the DDR is an active anti-R-loop mechanism operating at damaged transcribed sites which includes the exclusion of mRNA biogenesis factors such as SAF-A, FUS and TAF15.
INTRODUCTION
Deoxyribonucleic acid (DNA) double-strand break (DSB) is the most toxic type of DNA damage. If improperly repaired, DSBs can cause cell death or mutations and gross chromosomal rearrangements promoting cancer development (1–4). In mammalian cells, DSBs initiate a global DNA damage response (DDR) to overcome their toxicity and maintain genome stability. DDR includes lesions detection, checkpoint activation, modulation of gene expression and DNA repair (5–9). DDR defects manifest as a variety of human diseases, including neurodegenerative disorders, immunodeficiency, infertility and cancer (5).
Another component of the DDR is local transcription arrest triggered by DNA breaks (10–13). More generally, an expanding aspect of the DDR is its connection with ribonucleic acid (RNA) metabolism. Indeed, the DNA damage activated kinases ATM or ATR phosphorylate numerous proteins involved in RNA metabolism (14,15) and links with the DDR have been established for several members of the heterogeneous ribonucleoprotein (hnRNP) family (16), RNA-binding proteins (RBPs) (17–25) or pre-RNA processing factors (26,27). Moreover, RNA-processing factors are major mediators of genome stability, some of them by preventing interactions between the nascent RNA and template DNA (R-loops) (28–33) which are relevant source of DNA breaks (33,34).
We and another group have identified SAF-A/hnRNP U (hereinafter referred to as SAF-A), as a substrate for DNA-PK, a key protein kinase involved in DSB repair by non-homologous end-joining (NHEJ) (35,36). In NHEJ, DNA-PK operates together with the DSBs sensor Ku70/80 heterodimer and the XRCC4/DNA ligase IV ligation complex (37). SAF-A is an abundant nuclear protein found in hnRNP particles and contains both DNA-binding domain (DBD) and RNA-binding domain (RBD) (38,39) (Figure 1A). The HNRNPU gene coding for SAF-A is essential for cell viability (40) and the protein participates in chromatin organization and transcription repression in specialized territories (41,42). SAF-A is implicated in several aspects of RNA metabolism, including transcription elongation through interaction with nuclear actin and RNA polymerase II (43,44), RNA stability control (45) and alternative splicing through regulation of U2 snRNP maturation (46).
Figure 1.
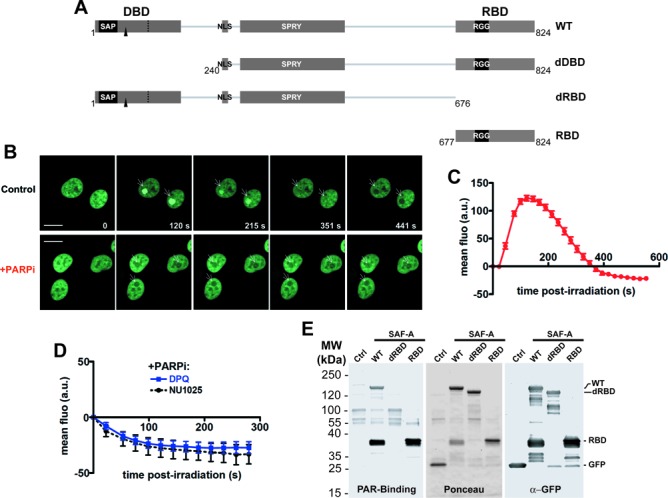
SAF-A dynamics in response to laser micro-irradation. (A) Map of SAF-A domains and of the truncations used. The main domains are as follows: the DNA-binding domain (DBD) that contains a SAP motif, a nuclear localization sequence (NLS), a SPRY (SPore lysis A and RYanodine receptor) domain and the RNA-binding domain (RBD) that contains an RGG motif. The phosphorylation site (S59) is indicated by a black arrow. WT: wild-type SAF-A; dDBD: deletion of the DNA-binding domain; dRBD: deletion of the RNA-binding domain; RBD: SAF-A RNA-binding domain only. (B) SAF-A-GFP behavior after 800-nm pulsed-laser nuclear irradiation assessed in HT1080 cells by live cell imaging at the indicated time post-irradiation in the presence or not of PARPi (DPQ). The white arrows mark the irradiated areas. Scale bar, 20 μm. (C) Dynamics of SAF-A-GFP at laser-damaged sites. Images were obtained at 22-s intervals, and fluorescence intensities at the damage sites were quantified. Mean values of the fluorescence intensities with SEM were calculated from 22 independent measurements. (D) Dynamics of SAF-A-GFP at laser-damaged sites in the presence of PARPi (DPQ or NU1025). Images were obtained at 22-s intervals, and fluorescence intensities at the damage sites were quantified. Mean values of the fluorescence intensities with SEM were calculated from 11 independent measurements under each condition. (E) PAR-binding assay. FLAG-GFP (Ctrl) or the indicated FLAG-GFP tagged forms of SAF-A were purified from transiently transfected HEK293T cells, separated on SDS-PAGE and transferred on membrane. The membrane was stained with Ponceau S to assess the quality of the purification. After incubation with purified PAR, retained PAR was detected using anti-PAR antibody. After stripping, an anti-GFP immunodetection was performed.
Here, we investigate further the involvement of SAF-A in the DDR. We document the local post-damage exclusion of an RBP complex including SAF-A and at least two of its partners FUS/TLS and TAFII68/TAF15, which is uncoupled from their initial poly(ADP-ribose) (PAR)-dependent recruitment at these sites. In addition, we present several results supporting that RBP exclusion is part of an anti-R-loop mechanism operating at DNA damage sites. Therefore, the present data further substantiate the links between RBPs, the DDR and genome stability.
MATERIALS AND METHODS
Cell culture
Human HT1080 fibrosarcoma cells, human U2OS osteosarcoma cells (ECCAC, Salisbury, UK) and human HEK293T were grown in Dulbecco's modified Eagle's medium medium (Fisher Scientific, Illkirch, France) supplemented with 10% foetal calf serum (Lonza, Basel, Switzerland), 2-mM glutamine, 125-U/ml penicillin and 125-μg/ml streptomycin. All cells were grown in a humidified atmosphere, at 37°C with 5% CO2.
Plasmids and DNA manipulations
A list of primers used in the study is provided in Supplementary Table S1. All DNA constructs were confirmed mutation free as tested by DNA sequencing. pEGFP-N1-FLAG plasmid containing a FLAG tag with a start codon and a strong kozak sequence between BamHI and MluI was generated by insertion of annealed FLAG-S and FLAG-AS oligonucleotides between BamHI and AgeI restriction sites of pEGFP-N1 (Clontech). For expression of SAF-A-FLAG-GFP, SAFA was sub-cloned into pEGFP-N1-FLAG between BamHI and MluI after amplification by polymerase chain reaction (PCR) with SAF-A-F and SAF-A-R primers and using pcDNA3-SAF-A-FLAG (36) as a matrix. Deletion mutants were derived from this plasmid. Plasmids expressing SAF-A-dDBD, SAF-A-dRBD and SAFA-RBD were generated by PCR using, respectively, SAF-A-dDBD-F and SAF-A-R, SAF-A-F and SAF-A-dRBD-R, and SAF-A-RBD-F and SAF-A-R as primer pairs. The resulting PCR products were cloned between BamHI and MluI into pEGFP-N1-FLAG. For FUS-FLAG-GFP expression, FUS was sub-cloned by PCR using FUS-F and FUS-R primers and pOTB7-FUS (IMAGE clone 7303359) as a matrix. The resulting PCR product was cloned between BamHI and MluI sites of pEGFP-N1-FLAG. For doxycycline-inducible expression of D10R E48R mutant of Escherichia coli mCherry-NLS-RNaseHI, a codon optimized sequence of the mutant RNase HI including a 5′ start codon in a strong kozak sequence and a 3′ in frame nuclear localization signal from SV40 large T antigen (NLS) were generated by gene synthesis (GeneArt, LIfe Technologies). The RNase HI-NLS sequences were recovered by HindIII and AgeI digestion and cloned together with AgeI and NotI-digested mCherry from pmCherry-C1 (Clontech) between HindIII and NotI restriction sites of pICE, a new synthetic plasmid allowing doxycline-inducible expression and conferring to human cells resistance to puromycin (47). A control plasmid expressing NLS-mCherry was generated by replacing RNase HI cDNA by annealed NLS-S and NLS-AS oligonucleotides cloned between HindIII and AgeI.
PAR-binding assay
For experiments carried in HEK293T, 140-mm dishes were seeded with 5 million cells 2 days before transfection with lipofectamine 2000 (Thermo scientific) according to manufacturer's instructions using 20 μg of plasmid DNA coding for each FLAG-GFP tagged constructs. Two days after transfection, cells were collected, washed in phosphate-buffered saline (PBS) and lysed 15 min on ice plus 5 min at room temperature in 300 μl of lysis buffer [10-mM Tris-HCl pH 7.8, 150-mM NaCl, 1-mM ethylenediaminetetraacetic acid (EDTA), 0.5% NP-40] containing 0.2-mg/ml RNase A and protease and phosphatase inhibitors (HALT, Thermo Scientific). Extracts were then clarified by 4-min centrifugation at 14 000 rpm at 4°C. Supernatant diluted with 400-μl dilution buffer (20-mM Tris-HCl pH 7.8, 150-mM NaCl, 1-mM EDTA, 0.05% NP-40 containing protease and phosphatase inhibitors (HALT, Thermo scientific) was incubated 4 h at 4°C on gentle shaking with 50 μl of GFP-trap magnetic beads (Chromotek). Beads were washed three times with 500-μl stringent washing buffer (20-mM Tris-HCl pH 7.8, 500-mM NaCl, 1-mM EDTA, 0.05% NP-40 buffer containing protease and phosphatase inhibitors (HALT) and once in dilution buffer. For experiments carried in HT1080, two 140-mm dishes were seeded with 2.5-million cells GFP-sorted for stable expression of FLAG-GFP, SAF-A-WT-FLAG-GFP, SAF-A-dRBD-FLAG-GFP and SAF-A-RBD-FLAG-GFP. After two days, cells were collected by scrapping and sonicated (24 pulses of 2 s and 30 of intensity, Vibra-cell sonicator) in 1 ml of GFP-trap lysis buffer [20-mM Tris-HCl pH 7.8, 1-mM EDTA, 150-mM NaCl, 0.1% NP-40 with HALT protease and phosphatase inhibitors Cocktail (Thermo scientific) and 1-mM fresh phenylmethylsulfonyl fluoride (PMSF)] containing 0.1-mg/ml RNase A. Extracts were clarified by a 20-min centrifugation at 4000 rpm at 4°C. Sixty microliter of GFP-trap magnetic beads (Chromotek) was incubated 3 h with supernatants on gentle shaking at 4°C. Beads were washed three times with 1-ml washing buffer (20-mM Tris-HCl pH7.8, 1-mM EDTA, 500-mM NaCl, 0.5% NP-40) and once in 1-ml lysis buffer (50-mM Tris-HCl pH7.8, 1-mM EDTA, 150-mM NaCl, 1% Triton X-100). For both for experiments in HEK293T and HT1080, beads were boiled after immunoprecipitation (IP) in lysis buffer, supernatant loaded on sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) gradient gels and transferred on polyvinylidene fluoride (PVDF, Millipore) or nitrocellulose (Protran, Whatman) membranes. Membranes were saturated with PBS-T containing 5% non-fat milk and incubated 1 h with 10-nM PAR polymer (Trevigen) in PBS-T. After extensive washes in PBS-T, the membrane was processed as a regular western blot membrane using 0.5 μg/ml of anti-PAR polyclonal antibody (1/2000 dilution, BD Pharmingen).
Transfection
HT1080 cells were seeded in 6-well plates until they reached 70–80% confluence. Transfection reactions were performed with 3 μg of plasmid using JetPEI (Polyplus Transfection, Illkirch, France) according to the manufacturer's recommendations. Cells were trypsinized and reseeded in 140-mm dishes 24 h after transfection and selected over 2 weeks by adding 500-μg/ml G418 (Fisher Scientific, Illkirch, France) or 0.5-μg/ml puromycin (InvivoGen, San Diego, CA, USA) for pEGFP-N1 or pICE vector backbones, respectively. Independent resistant clones were then picked separately, amplified and maintained in culture medium supplemented with 250-μg/ml G418 or 0.25-μg/ml puromycin. Expression of proteins of interest in the different clones was checked by direct GFP or mCherry fluorescence under the microscope when applicable and by western blot.
For inducible expression of RNaseHI-NLS-mCherry and NLS-mCherry, U2OS T-REx (Life Technologies) were transfected using Lipofectamine 2000 (LIfe Technologies) according to manufacturer's instructions with pICE-NLS-mCherry or pICE-RNaseHI-NLS-mCherry and selected with puromycin at 0.25 μg/ml. Individual clones were isolated and maintained in selective medium.
Live-cell microscopy, micro-irradiation and fluorescence recovery after photobleaching experiments
Cells were grown and observed in 35-mm glass-bottom culture dishes (MatTek). Experiments were carried out with a ZEISS LSM 710 confocal laser scanning microscope equipped with a coherent chameleon Vision II tunable laser (690–1080 nm) and a 40X/1.3 oil immersion objective. GFP was excited using a 488-nm Ar-laser line and mCherry with a 561-nm Ne-laser line. The microscope was equipped with a heated environmental chamber set at 37°C in 5% CO2 atmosphere.
Confocal image series were recorded with a frame size of 512 × 512 pixels. Nuclei micro-irradiation was carried out at 800 nm at 20% of max power (mean max power was 3600 mW) in spots of 5-μm diameter at 3X zoom during 22 s. Before and after micro-irradiation, confocal image series of one mid z-section were recorded at 60-s or 22-s time interval (typically two pre-irradiation and 30–40 post-irradiation frames). For evaluation of the recruitment kinetics, fluorescence intensities of the irradiated region were corrected for total nuclear loss of fluorescence over the time course and normalized to the pre-irradiation value. Data from micro-irradiation of individual cells obtained in several independent experiments performed on different days were averaged, analyzed and displayed using PRISM software. Images of fixed cells were recorded with the same microscope.
For immunofluorescence experiments, irradiation was carried out at 800 nm at X2 zoom, on multiple cell fields (tile scan 3 × 3); the laser line step was set up to 7 and the total irradiation time was 10 s. Images of fixed cells were recorded with the same microscope.
Fluorescence recovery after photobleaching (FRAP) experiments were performed with the Ar-laser line (GFP tag) or Ne-laser line (mCherry tag) at 100% intensity. A single optical section of a cell nucleus was acquired at 3× zoom. Fifteen iterations were used for the bleach pulse on a strip area (5 × 20 μm).
Fluorescence recovery was monitored using the same laser with 0.4% laser intensity (GFP tag) or 1% intensity (mCherry tag) to avoid bleaching during the time laps acquisition. Typically 20 images with an interval of 487 ms were taken before the bleach and during 50 or 100 s after.
Fluorescence intensities of the photo-bleached region, the total nuclear region and the background outside the nucleus were measured at each time point using the ‘mean intensity’ analysis function of the Zeiss software. For the quantitative evaluation of the fluorescence recovery, background value was first subtracted from raw mean fluorescence data series. Then the fluorescence signal measured in a region of interest normalized to the change in total fluorescence was determined as previously described (48). An additional normalization to the first post-bleach value allows comparison of independent experiments. Data from three to four independent experiments were averaged, analyzed and displayed using PRISM software. Recovery half-time was computed with Prism 5.0 through fitting of the normalized data to a one-phase exponential association curve of equation %Recovery(t) = Plateau x (1-exp (−K x t)) with Half-time ( = ln(2)/K).
DNA-damaging treatments and inhibitors
Calicheamicin γ1 (Cali), a generous gift from P.R. Hamann (Wyeth Research, Pearl River, NY, USA), was dissolved at 4 mM in ethanol. Cali treatment was carried in medium for 1 h at 37 °C. Infrared (IR) treatments were carried using a Faxitron RX-650 irradiator (Faxitron X-ray Corporation, Buffalo Grove, IL, USA) at a dose rate of 0.92 Gy/min. Inhibitors and concentrations used are listed in Supplementary Table S2.
Cell extraction
1.5 × 108 grown in culture plates were trypsinized, washed twice in PBS at 4°C. The cell pellet was resuspended in 2 packed cell volume (PCV) of lysis buffer (10-mM Tris-HCl pH7.5, 150-mM NaCl, 0.5-mM EDTA, 0.1% NP40) containing protease–phosphatase Halt Inhibitor Cocktail (Pierce, Thermo Scientific) and 0.1-mg/ml RNase A and processed by three freeze/thaw cycles with intermittent vortexing, followed by centrifugation at 14 000 rpm for 15 min at 4°C (Heraeus, Biofuge, primoR). Protein concentration was measured in the supernatant using the Bradford assay (Bio-Rad) and whole cell extracts were stored at −80°C.
Biochemical fractionation and immunoblotting
After drug exposure, cells were washed with PBS and harvested. Pellets of about 1.5 × 106 cells were fractionated as reported (49,50) with modifications as follows. Cells were first resuspended for 5 min on ice in 200 μl of fractionation buffer (50-mM Hepes pH 7.5, 150-mM NaCl, 1-mM EDTA) containing 0.05% Nonidet P-40 (NP-40) and supplemented with the Halt protease and phosphatase inhibitor cocktail (Pierce). Following centrifugation at 1000 x g for 5 min, the supernatant was collected (fraction I), and pellets were resuspended in 200 μl of the same buffer and centrifuged as above. The supernatant was collected (fraction 2), and the nuclear pellets were further extracted for 40 min on ice with 200 μl of fractionation buffer containing 0.5% NP-40. The extracts were clarified by centrifugation at 16 000 x g for 15 min (fraction 3). The pellets were resuspended in 200-μl extraction buffer supplemented with 1% Triton X-100 and 0.45-M NaCl and sonicated (Vibracel, Bioblock Scientific) (fraction 4).
Antibodies
Antibodies, origins, manufacturers, applications and dilutions used are listed in Supplementary Table S3.
Immunofluorescence
Cells were grown overnight at 37°C on glass coverslips (Marienfeld) or 35-mm glass-bottom dishes (MatTek). After chemical treatment or laser micro-irradiation, cells were fixed with 2 % paraformaldehyde (Electron Microscopy Sciences) and permeabilized with 0.2% Triton-X-100 (Euromedex) in PBS. For in situ detergent extraction experiments, cells grown on glass coverslips, mock-treated or treated with Cali at the dose indicated were pre-extracted before fixation. Briefly, the coverslips were incubated in extraction buffer containing 0.1% Triton X-100 diluted in PBS, on ice for 3 min. Non-pre-extracted control cells were incubated in PBS. Then fixation proceeded as above. In the particular case of pre-extraction with RNase A, the CSK+R method was used as described (47). After fixation, samples were blocked by incubating cells in 5 % bovine serum albumin (Euromedex) diluted in PBS, 1 h at room temperature. Cells were then stained with primary and secondary antibodies coupled to Alexa Fluor 488 nm, 594 nm or 647 nm (Life Technologies). DNA staining was performed by incubating cells with 4',6-diamidino-2-phenylindole (DAPI, 0.01 μg/ml, 8 min, room temperature) or propidium iodide (1.75 μg/ml, 5 min, room temperature). For RNA staining we used Click-It RNA Alexa Fluor 594-nm Imaging kit from LIfe Technologies. Staining of GFP was performed using GFP-booster (Chromotek). Cells were mounted in Vectashield (Vector Labs). Confocal images were captured on a Zeiss 510 or Zeiss 710 confocal microscope, using a 40x oil objective.
Immunoprecipitation and mass spectrometry analysis
For mass-spectrometry analysis, immunoprecipitation was performed for 14 h on 2 mg of whole cell extracts from HT1080 cells stably expressing SAF-A-RBD-FLAG-GFP or FLAG-GFP, in 1-ml lysis buffer on dry beads from 400-μl anti-FLAG magnetic beads suspension prepared with 14-μg anti-FLAG antibody (M2, Sigma) according to the manufacturer (M-280 anti-mouse magnetic beads, LIfe Technologies). After two washes for 15 min at 4°C in lysis buffer on a rotating wheel, FLAG-tagged proteins were eluted with 300-μg FLAG peptide (Sigma) in 0.4 ml for 90 min at room temperature on a wheel. Then, supernatants were incubated for 14 h at 4°C with 80-μl anti-GFP magnetic beads (GFP-Trap-M, Chromotek). After two washes for 15 min at 4°C in lysis buffer on a wheel, the beads were incubated for 10 min at 100°C in loading buffer and then for 30 min at 25°C in the dark after addition of 100-mM iodoacetamide, followed by neutralization with a small amount of 1.5-M Tris-HCl, pH 8.8. Samples were loaded on a 4–12% gradient precast-gel (NuPage, Life Technologies), and staining was performed with Coomassie Fast-blueR dye. Immunoprecipitated protein samples from HT1080 cells expressing recombinant SAF-A RDB (two biological samples) or FLAG-tagged GFP (Control) were then analyzed as described in Supplementary material. Specific proteins pulled-down by the RBD domain of SAFA in both immuno-precipitation experiments were queried in STRING (http://string-db.org) to retrieve known physical interactions between them and an interaction landscape was generated using Cytoscape (http://www.cytoscape.org). Additional editing was performed with Illustrator.
For validation of the interactions between SAF-A RBD, FUS and TAF15 proteins found by mass-spectrometry analysis, co-immunoprecipitations were performed as above but on a smaller scale in extracts from HT1080 cells stably expressing FLAG-GFP only or FLAG-GFP-tagged SAF-A RBD or FUS proteins.
Survival and proliferation assays
HT1080 cells were seeded at low density in 6-well plates and allowed to plate overnight. Cells were then exposed to X-ray irradiation, post-incubated 5 days and stained with 2% crystal violet solution for 10 min on an orbital shaker. After extensive washes with distilled water, the dye was dissolved with 10% acetic acid and absorbance at 570 nm was measured with a Titertek Multiskan Plus.
U2OS cells were seeded at low density in 60-mm plates and allowed to plate for 6 h before a 16-h induction with 2-μg/ml doxycycline followed by X-ray irradiation at 2, 4 or 6 Gy. After additional 6-day incubation, MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reagent (Sigma) at 1 mg/ml was added to each plate and incubated for 1 h at 37°C. Cells were incubated with a solution containing 0.1% NP40 (Euromedex) and 4-mM HCl (VWR) diluted in isopropanol (Fisher) and mixed 15 min on an orbital shaker. Absorbance was measured at 570 nm and 690 nm with a Titertek Multiskan Plus.
RESULTS
SAF-A exhibits a biphasic dynamics at site of laser-induced DNA damage including a PAR-dependent transient recruitment to damaged nuclear areas
To explore the link between SAF-A and the cell response to DSBs, SAF-A mobilization after laser DNA micro-irradiation was studied in nuclei of an HT1080 cell line stably expressing an SAF-A fused to a carboxy terminal FLAG-GFP tag. The laser micro-irradiation setup used here produces single- and double-strand breaks in DNA that promote accumulation in the irradiated areas of base excision repair proteins including PAR polymerase 1 (PARP-1) and XRCC1 together with Ku70/80 and XRCC4 of the NHEJ pathway (51,52). Live-cell imaging showed first a transient retention of SAF-A at the irradiated sites, with a fast accumulation peaking at around 90 s followed by a rapid decrease reaching the basal fluorescence level 5 min after irradiation (Figure 1B and quantification in Figure 1C; see Supplementary Movie 1). Surprisingly, the fluorescence decrease in the irradiated area further went on, although at a slower rate, and then stabilized around 10 min after irradiation at a value below the mean nuclear basal fluorescence so that a dark hole formed in the nuclear irradiated area (Figure 1B and C; see Supplementary Movie 1). A similar dynamics was obtained after pulsed-laser irradiation of another human cell line (MRC5SV, SV40-transformed human lung fibroblasts) expressing the same SAF-A GFP fusion (data not shown).
Although SAF-A is phosphorylated by DNA-PK in response to DSB induction (35,36), we observed no effect of separate or combined DNA-PK, ATM or ATR inhibition on SAF-A accrual at laser-damaged sites (data not shown).
The transient recruitment phase of SAF-A biphasic dynamics after micro-irradiation is typical for proteins accumulating at site of DNA damage through interaction with PAR. PAR is a polymer synthetized from nicotinamide adenine dinucleotide by PAR polymerases, mainly PARP-1, activated upon binding to DNA single-strand breaks (53,54). For example, the chromatin remodeler ALC1 showed a PAR-dependent recruitment to the irradiated site within seconds followed by a release mostly complete within 10 min (55,56). Therefore, cells were pretreated with a concentration of the PARP inhibitor (PARPi) 3,4-Dihydro-5-[4-(1-piperidinyl)butoxy]-1(2H)-isoquinolinone (DPQ) able to counteract efficiently PAR production in cells (Supplementary Figure S1A and data not shown). While the DSB repair protein XRCC4 was still recruited at the damaged site under PARP inhibition, as expected (52) (Supplementary Figure S1A), SAF-A-GFP fluorescence showed a rapid dropoff in the damaged region which already reached a minimal value 2 min after irradiation (Figure 1D and Supplementary Movie 2). An identical effect was obtained with another PARPi, NU1025 (Figure 1D). SAF-A has DNA- and RNA-binding properties, which rely on an amino-terminal DBD and a carboxy-terminal RBD, respectively (39,57) (Figure 1A). To check directly the PAR-binding capacity of SAF-A, various forms of SAF-A fused to GFP were immunoprecipitated from transiently transfected HEK293T cells, blotted on membrane and incubated with purified PAR. As shown in Figure 1E, full length SAF-A and the isolated carboxy-terminal domain bound PAR, while PAR binding by the carboxy-terminal-truncated form was not detected, indicating that the carboxy-terminal RBD is necessary and sufficient for PAR binding in vitro. Identical results were obtained with extracts from HT1080 cells stably expressing the various FLAG-GFP-tagged constructs (Supplementary Figure S1B).
DNA damage loosen SAF-A binding to nuclear insoluble structures
As shown above, SAF-A-GFP recruitment at the damaged nuclear sites was rapidly followed by a decrease of the local fluorescence below the basal nuclear level. This hereafter named exclusion of SAF-A-GFP clearly occurred at sites of DNA damage as shown by colocalization of the exclusion areas with the DSB marker γH2AX (Figure 2A). Exclusion was not a photobleaching artifact since (i) under standard conditions without PARPi, it was delayed by 5 min after the initial irradiation, (ii) it was detected with an antibody against the whole nuclear pool of SAF-A (Figure 2B) and (iii) it did not affect the nuclear distribution of RNA pol II (Supplementary Figure S1C). SAF-A degradation at the irradiated sites is unlikely since SAF-A exclusion was preserved in the presence of the proteasome inhibitor MG132, although recruitment of 53BP1 was impaired, as expected (58) (data not shown). Indeed, the local fluorescence loss was accompanied by a contemporary fluorescence gain outside the irradiated site, indicating that SAF-A was rather redistributed from the irradiated area to undamaged nuclear regions (Supplementary Figure S1D). In addition, SAF-A exclusion was uncoupled from the preceding recruitment phase since abolition of SAF-A mobilization to the irradiated area upon PARP inhibition revealed a strong exclusion, even earlier than without inhibitor (Figure 1D).
Figure 2.
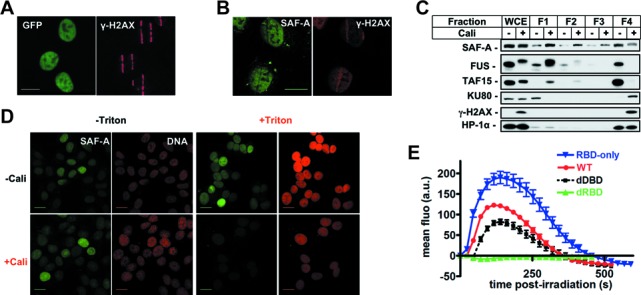
Analysis of SAF-A exclusion from chromatin following DNA damage. (A) Colocalization of SAF-A-GFP exclusion areas and γH2AX at stripes of laser damage 10 min after micro-irradiation in HT1080 cells. Scale bar, 20 μm. (B) Analysis by immunofluorescence of endogenous SAF-A and γH2AX 10 min after laser micro-irradiation in HT1080 cells. Scale bar, 20 μm. (C) HT1080 cells were mock-treated or treated with Cali for 1 h, fractionated as described in the Materials and Methods section, leading to fractions 1–4 (F1–F4) corresponding to proteins of decreasing solubility. Protein samples were denatured and separated on SDS-PAGE gel, followed by electrotransfer and blotting as indicated. (D) HT1080 cells grown on glass slides were mock-treated or treated with 10-nM Cali for 1 h at 37°C in medium. Cells were pre-extracted or not with Triton X-100 prior to fixation. Then cells were immunostained with anti-SAF-A primary and appropriate secondary antibodies and the DNA stained with propidium iodide. Scale bar, 20 μm. (E) Dynamics of various forms of SAF-A-GFP at laser-damaged sites. Images were obtained at 22-s intervals, and fluorescence intensities at the damage sites were quantified. Mean values of the fluorescence intensities with SEM were calculated from 26, 24, 29 and 16 independent measurements for RBD, WT, dDBD and dRBD forms, respectively. WT: wild-type SAF-A; RBD-only: SAF-A RNA-binding domain; dDBD: DNA-binding domain deletion; dRBD: RNA-binding domain deletion.
More importantly, SAF-A exclusion was not limited to laser irradiation but was also observed with a fractionation procedure based on successive detergent extractions following cell treatment with a clastogenic molecule (fractions 1–4) (49,59). We chose Cali as DNA breaking molecule because it yields a much higher ratio of DSBs to single-strand breaks in vivo, compared to ionizing radiation (60). Using this assay, a significant amount of SAF-A was associated with fraction 4 (F4) in untreated cells, corresponding to proteins tightly bound to nuclear structures such as heterochromatin protein HP-1α (Figure 2C). Strikingly, cell treatment with Cali induced a clear shift of SAF-A from F4 to F1, indicating conversion to a loosely bound nuclear protein (Figure 2C). In contrast, a significant amount of the major NHEJ protein Ku80 relocalized from F1 to F4 in response to Cali indicating a damage-induced attachment to chromatin, as already published (50). SAF-A release from F4 was dose- and time-dependent (Figure 3F and Supplementary Figure S1E). After a 1-h treatment with 10-nM Cali, exclusion of SAF-A from F4 lasted up to 5 h post-treatment (Supplementary Figure S1E). In contrast to SAF-A, RNA Pol II accumulated in the damaged chromatin (Figure 3F and Supplementary Figure S1E). In a similar way, when cells were pre-extracted with detergent before immunofluorescence staining on glass slides, Cali treatment induced a strong loss of SAF-A from the nucleus upon extraction (Figure 2D).
Figure 3.
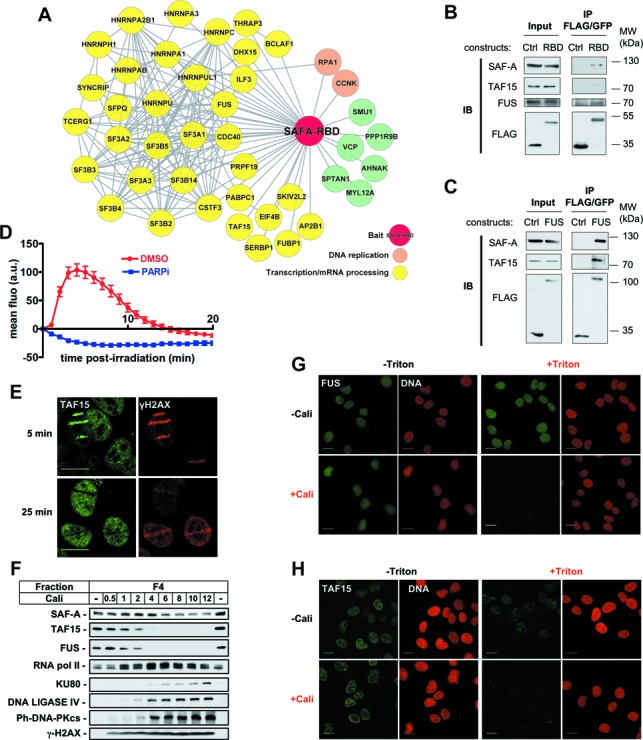
Identification of SAF-A RBD partners and analysis of their dynamics in response to DNA damage. (A) Interaction landscape representing proteins reproducibly co-immunoprecipitated with the RBD domain of SAF-A. (B,C) Co-immunoprecipitation analysis in extracts from HT1080 cells stably expressing FLAG-GFP, SAF-A-RBD-FLAG-GFP or FUS-FLAG-GFP. Immunoprecipitates were loaded on SDS-PAGE gel, followed by electrotransfer and blotting as indicated. (D) Effect of PARPi (DPQ) on the dynamics of FUS-GFP at laser-damaged sites. Images were obtained at 60-s intervals, and fluorescence intensities at the damage sites were quantified. Mean values of the fluorescence intensities with SEM were calculated from 30 and 20 independent measurements for conditions without and with PARPi, respectively. (E) Analysis by immunofluorescence of endogenous TAF15 and γH2AX 5 and 25 min after laser micro-irradiation in HT1080 cells. (F) HT1080 cells were mock-treated or treated with increase doses of Cali for 1 h, fractionated as described in the Materials and Methods section, leading to fractions 1–4 (F1–F4). Protein samples from fraction F4 were denatured and separated on SDS-PAGE gel, followed by electrotransfer and blotting as indicated. (G,H) HT1080 cells grown on glass slides were mock-treated or treated with 10-nM Cali for 1 h at 37°C in medium. Cells were pre-extracted or not with Triton X-100 prior to fixation. Then cells were immunostained with anti-FUS (G) or anti-TAF15 (H) primary and appropriate secondary antibodies and the DNA stained with propidium iodide. Scale bar, 20 μm.
SAF-A carboxy-terminal RBD domain recapitulates SAF-A dynamics in response to DNA damage
In order to delineate the SAF-A subdomains necessary for the dynamics described above, we tested by live imaging the dynamics at site of laser-induced DNA damage of various SAF-A truncations fused to GFP and stably expressed in cells (Figure 1A). As shown in Figure 2E, truncation of the amino-terminal DBD preserved the biphasic dynamics while truncation of the carboxy-terminal RBD strongly affected both the recruitment and exclusion phases. Moreover, the isolated RBD region was sufficient to recapitulate SAF-A biphasic dynamics in response to micro-irradiation (Figure 2E). Altogether these data show that the RBD of SAF-A is necessary and sufficient to promote its biphasic dynamics at sites of DNA damage. That the residue phosphorylated by DNA-PK (S59) is not located in the RBD but in the N-terminal DBD [Figure 1A; (36)] suggests that S59 phosphorylation is not responsible for SAF-A recruitment or exclusion. This was confirmed by studying the dynamics of a non-phosphorylatable form of SAF-A mutated on S59 which was identical to the one recorded for wild-type SAF-A (data not shown).
SAFA and its interactors FUS and TAF15 show similar dynamics at sites of DNA damage
Since the FLAG-GFP-tagged RBD domain of SAF-A recapitulated the dynamics of the full-length protein (Figure 2E), we used HT1080 cells stably expressing this construct, or FLAG-GFP as a control, to perform tandem immunoprecipitation and analysis by mass spectrometry of this domain and of associated proteins (Supplementary Figure S1F). Beside dimethylated arginine sites, as already described (61), no constitutive or induced post-translational modifications were detected (data not shown). More interestingly, analysis of the co-immunoprecipitated proteins revealed that most of them were involved in transcription and mRNA processing, in agreement with the RNA-binding property of the RDB domain (Figure 3A and Supplementary Table S1). Of note, full-length endogenous SAF-A was co-precipitated with SAF-A RDB. Among the coprecipitated proteins, FUS/TLS and TAF15/TAFII68 were chosen for further analysis.
First, results of the mass-spectrometry analysis were evaluated by western blotting after reciprocal immunoprecipitation of the SAF-A and FUS proteins. As shown in Figure 3B and C, both FUS and TAF15 were coprecipitated with the SAF-A RBD domain, together with endogenous full-length SAF-A. In addition, SAF-A and TAF15 were coprecipitated with FUS, thus validating results of the mass-spectrometry analysis and suggesting a tripartite complex of these proteins in cell extracts. Notably, the interaction between SAF-A, FUS and TAF15 with the SAF-A RBD was resistant to RNase A and DNAse I treatments (data not shown).
Second, in nuclei of HT1080 cells stably expressing an FUS GFP fusion, FUS showed a rapid recruitment at damage sites followed by an exclusion phase, mimicking the biphasic dynamic observed with SAF-A (Figure 3D and Supplementary Movie 3). As for SAF-A, PARP inhibition completely abolished the recruitment of FUS which then showed only the exclusion phase already detectable 60 s after irradiation, up to the last time point registered (Figure 3D, +PARPi and Supplementary Movie 4). For TAF15, immunodetection of the endogenous protein shows also a biphasic response to laser irradiation with an initial recruitment on laser-irradiated areas labeled by γH2AX, followed by local exclusion of the protein (Figure 3E).
Third, cell fractionation showed a striking shift of both TAF15 and FUS proteins from the F4 insoluble fraction in non-treated cells to the F1 soluble nuclear fraction after treatment with Cali (Figure 3F), similarly to SAF-A (Figure 2C). As for SAF-A, FUS and TAF15 release from F4 was dose- and time-dependent (Figure 3F and Supplementary Figure S1E) and after a 1-h treatment with 10-nM Cali, it lasted for up to 5 h (Supplementary Figure S1E). In agreement with these results, immunofluorescence staining after Cali treatment and detergent pre-extraction revealed a strong reduction in the amount of both FUS and TAF15 proteins present in the chromatin fraction compared to non-extracted or non-treated cells (Figure 3G and H).
Active transcription is necessary for SAF-A release from the damaged area
To further characterize how DNA damage impacted on SAF-A nuclear dynamics, we used FRAP (62). SAF-A-GFP fluorescence was bleached in a small region of the nucleus and the fluorescence recovery was subsequently monitored by time-lapse microscopy (Figure 4A). When fluorescence recovery was monitored in cells treated or not with Cali, we found that most of the original relative GFP signal recovered within 1 min. However, fluorescence recovery was much faster in cells treated with Cali, indicating an increased mobility of SAF-A after DNA damage. Similarly, when laser micro-irradiation was used as DNA damaging source, an increased mobility of SAF-A was observed in the damaged area (Supplementary Figure S2A).
Figure 4.
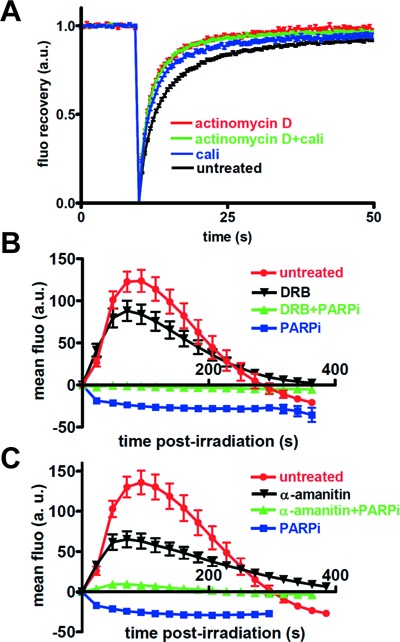
Effect of transcription inhibition on SAF-A mobility and dynamics in response to DNA damage. (A) Effect of DNA damage by calichemicin γ1 (Cali) and/or transcription inhibition (actinomycin D) on FRAP curve for SAFA-GFP. Images were obtained at 487-ms intervals. The data were normalized to the prebleach fluorescence level. The graph shows FRAP curves of mean values with SEM of 52, 52, 28 and 52 independent fluorescence measurements for conditions with actinomycin D, actinomycin D + Cali, Cali and no agent, respectively. (B) Effect of a PARPi (DPQ) and a transcription inhibitor (DRB) on the dynamics of SAF-A-GFP at laser-damaged sites. Images were obtained at 22-s intervals, and fluorescence intensities at the damage sites were quantified. Mean values of the fluorescence intensities with SEM were calculated from 12, 14, 17 and 17 independent measurements for conditions without inhibitor and with DRB, DRB+PARPi and PARPi, respectively. (C) Effect of a PARPi (DPQ) and a transcription inhibitor (α-amanitin) on the dynamics of SAF-A-GFP at laser-damaged sites. Images were obtained at 22-s intervals, and fluorescence intensities at the damage sites were quantified. Mean values of the fluorescence intensities with SEM were calculated from 12, 17, 17 and 25 independent measurements for conditions without inhibitor and with α-amanitin, α-amanitin+PARPi and PARPi, respectively.
The requirement of the RBD for exclusion post-DNA damage and the similar pattern of three proteins involved in RNA metabolism following DNA damage prompted us to explore the link between SAF-A dynamics and transcription by using transcription inhibitors. First, analysis of transcription by incorporation of the uridin analog 5-ethynyl-uridin (EU) into nascent RNA (63) showed that three different transcription inhibitors, actinomycin D, 5,6-dichloro-1-beta-ribo-furanosyl benzimidazole (DRB) and α-amanitin, efficiently inhibited transcription in our cellular model (Supplementary Figure S2B). Strikingly, FRAP analysis revealed that SAF-A mobility was significantly faster in the presence of actinomycin D and not further increased by Cali treatment (Figure 4A; recovery half-time was determined to be 2.916, 1.912, 1.724 and 1.657 s for conditions without treatment, and treatment with Cali, actinomycin D or Cali+actinomycin D, respectively). Similar results were obtained in the presence of DRB (Supplementary Figure S2C). Together, these data indicate that transcription is necessary to reveal a change in the mobility of SAF-A in the presence of DNA damage, suggesting that it concerns the protein pool associated with transcription. Thus, we checked the effect of transcription inhibitors on SAF-A dynamics after laser irradiation. DRB or α-amanitin did not prevent SAF-A PAR-dependent recruitment on the irradiated area but prolonged local residence time, showing that SAF-A recruitment is independent of transcription (Figure 4B and C). Accordingly, inclusion of RNaseA during pre-extraction did not affect SAF-A labeling on damaged stripes, although it efficiently removed the control nucleolin protein from nucleoli (Supplementary Figure S2D and E). In sharp contrast, DRB or α-amanitin completely abolished the exclusion phase of SAF-A from the damaged site, as revealed in the presence of PARPi (Figure 4B and C). Identical results were obtained with actinomycin D (Supplementary Figure S2F). Similarly, FUS-GFP exclusion from the laser-irradiated area was prevented by DRB (Supplementary Figure S2G). These data support that SAF-A and FUS exclusion concerns the pool of protein engaged in transcribed RNA metabolism.
Phosphatidylinositol 3-kinase-related kinases trigger SAF-A release from the damaged area
DDR is initiated by activation of the phosphatidylinositol 3-kinase-related kinases (PIKKs) ATM, ATR and DNA-PK that directly phosphorylate many substrates with a frequent redundancy (14,20). Therefore, we tested the effect of inhibition of these kinases on SAF-A exclusion. As shown in Figure 5A, treating cells with a combination of inhibitors of the three kinases largely prevented the release of SAF-A from the irradiated area, whereas single treatments yielded only partial inhibition of SAF-A exclusion (data not shown). Monitoring transcription through EU incorporation in the presence of these kinase inhibitors excluded a transcription defect at the time of irradiation which would mimic the DRB- or α-amanitin-mediated inhibition of SAF-A exclusion (Supplementary Figure S2H). Local transcription inhibition at endonuclease- or laser-induced DSB sites has been previously published (11–13). Since we observed that indeed de novo incorporation of EU was inhibited at damaged sites after laser irradiation (Figure 5B), a possibility was that exclusion of SAF-A may be a readout of local damage-induced reduction of transcription. However, we found that transcription was still inhibited despite inhibition of PIKKs, clearly indicating that SAF-A exclusion is uncoupled from transcription inhibition at damage sites (Figure 5B).
Figure 5.
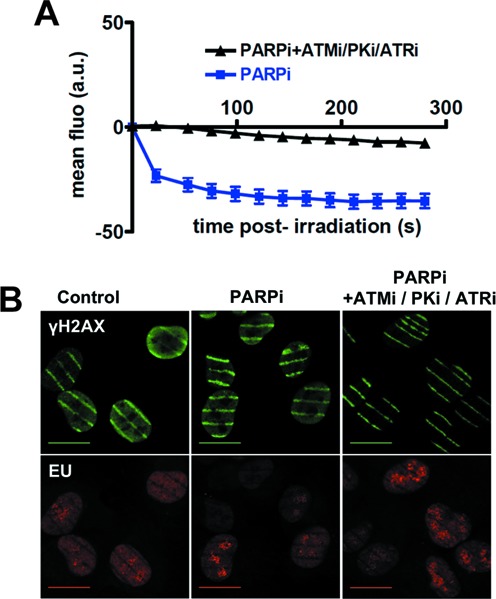
Effect of inhibitors of phosphatidylinositol 3-kinase-related kinases (PIKKs) on SAF-A exclusion from and transcription at laser damage sites. (A) Dynamics of SAF-A-GFP at laser-damaged sites was measured in the presence of a PARPi (DPQ) and without or with the combination of PIKKs inhibitors NU7441 (PKi, DNA-PK inhibitor), KU55933 (ATMi, ATM inhibitor) and VE821 (ATRi, ATR inhibitor). Images were obtained at 22-s intervals, and fluorescence intensities at the damage sites were quantified. Mean values of the fluorescence intensities with SEM were calculated from 16 and 39 independent measurements for conditions without and with PIKKs inhibitors, respectively. (B) Monitoring transcription following laser irradiation by incorporation of EU in HT1080 cells in the presence of PARPi (DPQ) and PIKKs inhibitors. Irradiation stripes were visualized by γH2AX immunostaining (note the strong decrease on PIKKs inhibition). Scale bar, 20 μm.
A mechanism antagonizing R-loop formation after DNA damage
Various abnormal transcription conditions from bacteria to mammals have been shown to promote interactions between the nascent RNA and template DNA (R-loops) which are prone to genomic instability (29–30,33). Thus, we checked whether there was a link between SAF-A dynamics post-damage and R-loop formation. Although we could visualize a faint staining at laser tracks by immunofluorescence with the DNA–RNA hybrid-specific monoclonal antibody S9.6 (64), the high background precluded a fine analysis (S. Britton, C. Delteil and P. Calsou, unpublished data). Therefore, we established an HT1080 cell line expressing the D10R E48R mutant of E. coli RNaseHI, fused to the mCherry fluorescent protein. This mutant RNaseHI displays a high affinity for RNA:DNA hybrids but is catalytically inactive (65). Laser micro-irradiation induced a recruitment of mutant RNaseHI at site of DNA damage, as shown by live-cell imaging (Figure 6A). This transient recruitment was enhanced upon PARP inhibition, indicating that, contrarily to SAF-A, FUS or TAF15, mutant RNaseHI recruitment at the irradiated site was not dependent on PARP activity (Figure 6B). Consistent with the idea that this recruitment reflects the transcription-dependent accumulation of R-loops at site of damage, mutant RNaseHI recruitment was abolished by actinomycin D treatment (Figure 6B). FRAP experiments performed on cells treated or not with actinomycin D showed an identical mobility of mutant RNaseHI under both conditions, ruling out that a side effect of actinomycin D on the protein mobility could account for these results (Supplementary Figure S3A). To further establish that recruitment of mutant RNaseHI reflects R-loops at damage sites, we used diospyrin D1. This natural compound has been shown to interfere with the assembly of the spliceosome (66) and thereby to induce replication defects and genomic instability that rely on R-loop production (67,68). Notably, mutant RNaseHI recruitment was strongly enhanced in the presence of diospyrin D1 (Supplementary Figure S3B).
Figure 6.
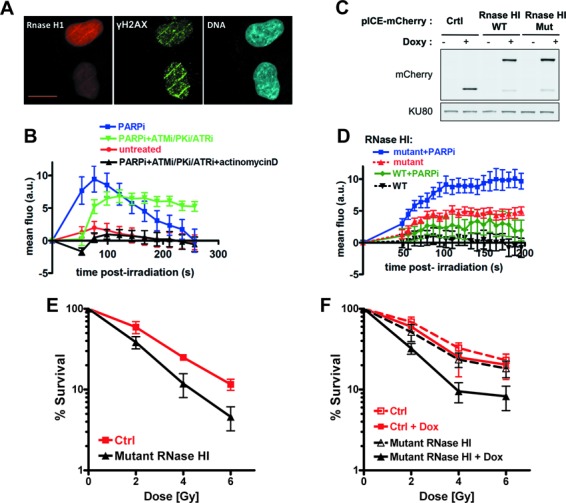
(A) Colocalization of mutant RNaseHI and γH2AX at stripes of laser damage 1 min after micro-irradiation in HT1080 cells. Scale bar, 20 μm. (B) Dynamics of mutant RNaseHI at laser-damaged sites in HT1080 cells was measured in the presence of a combination of PARPi (DPQ), PIKKs inhibitors and transcription inhibitor (actinomycin D) as indicated. Images were obtained at 22-s intervals, and fluorescence intensities at the damage sites were quantified. Mean values of the fluorescence intensities with SEM were calculated from 48, 35, 47 and 31 independent measurements for conditions with no agent, PARPi (DPQ), PARPi and PIKKs inhibitors (PARPi/ATMi/PKi/ATRi) and the combination of PARPi, PIKKs and transcription inhibitors (PARPi/ATMi/PKi/ATRi+ actinomycin D), respectively. (C) Analysis by western blot of the kinetics of expression of mCherry (Ctrl, control) and mCherry-wild-type (WT) or mutant (Mut) RNaseHI in U2OS cells treated with doxycycline for 24 h. Ku80 is used as loading control. (D) Dynamics of WT or mutant RNaseHI at laser-damaged sites was measured in the presence or not of PARPi (DPQ) as indicated, in U2OS cells pretreated with doxy for 20 h. Images were obtained at 7.75-s intervals, and fluorescence intensities at the damage sites were quantified. Mean values of the fluorescence intensities with SEM were calculated from 23, 15, 25 and 10 independent measurements for conditions with mutant RNaseHI + PARPi, mutant RNaseHI, WT RNaseHI + PARPi and WT RNaseHI, respectively. (E) Exponentially growing HT1080 cells expressing mutant RNaseHI or mCherry as control (Ctrl) were exposed to the indicated dose of X-rays. Surviving cell population was measured after 5 days by cell staining. Each point represents the mean of six experiments ± SD. (F) U2OS cells containing a construct for inducible expression of mutant RNaseHI or mCherry as control (Ctrl) were preincubated or not with doxycycline for 16 h (+Dox) and then irradiated with the indicated dose of X-rays. Cell viability was measured after 6 days with the MTT assay. Each point represents the mean of four experiments ± SD.
If there was a link between R-loops and SAF-A dynamics, R-loop levels may affect SAF-A behavior and vice versa. Strikingly, combined PIKK inhibitors that largely blocked release of SAF-A from the damage site after laser irradiation (Figure 5A) induced a prolonged local mutant RNaseHI recruitment (Figure 6B). Moreover, cell treatment with diospyrin D1 negatively impacted on SAF-A exclusion post-laser irradiation (Supplementary Figure S3C and D).
Inactive RNaseHI could itself interfere with resolution of laser-induced RNA:DNA hybrids. Therefore, we established and selected inducible constructs for equal level of expression of wt or mutated RNaseHI in U2OS cells (Figure 6C) and analyzed their dynamics at laser-damaged sites (Figure 6D). Similar to data in HT1080 cells, PARP inhibition enhanced the recruitment of either WT or mutant RNaseHI, confirming that it was not PARP mediated. Strikingly, the recruitment of mutant RNaseHI was constantly stronger than that of WT RNaseHI; indeed, WT RNaseHI accumulation at laser-induced damage was barely detectable without PARPi and, with PARPi, remained at a level still below that of mutant RNaseHI without PARPi (Figure 6D). These data further argue for the presence of R-loops at laser-damaged sites, the resolution of which might be helped by WT RNaseHI and impaired by mutant RNaseHI.
Finally, we tested whether interfering with R-loops resolution following DNA damage would impact cell survival. We used our two different human cell models (HT1080 and U2OS cell lines) for RNaseH overexpression. Despite several attempts, we could not assess the consequence of an overexpression of active RNaseHI on cell survival to IR due to the intrinsic cellular toxicity of an excess of this activity, as already reported in yeast (28). Nevertheless and as shown in Figure 6E and F, we found that overproduction of a catalytically inactive RNaseHI, which likely interfered with R-loops resolution as shown above, induced a significant radiosensitization in two different human cell models.
DISCUSSION
Our results point out two independent aspects of the involvement of a set of RBPs in DDR. First, we report the PAR-dependent recruitment of several RBPs to DNA damage sites and second, we characterize the exclusion of this set of RNA-binding proteins from the damaged areas. Additionally, we propose that exclusion relies on a local mechanism counteracting deleterious effects of R-loops.
Recruitment of RBPs to DNA damage areas
We observed that SAF-A, FUS and TAF15 were recruited to laser-irradiated areas in a PAR-dependent manner. We also observed an RNAse-resistant and PAR-dependent fraction of endogenous SAF-A in chromatin soon after H2O2 treatment (S. Britton and P. Calsou, unpublished results). Accordingly, in a large-scale analysis in human cells of PAR-binding proteins following treatment with a DNA alkylating agent, FUS and SAF-A were identified among proteins exhibiting the highest recruitment score upon 5-min exposure (69). Several RBPs including FUS were previously reported to be recruited on damaged DNA areas in a PAR-dependent manner (19,21–25). Interestingly, several of these proteins were isolated here by mass spectrometry in a complex with the carboxy-terminal domain of SAF-A (Supplementary Figure S1G). Furthermore, in addition to TAF15 and FUS as shown here, EWSR1, the third member of the TET family of RBPs, was very recently reported to be recruited at laser-damaged sites in a PAR-dependent manner (23).
We report a direct binding of the carboxy-terminal RBD of SAF-A to PAR in vitro. In addition, we showed that deletion of this domain abrogated SAF-A recruitment while, conversely, this domain alone was sufficient for recruitment to damaged DNA sites. This suggests that a direct interaction with PAR may at least contribute to SAF-A recruitment to PAR-enriched areas following DNA damage. Indeed, hnRNPs have been previously identified as a family of PAR-binding proteins (70). The characterization of the interaction of the SAF-A RNA-binding region with PAR is consistent with other studies on ASF/SF2 (71), hnRNPA1 (70), NONO (25) and FUS (23,24). Together, these results may indicate a more general trend for an affinity of RNA-binding motifs for PAR which may deserve further exploration.
Function of RBPs recruited at DNA damage sites
Given that PAR shares biochemical properties with nucleic acids, the potential competition for the same protein domain between RNA and PAR may serve biological purposes in the DDR for a subset of RBPs. For example, PAR binding to the splicing factor ASF/SF2 has been shown to regulate RNA splicing (71). Also, the mobilization of these proteins via PAR in close vicinity to PARP activity may favor their poly(ADP-ribosyl)ation and therefore may regulate their function in RNA metabolism. Indeed, poly(ADP-ribosyl)ation of hnRNPs in Drosophila modulates their RNA-binding ability and splicing function (72). Since SAF-A is a major regulator of alternative splicing (46), its PAR binding and possibly PAR-modification may contribute to the global change in splicing observed under stress conditions, including DNA damage (73). Indeed, EWSR1 that is also recruited to laser-damaged sites (23) is a key player in the regulation of alternative splicing following DNA damage (74,75). More generally, the recruitment onto PAR at sites of DNA damage of proteins involved in RNA biogenesis may regulate the balance between their role in RNA metabolism and a function in DDR and/or DNA repair (21–23,25,76–78). In that view, one repair function of SAF-A could rely on interaction with and stimulation of NEIL2, which has been shown to be particularly relevant to repair of oxidized base damage in transcribed genes (79).
Independent recruitment and exclusion of RBPs at DNA damage sites
We observed a biphasic dynamics of SAF-A, FUS and TAF15 after laser micro-irradiation, with a transient recruitment, as discussed above followed by a prolonged exclusion from the damaged chromatin areas. Despite requiring the same domain of SAF-A, several lines of evidence indicate that both phases are independent. First, SAF-A and FUS were excluded but not recruited upon PARP inhibition while they were recruited but no more excluded upon transcription inhibition. Second, the recruitment was enhanced and prolonged upon inhibition of the exclusion phase and conversely, the exclusion was detectable earlier upon inhibition of the recruitment phase, arguing for independent events superimposed under normal conditions. Third, the fraction of SAF-A recruited was not sensitive to RNaseA and preserved upon transcription inhibition while the exclusion concerned a fraction of SAF-A engaged in transcription. Fourth, the recruitment relied on PARP activity while exclusion depended on PIKKs. Notably, phosphorylation of the major DNA-PK target site in SAF-A that we identified previously (36) was not necessary for exclusion since a non-phosphorylatable mutant of SAF-A on this site was still excluded (data not shown). SAF-A is highly abundant (estimate 2 × 106 copies per nucleus) of which about half is soluble and half insoluble (57). Independent recruitment and exclusion post-DNA-damage most probably reflect differential effects of DNA damage on the two pools of SAF-A, the soluble fraction being recruited while the insoluble, mostly transcription-linked fraction was excluded. Interestingly this behavior is reminiscent of the independent exclusion and recruitment phases reported for hnRPUL1 which were similarly attributed to two protein pools (21,22). Finally, SAF-A protein mobilization to and from DNA damage sites both rely on its RNA binding domain. This sustains the notion that these responses may be mutually exclusive according to whether this domain is engaged in RNA transactions or not.
PIKKs-dependent exclusion of RBPs from damaged chromatin
Regarding SAF-A, TAF15 and FUS long-lasting exclusion from chromatin mediated by DNA damage as observed here, it was confined to sites of laser irradiation but was not limited to laser-induced DNA damage. Indeed, it was reproduced with the radiomimetic molecule Cali as shown by FRAP experiments, western blotting of the chromatin fraction and immunofluorescence after in situ cell extraction. Protein exclusion was immediate post-damage and prolonged up to several hours. Several data argue against a protein degradation as the basis of exclusion. First, exclusion persisted upon proteasome inhibition. Second, in laser irradiation experiments, SAF-A-GFP was redistributed from the irradiation sites to non-irradiated areas. Third, in western blot experiments, the fraction of endogenous SAF-A lost from chromatin accumulated in the supernatant without massive loss of overall protein amount.
Our FRAP experiments in the laser-damaged areas or in cells damaged with Cali showed that exclusion corresponded to a DNA damage-induced solubilization of the proteins otherwise attached to chromatin. Since after exclusion, SAF-A mobility as detected by FRAP mostly superimposed with the one obtained upon transcription inhibition, this strongly supports that exclusion concerns the RNA-associated pool of SAF-A. Indeed, exclusion of SAF-A and FUS was abolished upon transcription inhibition. Notably, RNA Pol II was not excluded from chromatin damaged with laser or Cali.
In addition to FUS and TAF15, several RBPs or proteins involved in RNA metabolism that we identified here in SAF-A co-precipitates were recently reported to exhibit a similar rapid and prolonged exclusion from laser-micro-irradiated sites distinct from protein degradation (Supplementary Figure S1G); these include hnRPUL1 (21), THRAP3 and BCLAF1 (20), hnRNPC (together with hnRNPK and RBMX/hnRNPG) (19). We observed that a combination of DDR PIKK inhibitors largely prevented SAF-A exclusion from the laser-damaged sites. Interestingly, Beli et al. also found that combined inhibition of PIKKs impaired THRAP3 localized exclusion after laser irradiation (20). Moreover, like for SAF-A as shown here, deletion of the RBD of hnRPUL1 and THRAP3 was reported to impair exclusion from the DNA damage regions (20,21). Together, these data likely uncover a general phenomenon in the DDR concerning the PIKK-dependent exclusion of a whole RBP complex but not RNA Pol II from damaged transcription areas.
A PIKKs-dependent anti-R-loop mechanism as a new component of the DDR
We report conditions (PIKK inhibition) under which exclusion of the RBPs SAF-A and FUS was prevented while RNA synthesis was still inhibited at damaged sites. This uncoupling between RBP exclusion and transcription arrest at DNA damage sites suggests that rather than reflecting damage-induced transcription arrest, exclusion accompanies a specialized mechanism mediated by DNA damage and leading to destabilization of ribonucleoprotein complexes (RNPCs). Moreover, we provide evidence that this localized post-damage destabilization of RNPCs is linked to R-loop avoidance. First, when an inactive form of bacterial RNaseHI was used as R-loop reporter in cells, a strong but transient recruitment at laser micro-irradiation tracks was observed, which was strictly dependent on transcription and enhanced under conditions favoring R-loop production (diospyrin D1). Second, the active form of the bacterial enzyme was less and more transiently recruited at laser damage sites when compared to the inactive RNaseHI. Third, recruitment of inactive RNaseHI at damage sites was significantly prolonged under PIKK inhibition that prevented RNPC destabilization and, conversely, R-loop overproduction negatively impacted on RNPC exclusion. Fourth, overproduction of catalytically dead RNaseHI in human cells negatively impacted on cell survival to IR, although we cannot rule out an indirect effect. Collectively, these data support that R-loops are produced upon DNA damage at transcription sites. If improperly resolved, they impair cell survival possibly by promoting genome instability.
Laser irradiation or radiomimetics produce nicks in DNA. Thus, R-loop occurrence following DNA damage may rely on the fact that nicks in the non-template strand greatly favor hybridization between the RNA transcript and the template DNA strand (80,81). Alternatively, damage-dependent inhibition of DNA topoisomerase I (82) may also be R-loop-prone. Nevertheless, the precise mechanism of R-loop formation following DNA damage deserves further investigations.
The mechanism removing R-loops secondary to DNA damage may be necessary in transcribed regions to ensure cell viability, as supported by our data, and to prevent genome instability, as described under other conditions (29,30,33,34). It is therefore a critical and new component of the DDR. We report that PIKKs are implicated in this mechanism. Although PiKKs have previously been implicated in splicing regulation following DNA damage (83,84), their precise function in R-loop removal and whether this removal relies on RNA degradation or active release remain to be established. A simple explanation could be the activation of endogenous RNaseH activity or of a dedicated helicase or nuclease through PIKK-mediated phosphorylation or PIKK-dependent chromatin remodeling. Nevertheless, our results uncover a bimodal response to DNA damage of a set of proteins involved in RNA biogenesis which emphasizes a complex intrication between RNA metabolism and the DDR that deserves further investigation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online, including [1–7].
Acknowledgments
We are indebted to P.R. Hamann (Wyeth Research) for the gift of calicheamicin γ1, to Pr B. Hazra (Jadavpur University, Calcutta, India) for the gift of Diospyrin, to Dr S.H. Leppla (National Institutes of Health, Bethesda, MD, USA) for the gift of the S9.6 antibody and to Dr G. Giglia-Mari (IPBS, Toulouse, France) for the gift of anti-RNA Pol II antibody. P. Calsou is a scientist from INSERM, France.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
Present address:
Bernard Salles, TOXALIM (Research Centre in Food Toxicology), UMR 1331 INRA/INP/UPS, 180 chemin de Tournefeuille, F-31027 Toulouse Cedex 3, France.
FUNDING
Ligue Nationale Contre le Cancer [Equipe Labellisée 2013]; Ligue Régionale Contre le Cancer [Comité Midi-Pyrénées]; Ministère de l'Enseignement Supérieur et de la Recherche [doctoral fellowship to E.D.] and the Fondation pour la Recherche Médicale [FDT20080913845; doctoral fellowship to S.B.]; Ligue Nationale Contre le Cancer [Equipe Labellisée 2003–2009 to B.S.]; Région Midi-Pyrénées (contrat de projets Etat-Région), the Grand Toulouse community, the Association pour la Recherche Contre le Cancer (Equipement 8505), the CNRS and the European Union through the Fonds Européen de Développement Régional program [to the Toulouse Reseau Imagerie (TRI)-RIO Optical Imaging Platform at IPBS (Genotoul, Toulouse, France)]. Funding for open access charge: Ligue Nationale Contre le Cancer [Equipe Labellisée 2013].
Conflict of interest statement. None declared.
REFERENCES
- 1.Jeggo P., Lavin M.F. Cellular radiosensitivity: how much better do we understand it? Int. J. Radiat. Biol. 2009;85:1061–1081. doi: 10.3109/09553000903261263. [DOI] [PubMed] [Google Scholar]
- 2.O'Driscoll M., Jeggo P.A. The role of double-strand break repair—insights from human genetics. Nat. Rev. Genet. 2006;7:45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y., Gostissa M., Hildebrand D.G., Becker M.S., Boboila C., Chiarle R., Lewis S., Alt F.W. The role of mechanistic factors in promoting chromosomal translocations found in lymphoid and other cancers. Adv. Immunol. 2010;106:93–133. doi: 10.1016/S0065-2776(10)06004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pardo B., Gomez-Gonzalez B., Aguilera A. DNA double-strand break repair: how to fix a broken relationship. Cell. Mol. Life Sci. 2009;66:1039–1056. doi: 10.1007/s00018-009-8740-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackson S.P., Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Polo S.E., Jackson S.P. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodarzi A.A., Jeggo P.A. The repair and signaling responses to DNA double-strand breaks. Adv. Genet. 2013;82:1–45. doi: 10.1016/B978-0-12-407676-1.00001-9. [DOI] [PubMed] [Google Scholar]
- 8.Thompson L.H. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: the molecular choreography. Mutat. Res. 2012;751:158–246. doi: 10.1016/j.mrrev.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 9.Ciccia A., Elledge S.J. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kruhlak M., Crouch E.E., Orlov M., Montano C., Gorski S.A., Nussenzweig A., Misteli T., Phair R.D., Casellas R. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature. 2007;447:730–734. doi: 10.1038/nature05842. [DOI] [PubMed] [Google Scholar]
- 11.Chou D.M., Adamson B., Dephoure N.E., Tan X., Nottke A.C., Hurov K.E., Gygi S.P., Colaiacovo M.P., Elledge S.J. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. U.S.A. 2010;107:18475–18480. doi: 10.1073/pnas.1012946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pankotai T., Bonhomme C., Chen D., Soutoglou E. DNAPKcs-dependent arrest of RNA polymerase II transcription in the presence of DNA breaks. Nat. Struct. Mol. Biol. 2012;19:276–282. doi: 10.1038/nsmb.2224. [DOI] [PubMed] [Google Scholar]
- 13.Shanbhag N.M., Rafalska-Metcalf I.U., Balane-Bolivar C., Janicki S.M., Greenberg R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell. 2010;141:970–981. doi: 10.1016/j.cell.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsuoka S., Ballif B.A., Smogorzewska A., McDonald E.R., III, Hurov K.E., Luo J., Bakalarski C.E., Zhao Z., Solimini N., Lerenthal Y., et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 15.Bensimon A., Schmidt A., Ziv Y., Elkon R., Wang S.Y., Chen D.J., Aebersold R., Shiloh Y. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Sci. Signal. 2010;3:rs3. doi: 10.1126/scisignal.2001034. [DOI] [PubMed] [Google Scholar]
- 16.Haley B., Paunesku T., Protic M., Woloschak G.E. Response of heterogeneous ribonuclear proteins (hnRNP) to ionising radiation and their involvement in DNA damage repair. Int. J. Radiat. Biol. 2009;85:643–655. doi: 10.1080/09553000903009548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ha K., Takeda Y., Dynan W.S. Sequences in PSF/SFPQ mediate radioresistance and recruitment of PSF/SFPQ-containing complexes to DNA damage sites in human cells. DNA Repair (Amst) 2010;10:252–259. doi: 10.1016/j.dnarep.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salton M., Lerenthal Y., Wang S.Y., Chen D.J., Shiloh Y. Involvement of matrin 3 and SFPQ/NONO in the DNA damage response. Cell Cycle. 2010;9:1568–1576. doi: 10.4161/cc.9.8.11298. [DOI] [PubMed] [Google Scholar]
- 19.Adamson B., Smogorzewska A., Sigoillot F.D., King R.W., Elledge S.J. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol. 2012;14:318–328. doi: 10.1038/ncb2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beli P., Lukashchuk N., Wagner S.A., Weinert B.T., Olsen J.V., Baskcomb L., Mann M., Jackson S.P., Choudhary C. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell. 2012;46:212–225. doi: 10.1016/j.molcel.2012.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Polo S.E., Blackford A.N., Chapman J.R., Baskcomb L., Gravel S., Rusch A., Thomas A., Blundred R., Smith P., Kzhyshkowska J., et al. Regulation of DNA-end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Mol. Cell. 2012;45:505–516. doi: 10.1016/j.molcel.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong Z., Jiang J., Ma J., Dai S., Xu T., Li H., Yasui A. The role of hnRPUL1 involved in DNA damage response is related to PARP1. PLoS ONE. 2013;8:e60208. doi: 10.1371/journal.pone.0060208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mastrocola A.S., Kim S.H., Trinh A.T., Rodenkirch L.A., Tibbetts R.S. The RNA binding protein fused in sarcoma (FUS) functions downstream of PARP in response to DNA damage. J. Biol. Chem. 2013;288:24731–24741. doi: 10.1074/jbc.M113.497974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rulten S.L., Rotheray A., Green R.L., Grundy G.J., Moore D.A., Gomez-Herreros F., Hafezparast M., Caldecott K.W. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 2013;42:307–314. doi: 10.1093/nar/gkt835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krietsch J., Caron M.C., Gagne J.P., Ethier C., Vignard J., Vincent M., Rouleau M., Hendzel M.J., Poirier G.G., Masson J.Y. PARP activation regulates the RNA-binding protein NONO in the DNA damage response to DNA double-strand breaks. Nucleic Acids Res. 2012;40:10287–10301. doi: 10.1093/nar/gks798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montecucco A., Biamonti G. Pre-mRNA processing factors meet the DNA damage response. Front. Genet. 2013;4:102. doi: 10.3389/fgene.2013.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dutertre M., Lambert S., Carreira A., Amor-Gueret M., Vagner S. DNA damage: RNA-binding proteins protect from near and far. Trends Biochem. Sci. 2014;39:141–149. doi: 10.1016/j.tibs.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 28.Paulsen R.D., Soni D.V., Wollman R., Hahn A.T., Yee M.C., Guan A., Hesley J.A., Miller S.C., Cromwell E.F., Solow-Cordero D.E., et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol. Cell. 2009;35:228–239. doi: 10.1016/j.molcel.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X., Manley J.L. Cotranscriptional processes and their influence on genome stability. Genes Dev. 2006;20:1838–1847. doi: 10.1101/gad.1438306. [DOI] [PubMed] [Google Scholar]
- 30.Aguilera A., Garcia-Muse T. R loops: from transcription byproducts to threats to genome stability. Mol. Cell. 2012;46:115–124. doi: 10.1016/j.molcel.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 31.Helmrich A., Ballarino M., Nudler E., Tora L. Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol. 2013;20:412–418. doi: 10.1038/nsmb.2543. [DOI] [PubMed] [Google Scholar]
- 32.Chan Y.A., Hieter P., Stirling P.C. Mechanisms of genome instability induced by RNA-processing defects. Trends Genet. 2014;30:245–253. doi: 10.1016/j.tig.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamperl S., Cimprich K.A. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA repair. 2014;19:84–94. doi: 10.1016/j.dnarep.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim N., Jinks-Robertson S. Transcription as a source of genome instability. Nat. Rev. Genet. 2012;13:204–214. doi: 10.1038/nrg3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berglund F.M., Clarke P.R. hnRNP-U is a specific DNA-dependent protein kinase substrate phosphorylated in response to DNA double-strand breaks. Biochem. Biophys. Res. Commun. 2009;381:59–64. doi: 10.1016/j.bbrc.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 36.Britton S., Froment C., Frit P., Monsarrat B., Salles B., Calsou P. Cell nonhomologous end joining capacity controls SAF-A phosphorylation by DNA-PK in response to DNA double-strand breaks inducers. Cell Cycle. 2009;8:3717–3722. doi: 10.4161/cc.8.22.10025. [DOI] [PubMed] [Google Scholar]
- 37.Wang C., Lees-Miller S.P. Detection and repair of ionizing radiation-induced DNA double strand breaks: new developments in nonhomologous end joining. Int. J. Radiat. Oncol. Biol. Phys. 2013;86:440–449. doi: 10.1016/j.ijrobp.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Romig H., Fackelmayer F.O., Renz A., Ramsperger U., Richter A. Characterization of SAF-A, a novel nuclear DNA binding protein from HeLa cells with high affinity for nuclear matrix/scaffold attachment DNA elements. EMBO J. 1992;11:3431–3440. doi: 10.1002/j.1460-2075.1992.tb05422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kiledjian M., Dreyfuss G. Primary structure and binding activity of the hnRNP U protein: binding RNA through RGG box. EMBO J. 1992;11:2655–2664. doi: 10.1002/j.1460-2075.1992.tb05331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roshon M.J., Ruley H.E. Hypomorphic mutation in hnRNP U results in post-implantation lethality. Transgenic Res. 2005;14:179–192. doi: 10.1007/s11248-004-8147-8. [DOI] [PubMed] [Google Scholar]
- 41.Ameyar-Zazoua M., Souidi M., Fritsch L., Robin P., Thomas A., Hamiche A., Percipalle P., Ait-Si-Ali S., Harel-Bellan A. Physical and functional interaction between heterochromatin protein 1alpha and the RNA-binding protein heterogeneous nuclear ribonucleoprotein U. J. Biol. Chem. 2009;284:27974–27979. doi: 10.1074/jbc.M109.037929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Helbig R., Fackelmayer F.O. Scaffold attachment factor A (SAF-A) is concentrated in inactive X chromosome territories through its RGG domain. Chromosoma. 2003;112:173–182. doi: 10.1007/s00412-003-0258-0. [DOI] [PubMed] [Google Scholar]
- 43.Kukalev A., Nord Y., Palmberg C., Bergman T., Percipalle P. Actin and hnRNP U cooperate for productive transcription by RNA polymerase II. Nat. Struct. Mol. Biol. 2005;12:238–244. doi: 10.1038/nsmb904. [DOI] [PubMed] [Google Scholar]
- 44.Obrdlik A., Kukalev A., Louvet E., Farrants A.K., Caputo L., Percipalle P. The histone acetyltransferase PCAF associates with actin and hnRNP U for RNA polymerase II transcription. Mol. Cell. Biol. 2008;28:6342–6357. doi: 10.1128/MCB.00766-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yugami M., Kabe Y., Yamaguchi Y., Wada T., Handa H. hnRNP-U enhances the expression of specific genes by stabilizing mRNA. FEBS Lett. 2007;581:1–7. doi: 10.1016/S0014-5793(07)01283-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiao R., Tang P., Yang B., Huang J., Zhou Y., Shao C., Li H., Sun H., Zhang Y., Fu X.D. Nuclear matrix factor hnRNP U/SAF-A exerts a global control of alternative splicing by regulating U2 snRNP maturation. Mol. Cell. 2012;45:656–668. doi: 10.1016/j.molcel.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Britton S., Coates J., Jackson S.P. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J. Cell Biol. 2013;202:579–595. doi: 10.1083/jcb.201303073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Phair R.D., Misteli T. High mobility of proteins in the mammalian cell nucleus. Nature. 2000;404:604–609. doi: 10.1038/35007077. [DOI] [PubMed] [Google Scholar]
- 49.Andegeko Y., Moyal L., Mittelman L., Tsarfaty I., Shiloh Y., Rotman G. Nuclear retention of ATM at sites of DNA double strand breaks. J. Biol. Chem. 2001;276:38224–38230. doi: 10.1074/jbc.M102986200. [DOI] [PubMed] [Google Scholar]
- 50.Drouet J., Delteil C., Lefrancois J., Concannon P., Salles B., Calsou P. DNA-dependent protein kinase and XRCC4-DNA ligase IV mobilization in the cell in response to DNA double strand breaks. J. Biol. Chem. 2005;280:7060–7069. doi: 10.1074/jbc.M410746200. [DOI] [PubMed] [Google Scholar]
- 51.Kong X., Mohanty S.K., Stephens J., Heale J.T., Gomez-Godinez V., Shi L.Z., Kim J.S., Yokomori K., Berns M.W. Comparative analysis of different laser systems to study cellular responses to DNA damage in mammalian cells. Nucleic Acids Res. 2009;37:e68. doi: 10.1093/nar/gkp221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mari P.O., Florea B.I., Persengiev S.P., Verkaik N.S., Bruggenwirth H.T., Modesti M., Giglia-Mari G., Bezstarosti K., Demmers J.A., Luider T.M., et al. Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc. Natl. Acad. Sci. U.S.A. 2006;103:18597–18602. doi: 10.1073/pnas.0609061103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hassa P.O., Haenni S.S., Elser M., Hottiger M.O. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going. Microbiol. Mol. Biol. Rev. 2006;70:789–829. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rouleau M., Patel A., Hendzel M.J., Kaufmann S.H., Poirier G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ahel D., Horejsi Z., Wiechens N., Polo S.E., Garcia-Wilson E., Ahel I., Flynn H., Skehel M., West S.C., Jackson S.P., et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science. 2009;325:1240–1243. doi: 10.1126/science.1177321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gottschalk A.J., Timinszky G., Kong S.E., Jin J., Cai Y., Swanson S.K., Washburn M.P., Florens L., Ladurner A.G., Conaway J.W., et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. U.S.A. 2009;106:13770–13774. doi: 10.1073/pnas.0906920106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fackelmayer F.O., Dahm K., Renz A., Ramsperger U., Richter A. Nucleic-acid-binding properties of hnRNP-U/SAF-A, a nuclear-matrix protein which binds DNA and RNA in vivo and in vitro. Eur. J. Biochem. 1994;221:749–757. doi: 10.1111/j.1432-1033.1994.tb18788.x. [DOI] [PubMed] [Google Scholar]
- 58.Mailand N., Bekker-Jensen S., Faustrup H., Melander F., Bartek J., Lukas C., Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 59.Cheng Q., Barboule N., Frit P., Gomez D., Bombarde O., Couderc B., Ren G.S., Salles B., Calsou P. Ku counteracts mobilization of PARP1 and MRN in chromatin damaged with DNA double-strand breaks. Nucleic Acids Res. 2011;39:9605–9619. doi: 10.1093/nar/gkr656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Elmroth K., Nygren J., Martensson S., Ismail I.H., Hammarsten O. Cleavage of cellular DNA by calicheamicin gamma1. DNA Repair (Amst) 2003;2:363–374. doi: 10.1016/s1568-7864(02)00235-5. [DOI] [PubMed] [Google Scholar]
- 61.Herrmann F., Bossert M., Schwander A., Akgun E., Fackelmayer F.O. Arginine methylation of scaffold attachment factor A by heterogeneous nuclear ribonucleoprotein particle-associated PRMT1. J. Biol. Chem. 2004;279:48774–48779. doi: 10.1074/jbc.M407332200. [DOI] [PubMed] [Google Scholar]
- 62.Lippincott-Schwartz J., Snapp E., Kenworthy A. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2001;2:444–456. doi: 10.1038/35073068. [DOI] [PubMed] [Google Scholar]
- 63.Jao C.Y., Salic A. Exploring RNA transcription and turnover in vivo by using click chemistry. Proc. Natl. Acad. Sci. U.S.A. 2008;105:15779–15784. doi: 10.1073/pnas.0808480105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hu Z., Zhang A., Storz G., Gottesman S., Leppla S.H. An antibody-based microarray assay for small RNA detection. Nucleic Acids Res. 2006;34:e52. doi: 10.1093/nar/gkl142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tsunaka Y., Haruki M., Morikawa M., Kanaya S. Strong nucleic acid binding to the Escherichia coli RNase HI mutant with two arginine residues at the active site. Biochim. Biophys. Acta. 2001;1547:135–142. doi: 10.1016/s0167-4838(01)00180-7. [DOI] [PubMed] [Google Scholar]
- 66.Tazi J., Bakkour N., Soret J., Zekri L., Hazra B., Laine W., Baldeyrou B., Lansiaux A., Bailly C. Selective inhibition of topoisomerase I and various steps of spliceosome assembly by diospyrin derivatives. Mol. Pharmacol. 2005;67:1186–1194. doi: 10.1124/mol.104.007633. [DOI] [PubMed] [Google Scholar]
- 67.Tuduri S., Crabbe L., Conti C., Tourriere H., Holtgreve-Grez H., Jauch A., Pantesco V., De Vos J., Thomas A., Theillet C., et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009;11:1315–1324. doi: 10.1038/ncb1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yuce-Petronczki O., West S.C. Senataxin, defective in the neurogenerative disorder AOA-2, lies at the interface of transcription and the DNA damage response. Mol. Cell. Biol. 2012;33:406–417. doi: 10.1128/MCB.01195-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gagne J.P., Pic E., Isabelle M., Krietsch J., Ethier C., Paquet E., Kelly I., Boutin M., Moon K.M., Foster L.J., et al. Quantitative proteomics profiling of the poly(ADP-ribose)-related response to genotoxic stress. Nucleic Acids Res. 2012;40:7788–7805. doi: 10.1093/nar/gks486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gagne J.P., Hunter J.M., Labrecque B., Chabot B., Poirier G.G. A proteomic approach to the identification of heterogeneous nuclear ribonucleoproteins as a new family of poly(ADP-ribose)-binding proteins. Biochem. J. 2003;371:331–340. doi: 10.1042/BJ20021675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Malanga M., Czubaty A., Girstun A., Staron K., Althaus F.R. Poly(ADP-ribose) binds to the splicing factor ASF/SF2 and regulates its phosphorylation by DNA topoisomerase I. J. Biol. Chem. 2008;283:19991–19998. doi: 10.1074/jbc.M709495200. [DOI] [PubMed] [Google Scholar]
- 72.Ji Y., Tulin A.V. Poly(ADP-ribosyl)ation of heterogeneous nuclear ribonucleoproteins modulates splicing. Nucleic Acids Res. 2009;37:3501–3513. doi: 10.1093/nar/gkp218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dutertre M., Sanchez G., Barbier J., Corcos L., Auboeuf D. The emerging role of pre-messenger RNA splicing in stress responses: sending alternative messages and silent messengers. RNA Biol. 2011;8:740–747. doi: 10.4161/rna.8.5.16016. [DOI] [PubMed] [Google Scholar]
- 74.Dutertre M., Sanchez G., De Cian M.C., Barbier J., Dardenne E., Gratadou L., Dujardin G., Le Jossic-Corcos C., Corcos L., Auboeuf D. Cotranscriptional exon skipping in the genotoxic stress response. Nat. Struct. Mol. Biol. 2010;17:1358–1366. doi: 10.1038/nsmb.1912. [DOI] [PubMed] [Google Scholar]
- 75.Paronetto M.P., Minana B., Valcarcel J. The ewing sarcoma protein regulates DNA damage-induced alternative splicing. Mol. Cell. 2011;43:353–368. doi: 10.1016/j.molcel.2011.05.035. [DOI] [PubMed] [Google Scholar]
- 76.Li S., Kuhne W.W., Kulharya A., Hudson F.Z., Ha K., Cao Z., Dynan W.S. Involvement of p54(nrb), a PSF partner protein, in DNA double-strand break repair and radioresistance. Nucleic Acids Res. 2009;37:6746–6753. doi: 10.1093/nar/gkp741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang W.Y., Pan L., Su S.C., Quinn E.J., Sasaki M., Jimenez J.C., Mackenzie I.R., Huang E.J., Tsai L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013;16:1383–1391. doi: 10.1038/nn.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rajesh C., Baker D.K., Pierce A.J., Pittman D.L. The splicing-factor related protein SFPQ/PSF interacts with RAD51D and is necessary for homology-directed repair and sister chromatid cohesion. Nucleic Acids Res. 2011;39:132–145. doi: 10.1093/nar/gkq738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Banerjee D., Mandal S.M., Das A., Hegde M.L., Das S., Bhakat K.K., Boldogh I., Sarkar P.S., Mitra S., Hazra T.K. Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J. Biol. Chem. 2011;286:6006–6016. doi: 10.1074/jbc.M110.198796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Belotserkovskii B.P., Neil A.J., Saleh S.S., Shin J.H., Mirkin S.M., Hanawalt P.C. Transcription blockage by homopurine DNA sequences: role of sequence composition and single-strand breaks. Nucleic Acids Res. 2013;41:1817–1828. doi: 10.1093/nar/gks1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Roy D., Zhang Z., Lu Z., Hsieh C.L., Lieber M.R. Competition between the RNA transcript and the nontemplate DNA strand during R-loop formation in vitro: a nick can serve as a strong R-loop initiation site. Mol. Cell. Biol. 2010;30:146–159. doi: 10.1128/MCB.00897-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pourquier P., Pommier Y. Topoisomerase I-mediated DNA damage. Adv. Cancer Res. 2001;80:189–216. doi: 10.1016/s0065-230x(01)80016-6. [DOI] [PubMed] [Google Scholar]
- 83.Katzenberger R.J., Marengo M.S., Wassarman D.A. ATM and ATR pathways signal alternative splicing of Drosophila TAF1 pre-mRNA in response to DNA damage. Mol. Cell. Biol. 2006;26:9256–9267. doi: 10.1128/MCB.01125-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shkreta L., Michelle L., Toutant J., Tremblay M.L., Chabot B. The DNA damage response pathway regulates the alternative splicing of the apoptotic mediator Bcl-x. J. Biol. Chem. 2011;286:331–340. doi: 10.1074/jbc.M110.162644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.