


Abstract
Hypoxia is associated with a variety of physiological and pathological conditions and elicits specific transcriptional responses. The elongation competence of RNA Polymerase II is regulated by the positive transcription elongation factor b (P-TEFb)-dependent phosphorylation of Ser2 residues on its C-terminal domain. Here, we report that hypoxia inhibits transcription at the level of elongation. The mechanism involves enhanced formation of inactive complex of P-TEFb with its inhibitor HEXIM1 in an HDAC3-dependent manner. Microarray transcriptome profiling of hypoxia primary response genes identified ∼79% of these genes being HEXIM1-dependent. Hypoxic repression of P-TEFb was associated with reduced acetylation of its Cdk9 and Cyclin T1 subunits. Hypoxia caused nuclear translocation and co-localization of the Cdk9 and HDAC3/N-CoR repressor complex. We demonstrated that the described mechanism is involved in hypoxic repression of the monocyte chemoattractant protein-1 (MCP-1) gene. Thus, HEXIM1 and HDAC-dependent deacetylation of Cdk9 and Cyclin T1 in response to hypoxia signalling alters the P-TEFb functional equilibrium, resulting in repression of transcription.
INTRODUCTION
Hypoxia (insufficient oxygen tension) is a fundamental stimulus for physiological processes and pathological conditions. Early embryonic organogenesis occurs in an oxygen-limited environment. Hypoxia is necessary to maintain undifferentiated states of embryonic, hematopoietic, mesenchymal and neural stem cell phenotypes (1). Inflamed tissues are often hypoxic, including those seen in rheumatoid arthritis, atherosclerotic plaques and healing wounds (2). In our previous publications, we addressed the question on how hypoxia modulates the basal and IL-1β-induced production of cytokines (3). We have also demonstrated that hypoxia repressed IL-1β-induced MCP-1 gene expression (4,5). Both hypoxia and inflammation are critical factors in tumor progression (6). A tumor hypoxic niche was recently proposed to harbor cancer stem cell populations (1). During hypoxia, cells activate a number of adaptive responses to match oxygen supply with metabolic demands. Limited energy resources under hypoxic stress lead to the global repression of protein and mRNA synthesis (7), while proangiogenic and survival-promoting genes induce their expression (8). Activation of specific transcription factors and chromatin remodeling with subsequent recruitment of the basic transcription machinery was thought to be the main mechanism for the selective expression of a subset of genes in response to stressors. Recent evidence, however, suggests that transcription of many genes, including primary response inflammatory genes and developmental control genes, is regulated primarily after the initiation step at the transition to productive elongation (9–11). Despite increasing knowledge about hypoxia responsive transcription factors, very little is known about the hypoxia-related signaling targeting transcription elongation.
One element of the regulation of productive elongation involves phosphorylation of the carboxy-terminal domain (CTD) of Rbp1, the largest subunit of RNA Polymerase II (Pol II). The Pol II CTD contains multiple heptapeptide repeats with the consensus amino acid sequence YSPTSPS. The number of these repeats varies among species and there are 52 such repeats in humans. The serines at positions 2 (Ser2) and 5 (Ser5) undergo dynamic phosphorylation, coinciding with the phases of the Pol II transcription cycle. Unphosphorylated Pol II is preferentially recruited to promoters to associate with both the preinitiation and mediator complexes (12). During promoter clearance, the Cdk7 kinase from the TFIIH general transcription factor phosphorylates CTD at the Ser5 position, facilitating promoter escape and stimulating binding of capping enzymes (13,14). Such an early elongation complex enters abortive elongation followed by pausing of Pol II (15). Promoter-proximal pausing has recently been found to be involved in transcriptional control of rapidly induced genes. The transition to productive elongation is determined by the subsequent phosphorylation of Ser2 by the Cdk9 kinase of the positive transcription elongation factor b (P-TEFb), which also directs co-transcriptional processing of primary transcripts (capping, splicing and polyadenylation) (16). When transcription terminates, Fcp1 phosphatase dephosphorylates the Ser2 of the CTD, stimulating Pol II recycling into initiation-competent complexes (17,18).
P-TEFb is the only factor known to release poised Pol II to promote productive elongation (10). The activity of P-TEFb can be controlled in a number of different ways. It has been suggested that signal-dependent recruitment of P-TEFb to promoters may be a key role of transcriptional activators (19). In addition to regulating its recruitment, P-TEFb itself is directly controlled through sequestering into an inactive complex. Catalytically active P-TEFb consists of a catalytic subunit, Cdk9, and a regulatory subunit, which can be Cyclin T1 or Cyclin T2 (20). Besides the active, heterodimeric form, P-TEFb exists in a larger, catalytically inactive complex that contains 7SK small nuclear RNA (7SK snRNA) and HEXIM1 (21–23). In human HeLa cells, more than half of the P-TEFb is associated in large ribonucleoprotein (RNP) complexes and represents a major reservoir of activity from which P-TEFb can be rapidly mobilized (21). The kinase activity of Cdk9 and formation of an inactive P-TEFb complex can also be regulated by acetylation of Cdk9 and Cyclin T1 proteins by p300 and P/CAF (24–26). In addition to acetylation, the activity of P-TEFb can be regulated by reversible phosphorylation of Cyclin T1, Cdk9 and Hexim1 and by polyubiquitination of Cdk9 and Hexim1 (27). Although it was demonstrated that chemical agents or ultraviolet irradiation disrupted the P-TEFb/7SK snRNA complex (21,22), the physiological stimuli that control cellular levels of active and inactive P-TEFb and details of the pathway have yet to be elucidated.
This work describes the mechanism of transcriptional repression under hypoxic conditions by interfering with P-TEFb functions. We demonstrate that hypoxia affects the physiological equilibrium between active and inactive P-TEFb in cells. The pathway involves histone deacetylase HDAC3 and its co-factor N-CoR. Consequently, acetylation levels of Cdk9 and Cyclin T1 subunits of P-TEFb were markedly reduced in hypoxic cells. The described mechanism may lead to rapid changes in gene expression under energy-stressed conditions.
MATERIALS AND METHODS
Reagents and antibodies
The following antibodies were used in this study: anti-RNA Polymerase II (H-224), Fcp1 (H-300), Cdk9 (H-169) rabbit polyclonal, Cdk9 (D-7) mouse monoclonal, cyclin T1 (T-18), c-Myc (9E10) antibodies were purchased from Santa Cruz Biotechnology. RNA Polymerase II H5 and H14 antibodies which recognize phosphoserine 2 and phosphoserine 5 versions of Pol II were from Covance. HEXIM1 sheep (ab28016) and rabbit (ab25388) polyclonal antibodies were purchased from Abcam. Anti-HDAC1 (SA-401), anti-HDAC2 (SA-402) and anti-HDAC3 (SA-403) rabbit polyclonal antibodies were obtained from BIOMOL (currently Enzo Life Sciences). Anti-SMRT (PA1–842) and anti-N-CoR (PA1–844A) antibodies were purchased from Affinity BioReagents. Anti-goat Alexa Fluor 488 (A21467) and Alexa Fluor 568 (A11057), anti-rabbit Alexa Fluor 546 (A11010) and anti-mouse Alexa Fluor 633 (A21052) IgGs, as well as TO-PRO-3 and SYTO 16 fluorescent nucleic acid stains, were obtained from Molecular Probes. Anti-acetyl-Lysine clone 4G12 mouse monoclonal IgG was from Upstate. Trichostatin A, hexamethylene bis(acetamide) (HMBA) and Ribonuclease A (RNase A) were purchased from Sigma-Aldrich.
Cell culture and hypoxia treatment
HeLa cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Exposure to normoxia or hypoxia was performed either with or without 50 ng/ml of recombinant human IL-1β (PeproTech). For short-term hypoxia exposures (15 or 30 min), cells were incubated in a hypoxic conditioned medium. For 1 or 2 h hypoxic treatments, cells were placed in a modular incubator chamber (Billus-Rothenberg Inc.) filled with a hypoxic gas mixture (5% CO2, 0.5% O2, balanced with N2) with routine checks of O2 concentrations by an Oxygen Monitor JKO-25S.
Reverse transcription and quantitative real-time PCR
Total RNA was extracted with TRI-zol reagent (Invitrogen) followed by cDNA synthesis in a reaction containing 5 μg of RNA as a template using a ReverTraAce kit (TOYOBO). MCP-1 and IL-8 mRNA expressions and levels of 18S rRNA were analyzed by quantitative real-time PCR (Q-PCR) in a LightCycler instrument (Roche). Details of the method and primer sequences for MCP-1 and 18S rRNA were described previously (4).
Chromatin immunoprecipitation analysis
The chromatin immunoprecipitation (ChIP) assay was performed using a Chromatin Immunoprecipitation Assay Kit from Upstate biotechnology (currently Millipore) as described previously (3). Chromatin from 3 × 106 HeLa cells was used for each reaction. Immunoprecipitated DNA was quantified using TaqMan probes labeled with a 5’ FAM reporter and a 3’BHQ1 nonfluorescent quencher and normalized by 10% input DNA. Each cycle threshold (Ct) value was determined from three parallel PCRs. Promoter sequences of MCP-1 and IL-8 subcloned into the pGL3-Basic vector (Promega) were used to obtain the calibration curve.
SiRNA transfections
For knock-down experiments, 2 × 105 HeLa cells were seeded 24 h prior to transfection. Delivery of double-stranded siRNA oligonucleotides was performed using X-tremeGENE siRNA Transfection Reagent (Roche) according to the manufacturer's protocol. After 48 h, cells were treated with IL-1β and hypoxia as designated, sampled for total RNA or cell lysate preparations and processed for Q-PCR or immunoprecipitation, respectively. The following siRNA oligonucleotides were used for transfection: HDAC1, NCoR2/SMRT, NCoR, HEXIM1 and CTDP1/Fcp-1 siRNAs were obtained from Dharmacon RNA technologies. Control siRNA-A (sc-37007), HDAC3 siRNA (sc-35538) and HDAC2 siRNA (sc-29345) were purchased from Santa Cruz Biotechnology.
Cdk9 expression vector construct and transfections
A full length human cDNA clone of Cdk9 (accession NM_001261) was obtained from the Thermo Scientific Open Biosystems library in a pSPORT1 vector. Cdk9 cDNA was subcloned into a pcDNA3.1/myc-His C vector and used for transient transfections with subsequent immunoprecipitation. For transient transfections, Hela cells were plated at a concentration of 5 × 105 cells/well and 24 h later transfected with 2.5 μg of the plasmid DNA using a Lipofectamine 2000 transfection reagent (Invitrogene). The next day, cells were treated with or without IL-1β and incubated under normoxia or hypoxia for 1 h.
Co-immunoprecipitation and western blotting
Immunoprecipitation was carried out according to the protocol recommended by Millipore Technical Library using an EBC buffer. Cell lysates were diluted to 2 μg/μl total cell protein and immunoprecipitated overnight with either a Cdk9 or Cyclin T1 antibody. The inactive P-TEFb complex was analyzed by western blot with a HEXIM1, Cyclin T1 or Cdk9 antibody. Acetyl-Lysine antibodies were used to analyze the acetylation status of Cdk9. Details of immunoprecipitation and western blot protocols were described previously (3).
Immunocytochemistry and proximity-dependent DNA ligation in situ assay (P-LISA)
HeLa cells were seeded on glass bottom dishes at a concentration of 1.2 × 105/dish two days prior to hypoxia exposure. After the treatment as designated, cells were fixed with a 10% formaldehyde solution for 15 min at room temperature, permeabilized for 20 min using 0.2% Triton X-100 and incubated with blocking solution (1% FBS in PBS). Incubation with the primary antibody was carried out overnight at 4°C and the next day cells were processed for standard immunocytochemistry or a P-LISA assay. Cellular fluorescence was monitored by ZEISS 510 META laser scanning confocal microscopy.
For immunocytochemistry, incubation with the primary antibody was followed by incubation for 2 h with appropriate Alexa Fluor fluorescent secondary antibodies. Nucleic acids were visualized by TO-PRO-3 or SYTO 16.
Cdk9 and Cyclin T1 interactions with HEXIM1 and specific posttranslational acetylations of Cdk9 and Cyclin T1 in their subcellular localizations were analyzed using a P-LISA assay. In this assay, proximity probes—oligonucleotides attached to specific antibodies—guide the formation of circular DNA strands when bound to the sample in close proximity. In subsequent steps, the DNA cycles are amplified by rolling-circle amplification and visualized by fluorescently labeled oligonucleotides. The P-LISA assay was performed after the incubation with primary antibodies using a Duolink In Situ (Figures 1C and 4) or a Duolink II (Figure 3) Fluorescence kits from Olink Bioscience following the recommended protocols.
Figure 1.
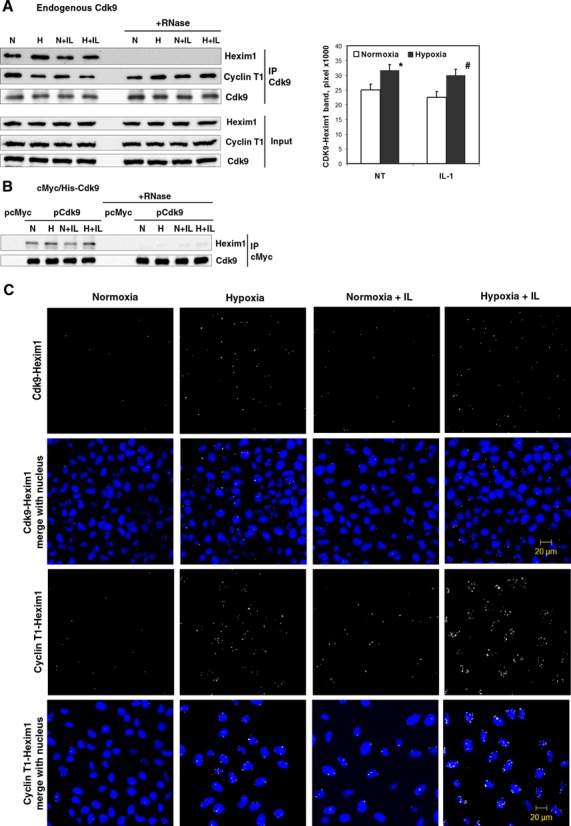
Formation of an inactive P-TEFb complex in response to hypoxia. (A) Association of endogenous Cdk9 with HEXIM1 and Cyclin T1 in cells treated with normoxia (N) and hypoxia (H) in the presence or absence of IL-1β for 1 h. Whole cell lysates were immunoprecipitated (IP) with a rabbit anti-Cdk9 antibody followed by western blotting with goat anti-Cyclin T1 and a sheep anti-HEXIM1 antibody. Western blot with a monoclonal anti-Cdk9 antibody served as a control of total immunoprecipitated protein. To disrupt 7SK snRNA in ribonucleoprotein complexes, one set of cell lysates was treated with RNase A for 2 h prior to immunoprecipitation. Columns represent mean of densitometric quantification of 6 western blots obtained from independent immunoprecipitations (n = 6); bars, SE; NT, without IL-1β treatment. *P < 0.05 as compared with normoxia; #P < 0.05 as compared with IL-1β-treated normoxic samples. (B) Association of cMyc/His-taged Cdk9 with HEXIM1 under normoxia (N) and hypoxia (H). Samples were immunoprecipitated using a monoclonal anti-cMyc antibody and analyzed as in (A). Western blot with a rabbit anti-Cdk9 antibody served as a control of total immunoprecipitated protein. Data shown represent one of two independent experiments. (C) Interactions of Cdk9-HEXIM1 and Cyclin T1-HEXIM1 were analyzed by P-LISA assay using rabbit anti-HEXIM1, mouse anti-Cdk9 and goat anti-Cyclin T1 antibodies. HeLa cells were exposured to hypoxia in the presence or absence of IL-1β for 1 h. PLA-signals of protein–protein interactions are shown as white speckles. Nuclei stained with SYTO 16 Nucleic Acid Stain in blue.
Figure 4.
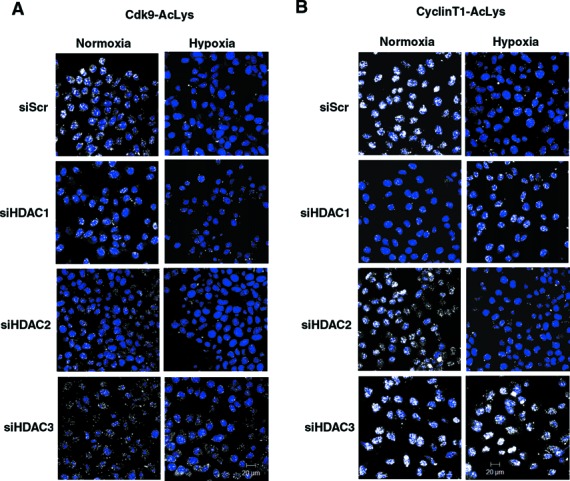
The role of HDACs in hypoxic deacetylation of Cdk9 and Cyclin T1. HeLa cells were exposed for 1 h under normoxia or hypoxia 48 h after transfection with siRNA directed against HDAC1, HDAC2 or HDAC3. (A) Acetylated Cdk9 was analyzed by P-LISA assay using rabbit anti-AcLys and mouse anti-Cdk9 antibodies. (B) Acetylated Cyclin T1 was analyzed by P-LISA assay using rabbit anti-AcLys and goat anti-Cyclin T1 antibodies. P-LISA signals are shown in white and the nuclei in blue.
Figure 3.
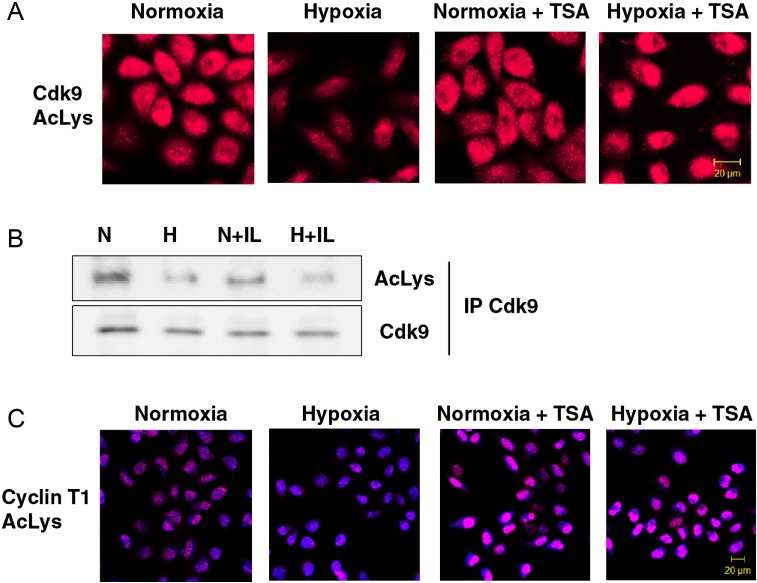
Hypoxic repression of Cdk9 and Cyclin T1 acetylation. (A) Intracellular distribution of Cdk9 acetylated at lysine residues in response to hypoxia. HeLa cells were pretreated with or without 500 nM TSA for 1 h and subsequently exposed to hypoxia for 1 h. Acetylated Cdk9 was analyzed by P-LISA assay using rabbit anti-AcLys and mouse anti-Cdk9 antibodies. (B) Acetylation of Cdk9 in response to 1 h exposure under normoxic (N) or hypoxic (H) gas mixtures with or without IL-1β. Immunoprecipitated Cdk9 was examined by western blotting with antibodies against acetyl-Lysin (AcLys) or Cdk9. All buffers used in this assay were supplemented with 100 ng/ml TSA to block endogenous HDAC activity. (C) Intracellular distribution of acetylated Cyclin T1 was analyzed by P-LISA assay using rabbit anti-AcLys and goat anti-Cyclin T1 antibodies. HeLa cells were pretreated with or without 500 nM TSA for 1 h followed by exposure to hypoxia for 1 h. Nucleic acids were stained using TOPRO-3 (blue).
cDNA microarray and functional analysis
Total RNA was extracted with TRI-zol reagent (Invitrogen). Pooled RNA from two replicates was obtained from HeLa cells transfected with double-stranded siRNA oligonucleotides directed against HEXIM1 (Thermo Scientific Dharmacon) or control siRNA (Santa Cruz) and 48 h later treated for 1 h with IL-1β in normoxic or hypoxic environment. Sample preparation, hybridization to SurePrint G3 Human GE microarray (G4851A, Agilent Technologies) and scanning were performed at the Oncomics facility, Nagoya, Japan. Microarray data and details of the protocol have been submitted to the Gene Expression Omnibus (GEO) database (accession number GSE41023; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE41023)
Data was analyzed using Subio Platform software (http://www.subio.jp/products/platform). The log2 transformed intensities were normalized on 75th percentile in each sample, and then turned into log ratios against the control sample. Genes with very low signals (gIsWellAboveBG = 0 at all samples) were filtered out. Those genes with value grater than 1 or less than −1 (2-fold difference) were selected for further analysis. Differently expressed genes underwent functional analysis manually and with web-based resources DAVID functional annotations (The Database for Annotation, Visualization and Integrated Discovery; http://david.abcc.ncifcrf.gov/) and GOrilla (http://cbl-gorilla.cs.technion.ac.il/).
RESULTS
Hypoxia induces formation of a large inactive complex of P-TEFb
Pol II productive elongation is highly dependent on the P-TEFb factor. To determine whether hypoxia disturbs the equilibrium between active and inactive forms of P-TEFb, we performed immunoprecipitation of the P-TEFb Cdk9 subunit with subsequent detection of the co-immunoprecipitated HEXIM1 and Cyclin T1 by western blotting (Figure 1A). IgG controls are shown in Supplementary Figure S1. There was a clear shift in the P-TEFb equilibrium toward the inactive complex with HEXIM1 in cells incubated in a hypoxic environment for 1 h in both IL-1β-treated and non-treated conditions (Figure 1A). For Cdk9-Hexim1 protein–protein interactions, this was quantified as a 35% increase in IL-1 treated and 30% increase in non-treated cells and these increases were statistically significant. Interestingly, the amount of Cyclin T1 co-precipitating with Cdk9 was lower in the hypoxic state (Figure 1A). The observed changes in protein interactions were not due to altered expressions of Hexim1, Cyclin T1 or Cdk9 proteins that were not changed in the inputs after 1 h of hypoxic treatment (Figure 1A). Incubation of the Cdk9 immune complex with RNase A prior to precipitation disrupted the binding of HEXIM1 to the immobilized P-TEFb in both hypoxic and normoxic cells, but did not have an impact on the formation of the Cdk9/Cyclin T1 heterodimer, reflecting the role of 7SK snRNA in inactive complex formation (Figure 1A).
To support the observation of induced association between endogenous P-TEFb and HEXIM1 in hypoxic cells, we constructed a vector containing cMyc/His-tagged human Cdk9 cDNA. Immunoprecipitation of cMyc-Cdk9 expressed in transfected HeLa cells using anti-cMyc antibody revealed complex formation with endogenous HEXIM1 (Figure 1B). The association between cMyc-Cdk9 and HEXIM1 was enhanced in cells exposed to hypoxia for 1 h. The binding of transiently expressed cMyc-Cdk9 to HEXIM1 was disrupted when the immune complexes were preincubated with RNase A. The data in Figure 1B indicate that cMyc produced from the transfected Cdk9 cDNA, but not an empty vector, co-immunoprecipitated with HEXIM1 in HeLa protein extracts.
To study the increase in HEXIM1 and P-TEFb functional equilibrium in hypoxic cells, we performed P-LISA assay of Cdk9-Hexim1 and Cyclin T1-HEXIM1 interactions (Figure 1C). In this assay, the signal from each detected protein–protein interaction is visualized at single-molecule resolution by a pair of oligonucleotide labeled secondary antibodies (PLA probes) as an individual fluorescent dot (28). In our studies, it is clear that HEXIM1 binding to Cdk9 and Cyclin T1 is enhanced in hypoxic cells. Cyclin T1 interaction with HEXIM1 was further enhanced when hypoxia was used in combination with IL-1β. Notably, the size of PLA-signals was also increased in hypoxic cells, suggesting that interacting proteins were accumulating in certain nuclear structures. The speckles of Cdk9 and HEXIM1 interactions were observed in both cytoplasm and nucleus, whereas Cyclin T1 interacted with Hexim1 only within nucleus (Figure 1C). This suggests that Cdk9 and HEXIM1 may interact in the cytoplasm without Cyclin T1 and can partly explain decreased association of Cdk9 with Cyclin T1 in hypoxic cells. Collectively, these data show that hypoxia enhances formation of an inactive complex of P-TEFb with its endogenous inhibitors HEXIM1 and 7SK snRNA.
HDAC3 is required for hypoxia-mediated formation of an inactive P-TEFb complex
We have previously shown that hypoxia enhances HDAC activity and that HDACs are required for the transcriptional repression in response to hypoxia (3,29). To determine whether class I HDACs were involved in the hypoxia-mediated formation of inactive P-TEFb complex, we disrupted HDAC1, HDAC2 and HDAC3 proteins in HeLa cells using double-stranded siRNA (Figure 2A). Input levels of Cdk9 and Cyclin T1 protein expressions were not affected by HDACs knockdown. Expressions of HEXIM1 were slightly higher in knocked-down cells, but there were no differences between normoxic and hypoxic samples. Immunoprecipitation of the Cyclin T1 regulatory subunit of P-TEFb with subsequent immunodetection of its associated HEXIM1 revealed that different isoforms of class I HDACs played distinct roles in P-TEFb regulation. Silencing of HDAC2 enhanced formation of the inactive P-TEFb complex with HEXIM1 under hypoxia. Therefore, HDAC2 is likely to be a positive regulator of P-TEFb. Silencing of HDAC3 induced 7SK snRNP formation in normoxia, completely reversed hypoxic inhibition of P-TEFb and shifted the P-TEFb equilibrium in hypoxic cells toward its active form (Figure 2A). Our data revealed that HDAC3 contributes to the shifting of P-TEFb equilibrium toward its association with HEXIM1 in response to hypoxia. Silencing of HDACs did not affect Cyclin T1 complex formation with its catalytic partner Cdk9 (Figure 2A).
Figure 2.
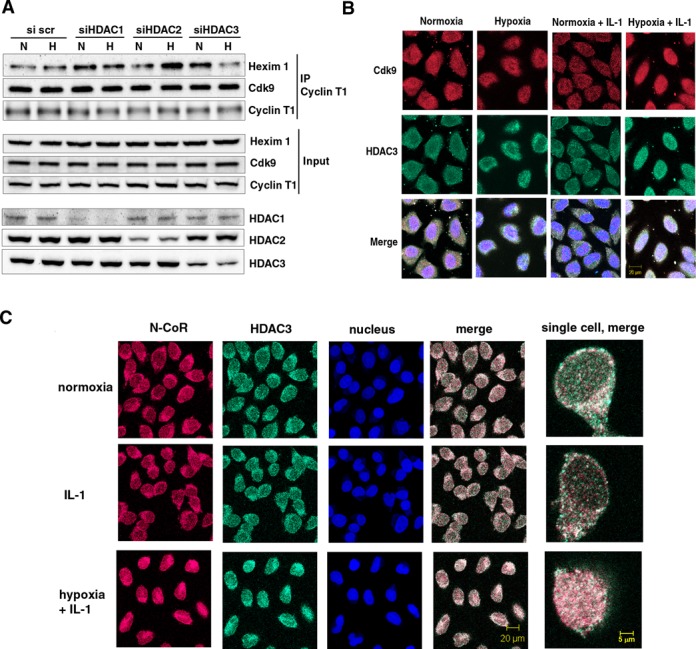
Effect of depletion of class I HDACs on 7SK snRNA and cellular distribution of N-CoR/HDAC3 and Cdk9 in normoxic and hypoxic HeLa cells. (A) Effect of disruption of class I HDACs on the formation of an inactive endogenous P-TEFb complex in response to hypoxia. HeLa cells were exposed for 1 h to a normoxic (N) or hypoxic (H) gas mixture 48 h after transfection with siRNA. Whole cell lysates were immunoprecipitated with a goat anti-Cyclin T1 antibody and analyzed by western blot using rabbit anti-HEXIM1 and an anti-Cdk9 antibody. Direct western blots of HDAC1, HDAC2 and HDAC3 served as a control for siRNA-mediated knockdown. Western blot with an anti-Cyclin T1 antibody served as a control of total immunoprecipitated protein. (B) Localization of endogenous Cdk9 (red) and HDAC3 (green). Cells were treated with hypoxia, IL-1β or their combination for 2 h and analyzed by immunofluorescence staining. (C) Immunofluorescence showing cellular localization of endogenous HDAC3 (green) and N-CoR (red) in response to 2 h exposure to IL-1β alone or in combination with hypoxia. Nucleic acids were stained using TO-PRO-3 (blue).
Cdk9 and HDAC3 are translocated to the nucleus and co-localized in response to hypoxia
To study the cellular localization of Cdk9 and its relation to HDAC3, we performed immunofluorescent analysis of Cdk9 and HDAC3. Cdk9 was observed not only in the nucleus but also occupied the entire cytoplasmic compartment of HeLa cells (Figure 2B). Speckles of Cdk9 co-localization with HDAC3 were detected mainly in the cytosol of normoxic cells. Treatment with IL-1β did not strongly affect distribution of Cdk9, although speckles of its co-localization with HDAC3 appeared to be slightly enhanced in the nucleus. Exposure to hypoxia caused cellular redistribution of Cdk9 toward the nucleus and perinuclear area (Figure 2B). Although in ∼5% of hypoxic cells Cdk9 and HDAC3 co-localization was observed in the nucleus, in other cells it was observed in the perinuclear area. Under the simultaneous treatment of hypoxia and IL-1β, both Cdk9 and HDAC3 were located in the nucleus in nearly 85% of cells. (Figure 2B). Speckles of their co-localization were grouped in the center and occupied the periphery of the nucleus.
Unlike other members of the Class I HDAC family, HDAC3 contains nuclear localization and export signals and requires co-repressors N-CoR (nuclear receptor co-repressor) and SMRT (silencing mediator for retinoid and thyroid hormone receptors) for deacetylase enzymatic activity (30). N-CoR and HDAC3 were both localized in the nucleus and cytoplasm in normoxic HeLa cells with predominant localization in the cytoplasm (Figure 2C). While stimulation by IL-1β alone did not affect cellular distribution of these repressors, simultaneous exposure to hypoxia and IL-1β shifted localization of N-CoR and HDAC3 to the nucleus. Merged images revealed co-localization in the same structures (Figure 2C). SMRT was predominantly localized in the nucleus and did not change localization under any condition examined (Supplementary Figure S2). Cellular localization of N-CoR in hypoxic cells without IL-1β treatment is shown in Supplementary Figure S3.
Acetylation levels of Cdk9 and Cyclin T1 are reduced in hypoxic cells in a HDAC3-dependent manner
Because of hypoxic inhibition of P-TEFb was mediated by HDAC, we examined whether the acetylation status of endogenous Cdk9 and Cyclin T1 can be affected by hypoxia. For this purpose, we used a proximity-dependent DNA ligation in situ assay (P-LISA) (28). A considerable reduction in intracellular levels of acetylated Cdk9 was observed in HeLa cells treated with hypoxia for 1 h (Figure 3A). Pretreatment with HDAC inhibitor Trichostatin A (TSA) prevented hypoxic deacetylation of Cdk9. Notably, in the presence of TSA, acetylated Cdk9 was located primarily in the nucleus of hypoxic cells, indicating that nuclear translocation of Cdk9 in response to hypoxia is acetylation-independent and does not require HDAC activity.
To further substantiate the effect of hypoxia, we performed immunoprecipitations of endogenous Cdk9 followed by western blotting analysis with pan-AcLys antibodies (Figure 3B). The identity of the acetyl-lysine bands was confirmed by striping and re-exposure of the same membrane to mouse Cdk9 antibodies. Treatment with IL-1β did not impact the acetylation level of Cdk9. Consistent with the study using P-LISA, exposure to hypoxia for 1 h led to a significant reduction in acetyl-Cdk9 in both untreated cells and in the background of IL-1β (Figure 3B).
Acetylated Cyclin T1 was clearly detected in normoxic cells using P-LISA, and treatment with hypoxia for 1 h led to a significant reduction in its acetylated levels (Figure 3C). The levels of acetylated Cyclin T1 were increased by treatment with TSA. Inhibition of HDAC activity abolished hypoxic reduction of Cyclin T1 acetylation. These results collectively show that hypoxic signaling to P-TEFb leads to deacetylation of its core components Cdk9 and Cyclin T1.
To identify which HDAC was responsible for the reduced acetylation of Cdk9 and Cyclin T1 under hypoxic conditions, we tested the levels of acetylations upon siRNA-mediated knockdown of HDAC1, HDAC2 and HDAC3 isoforms. We found that knockdown of HDAC3 inhibited hypoxia-mediated deacetylation of Cdk9, while knockdown of HDAC1 or HDAC2 did not have significant impact on the levels of Cdk9 acetylation in normoxic cells and their response to hypoxia (Figure 4A). This observation was in agreement with previous report that N-CoR/HDAC3 is likely to repress P-TEFb activity through deacetylation (25). The role of HDACs in the regulation of Cyclin T1 acetylations has not been described previously. Unexpectedly, we found that knockdown of HDAC1 resulted in reduced levels of Cyclin T1 acetylations in the cells (Figure 4B), suggesting that Cyclin T1 acetylation is positively regulated by HDAC1. Interestingly, acetylation of Cyclin T1 was induced by hypoxia in HDAC1-depleted cells. Depletion of HDAC2 did not affect acetylation of Cyclin T1 in normoxia and its reduction after 1 h exposure to hypoxia. When HDAC3 was knocked down in the cells, we found that Cyclin T1 acetylation was not affected by hypoxia (Figure 4B). These data supported the proposed role of HDAC3 in hypoxia-mediated repression of P-TEFb. Acetylation of both Cdk9 and Cyclin T1 can be targeted for deacetylation in hypoxic cells by HDAC3.
Hypoxia represses phosphorylation of Ser2 residues at Pol II CTD
Next, we questioned whether hypoxic inhibition of P-TEFb is actually involved in the transcriptional regulation of gene targets. Previously, we observed induction of the IL-8 gene and HDAC-dependent repression of the MCP-1 gene after 6 h or 24 h exposure to hypoxia (3,4). Firstly, we proved that these changes can already be observed 1 h after initiation of hypoxia (Figure 5A). Moreover, the cumulative effect of IL-1β and hypoxia was much greater than that seen after prolonged exposure. To determine whether hypoxia interferes with pre-initiation complex (PIC) assembly on MCP-1 and IL-8 genes, we performed a ChIP assay using an antibody against the Pol II Rbp1 subunit. Pol II occupancy of the IL-8 promoter was unaffected by hypoxia alone (Figure 5A). Simultaneous treatment with hypoxia and IL-1β caused a sharp increase in Pol II recruitment to the IL-8 promoter, which was much stronger than that under IL-1β alone (Figure 5B). Repression of the MCP-1 gene by hypoxia was not associated with considerable changes in the total Pol II occupancy of its promoter (Figure 5B). Thus, hypoxia reduces MCP-1 expression by interfering with a step after PIC assembly.
Figure 5.
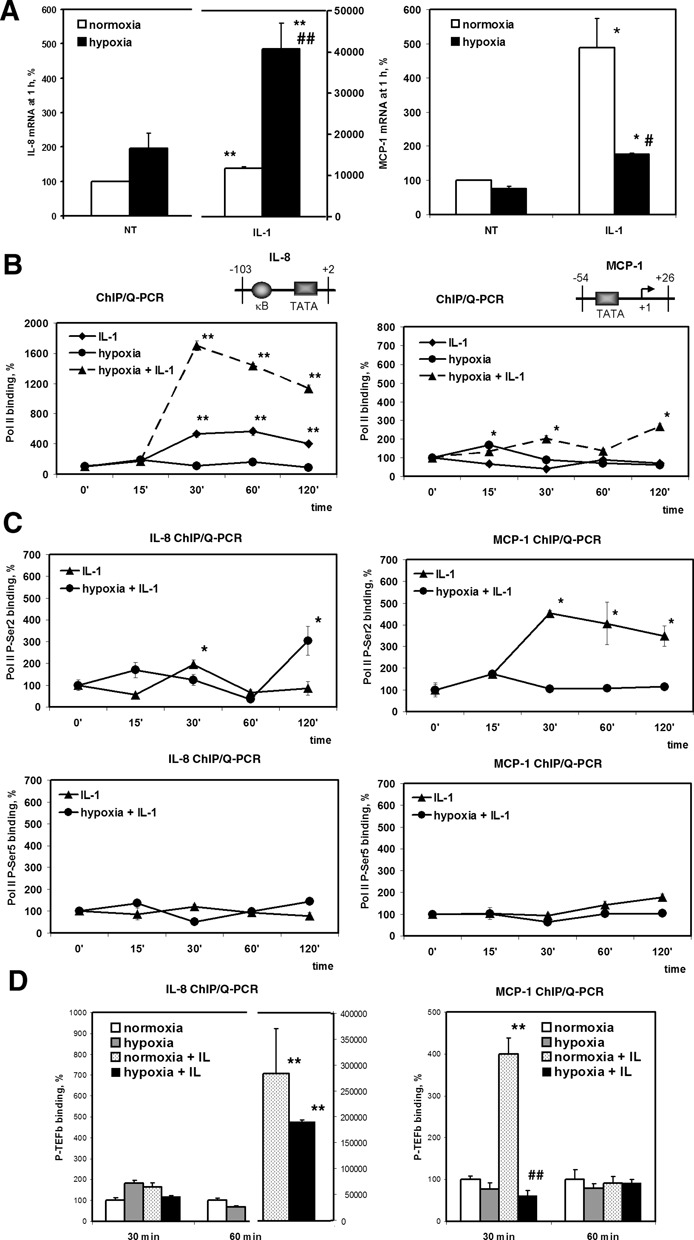
Hypoxia represses MCP-1 mRNA synthesis by inhibiting IL-1β-induced Ser2 phosphorylation of Pol II CTD. (A) Effect of hypoxia on MCP-1 and IL-8 mRNAs expressions. HeLa cells were exposed to normoxia or hypoxia without (NT) or with IL-1β for 1 h. mRNA levels were quantitated by Q-PCR in triplicate and normalized by 18S rRNA levels. Data are expressed as a percentage of expression under normoxia. *P < 0.05; **P < 0.01, as compared with normoxia; #P < 0.05; ##P < 0.01, as compared with IL-1β-treated normoxic samples. (B) Pol II Rbp1 promoter occupancy of the IL-8 and MCP-1 genes analyzed by ChIP assay after treatment with IL-1β and/or hypoxia for the indicated periods of time. (C) The phosphorylation status of Ser5 and Ser2 residues of Pol II CTD set on the MCP-1 and IL-8 gene promoters in the cells treated with IL-1β under normoxia or hypoxia for the indicated time periods. (B-C) Immunoprecipitated DNA was quantified by Q-PCR in triplicate and after normalization by 10% input expressed as a percentage of binding at a 0 min time point. *P < 0.05; **P < 0.01, as compared with binding at 0 min. (D) ChIP assay analysis of P-TEFb enrichment on the MCP-1 and IL-8 promoters. HeLa cells were treated with IL-1β and/or hypoxia for the indicated periods of time. DNA was immunoprecipitated using Cdk9 and Cyclin T1 antibodies and quantified by Q-PCR Data expressed as a percentage of binding in normoxic cells at 30 min or 60 min time point after normalization by 10% input. **P <0.01 as compared with normoxia; ##P < 0.01, as compared with IL-1β-treated normoxic samples.
Pol II is recruited to the promoters in an unphosphorylated state; initiation and productive elongation are accompanied by phosphorylation of CTD on Ser5 and Ser2 residues of heptapeptide repeats (31). In ChIP assays using monoclonal antibodies specific for phospho-serine-5 (P-Ser5) and phospho-serine-2 (P-Ser2) Pol II CTD, we found that IL-1β alone or with simultaneous hypoxia exposure had little effect on the occupancy of the MCP-1 and IL-8 promoters with Pol II phosphorylated at Ser5 (Figure 5C). Treatment with IL-1β alone caused a 4-fold induction in the phosphorylation of Ser2 on Pol II bound to the MCP-1 promoter, whereas simultaneous treatment with hypoxia completely abolished Ser2 phosphorylation (Figure 5C). Therefore, hypoxic repression occurs via inhibition of the elongation competence of Pol II on the MCP-1 gene promoter by selectively reducing the levels of phosphorylation of Ser2 residues on its CTD.
Phosphorylation of Ser2 of the CTD is highly dependent on P-TEFb. To verify that IL-1β induction of the MCP-1 and IL-8 genes is dependent on P-TEFb, we carried out ChIP assays using Cdk9 and Cyclin T1 antibodies. We found that IL-1β enhanced the recruitment of P-TEFb to both MCP-1 and IL-8 genes with different kinetics. P-TEFb was recruited to IL-8 gene at 60 min, but was not observed after 30 min of IL-1β treatment, when Pol II was just recruited to the promoter (Figure 5D). As for MCP-1 gene, P-TEFb was recruited to the transcription start site at 30 min time point, simultaneously with the increase in Ser2 phosphorylation (Figure 5D). Hypoxia alone did not affect P-TEFb binding to MCP-1 and IL-8 promoters at any conditions checked. P-TEFb was also recruited to IL-8 promoter in HeLa cells treated with IL-1β in combination with hypoxia and the amount of P-TEFb present on the IL-8 promoter was not significantly different from that seen under the treatment with IL-1β only. Consistent with observations on MCP-1 mRNA expressions, recruitment of P-TEFb to MCP-1 promoter was abolished under simultaneous treatment with IL-1β and hypoxia (Figure 5D). These data suggest that hypoxia represses IL-1β-induced MCP-1 mRNA expression via the mechanism that involves inhibition of elongation.
Given that HEXIM1 negatively regulates P-TEFb we performed ChIP assays using antibodies against HEXIM1. We observed that HEXIM1 occupancy of the MCP-1 gene transcription start site was ∼50 times higher than that of the distant NF-κB binding site (Supplementary Figure S4A). The amount of HEXIM1 set on MCP-1 transcription start site was not changed in cells treated with IL-1β and/or hypoxia (Supplementary Figure S4B). Although it is possible that hypoxia modulates HEXIM1 occupancy at other regions of the MCP-1, these data suggest that P-TEFb recruitment to the transcription start site of MCP-1 gene is not affected by the amounts of HEXIM1 at the same area. There is a possibility that HEXIM1 post-translational modifications rather than protein occupancy might be involved in the repression of elongation of MCP-1 gene under hypoxia.
Hypoxia represses MCP-1 mRNA synthesis via a HEXIM1-dependent mechanism
The phosphorylation status of Ser2 residues on Pol II CTD is determined by opposing activities of the P-TEFb kinase complex and Fcp-1 phosphatase (32). To examine the role of these factors in the regulation of hypoxia-responsive gene expression, we used double-stranded siRNA oligonucleotides directed against HEXIM-1, the negative regulator of P-TEFb and Fcp-1 (Figure 6A). Knocking down of either HEXIM1 or Fcp-1 barely affected the hypoxic response of IL-1β-induced IL-8 mRNA expression (Figure 6A). Depletion of HEXIM1 by siRNA completely abolished hypoxic inhibition of MCP-1 mRNA expression, while Fcp-1 siRNA had no effect on the hypoxic response of MCP-1 (Figure 6A). Thus, HEXIM1, but not Fcp1, is involved in transcriptional repression of the MCP-1 gene by hypoxia.
Figure 6.
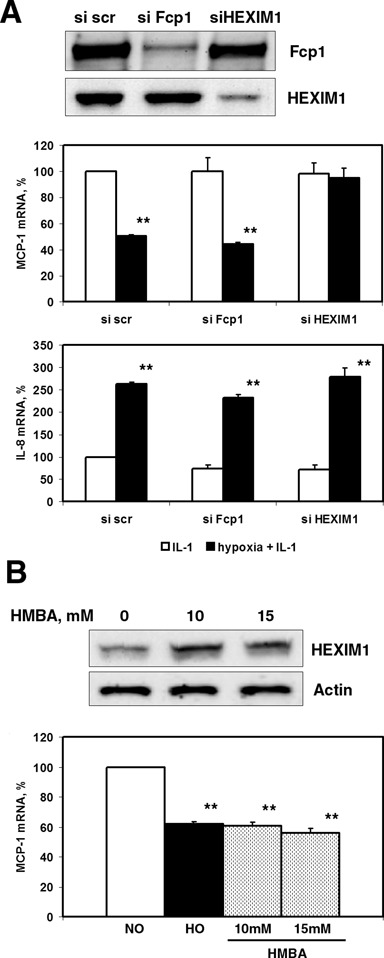
HEXIM1 is required for hypoxia-mediated MCP-1 gene repression. (A) Effect of Fcp-1 and HEXIM1 depletion by siRNA on the responsiveness of MCP-1 and IL-8 mRNAs expressions to hypoxia. Forty-eight hours after transfection with siRNA, cells were treated with IL-1β in combination with normoxia or hypoxia for 6 h. Total RNA was isolated and analyzed by RT-qPCR. Data were normalized by 18S rRNA expressions and presented as a percentage of expression of normoxic samples transfected with scrambled control siRNA. Data are representative of two independent experiments. (B) HMBA treatment mimicked the effect of hypoxia on MCP-1 mRNA expression. HeLa cells were treated with IL-1β alone or in combination with hypoxia or the indicated concentrations of HMBA for 6 h. The levels of MCP-1 mRNA were normalized by 18S rRNA. The expression level in normoxic cells was set as 100%. **P < 0.01, as compared with normoxia.
It was found that HEXIM1 is the only protein that is induced after the administration of HMBA to smooth muscle cells (33). Next, we checked whether the administration of HMBA could mimic hypoxic repression of MCP-1. As shown in Figure 6B, treatment of normoxic HeLa cells with HMBA caused inhibition of MCP-1 mRNA expression, and the level of such inhibition was similar to that observed for hypoxia.
Nuclear receptor co-repressor N-CoR is essential for active repression by hypoxia
Next, we checked the role of HDAC3 co-repressors N-CoR and SMRT in hypoxic gene repression. In cells with disrupted N-CoR expression, hypoxic inhibition of MCP-1 mRNA expression was eliminated, whereas disruption of SMRT had no significant impact on hypoxic response (Figure 7A and Supplementary Figure S5). IL-8 mRNA expression was not significantly affected by SMRT or N-CoR-directed siRNA in any condition examined (Figure 7A). Using a ChIP assay, we examined recruitment of N-CoR and SMRT to the regulatory regions of the MCP-1 and IL-8 genes. Associations of N-CoR with the distal enhancer and with the proximal basic promoter of the MCP-1 gene were observed after 15 min of hypoxia in the presence of IL-1β (Figure 7B). In contrast, we could not detect recruitment of N-CoR to the transcription initiation region of the IL-8 promoter. SMRT was not detected within the MCP-1 proximal basic promoter. A reduced association of SMRT with NF-κB binding sites of both MCP-1 and IL-8 genes was detected after 15 min of hypoxia in the presence of IL-1β and was induced at later times (Figure 7B). Because of SMRT is responsible for basal repression of NF-κB binding (34,35), these observations may reflect oscillation of de-repression and repression of the NF-κB binding sites. Together, these results indicate that MCP-1 gene repression in response to hypoxia was mediated by a HEXIM1-dependent mechanism that involved recruitment of the N-CoR co-repressor. Previously, purification of the N-CoR complexes identified P-TEFb and HEXIM1 as their associated proteins (25).
Figure 7.
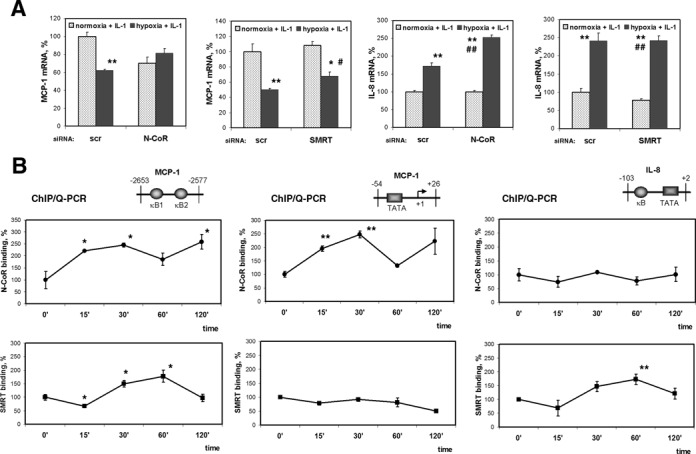
Role of nuclear receptor co-repressors in hypoxic repression of MCP-1 expression. (A) Cells were transfected with 200 pmol of N-CoR or SMRT siRNA 48 h prior to the treatment with IL-1β under normoxia or hypoxia for 6 h. MCP-1 and IL-8 mRNAs were measured by Q-PCR in triplicate. Expression in the cells transfected with control siRNA and treated with IL-1β under normoxia was set as 100%. *P < 0.05; **P < 0.01, as compared with normoxia + IL-1, siScr; #P < 0.05; ##P < 0.01, as compared with normoxia + IL-1, siN-CoR or siSMRT. (B) Relative N-CoR and SMRT binding to the indicated regions of MCP-1 and IL-8 promoters in cells treated with IL-1β and hypoxia for the indicated periods. Immunoprecipitated DNA was quantified by Q-PCR and after normalization by 10% input expressed as a percentage of binding at a 0 min time point. *P < 0.05; **P < 0.01, as compared with binding at 0 min.
Transcriptional profiling of hypoxic cells and identification of Hexim1-dependent genes
To identify primary response genes (PRGs) that were differently expressed (a cutoff of 2-fold changes) within 1 h of hypoxia exposure, we performed global transcriptional profiling using the Agilent Technologies SurePrint G3 Human GE Microarray. Comparison of the transcriptome of normoxic and hypoxic HeLa cells resulted in the identification of 3292 differentially expressed probes. Among them, 1052 (32%) were increased and 2240 (68%) were decreased as a result of 1 h hypoxia exposure. To identify HEXIM1-dependent genes, we compare profiles of hypoxia PRGs in cells transfected with control or Hexim1 targeting double-strand siRNA (Supplementary Figure S6). To our surprise, majority of probes that showed early response to hypoxia (2602 out of total 3292 or 79%) appeared to be HEXIM1-dependent.
We performed functional analysis of all hypoxia-responsive HEXIM1-dependent probes using GOrilla web-based application (36). Among these probes GOrilla system has recognized 1488 identified genes and 1072 of these genes were associated with a gene ontology (GO) term within GOrilla. Functional analysis of terms related to cellular localization revealed that the products of hypoxia-responsive HEXIM1-dependent genes were predominantly associated with the membrane, mitochondrial matrix, macromolecular complexes and extracellular matrix (Supplementary Figure S7 and Table S1). The most significant enrichment of terms relating to molecular function was for catalytic activity (metallopeptidise and ATPase), signal transducer and receptor activity (G-protein coupled receptors), cytokine activity and chemokine receptor binding (Supplementary Figure S8 and Table S1). Functional analysis of biological processes showed that hypoxia-responsive HEXIM1-dependent genes were enriched for terms related to proteolysis and cell surface receptor signalling pathway, regulation of apoptotic process, negative regulation of cell migration, positive regulation of protein phosphorylation and hydrolase activity (Supplementary Figure S9 and Table S1).
Further, using DAVID bioinformatics resources (37) and extensive manual GO study of gene lists, we performed detailed functional annotation clustering of the genes up-regulated and down-regulated by hypoxia separately. Among the 2240 probes that were repressed by hypoxia, the response of 499 probes or 22% was unaffected by HEXIM1 knockdown, 1661 probes or 74% lost their responsiveness to hypoxia as a result of HEXIM1 targeting siRNA transfection (Figure 7A and C). The hypoxic response of the rest 80 probes (4%) was reversed from inhibition to induction in HEXIM1 depleted cells (Figure 8A and C).
Figure 8.
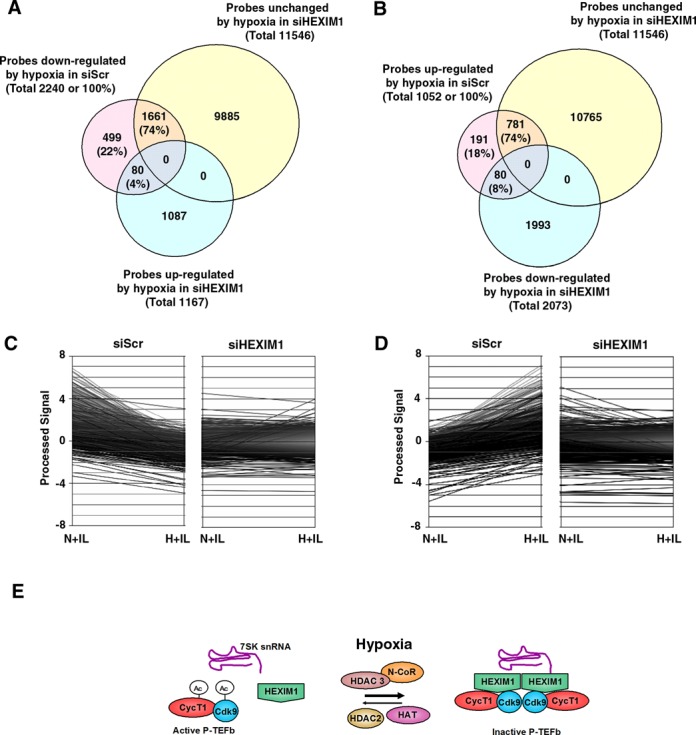
Microarray analysis of primary responses to hypoxia and identification of HEXIM1-dependent genes. HeLa cells were transfected with control (siScr) and HEXIM1-targeted (siHEXIM) double-strand siRNA and 48 h after treated with IL-1β in combination with normoxia (N+IL) or hypoxia (H+IL) for 1 h. (A) Venn diagram of genes down-regulated by hypoxia and their dependence on HEXIM1. (B) Venn diagram of genes up-regulated by hypoxia and their dependence on HEXIM1. (C) Microarray identification of HEXIM1-dependent genes among the probes with decreased expression in hypoxia. (D) Microarray identification of HEXIM1-dependent genes among the probes with increased expression in hypoxia. (E) The model of hypoxic inhibition of P-TEFb. Hypoxia enhances formation of an inactive P-TEFb complex with its endogenous inhibitors Hexim1 and 7SK snRNA. This process requires the HDAC3/N-CoR repressor complex. Inhibition of P-TEFb is mediated by HDAC-dependent deacetylation of P-TEFb subunits Cdk9 and Cyclin T1. HDAC2 is required for active P-TEFb via an unknown mechanism.
Although HEXIM1 is thought to be an inhibitor of P-TEFb and therefore negative regulator of transcription, the large proportion of hypoxia up-regulated probes also showed HEXIM1-dependent activation (Figure 8 B and D). Out of total 1052 probes that were induced by hypoxia, the response of 781 or 74% was unchanged and the response of 80 or 8% was reversed to repression as a result of HEXIM1-specific knockdown. Hypoxic induction of 191 (18%) probes was unaffected in cells transfected with HEXIM1 siRNA.
We categorized HEXIM1-dependent genes into two classes (hypoxia down-regulated and hypoxia up-regulated) and listed selected genes in different functional categories (Supplementary Tables S2 and S3). Among hypoxia down-regulated HEXIM1-dependent probes there were 1034 identified genes, 964 of which were associated with DAVID IDs. The class of hypoxia up-regulated HEXIM-dependent probes contained 486 identified genes. Among them 454 genes were associated with DAVID IDs. Hypoxia largely affected immunity and pro-inflammatory response genes (such as cytokines, interferones and chemokines) in both classes, though the genes with the decreased expression in hypoxia were prevailing. These categories include inflammatory response (9 decreased and 2 increased), T-cell mediated immunity (12 decreased and 2 increased), defence response to bacterium (12 decreased and 2 increased), interleukin and chemokine receptors (6 decreased and 1 increased) (Supplementary Table S3). Notably, both CCL2/MCP-1 chemokine and its CCR2 receptor were repressed by hypoxia. Certain regulators of angiogenesis were also repressed by 1 h hypoxia exposure (Supplementary Table S3). The largest gene functional categories in both primary hypoxia down-regulated and hypoxia up-regulated classes were for signalling molecules and signal transduction as well as for regulators of transcription (Supplementary Table S3), suggesting the importance of transcriptome remodeling in the hypoxia primary adaptive responses and inflammation. Although genes involved in chromatin and histone modifications were presented in both hypoxia down-regulated and hypoxia up-regulated classes, genes associated with histones and DNA packaging (such as nucleosome core histone H2A) exhibited increased expression in response to hypoxia (Supplementary Table S3). The microarray data revealed strong enrichment of genes related to epidermal growth factor (EGF) and proteolysis, with the large number of affected metallopeptidases, among hypoxia down-regulated genes (Supplementary Table S2). Other gene functional groups affected by hypoxia, with the prevalence of hypoxia down-regulated genes, were related to ATPase activity and mitochondrial function (Supplementary Table S3), which suggests changes in energy metabolism.
DISCUSSION
Promoter-proximal pausing is a general feature of transcription by Pol II in mammalian cells and an additional step in which regulation of gene expression can occur (11). In this case, the signal-dependent step is not formation of PIC, but the transition from Pol II initiation to Pol II elongation by the signal-induced recruitment of P-TEFb and subsequent phosphorylation of Ser2 of Pol II CTD. Although, basic molecular characteristics of P-TEFb have been described recently, hypoxic signalling to target transcription elongation have not been studied yet. The important finding of this work is that hypoxia accomplishes repression of elongation by targeting two functional characteristics of P-TEFb: formation of a large inactive complex with its endogenous inhibitors and posttranslational deacetylation of P-TEFb subunits (Figure 8E). We demonstrated that inhibition of Pol II Ser2 phosphorylation in response to hypoxia is gene-dependent and can be observed on the hypoxia-repressed MCP-1 promoter as early as 30 min after treatment initiation. Our results indicate that hypoxia plays a role in pausing Pol II productive elongation rather than inhibition of its recruitment to the repressed MCP-1 promoter.
We presented evidence that the equilibrium of P-TEFb in hypoxic cells was shifted toward the inactive complex with HEXIM1/7SK snRNA. Moreover, a HEXIM1-dependent mechanism was shown for hypoxic inhibition of the MCP-1 gene. Among other factors that are known to perturb the functional equilibrium of P-TEFb are inhibitors of transcription such as DRB and actinomycin D, and the DNA-damaging agent UV (21,22,38). Both convert P-TEFb into the active Brd4-bound form. In cardiomyocytes, activation of hypertrophic signals leads to release of P-TEFb from 7SK snRNA with a subsequent increase in cellular protein and RNA contents (39). It has also been reported that increased HEXIM1 expression inhibits VEGF transcription under hypoxic conditions and leads to a reduction in estrogen-induced HIF-1α protein expression in breast cancer cells (40). However, these effects were shown to be independent of P-TEFb.
In this study, we examined the role of multiple HDACs in the regulation of P-TEFb functional equilibrium and identified that HDAC3 is suitable target for inhibition of P-TEFb in hypoxic signaling. We showed that exposure to hypoxia led to the redistribution and co-localization of N-CoR, HDAC3 and Cdk9 in the nucleus. Moreover, in cells with disrupted N-CoR expression, hypoxic inhibition of the MCP-1 gene was eliminated. Previous report showed that HDAC3/N-CoR interacts with and deacetylates Cdk9 at the K44 residue, which inhibited Cdk9 kinase activity, however these did not affect the association of P-TEFb with Cyclin T1 and HEXIM1 (25). The dissociation of P-TEFb from HEXIM1 and 7SK snRNA was shown to be affected by acetylation of other P-TEFb subunit, cyclin T1 (24). Cardiac lineage protein 1 (CLP-1), the mouse homolog of human HEXIM1, was shown to be associated with HDAC1, HDAC3 and HDAC5 in skeletal muscle and this association was specifically enhanced in the early differentiation phase (41). According to our observations, HDAC2 is likely to be an inhibitor of P-TEFb and HEXIM1 interaction; however, the mechanism behind such an effect requires further investigation. Here, we demonstrated a considerable reduction in intracellular levels of acetylated Cdk9 and acetylated Cyclin T1 in HeLa cells treated with hypoxia. Cdk9 and Cyclin T1 acetylations were suppressed in hypoxic cells by the action of HDAC3. Based on previous reports (24,25), we speculate that deacetylation of Cyclin T1 in hypoxic cells likely contributes to the shift in P-TEFb equilibrium toward its inactive form, while deacetylation of Cdk9 may lead to reduced kinase activity. Indeed, Charles et al. demonstrated that Cdk9/Cyclin T1 heterocomplex formation and Cdk9 kinase activity were markedly reduced in 293T cells cultured at 3% O2 for 18 h (42). In agreement with this report, we demonstrated that reduced association of Cdk9 with Cyclin T1 can already be observed in cells treated with hypoxia for as little as 1 h.
We investigated the effect of hypoxia on global gene expression profile and identified HEXIM1-dependent genes among hypoxia PRGs (changed within 1 h of stimulation). Gene functional analysis revealed crucial differences in gene categories affected by short time hypoxia exposures and prolonged treatments used in previous reports (8,43,44). Exposure to hypoxia for 24 h has profound effects on general cellular metabolism such as glucose utilization, gluconeogenesis, lipid oxidation and lipolysis (43). In our study, we could not find enrichment of these functional categories among hypoxia PRGs. Primary transcriptional hypoxia responses affected rather genes related to signal transduction and regulation of gene expression. Unlike increased expression of inflammation related genes after 24 h of hypoxia exposure (43), primary hypoxia responses prevail in decreased expression of genes related to inflammation and immunity.
The microarray analysis identified ∼79% of genes within the cohort of hypoxia PRGs as being regulated in a HEXIM1-dependent manner. Interestingly, the response of the large number of hypoxia up-regulated genes also found to be HEXIM1-dependent. These changes occurred within 1 h of hypoxia exposure, suggesting that this response is not the result of feedback of any other HEXIM1-dependently expressed upstream factor. HEXIM1-mediated regulation of P-TEFb usually leads to transcriptional pausing and inhibition of elongation. Therefore, we can not exclude that HEXIM1-dependent gene activation in response to hypoxia occurred due to previously unidentified P-TEFb-independent mechanism.
P-TEFb plays a significant role in normal and pathological conditions, such as differentiation of stem cells and development, cardiac hypertrophy, cancer and innate immune responses in inflammation (45). Interestingly, the hypoxic microenvironmental component is present in many of the above mentioned conditions. Regarding the control of inflammatory gene expression, the study of LPS-inducible gene expression in macrophages revealed that PRGs (induced within an hour of stimulation) have preassembled Pol II and positive histone modifications at their promoters in the basal state (9,46). Induction of such genes is controlled at the level of transcriptional elongation through the signal-dependent recruitment of P-TEFb. MCP-1 (CCL2) was identified among PRGs in macrophages. Therefore, regulation of P-TEFb ensures coordinated, timely activation of the inflammatory gene expression program.
Similar to proinflammatory PRGs, it was shown that in human embryonic stem cells (hESC) genes encoding most developmental regulators experience transcription initiation, but show no evidence of expression, suggesting that much of development regulation occurs at the level of transcription elongation (47). Thus, the selective function of P-TEFb is logically required to appropriately activate developmental control genes (10). C-Myc promotes self-renewal and proliferation of ESC through its ability to recruit P-TEFb and efficiently release Pol II from poised promoters at one-third of the active genes (11,48). Numerous reports have described the protective effects of reducing oxygen concentrations on a broad spectrum of stem cells that includes embryonic, hematopoietic, mesenchymal and neural stem cells, cancer stem cells and induced pluripotent stem cells (1). Culture under 5% O2 aids in maintaining pluripotency and suppressing spontaneous differentiation of hESC (49). It is very likely that the critical role of oxygen in stem cell biology is mediated by hypoxic inhibition of P-TEFb-dependent activation of elongation. Such an assumption requires further investigation.
Collectively, our findings support a new aspect of PRG regulation in hypoxic cells via signalling to P-TEFb and productive elongation of preassembled Pol II. These highlight the important physiological significance in oxygen concentrations in signal-dependent transcriptional elongation in a wide range of physiological and pathological conditions, such as development, stem cell maintenance, inflammation and cancer. Hypoxia-mediated repression of transcription elongation may provide rapid control of gene expression in contrast to more energy-consuming gene repression which involves chromatin remodeling.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors thank Warren Dela Cruz and Marketa Schinkmanova for technical help with several of the experiments and Akio Tanabe (Subio Inc.) for the support with the use of Subio Platform software.
Research related to this work was supported by a grant from the Center of Excellence Program, International Research Center for Molecular Science in Tooth and Bone Diseases, at Tokyo Medical and Dental University. This work was also supported by national funding by a Grant-in-Aid for Scientific Research and by the Japan Society for the Promotion of Science (15.52341 and 17.05243).
Conflict of interest statement. None declared.
REFERENCES
- 1.Mohyeldin A., Garzon-Muvdi T., Quinones-Hinojosa A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell. 2010;7:150–161. doi: 10.1016/j.stem.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 2.Safronova O., Morita I. Transcriptome remodeling in hypoxic inflammation. J. Dent. Res. 2010;89:430–444. doi: 10.1177/0022034510366813. [DOI] [PubMed] [Google Scholar]
- 3.Safronova O., Pluemsampant S., Nakahama K., Morita I. Regulation of chemokine gene expression by hypoxia via cooperative activation of NF-kappaB and histone deacetylase. Int. J. Biochem. Cell Biol. 2009;41:2270–2280. doi: 10.1016/j.biocel.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 4.Aoi Y., Nakahama K., Morita I., Safronova O. The involvement of DNA and histone methylation in the repression of IL-1beta-induced MCP-1 production by hypoxia. Biochem. Biophys. Res. Commun. 2011;414:252–258. doi: 10.1016/j.bbrc.2011.09.066. [DOI] [PubMed] [Google Scholar]
- 5.Safronova O., Nakahama K., Onodera M., Muneta T., Morita I. Effect of hypoxia on monocyte chemotactic protein-1 (MCP-1) gene expression induced by Interleukin-1beta in human synovial fibroblasts. Inflamm. Res. 2003;52:480–486. doi: 10.1007/s00011-003-1205-5. [DOI] [PubMed] [Google Scholar]
- 6.Bertout J.A., Patel S.A., Simon M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer. 2008;8:967–975. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casey T.M., Pakay J.L., Guppy M., Arthur P.G. Hypoxia causes downregulation of protein and RNA synthesis in noncontracting Mammalian cardiomyocytes. Circ. Res. 2002;90:777–783. doi: 10.1161/01.res.0000015592.95986.03. [DOI] [PubMed] [Google Scholar]
- 8.Leonard M.O., Cottell D.C., Godson C., Brady H.R., Taylor C.T. The role of HIF-1 alpha in transcriptional regulation of the proximal tubular epithelial cell response to hypoxia. J. Biol. Chem. 2003;278:40296–40304. doi: 10.1074/jbc.M302560200. [DOI] [PubMed] [Google Scholar]
- 9.Hargreaves D.C., Horng T., Medzhitov R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell. 2009;138:129–145. doi: 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Price D.H. Poised polymerases: on your mark…get set…go. Mol. Cell. 2008;30:7–10. doi: 10.1016/j.molcel.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 11.Rahl P.B., Lin C.Y., Seila A.C., Flynn R.A., McCuine S., Burge C.B., Sharp P.A., Young R.A. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prelich G. RNA polymerase II carboxy-terminal domain kinases: emerging clues to their function. Eukaryot Cell. 2002;1:153–162. doi: 10.1128/EC.1.2.153-162.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Komarnitsky P., Cho E.J., Buratowski S. Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 2000;14:2452–2460. doi: 10.1101/gad.824700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schroeder S.C., Schwer B., Shuman S., Bentley D. Dynamic association of capping enzymes with transcribing RNA polymerase II. Genes Dev. 2000;14:2435–2440. doi: 10.1101/gad.836300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marshall N.F., Price D.H. Control of formation of two distinct classes of RNA polymerase II elongation complexes. Mol. Cell. Biol. 1992;12:2078–2090. doi: 10.1128/mcb.12.5.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peterlin B.M., Price D.H. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell. 2006;23:297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 17.Cho E.J., Kobor M.S., Kim M., Greenblatt J., Buratowski S. Opposing effects of Ctk1 kinase and Fcp1 phosphatase at Ser 2 of the RNA polymerase II C-terminal domain. Genes Dev. 2001;15:3319–3329. doi: 10.1101/gad.935901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mandal S.S., Cho H., Kim S., Cabane K., Reinberg D. FCP1, a phosphatase specific for the heptapeptide repeat of the largest subunit of RNA polymerase II, stimulates transcription elongation. Mol. Cell. Biol. 2002;22:7543–7552. doi: 10.1128/MCB.22.21.7543-7552.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nechaev S., Adelman K. Pol II waiting in the starting gates: Regulating the transition from transcription initiation into productive elongation. Biochim Biophys Acta. 2011;1809:34–45. doi: 10.1016/j.bbagrm.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Price D.H. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol. Cell. Biol. 2000;20:2629–2634. doi: 10.1128/mcb.20.8.2629-2634.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nguyen V.T., Kiss T., Michels A.A., Bensaude O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature. 2001;414:322–325. doi: 10.1038/35104581. [DOI] [PubMed] [Google Scholar]
- 22.Yang Z., Zhu Q., Luo K., Zhou Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature. 2001;414:317–322. doi: 10.1038/35104575. [DOI] [PubMed] [Google Scholar]
- 23.Yik J.H., Chen R., Nishimura R., Jennings J.L., Link A.J., Zhou Q. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell. 2003;12:971–982. doi: 10.1016/s1097-2765(03)00388-5. [DOI] [PubMed] [Google Scholar]
- 24.Cho S., Schroeder S., Kaehlcke K., Kwon H.S., Pedal A., Herker E., Schnoelzer M., Ott M. Acetylation of cyclin T1 regulates the equilibrium between active and inactive P-TEFb in cells. EMBO J. 2009;28:1407–1417. doi: 10.1038/emboj.2009.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu J., Yoon H.G., Qin J., Wong J. Regulation of P-TEFb elongation complex activity by CDK9 acetylation. Mol. Cell. Biol. 2007;27:4641–4651. doi: 10.1128/MCB.00857-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sabo A., Lusic M., Cereseto A., Giacca M. Acetylation of conserved lysines in the catalytic core of cyclin-dependent kinase 9 inhibits kinase activity and regulates transcription. Mol. Cell. Biol. 2008;28:2201–2212. doi: 10.1128/MCB.01557-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho S., Schroeder S., Ott M. CYCLINg through transcription: posttranslational modifications of P-TEFb regulate transcription elongation. Cell Cycle. 2010;9:1697–1705. doi: 10.4161/cc.9.9.11346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soderberg O., Gullberg M., Jarvius M., Ridderstrale K., Leuchowius K.J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L.G., et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 29.Pluemsampant S., Safronova O.S., Nakahama K., Morita I. Protein kinase CK2 is a key activator of histone deacetylase in hypoxia-associated tumors. Int. J. Cancer. 2008;122:333–341. doi: 10.1002/ijc.23094. [DOI] [PubMed] [Google Scholar]
- 30.Guenther M.G., Barak O., Lazar M.A. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol. Cell. Biol. 2001;21:6091–6101. doi: 10.1128/MCB.21.18.6091-6101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phatnani H.P., Greenleaf A.L. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 2006;20:2922–2936. doi: 10.1101/gad.1477006. [DOI] [PubMed] [Google Scholar]
- 32.Sims R.J., 3rd, Belotserkovskaya R., Reinberg D. Elongation by RNA polymerase II: the short and long of it. Genes Dev. 2004;18:2437–2468. doi: 10.1101/gad.1235904. [DOI] [PubMed] [Google Scholar]
- 33.Kusuhara M., Nagasaki K., Kimura K., Maass N., Manabe T., Ishikawa S., Aikawa M., Miyazaki K., Yamaguchi K. Cloning of hexamethylene-bis-acetamide-inducible transcript, HEXIM1, in human vascular smooth muscle cells. Biomed. Res-Tokyo. 1999;20:273–279. [Google Scholar]
- 34.Hoberg J.E., Popko A.E., Ramsey C.S., Mayo M.W. IkappaB kinase alpha-mediated derepression of SMRT potentiates acetylation of RelA/p65 by p300. Mol. Cell. Biol. 2006;26:457–471. doi: 10.1128/MCB.26.2.457-471.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoberg J.E., Yeung F., Mayo M.W. SMRT derepression by the IkappaB kinase alpha: a prerequisite to NF-kappaB transcription and survival. Mol. Cell. 2004;16:245–255. doi: 10.1016/j.molcel.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 36.Eden E., Navon R., Steinfeld I., Lipson D., Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 38.Yang Z., Yik J.H., Chen R., He N., Jang M.K., Ozato K., Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 39.Sano M., Abdellatif M., Oh H., Xie M., Bagella L., Giordano A., Michael L.H., DeMayo F.J., Schneider M.D. Activation and function of cyclin T-Cdk9 (positive transcription elongation factor-b) in cardiac muscle-cell hypertrophy. Nat Med. 2002;8:1310–1317. doi: 10.1038/nm778. [DOI] [PubMed] [Google Scholar]
- 40.Ogba N., Doughman Y.Q., Chaplin L.J., Hu Y., Gargesha M., Watanabe M., Montano M.M. HEXIM1 modulates vascular endothelial growth factor expression and function in breast epithelial cells and mammary gland. Oncogene. 2010;29:3639–3649. doi: 10.1038/onc.2010.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galatioto J., Mascareno E., Siddiqui M.A. CLP-1 associates with MyoD and HDAC to restore skeletal muscle cell regeneration. J. Cell Sci. 2010;123:3789–3795. doi: 10.1242/jcs.073387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Charles S., Ammosova T., Cardenas J., Foster A., Rotimi J., Jerebtsova M., Ayodeji A.A., Niu X., Ray P.E., Gordeuk V.R., et al. Regulation of HIV-1 transcription at 3% versus 21% oxygen concentration. J. Cell. Physiol. 2009;221:469–479. doi: 10.1002/jcp.21882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mazzatti D., Lim F.L., O'Hara A., Wood I.S., Trayhurn P. A microarray analysis of the hypoxia-induced modulation of gene expression in human adipocytes. Arch. Physiol. Biochem. 2012;118:112–120. doi: 10.3109/13813455.2012.654611. [DOI] [PubMed] [Google Scholar]
- 44.Kirwan R.P., Felice L., Clark A.F., O'Brien C.J., Leonard M.O. Hypoxia regulated gene transcription in human optic nerve lamina cribrosa cells in culture. Invest. Ophthalmol. Vis. Sci. 2012;53:2243–2255. doi: 10.1167/iovs.11-6729. [DOI] [PubMed] [Google Scholar]
- 45.Kohoutek J. P-TEFb- the final frontier. Cell Div. 2009;4:19. doi: 10.1186/1747-1028-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adelman K., Kennedy M.A., Nechaev S., Gilchrist D.A., Muse G.W., Chinenov Y., Rogatsky I. Immediate mediators of the inflammatory response are poised for gene activation through RNA polymerase II stalling. Proc. Natl Acad. Sci. U.S.A. 2009;106:18207–18212. doi: 10.1073/pnas.0910177106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guenther M.G., Levine S.S., Boyer L.A., Jaenisch R., Young R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mellor J. Transcription: from regulatory ncRNA to incongruent redundancy. Genes Dev. 2010;24:1449–1455. doi: 10.1101/gad.1950310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lengner C.J., Gimelbrant A.A., Erwin J.A., Cheng A.W., Guenther M.G., Welstead G.G., Alagappan R., Frampton G.M., Xu P., Muffat J., et al. Derivation of pre-X inactivation human embryonic stem cells under physiological oxygen concentrations. Cell. 2010;141:872–883. doi: 10.1016/j.cell.2010.04.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.