


Abstract
The multicopy sRNA LhrC of the intracellular pathogen Listeria monocytogenes has been shown to be induced under infection-relevant conditions, but its physiological role and mechanism of action is not understood. In an attempt to pinpoint the exact terms of LhrC expression, cell envelope stress could be defined as a specific inducer of LhrC. In this process, the two-component system LisRK was shown to be indispensable for expression of all five copies of LhrC. lapB mRNA, encoding a cell wall associated protein that was recently identified as an important virulence factor, was disclosed to be directly bound by LhrC leading to an impediment of its translation. Although LhrC binds to Hfq, it does not require the RNA chaperone for stability or lapB mRNA interaction. The mechanism of LhrC-lapB mRNA binding was shown to involve three redundant CU-rich sites and a structural rearrangement in the sRNA. This study represents an extensive depiction of a so far uncharacterized multicopy sRNA and reveals interesting new aspects concerning its regulation, virulence association and mechanism of target binding.
INTRODUCTION
Throughout the last decade it has been recognized that small non-coding RNAs in bacteria are not rare and singular cases, but rather play an important and extensive regulatory role (1,2). These small RNAs (sRNAs) are often involved in the adaptation to changing environmental conditions, and several studies even proved their importance for the virulence of pathogenic bacteria (3–5). A non-coding sRNA can be encoded antisense, i.e. on the opposite DNA strand of its perfect complementary mRNA target, or it may be transcribed from a distant location (trans-encoded sRNAs) and then often regulates more than one target. It can be attributed mainly to novel high-throughput technologies (high-resolution genome tiling arrays and deep sequencing), and improved bioinformatics tools, that virtually catalogs of bacterial sRNAs (RNomes) have been published throughout the last years (5–8). However, the actual challenge starts only now. What are the functions of all these newly identified sRNAs? How exactly do they act on a molecular basis? Are they important for the pathogenicity of a bacterium, and if so, could this be exploited to develop new antimicrobial strategies?
The first sRNAs of the Gram-positive pathogen Listeria monocytogenes were discovered in 2006 (9) and ever since comprehensive studies in the bacterium uncovered not only an enormous number of new non-coding RNAs, but also revealed specific details on their expression under infection-relevant conditions (6,10–12). Because listeriosis represents a severe health danger especially for immunocompromised people and pregnant women and their offspring (13), research on how the pathogen regulates virulence is of high public interest. New knowledge on sRNAs that are likely to be involved in pathogenesis could be a starting point of a deeper understanding in this respect.
While in Gram-negative bacteria the RNA chaperone Hfq contributes to virtually every interaction of a trans-encoded sRNA to its mRNA target (3,14), the situation is different in Gram-positive species. Several detailed studies on sRNAs in the model organism Bacillus subtilis revealed dispensability of Hfq for mRNA interaction, although the RNAs were mostly capable of Hfq binding (15–18), as shown for numerous other sRNAs in this organism (19). Instead, alternative proteins of B. subtilis are assumed to act as RNA chaperones and to facilitate sRNA-mRNA interaction in certain cases (16,20). The function and significance of Hfq for sRNA-mRNA interactions in other Gram-positive bacteria had only rarely been shown (21), while other reports are contradictory as for Staphylococcus aureus (22,23). Some, like Streptococci and Lactobacilli, do not even possess an Hfq homolog (14). However, the significance of the protein in L. monocytogenes during specific stress conditions and for virulence has been demonstrated (24), and as a paradigm the small listerial sRNA LhrA (for Listeria Hfq-binding RNA A) was proven to depend on Hfq in terms of stability (9) and binding to its mRNA targets (25,26). In addition to LhrA, the sRNAs LhrB and LhrC were initially identified via co-immunoprecipitation with Hfq (9). LhrC is conserved among all Listeria species and present in five almost identical copies that vary from 111 to 114 nt in length (Supplementary Figure S1). A putative role of the LhrC sRNAs during listerial infections can be anticipated from later studies where they were reported to be highly expressed in blood (6) and during intracellular growth in macrophages (11).
In this study we provide evidence that the LhrC sRNAs are highly induced in response to cell envelope stress and find that expression of all five lhrC copies strictly depends on the two-component system (TCS) LisRK. Using bioinformatics tools, the lapB gene, encoding a cell-wall anchored virulence adhesin, was predicted as a target for all five LhrCs. Analyses of its regulation show that expression of lapB is targeted by the sRNAs at the post-transcriptional level. Interestingly, interaction of LhrC to lapB mRNA does not follow the predicted binding scheme involving two complementary sequences in sRNA and mRNA. Apparently, the LhrC molecule contains three redundant CU-rich motifs, residing in a stem-loop structure, a single-stranded region and the terminator structure, respectively, which are all capable of target recognition. Although LhrC was originally identified as an Hfq-binding sRNA, we find that it dispenses with Hfq for the regulatory effect on lapB.
MATERIALS AND METHODS
Bacterial strains and growth conditions
The wild type strain used in this study was L. monocytogenes serotype 1/2c strain LO28 (27). Isogenic mutant derivatives of this strain were constructed as described previously (24). Primers used for in-frame deletions are listed in Supplementary Table S1. All strains used in the study are listed in Supplementary Table S2. In order to grow L. monocytogenes strains, overnight cultures were diluted 100-fold into Brain Heart Infusion medium (BHI, Oxoid) and vigorously shaken at 37°C; when appropriate, kanamycin (50 μg/ml) was added. For northern blot experiments, the following concentrations were used to induce cell envelope stress: 4 μg/ml cefuroxime (Sigma-Aldrich), 0.03% bile salts (DIFCO, No. 3), 4% NaCl, pH5 adjusted with HCl and 2% ethanol. In stress tolerance assays overnight cultures were diluted 1000-fold into BHI adjusted with cefuroxime (4 μg/ml), bile salts (0.07%), NaCl (8%), pH5 and ethanol (5%); growth was monitored for 24 h.
For cloning purposes and green fluorescent protein (GFP) reporter experiments, Escherichia coli TOP10 and E. coli DH5α (Invitrogen) were used and grown in Luria Broth (LB, Oxoid) at 37°C. When appropriate, LB was supplemented with kanamycin (50 μg/ml), ampicillin (30 μg/ml), chloramphenicol (25 μg/ml) or erythromycin (150 μg/ml).
RNA techniques
Northern blot analyses
For testing the induction of LhrC in response to cell envelope stress, L. monocytogenes was grown to OD600 = 0.35, cultures were split and one of them stressed. Samples were taken from stressed and control cultures after 30 min. To assimilate an induction profile of LhrC several time points of stress were sampled (10, 20, 30, 60 and 120 min). In order to test RNA stability cells were treated with rifampicin (10 μg/ml) after 30 min of stress and samples collected at indicated time points. Cells were disrupted using the FastPrep instrument (Bio101, Thermo Scientific Corporation) and total RNA was extracted using TRI Reagent (Ambion) and purified as described previously (25). Total RNA from E. coli was prepared by resuspending cell pellets in 150 μl of 10 mM Na-citrate, 10 mM Na-acetate, 2 mM ethylenediaminetetraacetic acid, pH4.5. The solution was mixed with 600 μl acidic phenol and 150 μl of 10 mM Na-acetate pH4.5, 2% sodium dodecyl sulphate (SDS) and incubated at 90°C for 4 min. RNA was subsequently extracted with acidic phenol (pH4.5) and chloroform using Phase Lock Gel Heavy 2.0 ml tubes (5Prime). Supernatants were precipitated with 2.5 volumes of ethanol (96%) and 1/10 volume 3M Na-acetate (pH4.5). RNA integrity was confirmed by agarose gel electrophoresis; concentration and purity determined on a NanoDrop ND-1000 (Saveen Werner). Northern blotting of 10 μg total RNA separated on an 8% denaturing polyacrylamide gel was performed as previously described (25). DNA primers used as probes for detection of LhrC, mutated versions of LhrC, LhrA, 5S RNA of L. monocytogenes and E. coli are listed in Supplementary Table S1. RNA bands were visualized using a Typhoon Trio (GE Healthcare) and analyzed with IQTL 8.0 quantification software (GE Healthcare).
Reverse transcriptase-polymerase chain reaction (RT-qPCR)
Cultures were grown to OD600 = 0.35, split, and one stressed with 4 μg/ml cefuroxime. After 30 and 60 min samples were taken from stressed and control cultures and swiftly pelleted by centrifugation. Pellets were shock-frozen in liquid nitrogen and stored at −80°C. Total RNA was extracted according to (25). Note that 50 μg of RNA was treated with 2 μl RNAsin® (Promega) and DNase-treated according to the manufacturer's instructions (Roche). The Fermentas Maxima First Strand cDNA synthesis kit was used for cDNA synthesis on 3 μg of RNA per sample adding 0.5 μl RNAsin®. The qPCR reactions were performed using primer sets resulting in ∼100 bp long amplicons and SYBR Green PCR Master Mix (Fermentas) on a MX3000 quantitative PCR thermocycler (Stratagene) (initial step at 95°C for 10 min; 40 cycles of 15 s 95°C, 30 s 60°C and 30 s 72°C). Primers were designed using the primer design tool of Eurofins MWG Operon and are listed in Supplementary Table S1. Quantitative RT-PCR data were analyzed using the Relative Expression Software Tool—Multiple Condition Solver (REST©) version 2 (28,29). RNAs of tpi and rpoB served as reference genes. Experiments were carried out in three biological replicates having two technical replicates of each sample. Results were analyzed using two tailed Student's t-test. The differences reported were statistically significant with at least 95% confidence.
Electrophoretic mobility shift assays (EMSA)
Templates for in vitro transcription contained a T7 RNA Polymerase binding site at their 5′ site. In general, LhrC4 and its mutant templates were produced by PCR using overlapping primers. The template for lapB was prepared using primer pair T7_lapB_fw/rev on LO28 chromosomal DNA. Mutations in lapB were introduced by running three PCRs. PCR 1 and 2 were run on LO28 chromosomal DNA resulting in an upstream and downstream fragment of the final template, respectively. The third PCR was carried out on the products of PCR 1 and 2 using primer pair T7_lapB_fw/rev. All primers and details are listed in Supplementary Table S1. In vitro transcription, RNA cleaning, dephosphorylation and labeling was carried out according to (26) with minor deviations: RNA was extracted from gel by simple diffusion into 2 M NH4-acetate without electro elution. Dephosphorylation was achieved using shrimp alkaline phosphatase (Affymetrix Inc., USB) and labelling by polynucleotide kinase (New England Biolabs (NEB)), all in PNK buffer (NEB). Labeled transcripts were purified before gel shift employing Nucleospin miRNA kit (Macherey–Nagel) according to the manufacturer's instructions. Secondary structures of mutated LhrC4 molecules were predicted to resemble the one of wild type according to Mfold (30). The gel shifts were done according to Nielsen et al. (25). Briefly, 5′-end labeled LhrC4 was incubated with an indicated fold excess of non-labeled lapB RNA for 20 min at 37°C and additional 10 min on ice (in the presence of non-specific tRNA). For experiments testing mut_9 and mut_10, RNAs were mixed and incubated 5 min at 70°C, slowly cooled to 37°C and incubated for 40 min at 37°C before putting them on ice. Samples were loaded with current running and separated at 4°C in a 5% non-denaturing polyacrylamide gel.
In vitro structure probing
In vitro synthesized 5′ end-labeled RNAs were prepared in the same way as for EMSA experiments. Alkaline hydrolysis buffer, structure buffer, loading buffer type II, yeast tRNA and RNase T1 were from Ambion T1 RNase kit (AM2283). For an alkaline hydrolysis ladder 0.2 pmol labeled RNA was incubated in the presence of 10 μg yeast tRNA and alkaline hydrolysis buffer at 95°C for 5 min. The reaction was placed on ice and stopped by the addition of loading buffer type II. For RNase T1 sequencing ladders 0.1 pmol of labeled RNA was denatured (95°C, 1 min) in the presence of yeast tRNA (10 μg) and structure buffer. The reaction was placed at 37°C for 1 min, provided with 0.1 U of T1 RNase and incubated for another 5 min at 37°C. Structure probing of RNA-RNA interaction generally started with denaturation of 0.1 pmol labeled RNA (95°C, 1 min) which was afterwards placed on ice. In T1 RNase and lead(II)acetate experiments the labeled RNA was mixed with yeast tRNA (10 μg) and non-labeled RNA in the presence of structure buffer and kept at 37°C for 10 min. To provoke RNA cleavage the mixture was subsequently incubated with 0.1 U T1 RNase and fresh lead(II)acetate (5 mM) (Fluka), respectively. For RNase V1 or RNase A digestion, labeled denatured RNA was incubated in 10× structure buffer (37°C, 10 min) before the addition of yeast tRNA, non-labeled RNA and water. After another 10 min at 37°C, 0.05 U RNAse V1 (Ambion, AM2275) or RNase A (AM2274) was added and samples were incubated for 2 or 5 min, respectively. Details on fold excess of non-labeled RNA, amount of RNase and differing incubation times of cleaving reactions are indicated specifically. Samples were mixed with 2× loading buffer type II, denatured at 95°C for 3 min and placed on ice. A total of 2 μl of each sample was separated on an 8% polyacrylamide gel in 7 M urea at 45 W.
Bioinformatics target prediction
The RNApredator software (31) was used to predict putative targets of LhrC1–5. All five LhrC sequences were searched against the L. monocytogenes EGD-e chromosome. The top five hits of each of the five result lists were chosen and merged into one final table comprising 13 putative targets (Supplementary Table S3).
lacZ-fusions and β-galactosidase assays
For testing LhrC promoter activity, DNA fragments incorporating each of the five promoters were synthesized by PCR on LO28 chromosomal DNA. Resulting fragments were digested with EcoRI and BamHI and ligated as transcriptional fusions into pTCV-lac (32). The lhrA core promoter, as well as upstream regions of lapB and lmo1669, were also ligated into pTCV-lac, respectively. For an in-frame translational lacZ fusion pCK-lac was used (25). A fusion of the moderate core promoter of lhrA and the upstream region of lapB was digested with EcoRI and BamHI and ligated into the vector (resulting construct called pC-lapB-lacZ). All primers and details for transcriptional and translational fusions are listed in Supplementary Table S1. Beta-galactosidase assays were carried out as previously described (24). Negative β-galactosidase units were put on reference level (0 units).
GFP reporter experiments
The RNA sequences notionally interacting were expressed in E. coli from two compatible plasmids, pXG-10 and pNDM220, as described in detail in (33). The lapB sequence was generated by PCR using primer pairs lapB_pXG10_fw/rev on chromosomal DNA of LO28, and translationally fused under constitutive expression to a gfp reporter gene into pXG-10 (34). LhrC4 expression was Isopropyl β-D-1-thiogalactopyranoside (IPTG) inducible from pNDM220 (35). The PCR fragment of LhrC4 was made by using primers lhrC4_pNDM220_fw/rev on chromosomal DNA. The mutated lapB and lhrC4 sequences were generated in three PCRs. Details and primers are given in Supplementary Table S1. Overnight cultures of E. coli strains carrying both plasmids were inoculated to OD450 = 0.006 into fresh LB also containing ampicillin and chloramphenicol, and in case of LhrC4 induction 1 mM IPTG. Cells were grown to OD450 = 0.4 and harvested for western blot and northern blot analyses. One-dimensional SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequent western blot analysis were performed as described previously (33). The GFP (Roche) and GroEL (Sigma) monoclonal antibodies were diluted 1:10 000 and 1:50 000, respectively. Secondary mouse and rabbit horseradish peroxidase-conjugated antibodies (DakoCytomation) were diluted 1:2000. Western blots were developed using western lightning reagent (PerkinElmer) and exposed to a film (GE Healthcare). GFP signal intensity was estimated with the help of ImageJ software (36) and normalized by the corresponding GroEL signal intensities.
LapB protein detection
LO28 wild type and ΔlhrC1–5 were grown up to OD600 = 0.35, cultures split and one of each stressed with 4 μg/ml cefuroxime. After 2 h the four cultures were harvested and cell pellets washed twice in ice-cold 20 mM HEPES buffer (pH7.5). Cells were lysed in HEPES buffer containing complete protease inhibitor (Roche) using a FastPrep instrument (2 cycles of 40 s, speed 6 m/s with intermediate cooling). Extracts were centrifuged (5 min, 10 000 × g) and supernatants ultracentrifuged at 100 000 × g for 1 h. Pellets comprising the membrane protein fraction were resuspended in HEPES buffer with the help of a hand homogenizer (VWR) and protein concentration determined via a BCA assay (PIERCE) according to the manufacturer's instructions. Western blot analysis was performed as described earlier (33) with the following modifications: 60 μg of proteins were separated via 1-D SDS-PAGE which was run at 100 V for 3 h to ensure the large LapB protein (185 kDa) migrated into the gel. Proteins on the polyvinylidene difluoride (PVDF) membrane were stained with amido black staining solution (1% acetic acid, 10% methanol, 0.1% (w/v) Naphthol Blue Black (Santa Cruz Biotechnology, Inc.) which served as a loading control. After destaining, the membrane was incubated with polyclonal α-LapB antibody (1:3000) generously provided by Didier Cabanes (37). Rabbit horseradish peroxidase-conjugated secondary antibody was diluted 1:2000 (DakoCytomation). The relative amount of LapB was estimated by using ImageJ (36). LapB signals were normalized by means of a representative band of the amido black staining.
RESULTS
The role of LhrC1–5 during cell envelope stress
Induction of the five LhrC sRNAs (LhrC1–5) was recently reported for L. monocytogenes residing in human blood (6) as well as inside macrophages (11). In order to determine the exact physical conditions leading to LhrC1–5 expression, L. monocytogenes was exposed to subinhibitory concentrations of numerous stresses, and LhrC1–5 levels determined via northern blot analysis. The sRNAs were found to be induced in LO28 wild type by a whole range of stress agents affecting the integrity of the cell envelope, including the β-lactam antibiotic cefuroxime, bile salts, high osmolarity, low pH and ethanol (Figure 1A, Supplementary Figure S2A). Due to their virtual sequence identity, the probe used for northern blotting binds to all five LhrC copies. An LhrC signal was still detected in a mutant lacking lhrC5 as well as in one lacking the lhrC1–4 locus meaning that both loci were expressed during cell envelope stress (Figure 1A). The signal was lost after knockout of all five lhrC copies (Supplementary Figure S2B) proving the specificity of the northern blot probe and a full lhrC mutant to be actually on hand.
Figure 1.
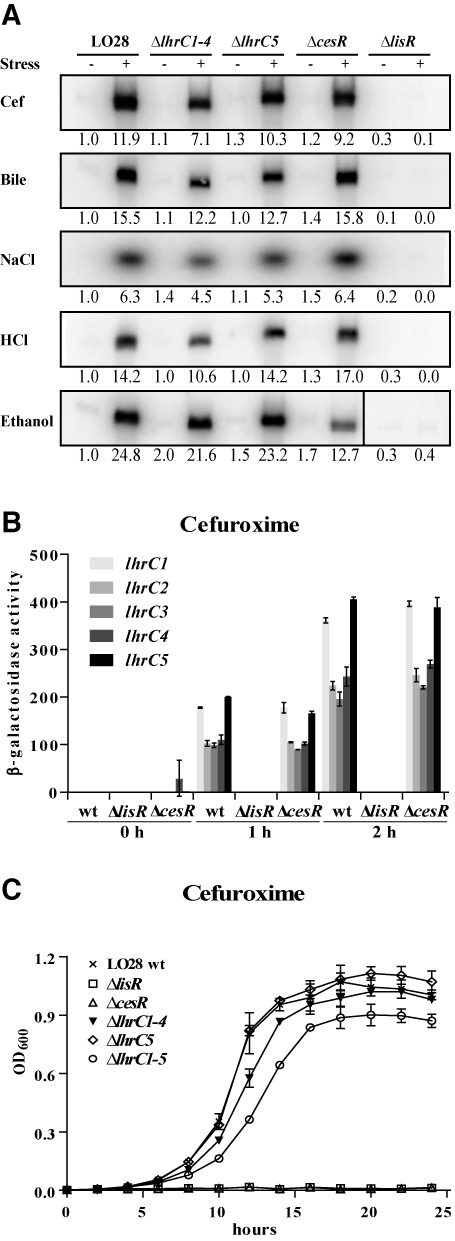
The role of LhrC1–5 during cell envelope stress. (A) Northern blot analyses of LhrC expression. Samples were taken from L. monocytogenes LO28 wild type (lane 1, 2), ΔlhrC1–4 (3,4), ΔlhrC5 (5,6), ΔcesR (7,8) and ΔlisR (9,10) from cultures stressed with various agents acting on the cell envelope (+) as well as from control samples without stress (−). Northern blots were probed for LhrC1–5 and 5S rRNA as a loading control (Supplementary Figure S2A). Relative levels of LhrC (normalized to 5S) are shown below each lane. (B) Transcriptional reporter gene fusions of lhrC promoters. Promoter regions of all five lhrC copies were each cloned into vector pTCV-lac and plasmids transformed into LO28 wild type, ΔlisR and ΔcesR. The resulting 15 strains were grown up to OD600 = 0.35 and stressed with cefuroxime (4 μg/ml), after control samples had been taken (0 h). Further samples for a following β-gal assay were withdrawn after 1 and 2 h of ongoing stress. Results are the average of two biologically independent experiments, each carried out in technical duplicates. (C) Stress tolerance assay. Wild type, ΔlhrC1–4, ΔlhrC5, ΔlhrC1–5, ΔlisR and ΔcesR were grown in BHI containing 4 μg/ml cefuroxime. Bacterial growth was monitored for 24 h. The average of three independent biological replicates is shown.
The promoter activity of the single lhrC copies was determined using transcriptional fusions of each of the five lhrC promoters to the reporter gene lacZ in vector pTCV-lac (32). Prior to the addition of cefuroxime, no β-galactosidase (β-gal) activity was detected in wild type cells harboring any of the five lhrC-lacZ fusion plasmids, i.e. all five lhrC promoters were inactive or transcription was beneath the detection limit of the assay (Figure 1B). The activity of all five promoters increased dramatically when cell surface integrity was disturbed by cefuroxime with promoters lhrC1 and lhrC5 being most strongly induced, relative to the pre-stressed condition (Figure 1B). While the extent of induction was different, the induction pattern among the five promoters was the same when exposing the cells to ethanol or bile salts stress (Supplementary Figure S3).
In order to determine the importance of LhrC1–5 for stress tolerance, growth of the wild type and mutant strains ΔlhrC1–5, ΔlhrC1–4 and ΔlhrC5 was assessed under stress conditions known to affect the integrity of the cell envelope (cefuroxime, ethanol, bile salts, low pH, high osmolarity). Previous studies have shown that L. monocytogenes defective in the TCSs LisRK or CesRK shows growth defects in the presence of several such stressors (38–42). As a control, mutants lacking the response regulators LisR or CesR were carried along in the stress tolerance assays. No difference in growth could be observed between wild type and the three lhrC mutant strains when exposed to acid, ethanol or osmotic stress conditions (Supplementary Figure S4). However, in the presence of 4 μg/ml cefuroxime the ΔlhrC1–5 mutant strain exhibited a defect in growth compared to wild type (Figure 1C). Interestingly, also ΔlhrC1–4 was hampered, yet not to the same extent as the full mutant. As expected, mutants lacking the response regulators LisR or CesR were not able to grow in 4 μg/ml cefuroxime (Figure 1C). A similar tendency was observed when cells were exposed to 0.07% bile salts (Supplementary Figure S4).
The TCS LisRK is mandatory for LhrC expression
Because LhrC proved to be strongly induced by a series of cell envelope acting agents, and a knockout of lhrC1–5 resulted in phenotypes that to some extent resembled the ones observed for mutants in the TCSs LisRK and CesRK, we considered the possibility that the sRNAs LhrC1–5 are controlled by at least one of these TCSs. Therefore, mutants lacking the response regulators LisR and CesR, respectively, were investigated for the presence of LhrC1-5 during cell envelope stress in northern blot analyses (Figure 1A). In a ΔlisR mutant background, no LhrC1–5 could be detected at any of the stress conditions tested, meaning that a functional LisRK TCS is mandatory for LhrC expression. The picture of ΔcesR resembled the one of wild type with the exception of ethanol stress. Here, the LhrC signal was weaker in ΔcesR compared to wild type indicating an involvement of the CesRK TCS in LhrC1–5 expression for this specific stress condition. However, LisRK was still essential for LhrC1–5 expression during ethanol stress (Figure 1A). Further, activity of the five lhrC promoters was tested in a ΔlisR and ΔcesR background using reporter gene fusions. In a ΔlisR background, all five lhrC promoters were inactive for the tested conditions (cefuroxime, bile salts and ethanol) (Figure 1B, Supplementary Figure S3). The ΔcesR strain resembled wild type in the presence of cefuroxime and bile salts, but stressed with ethanol, ΔcesR cells expressed a reduced level of all five LhrC copies, confirming the observation made on northern blots. In conclusion, the TCS LisRK is mandatory for transcription of all five LhrC copies, whereas CesRK is also involved in LhrC1–5 induction during ethanol stress.
Many sRNAs have been reported to be regulated by TCSs in Gram-negative bacteria (43), and also for some Gram-positive bacteria with a low GC-content, an interconnection of TCSs and sRNAs has been described (44–46). LhrC represents the first sRNA in L. monocytogenes described to be regulated by a TCS. Despite intensive research on the LisRK system (38,40,42), no consensus sequence for LisR binding in promoter regions of LisRK-regulated genes has been identified yet. In order to pinpoint the regions in the five lhrC promoters that are important for LisR-dependent induction, promoter sequences of the five lhrC copies were aligned using ClustalW 2.0 (47), and conserved nucleotides, which might constitute a LisR-responsive element, were determined (Figure 2A). In a next step, regions of the lhrC5 promoter were successively deleted to narrow down the LisR-responsive element. The cropped versions of the lhrC5 promoter were analyzed in transcriptional fusion experiments as depicted in Figure 2B. Results revealed the promoter region upstream of -53 from the transcriptional start site to be important for LhrC induction. In the following, conserved nucleotides upstream of −53 were substituted and the ability of the mutated promoters to be induced tested via transcriptional fusions (Figure 2C). Substitution of nucleotides −57 to −59 almost entirely abolished induction of lhrC5-lacZ, but also a single substitution of position −55 and a concurrent change in sites −72, −74 and −75 had a negative effect on induction. These residues most likely reside within the LisR-responsive element important for induction of LhrC1–5 in response to cell envelope stress.
Figure 2.
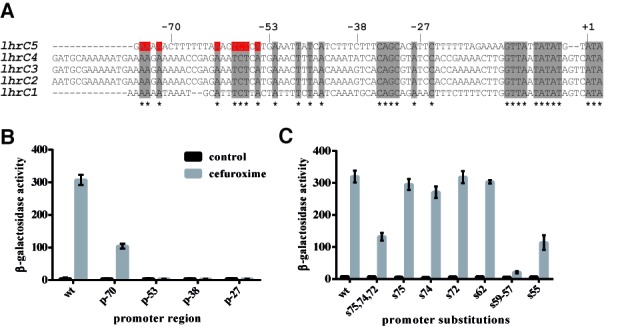
The promoter region of lhrC5 contains a LisR-responsive element. (A) Alignment of the five lhrC promoters. The promoter sequences were aligned using ClustalW 2.0 (47). Conserved nucleotides are marked in gray. Conserved nucleotides substituted in mutant derivatives of the lhrC5 promoter are shown in red. Numbers refer to lhrC5; +1 corresponds to the transcriptional start site. (B) Transcriptional reporter gene fusions of truncated lhrC5 promoter. Promoter regions from −70, −55, −38 and −28 to + 87 relative to the lhrC5 transcriptional start site were fused to a promoter-less lacZ gene in pTCV-lac. The complete intergenic region between the upstream gene (lmo0946) and lhrC5 was used as a positive control (wt). Samples for β-gal assays were taken from cultures stressed with cefuroxime for 1 h and from corresponding control samples. The presented β-gal activities are the average of three independent experiments each conducted in duplicates. (C) Transcriptional reporter gene fusions of lhrC5 promoter mutants. The lhrC5 promoter was substituted at indicated positions and such mutated promoters tested as described in (B).
The lapB mRNA is target of LhrC
A bioinformatics approach was pursued to search for putative direct RNA-targets of LhrC1–5 using the software RNApredator (31). All five copies of LhrC were included in this search. A list of targets containing the five top hits for each LhrC copy was built resulting in a number of 13 putative targets in total (Supplementary Table S3). Assuming that action of LhrC1–5 on an mRNA target will change the abundance of the latter one, real-time RT-qPCR was employed to survey for changed mRNA levels of the 13 putative targets in ΔlhrC1–5 after cefuroxime stress. For this purpose cultures of wild type and ΔlhrC1–5 cells were grown up to OD600 = 0.35, split, and one of them stressed with cefuroxime. Samples for RNA purification were taken from unstressed and stressed cultures 30 min and 1 h after onset of stress. These time points were chosen in conclusion from a time course experiment disclosing the strongest LhrC1–5 induction 1 h after cefuroxime had been added (Supplementary Figure S5). Detailed RT-qPCR results of two biological replicates on all 13 targets are provided in Supplementary Table S4. A third biological replicate was performed on four of the targets: lapB (formerly known as lmo1666), lmo1041, lmo1993 and actA (Figure 3A, Supplementary Figure S6A). Figure 3A shows the ratio of mutant and wild type for the target with the most significant difference between control conditions (LhrC1–5 absence) and cefuroxime stress (LhrC1–5 presence): The lapB mRNA encoding a cell wall protein recently identified as a virulence determinant in L. monocytogenes (37). lapB mRNA was approximately three times as abundant in the mutant after 1 h of cefuroxime stress, but not under control conditions. Notably, of the initially predicted 13 targets, lapB was the only one ranked in top 5 of all five LhrC copies (Supplementary Table S3).
Figure 3.
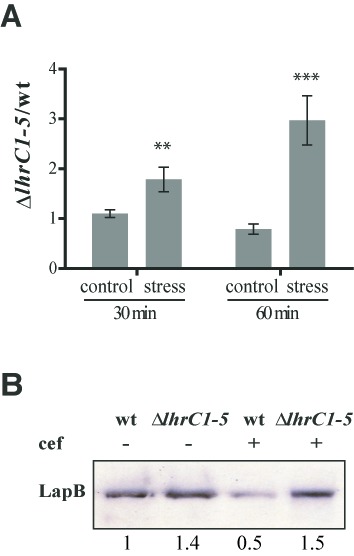
Expression of lapB is controlled by LhrC1–5. (A) LhrC-mediated down-regulation of lapB mRNA. lapB mRNA was quantified in ΔlhrC1–5 relative to wild type (wt) by RT-qPCR. The ratio of ΔlhrC1–5/wt was determined at the time points 30 and 60 min for both non-stressed (control) and cefuroxime-stressed samples. The result shown is the average of three independent experiments each conducted in duplicate. Two and three asterisks indicate a significant increase of the ratio under stress conditions compared to the corresponding control with P < 0.005 and P < 0.001, respectively. (B) LhrC-mediated down-regulation of LapB protein. For western blot analysis of LapB protein, samples were taken 2 h post-treatment with cefuroxime (4 μg/ml) from wild type (wt) and ΔlhrC1–5, but also from unstressed cultures. Surface protein enriched extracts were separated via 1D-PAGE, and LapB protein was detected using an α-LapB antibody. As a loading control, all proteins on the membrane were stained with amido black (Supplementary Figure S6B). Relative levels of LapB (normalized to control) are shown below each lane.
As an implication of the results obtained from RT-qPCR experiments, one would assume reduced amounts of LapB protein in times of high LhrC1–5 expression. In order to evaluate the effect of LhrC1–5 on LapB protein level, a western blotting experiment was performed. Surface proteins of L. monocytogenes were prepared from wild type and ΔlhrC1–5, both from cultures stressed with cefuroxime and from unstressed samples. Cefuroxime stress was exerted on the cells for 2 h justified by the peak expression of LhrC1–5 not before 1 h of stress (Supplementary Figure S5). Whereas LapB levels in unstressed samples were comparable between the wild type and ΔlhrC1–5, there was a clear down-regulation of the protein in the wild type strain in response to cefuroxime stress but not in the LhrC mutant (Figure 3B, Supplementary Figure S6B). Thus, induction of LhrC during cell envelope stress indeed has a verifiable impact on LapB quantity.
Direct interaction of LhrC4 and lapB mRNA
A reporter gene fusion strategy was employed to provide evidence for a post-transcriptional effect of LhrC1–5 on lapB. The RNApredator software predicted an interaction of a single stranded stretch of LhrC1–5 and the ribosome binding site of lapB mRNA (Figure 4A). The gene encoding LapB is the last in an operon of four genes (lmo1669, lmo1668, lmo1667 and lmo1666 (lapB)) according to the L. monocytogenes operon structure (6). In addition, Reis et al. reported PrfA-dependent lapB transcription from a promoter directly upstream of lapB (37). However, neither a fusion of the 400 bp region upstream of lapB to a promoter-less lacZ, nor a fusion of the upstream region of lmo1669 (300 bp) to lacZ, yielded any activity in β-gal assays in our hands (Supplementary Table S5). Notably, lapB mRNA could be detected in RT-qPCR experiments, so the β-gal assay was assumed to be not sensitive enough to report transcription of lapB from its natural promoters at the tested conditions. To still make use of a reporter gene fusion strategy, the intergenic lmo1667-lapB sequence (100 bp) and additional 32 bp of lapB's coding region were fused downstream to a moderate promoter, and in-frame to lacZ in the translational reporter vector pCK-lac (26) (Supplementary Figure S7). Comparison of this construct (pC-lapB-lacZ) in wild type and ΔlhrC1–5 cells showed increased β-gal activity in the mutant after cefuroxime stress (more than 3-fold), but not under non-stress conditions (Figure 4B and C). This clearly indicates that the region −100 to +32 of lapB includes a sequence mediating decreased expression of lapB after cefuroxime stress, but only when LhrC1–5 is present. A difference in transcription of the moderate promoter in wild type and ΔlhrC1–5 was excluded by carrying along its transcriptional fusion to lacZ (Supplementary Figure S8). The ΔlhrC1–4 and ΔlhrC5 cells were also transformed with pC-lapB-lacZ, and β-gal activity measured under control conditions and after cefuroxime stress (Supplementary Figure S9). None of the mutants showed an increase in translation as observed for ΔlhrC1–5, but in both mutants β-gal activity increased compared to the wild type after cefuroxime stress, only slightly in ΔlhrC5, but clearly in ΔlhrC1–4.
Figure 4.
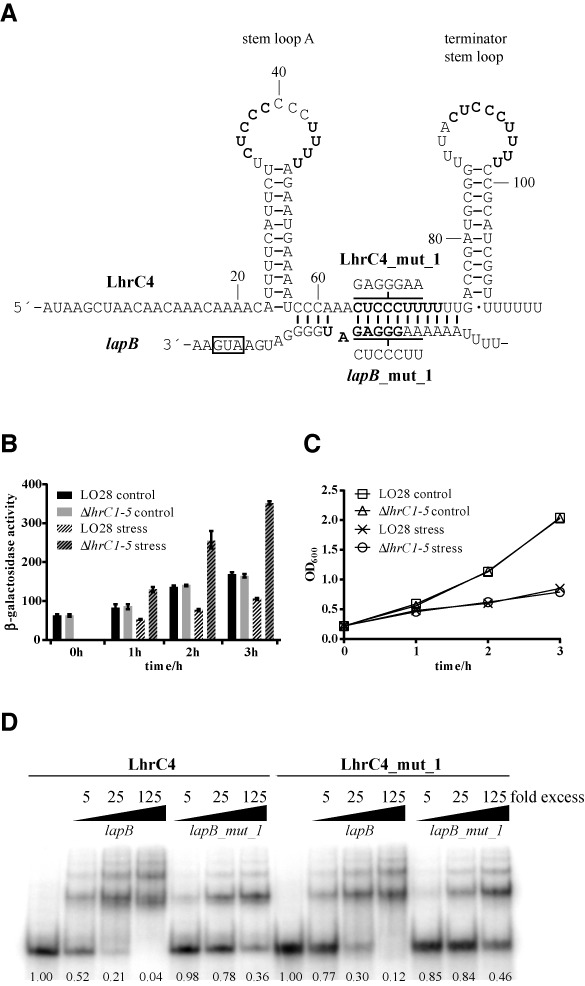
Predicted LhrC and lapB mRNA interaction. (A) According to the RNApredator software the single-stranded stretch between the two stem loops of LhrC binds to lapB mRNA thereby blocking the Shine–Dalgarno sequence (shown in bold). LhrC4 is shown as a representative of the five LhrC copies. Three similar sequences CUCCC(…)UUUU in loop A, the single-stranded stretch and the terminator loop, repectively, are marked in bold. The sequences mutated in LhrC4_mut_1 and lapB_mut_1 are shown. Lines above and below LhrC4 and lapB, respectively, indicate the regions mutated in the mut_1 variants. (B) LhrC-mediated down-regulation of lapB-lacZ expression. β-gal activities were assessed in wild type and ΔlhrC1–5 strains carrying pC-lapB-lacZ. Under non-stress conditions β-gal activity of wild type and mutant was comparable at all tested time points. After cefuroxime stress, β-gal activity in the wild type strain decreased whereas in ΔlhrC1–5 an increase was observed. After 2 and 3 h of stress, β-gal activity in the mutant was more than three times as high as in the wild type, pointing to a released negative regulation in ΔlhrC1–5. Results of the β-gal assay are the average of four biological replicates each conducted in technical duplicates. (C) Growth of ΔlhrC1–5 and wild type strains, tested in the β-gal assay shown in Figure 4(B), was comparable at all tested time points. (D) Gel mobility shift assay of the interaction between lapB mRNA and LhrC4. Labeled LhrC4 was shifted with increasing concentrations of lapB mRNA. Mutation of 7 nt in the lapB sequence predicted to be involved in the interaction (lapB_mut_1) reduced the interaction. Correspondingly mutated LhrC4 (LhrC4_mut_1) was still capable of lapB mRNA binding and could not compensate for the mutation in lapB_mut_1. The fraction of unbound LhrC is shown below each lane.
To further substantiate the predicted direct interaction of lapB mRNA and LhrC, without the involvement of any other factors in Listeria, both RNAs were expressed from two compatible plasmids in E. coli (33). The lapB sequence (from −100 to +32 relative to translational start) was expressed from a constitutive promoter in pXG-10, a vector featuring GFP as reporter (34). Expression of the sRNA was induced by IPTG from plasmid pNDM220 (35). Of the five existing LhrC copies, LhrC4 was predicted to bind lapB mRNA with the highest energy of interaction (−18.48 kJ/mol) according to RNApredator, and was therefore chosen for cloning into pNDM220. Induction of LhrC4 led to a reduction of the GFP signal on a western blot (Supplementary Figure S10A, lane 4) indicating an interaction of the two RNAs in this E. coli reporter system. In order to ensure that reduction of the GFP signal indeed resulted from binding of LhrC4 to lapB mRNA, the lapB sequence was mutated to disrupt interaction with LhrC4 (Supplementary Figure S10B). The mutation reduced the negative effect LhrC4 had on GFP expression (Supplementary Figure S10B, lane 5). Curiously, the interaction of the two RNAs could not be disrupted by expressing wild type lapB and correspondingly mutated LhrC4 (Supplementary Figure S10B, lane 6). A similar expression level of LhrC4 and mutated LhrC4 in E. coli was assured by detecting both versions of the sRNA via northern blot analysis (Supplementary Figure S10B). A gel mobility shift experiment was consulted as another independent method to clarify the interesting discrepancy that relieving the proposed RNA-RNA interaction worked only unidirectionally. The gel mobility shift (Figure 4D) validated this observation leading to the conclusion that lapB was indeed mutated in the sequence interacting with LhrC4, but that another region than the predicted one of LhrC4 must be involved in lapB mRNA binding.
In vitro binding experiments were also employed to test whether Hfq presence could stimulate the association of the two RNAs (Supplementary Figure S11). These gel shift experiments demonstrated the ability of LhrC4 to bind Hfq, but with an increasing concentration of lapB mRNA, the presence of Hfq had no effect on sRNA-mRNA complex formation. Moreover, the translational fusion vector pC-lapB-lacZ in a Δhfq background gave rise to a β-gal activity equivalent to wild type (Supplementary Figure S12). Finally, LhrC did not require Hfq in terms of stability in strain LO28 (Supplementary Figure S13). Collectively, these results demonstrated that Hfq is dispensable for the regulatory action of LhrC on lapB.
The multiple faces of LhrC
A closer inspection of the sequence of LhrC4 revealed similarity of the sequences of the two loops and the proposed site of interaction in the single-stranded stretch between the two stem loops (Figure 4A). The sequence 5’-CUCCC and a series of four Us can be found at all three sites in the sRNA, holding perfect complementarity with lapB. As loops have been extensively described as being best-suited for an initial interaction of two RNAs (48–50), we took both loops (loop A and terminator loop) into consideration to be the actual site of interaction with lapB. Several different LhrC4 mutants were screened for their ability to bind lapB mRNA in gel mobility shift experiments (Figure 5). The sequences of these LhrC mutants are shown in Supplementary Figure S14. Mutation of the single-stranded stretch (LhrC4_mut_2) did not reduce any of the interaction ability of the two RNAs. In contrast, mutation of the terminator loop (LhrC4_mut_3) or loop A (LhrC4_mut_4) indeed had a negative effect on LhrC4-lapB mRNA binding. Mutating both the terminator loop and the single-stranded stretch (LhrC4_mut_5) disturbed sRNA-mRNA interaction even more than the sole mutations of the terminator loop (LhrC4_mut_3) and the single-stranded stretch (LhrC4_mut_2). Mutating both loop A and the single-stranded stretch (LhrC4_mut_6) resulted in a binding resembling that of the sole loop A mutant to lapB mRNA (LhrC4_mut_4). When both loops of LhrC4 were mutated in combination (LhrC4_mut_8) there was almost no detectable binding to lapB mRNA. Eventually, the concerted mutation of all three homologous sites in LhrC4 (LhrC4_mut_7) entirely abolished interaction with the target RNA. In conclusion, both loops of LhrC4 as well as the single-stranded stretch are capable of mediating binding to lapB mRNA, however, the capability of the latter seems to be much smaller compared to the loops.
Figure 5.
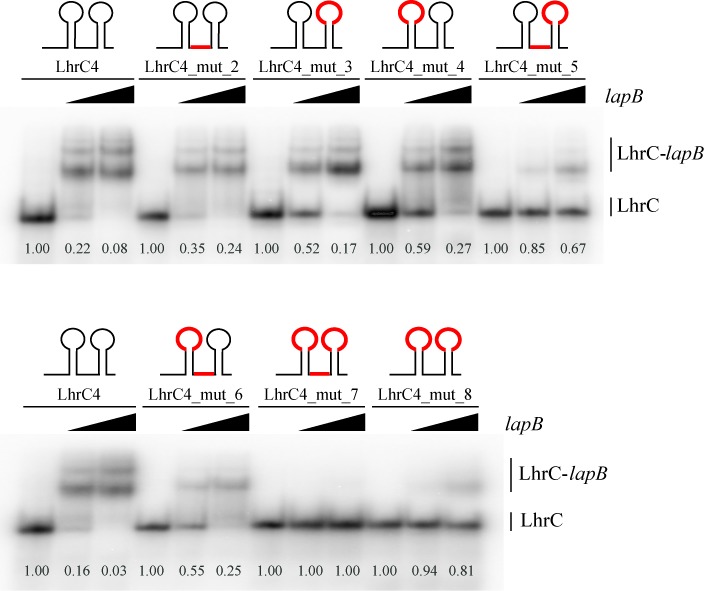
LhrC4 mutant screening of loop A, single-stranded stretch, and the terminator loop. Labeled LhrC4 and several mutant derivatives were tested for their ability to interact with lapB RNA (25 and 125 times excess). In the LhrC sketches, mutated regions are shown in red. LhrC4: wild type LhrC4. LhrC4_mut_2: mutation in single-stranded stretch. LhrC4_mut_3: mutation in terminator loop. LhrC4_mut_4: mutation in loop A. LhrC4_mut_5: mutation in single-stranded stretch and terminator loop. LhrC4_mut_6: mutation in single-stranded stretch and loop A. LhrC4_mut_7: mutation in single-stranded stretch, loop A and terminator loop. LhrC4_mut_8: mutation in loop A and terminator loop. The sequences of the LhrC mutants are shown in Supplementary Figure S14. The fraction of unbound LhrC is shown below each lane.
In order to further explore the binding of LhrC4 to lapB mRNA, structural probing was employed on 5′ labeled lapB mRNA as well as on 5′ labeled LhrC4. RNA cleavage of lapB mRNA was provoked by RNase T1 at single-stranded G residues or lead(II) acetate treatment at single-stranded sequences in general. The lapB mRNA sequence originally predicted to be involved in LhrC4 binding (−21 to −7 relative to translational start) could be confirmed by RNA probing, since it was partially protected from cleavage in the presence of LhrC4 (Figure 6A; from −21 to −11). But surprisingly, the interaction was not limited to this site. Protection from cleavage stretched out down to position +7, into the coding region of lapB (Figure 6A; from −8 to +7).
Figure 6.

(A), (B) and (C): Structure probing of LhrC4 and lapB mRNA interaction. (A) 5′ labeled lapB mRNA was partially digested with RNase T1 (5 min) or cleaved by lead(II) (2 min), either in the absence (−) or in the presence of 25- or 125-fold excess of LhrC4. As a control, untreated lapB mRNA was separated (C, lane 1). An RNase T1 ladder (T1) was separated in lane 2, and cleaved G residues are labeled along the left side of the gel. An alkaline ladder (OH) is shown in lane 3. The lapB sequence found to be protected from cleavage during LhrC4 presence is indicated on the right side of the gel. (B) Partial cleavage of 5′ labeled LhrC4 with lead(II) for 1 min (lanes 4–6) or RNase V1 for 2 min (lanes 7–9). LhrC4 was either digested alone (−) (lanes 4 and 7) or in the presence of 125- or 500-fold excess of non-labeled lapB mRNA. Untreated LhrC4 (C), an alkaline ladder (OH) and an RNase T1 ladder (T1) are shown in lane 1, 2 and 3, respectively. For an overview, selected nucleotides are labeled on the left side. Sequences showing structural changes upon lapB binding are marked on the right side of the gel: Stem loop A (lower and middle brackets; the 3′ side of stem A is marked by an asterisk) and the single-stranded stretch (upper bracket). (C) Labeled LhrC4 was partially cleaved with RNase T1 for 3 min (lanes 4–6) and lead(II) for 1 min (lanes 7–9) in the absence (−)(lanes 4 and 7) or presence of increasing amounts of non-labeled lapB mRNA (25-fold excess in lanes 5 and 8; 125-fold excess in lanes 6 and 9). Untreated LhrC4 (C), an RNase T1 (T1) and alkaline ladder (OH) are shown in lane 1, 2 and 3, respectively. On the left, the location of selected nucleotides is labeled. On the right, the observed change in secondary structure in the 3′ side of stem A upon lapB mRNA binding is marked (arrows: G48 and G52). (D) and (E) Gel mobility shift assays. In the LhrC sketches, mutated regions are shown in red. (D) lapB_mut_9 was mutated in the sequence interacting with LhrC4 and lost almost all of its capability to shift LhrC4. LhrC4_mut_9, the sRNA compensatory mutated in loop A, single-stranded stretch and terminator loop, was not shifted by lapB mRNA, but could partially compensate for the mutation in lapB_mut_9. An overview of the substitutions in lapB_mut9 and LhrC_mut_9 is shown in Supplementary Figure S17. Numbers in the bottom indicate the fraction of LhrC4 that was not shifted. (E) Mutation of only three sites in the lapB sequence (lapB_mut_10) nearly abolished all of the binding between the two RNAs. LhrC4_mut_10 which carried a corresponding mutation only in the terminator loop could still be shifted by lapB, but was also able to partially compensate for the mutation in lapB_mut_10. (F), (G) and (H) Deduced base pairing of lapB mRNA and loop A, the single-stranded stretch and the terminator loop of LhrC4, respectively. The lapB mRNA and LhrC4 sequences found to be bound in structure probing experiments is printed in bold. The start codon is marked in red. The UCCC motif is boxed. In figure (H) the three point mutations of mut_10 are indicated.
Because the sRNA LhrC is poor in guanosine residues, cleavage of 5′ labeled LhrC4 was not only induced by RNase T1 (cuts single-stranded G) and lead(II) treatment, but additionally by RNase V1 cleaving double-stranded RNA and single-stranded stacked RNA. Structural probing of unbound LhrC4 revealed that loop A might exist in the form proposed in Figure 4A, but an alternative structure is also conceivable, as cleavage by RNase V1, but not lead(II), was observed in the UCCC sequence in loop A (Figure 6B and C). Interestingly, a change in secondary structure of stem A was detected when the sRNA was incubated with lapB mRNA (Figure 6B and C): In the 3′ side of stem A, position 48–56 changed from bound- to single-stranded status, whereas position 24–32 in the 5′ side of stem A remained in a double-stranded structure. Remarkably, position 24–32 in stem A exhibits perfect complementarity to the lapB sequence from −2 to +7 leading to the conclusion that these two sequences interact, as illustrated in Figure 6F. Regarding the three sites of LhrC4 that were determined by gel shift experiments to be important for lapB mRNA interaction: Lead(II) treatment did not show a change in secondary structure in any of the two loops or in the single-stranded stretch of LhrC4, however, RNase V1 could clearly reveal loop A to change from unbound- to double-stranded state after addition of lapB mRNA, and also the stretch between the loops was subject to a change in secondary structure (Figure 6B). The matching interactions between these two sites in LhrC4 and lapB mRNA are illustrated in Figure 6F and G, respectively. The sequence of the terminator loop was not covered in this 5′ labeling experiment, however, its role in binding to lapB mRNA will be specifically addressed in gel shift experiments presented below. From the results obtained until this point we assumed that any of the three homologous sites in LhrC4 can make the first contact with the complementary sequence in lapB mRNA (−19 to −11) before a structural rearrangement in the sRNA takes place and the final sRNA-mRNA duplex forms involving stem loop A in LhrC4 and the sequence −21 to +7 of lapB (Figure 7, Supplementary Figure S15). For an intended mutation of LhrC4 to destroy interaction of the two RNAs with a subsequent compensatory mutation in lapB, the stem A sequence of LhrC4 seemed to be best suited and more straightforward than picking a sequence in lapB mRNA that is capable of an interaction with three different sites in LhrC4. Hence, stem A was mutated by inversion of the sequence (5′ ≥ 3′ to 3′ ≥ 5′) to keep the structure of LhrC4 (Supplementary Figure S16). However, mutation of stem A did not result in any reduction of binding efficiency of the two RNAs in vitro (Supplementary Figure S16). This observation endorsed our theory that the stem opening and binding represents a later event in the course of sRNA-mRNA complex formation, meaning that the two RNAs are still capable of binding, but the final duplex cannot be formed. In an attempt to still mutate and compensate, lapB was mutated (lapB_mut_9) to prevent most of its interaction with LhrC4. Likewise, all three putative interaction sites in LhrC4 were substituted to match lapB_mut_9 (LhrC4_mut_9) (Supplementary Figure S17). The interaction capability of lapB_mut_9 and LhrC4_mut_9 could not be fully restored to wild type levels, but the lapB mutation could indeed be compensated for partially by the mutations in all three LhrC4 interaction sites (Figure 6D).
Figure 7.
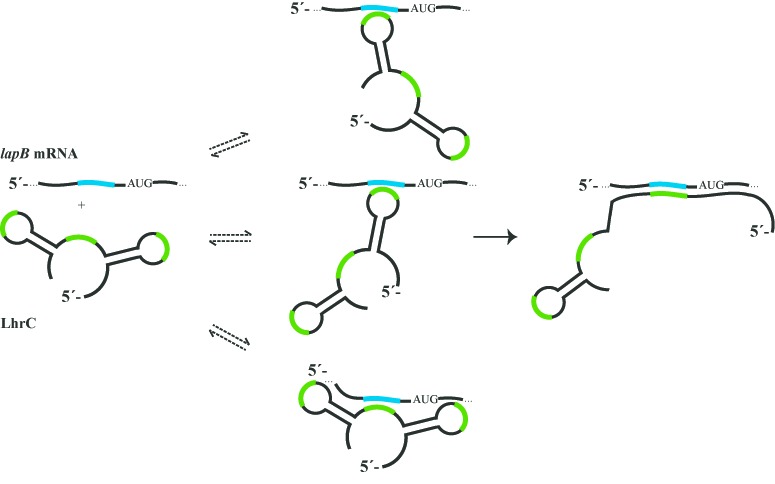
Model of LhrC4-lapB mRNA interaction. Three different sites in LhrC4, all containing a UCCC motif (green), are capable of binding to the G-rich sequence (blue) upstream of the start codon in lapB. Formation of the final sRNA-mRNA duplex involves opening of stem loop A, i.e. the 5′ side of stem A and loop A are bound to lapB mRNA. One LhrC4 molecule could potentially bind two or three lapB mRNA molecules simultaneously (not illustrated).
The three sites in LhrC4 important for lapB mRNA binding all contain a UCCC motif which was reported in the literature to be a common loop motif for target recognition in sRNAs of several Gram-positive bacteria (51). We wondered whether this motif mediates first contact between LhrC4 and lapB mRNA as well. In order to test this, the target lapB was mutated in two nucleotides in the site where the UCCC motif of LhrC4 is predicted to bind, in addition to a single nucleotide flanking this site (5′-GGGAG to 5′-CCGAC), which was mutated to keep the secondary structure of the compensatory LhrC4 mutant mentioned below. Strikingly, this minimal mutant (lapB_mut_10) lost almost all of its binding capability to LhrC4 demonstrating the importance of the implied motif for target recognition (Figure 6E and H). A compensatory LhrC4 mutant (LhrC4_mut_10) was constructed, but this time mutating only the according three nucleotides in the terminator loop (5′-CUCCC to 5′-GUCGG) to unequivocally demonstrate its involvement in lapB mRNA binding (Figure 6H). LhrC4_mut_10 lost only part of its ability to bind lapB mRNA, once more indicating that there are two more possible sites in the sRNA capable of an interaction with one important binding site in the mRNA (Figure 6E). The compensatory terminator loop mutations indeed regained the ability of LhrC4 to shift lapB_mut_10, although not to the same level of binding as seen for lapB wild type and LhrC4 wild type (Figure 6E), since LhrC4_mut_10 features only a single site that can bind to lapB_mut_10 instead of three sites (corresponding to the wild type situation).
DISCUSSION
Induction of LhrC
This study aimed to characterize the five homologs of LhrC which were repeatedly found to be highly induced under infection-relevant conditions (6,11). We show that disturbance of the cell envelope integrity is the actual signal for LhrC1–5 induction, and that the presence of the sRNAs contributes to the growth of L. monocytogenes during cell envelope stress. An involvement of sRNAs when dealing with cell envelope stress has been frequently described for Gram-negative bacteria. In fact, a disproportionately high number of trans-encoded sRNAs regulate proteins of the outer membrane (OMP) or transporters (1,52) which can to some extent be explained by the relatively easy detection of these highly abundant proteins. Although the cell envelope stress response of Gram-positive bacteria is a long and intensively studied research area (53), reports on an involvement of sRNAs are rare in this respect. Many of the numerous OMP regulating sRNAs in E. coli are aptly regulated by cell envelope stress sensing systems (54,55). Analogously, we checked the possibility of LhrC1–5 being regulated by one of the stress sensing systems of L. monocytogenes and could demonstrate that the TCS LisRK is mandatory for LhrC induction. This poses LhrC as the first sRNA in L. monocytogenes to be regulated by a TCS. A role of LisRK in the stress response to low pH and high ethanol concentrations has been described long ago (38), just as its importance for L. monocytogenes’ virulence (38,39). Later, LisRK was reported to also be involved in osmoregulation (56) and in dealing with the lantibiotic nisin as well as cephalosporin antibiotics (40). Finally, a comprehensive study by Nielsen et al. unveiled LisR as a central regulator controlling a whole series of genes after cell envelope stress (42). With LhrC being absolutely dependent on the LisRK system, one could speculate about the sRNA to be a hitherto unknown regulatory component between LisR and its responsive genes. Small RNAs being regulated by a TCS are capable of controlling a major fraction of the TCS’ regulon which is maxed out by the GacAS system of P. aeruginosa acting exclusively through the sRNAs RsmY and RsmZ (57).
LapB mRNA is target of LhrC
In the scope of our work we could identify lapB mRNA as the first direct target of the sRNAs LhrC1–5. The gene lapB was predicted to have a σB-dependent promoter 67 bp upstream of its translational start site (58) and its expression was reported to be positively influenced by the major listerial transcriptional activator of virulence genes PrfA (37,59). Both σB regulation as well as positive control by PrfA point to an involvement of LapB in stress response and/or virulence. This was proven by Reis et al. who coined the name LapB (Listeria adhesion protein B) and demonstrated lapB, which is absent from non-pathogenic Listeria species, to encode a sortase-dependent, cell wall-anchored adhesin required for entry into eukaryotic cells (37). Our observations and previous studies of others disclosed a regulation of lapB inverse to the one of LhrC1–5. Quillin et al. determined lapB to be down-regulated by more than 2-fold during bile stress (60), and 1 h of cefuroxime stress resulted in lapB down-regulation by a factor of about 2.5 (42). Both conditions cause strong induction of LhrC1–5 affirming our data that the sRNA negatively regulates lapB expression. In a comprehensive study on gene regulation in L. monocytogenes, all five copies of LhrC were found to be highly up-regulated in blood, and at the same time the amount of lapB mRNA was decreased (6). The adhesin was furthermore shown to be up-regulated in stationary phase and slightly down-regulated at 30°C. This correlates with results of the first report on LhrC in which it was described to be more highly expressed in exponential growth phase than in stationary phase and more prevalent under cold stress conditions (9).
LapB was demonstrated to play an important role for L. monocytogenes’ virulence by mediating the adhesion to and invasion of non-phagocytic cells in a cell type-dependent manner (37). LhrC1–5 are possibly the critical regulators which ensure a coordinated expression of lapB, such as its repression in blood (6) to escape from the hosts immune response. Reis et al. assigned lapB to a whole group of genes that are down-regulated in blood but up-regulated in the spleen (37). This group of genes is composed of 11 others all associated with virulence or cell wall metabolism. It will be interesting to explore the assumption that more members of this group of genes represent targets of LhrC1–5 which could turn the sRNAs into an important regulatory hub controlling the proper site- and time-specific expression of several virulence factors during the process of listerial infection.
Although involvement of sRNAs in the regulatory network of virulence is beyond question (21,61), lapB represents one of the first virulence factors regulated by a trans-encoded sRNA in L. monocytogenes. To our knowledge only two further examples have been described so far. These are the expression of the listerial master regulator of virulence PrfA which is controlled by a trans-acting riboswitch (62), and the regulation of chiA mRNA, encoding for a virulence-associated chitinase (63), which is directly bound by the sRNA LhrA (26).
Multiple copies of LhrC
Multicopy sRNAs have been reported for several bacteria (1,43), either being redundant (64), acting additively (65,66) or following a hierarchical mode of action (67,68). One of the few closer characterized examples in Gram-positive species is the five highly similar csRNAs (cia-dependent sRNAs) of Streptococcus pneumonia recently shown to be involved in competence regulation (69). Analogous to LhrC they are controlled by a TCS, CiaRH. For several of their targets an additive mode of action was proven. With respect to LhrC, β-gal assays revealed that all five lhrC promoters are active upon cell surface stress, showing the same induction pattern regardless of which kind of stress. This observation could lead to the assumption that the five LhrC copies act redundantly, i.e. one is capable of substituting for all others. However, a stress tolerance assay with cefuroxime and bile salts hampered not only the full lhrC mutant but also ΔlhrC1–4 compared to wild type which rather indicates an additive action of the five copies. An additive mechanism can also be concluded from results of translational fusion experiments for the target lapB. The observed β-gal activity in the partial lhrC mutants ΔlhrC1–4 and ΔlhrC5 was measured to be in between the ones of the wild type and of the full lhrC mutant (ΔlhrC1–5). Although all five LhrC molecules are quite similar, they still show slight differences in their C-rich segments and flanking regions which could supply LhrC with a lot of flexibility in terms of target recognition as shown for the Qrr sRNAs in V. harveyi (66). Most likely, LhrC has more than only one target, and possibly its mechanism of action is different depending on the target. It is not always possible to distinctly categorize multicopy sRNAs as was exemplified in Pseudomonas aeruginosa. The sRNAs RsmY and RsmZ act redundantly in controlling type III secretion system, but an additive mechanism was found for the regulation of genes involved in biofilm formation (70). An identification of more LhrC targets and detailed studies on the single LhrC copies will certainly shed more light on the purpose of multiplicity and on how the five sRNA homologs interplay.
A redundant CU-rich motif
LhrC is different from other multiple sRNAs described so far by featuring an additional dimension of multiplicity. The sRNA not only exists in five homologs, but each homolog exhibits three redundant CU-rich motifs that were shown to be actual sites of target recognition. It has been proposed that multiplicity of sRNAs permits a very sensitive reaction and helps to turn a small input signal into a large output response (1). Keeping five copies of LhrC with three internal copies of the regulatory motif, leads to the assumption, that these sRNAs control important adaptations that require ultra-sensitivity.
Repeated sequence elements in loops or single-stranded stretches of sRNA sites are reminiscent of protein-binding RNAs of the Rsm (repressor of secondary metabolites)/Csr (carbon storage regulator) family which have been discovered and characterized in several γ-proteobacteria (71). Originally, this elaborate mechanism of post-transcriptional regulation was discovered in E. coli (72). CsrA, a small protein which globally regulates gene expression by binding to mRNA targets can be sequestered by CsrB, a sRNA that hence antagonizes the effects of CsrA. Often several copies of the antagonizing sRNA exist. Some Pseudomonads exhibit three sRNAs (RsmZ, RsmY and RsmX) to antagonize the RNA binding proteins RsmA and RsmE, with RsmX existing in even five copies in Pseudomonas syringae (73). The fact that these sRNAs of Pseudomonads are all regulated by a TCS (GacAS) extends the observed analogy to listerial LhrC1–5. Even though the redundant motif of LhrC1–5 is different from the GGA motif that characterizes antagonizing sRNAs in γ-proteobacteria, one could speculate that LhrC1–5 could also function as protein-binding sRNAs, especially, since the dogma of an sRNA to be either protein-binding or RNA-binding was recently disproven by Jørgensen et al. who showed that McsA in E. coli not only directly binds to mRNAs but is also capable of capturing at least two copies of the global RNA-binding protein CsrA (74).
Strikingly, the redundant motif in LhrC comprises a sequence of UCCC. This C-rich motif is assumed to be a characteristic feature of sRNAs of low GC content bacteria that block translation initiation (51,75) by binding to the SD sequence. The involvement of the UCCC motif in mRNA target binding was proven for RNAIII (76), RsaE (51) and SprD (77) in S. aureus and for FsrA in B. subtilis (20). Indeed, also some of the sRNAs discovered in L. monocytogenes hold the UCCC motif in their structure (6,10). However, an involvement of the motif in target binding in L. monocytogenes is shown for the first time in this study.
The three sites containing the UCCC motif are located in both loops and in the single-stranded stretch between the two stem loops of the LhrC molecule. This appears plausible as loops and single stranded regions are known to be best suited for an initial contact of two RNA molecules (48–50). Besides the involvement of three locally separated sites of LhrC4 which all target the same specific site in lapB, LhrC4-lapB mRNA interaction holds another unexpected result as disclosed by structural probing experiments. Binding of LhrC4 to lapB mRNA entails a structural rearrangement in the sRNA, more specifically the 5′ site of stem A is engaged in a duplex with lapB mRNA extending the site of interaction into the coding region of the mRNA. Mutation of LhrC4 stem A did not have an effect on LhrC4-lapB mRNA binding efficiency in gel shift experiments. This observation is reminiscent of what has been described in the literature for RNA-OUT of IS10 transposition (48) and for FinP in the FinOP repressor system of plasmid R1 (78). In both cases mutations in a stem of an antisense RNA did not have an effect on target binding in vitro. As an explanation the authors argued that the stem was first involved in a secondary step of RNA-RNA interaction after the two RNAs had already made initial contact. One could speculate that the mechanism of LhrC4-lapB mRNA binding works in a similar way, i.e. the antisense RNA makes first contact with its RNA target in a very fast reaction restricted to only a few nucleotides most likely located in a loop structure (kissing), and subsequently this first complex is transferred into the ultimate RNA–RNA duplex in an irreversible and mostly not as rapid reaction (48,79). In the case of LhrC4, three sites could mediate first contact with lapB mRNA already inhibiting translational initiation (48–50). The irreversible step of final complex formation involving stem A can only occur starting from an initial interaction of lapB mRNA with loop A of LhrC4, as illustrated in Figure 7 and Supplementary Figure S15. Removal of this final RNA-RNA complex by RNases could work like a sink implicating that eventually all lapB molecules will take this path. In our study we were able to abolish binding of LhrC4 and lapB mRNA by mutating the three sites in LhrC4 capable of an interaction with lapB mRNA. A compensatory mutation of the lapB sequence could only partially restore interaction which we assume is caused by structural issues proven to be highly relevant for such RNA–RNA interactions (48–50,79). Future experiments with corresponding mutants in vivo have to be performed to corroborate the suggested interactions between LhrC and lapB mRNA.
A plethora of non-coding sRNAs in scores of bacterial species have been identified throughout the last decade. With respect to pathogenic bacteria one hopes to exploit these regulatory sRNAs in anti-microbial strategies one day. However, there is still a long way to go since most of them are completely uncharacterized. This study unveiled details on the sRNAs LhrC1–5 concerning conditional and regulatory requirements for their expression, presents the mRNA of a virulence-associated adhesin as their first direct target, and shows interesting new mechanistic aspects on sRNA-mRNA interaction. The association with L. monocytogenes virulence and the sophisticated way of target binding calls for further in-depth analyses on these five homologous sRNAs.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We are grateful to Didier Cabanes for sending us the LapB antibody.
Footnotes
Present addresses:
Susanne Sievers, Institute for Microbiology, Ernst-Moritz-Arndt-University Greifswald, Greifswald, Germany.
Kirstine Jacobsen, Institute of Molecular Medicine, University of Southern Denmark, Odense, Denmark.
Pia Kiil Nielsen, Work environment and Work life, Orbicon A/S, Roskilde, Denmark.
FUNDING
German Research foundation [Si 1678/1–1 to S.S.]; The Danish Council for Independent Research │Natural Sciences [12–124735 to B.H.K.]; The Lundbeck Foundation [R100-A9235 to B.H.K.]; The Novo Nordisk Foundation [to B.H.K.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Waters L.S., Storz G. Regulatory RNAs in bacteria. Cell. 2009;136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Storz G., Vogel J., Wassarman K.M. Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell. 2011;43:880–891. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chao Y., Vogel J. The role of Hfq in bacterial pathogens. Curr. Opin. Microbiol. 2010;13:24–33. doi: 10.1016/j.mib.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Papenfort K., Vogel J. Regulatory RNA in bacterial pathogens. Cell Host. Microbe. 2010;8:116–127. doi: 10.1016/j.chom.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Felden B., Vandenesch F., Bouloc P., Romby P. The Staphylococcus aureus RNome and its commitment to virulence. PLoS. Pathog. 2011;7:e1002006. doi: 10.1371/journal.ppat.1002006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toledo-Arana A., Dussurget O., Nikitas G., Sesto N., Guet-Revillet H., Balestrino D., Loh E., Gripenland J., Tiensuu T., Vaitkevicius K., et al. The Listeria transcriptional landscape from saprophytism to virulence. Nature. 2009;459:950–956. doi: 10.1038/nature08080. [DOI] [PubMed] [Google Scholar]
- 7.Sharma C.M., Hoffmann S., Darfeuille F., Reignier J., Findeiss S., Sittka A., Chabas S., Reiche K., Hackermuller J., Reinhardt R., et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;464:250–255. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- 8.Acebo P., Martin-Galiano A.J., Navarro S., Zaballos A., Amblar M. Identification of 88 regulatory small RNAs in the TIGR4 strain of the human pathogen Streptococcus pneumoniae. RNA. 2012;18:530–546. doi: 10.1261/rna.027359.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christiansen J.K., Nielsen J.S., Ebersbach T., Valentin-Hansen P., Sogaard-Andersen L., Kallipolitis B.H. Identification of small Hfq-binding RNAs in Listeria monocytogenes. RNA. 2006;12:1383–1396. doi: 10.1261/rna.49706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mandin P., Repoila F., Vergassola M., Geissmann T., Cossart P. Identification of new noncoding RNAs in Listeria monocytogenes and prediction of mRNA targets. Nucleic Acids Res. 2007;35:962–974. doi: 10.1093/nar/gkl1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mraheil M.A., Billion A., Mohamed W., Mukherjee K., Kuenne C., Pischimarov J., Krawitz C., Retey J., Hartsch T., Chakraborty T., et al. The intracellular sRNA transcriptome of Listeria monocytogenes during growth in macrophages. Nucleic Acids Res. 2011;39:4235–4248. doi: 10.1093/nar/gkr033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mellin J.R., Cossart P. The non-coding RNA world of the bacterial pathogen Listeria monocytogenes. RNA. Biol. 2012;9:372–378. doi: 10.4161/rna.19235. [DOI] [PubMed] [Google Scholar]
- 13.Ramaswamy V., Cresence V.M., Rejitha J.S., Lekshmi M.U., Dharsana K.S., Prasad S.P., Vijila H.M. Listeria–review of epidemiology and pathogenesis. J. Microbiol. Immunol. Infect. 2007;40:4–13. [PubMed] [Google Scholar]
- 14.Valentin-Hansen P., Eriksen M., Udesen C. The bacterial Sm-like protein Hfq: a key player in RNA transactions. Mol. Microbiol. 2004;51:1525–1533. doi: 10.1111/j.1365-2958.2003.03935.x. [DOI] [PubMed] [Google Scholar]
- 15.Silvaggi J.M., Perkins J.B., Losick R. Genes for small, noncoding RNAs under sporulation control in Bacillus subtilis. J. Bacteriol. 2006;188:532–541. doi: 10.1128/JB.188.2.532-541.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaballa A., Antelmann H., Aguilar C., Khakh S.K., Song K.B., Smaldone G.T., Helmann J.D. The Bacillus subtilis iron-sparing response is mediated by a Fur-regulated small RNA and three small, basic proteins. Proc. Natl. Acad. Sci. U.S.A. 2008;105:11927–11932. doi: 10.1073/pnas.0711752105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heidrich N., Moll I., Brantl S. In vitro analysis of the interaction between the small RNA SR1 and its primary target ahrC mRNA. Nucleic Acids Res. 2007;35:4331–4346. doi: 10.1093/nar/gkm439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jahn N., Preis H., Wiedemann C., Brantl S. BsrG/SR4 from Bacillus subtilis–the first temperature-dependent type I toxin-antitoxin system. Mol. Microbiol. 2012;83:579–598. doi: 10.1111/j.1365-2958.2011.07952.x. [DOI] [PubMed] [Google Scholar]
- 19.Dambach M., Irnov I., Winkler W.C. Association of RNAs with Bacillus subtilis Hfq. PLoS. One. 2013;8:e55156. doi: 10.1371/journal.pone.0055156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smaldone G.T., Antelmann H., Gaballa A., Helmann J.D. The FsrA sRNA and FbpB protein mediate the iron-dependent induction of the Bacillus subtilis lutABC iron-sulfur-containing oxidases. J. Bacteriol. 2012;194:2586–2593. doi: 10.1128/JB.05567-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Romby P., Charpentier E. An overview of RNAs with regulatory functions in gram-positive bacteria. Cell Mol. Life Sci. 2010;67:217–237. doi: 10.1007/s00018-009-0162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bohn C., Rigoulay C., Bouloc P. No detectable effect of RNA-binding protein Hfq absence in Staphylococcus aureus. BMC. Microbiol. 2007;7:10. doi: 10.1186/1471-2180-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y., Wu N., Dong J., Gao Y., Zhang X., Mu C., Shao N., Yang G. Hfq is a global regulator that controls the pathogenicity of Staphylococcus aureus. PLoS. One. 2010;5:e13069. doi: 10.1371/journal.pone.0013069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christiansen J.K., Larsen M.H., Ingmer H., Sogaard-Andersen L., Kallipolitis B.H. The RNA-binding protein Hfq of Listeria monocytogenes: role in stress tolerance and virulence. J. Bacteriol. 2004;186:3355–3362. doi: 10.1128/JB.186.11.3355-3362.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nielsen J.S., Lei L.K., Ebersbach T., Olsen A.S., Klitgaard J.K., Valentin-Hansen P., Kallipolitis B.H. Defining a role for Hfq in Gram-positive bacteria: evidence for Hfq-dependent antisense regulation in Listeria monocytogenes. Nucleic Acids Res. 2010;38:907–919. doi: 10.1093/nar/gkp1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nielsen J.S., Larsen M.H., Lillebaek E.M., Bergholz T.M., Christiansen M.H., Boor K.J., Wiedmann M., Kallipolitis B.H. A small RNA controls expression of the chitinase ChiA in Listeria monocytogenes. PLoS. One. 2011;6:e19019. doi: 10.1371/journal.pone.0019019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vazquez-Boland J.A., Kocks C., Dramsi S., Ohayon H., Geoffroy C., Mengaud J., Cossart P. Nucleotide sequence of the lecithinase operon of Listeria monocytogenes and possible role of lecithinase in cell-to-cell spread. Infect. Immun. 1992;60:219–230. doi: 10.1128/iai.60.1.219-230.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pfaffl M.W., Horgan G.W., Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eggenhofer F., Tafer H., Stadler P.F., Hofacker I.L. RNApredator: fast accessibility-based prediction of sRNA targets. Nucleic Acids Res. 2011;39:W149–W154. doi: 10.1093/nar/gkr467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poyart C., Trieu-Cuot P. A broad-host-range mobilizable shuttle vector for the construction of transcriptional fusions to beta-galactosidase in gram-positive bacteria. FEMS Microbiol. Lett. 1997;156:193–198. doi: 10.1111/j.1574-6968.1997.tb12726.x. [DOI] [PubMed] [Google Scholar]
- 33.Boysen A., Moller-Jensen J., Kallipolitis B., Valentin-Hansen P., Overgaard M. Translational regulation of gene expression by an anaerobically induced small non-coding RNA in Escherichia coli. J. Biol. Chem. 2010;285:10690–10702. doi: 10.1074/jbc.M109.089755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Urban J.H., Vogel J. Translational control and target recognition by Escherichia coli small RNAs in vivo. Nucleic Acids Res. 2007;35:1018–1037. doi: 10.1093/nar/gkl1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gotfredsen M., Gerdes K. The Escherichia coli relBE genes belong to a new toxin-antitoxin gene family. Mol. Microbiol. 1998;29:1065–1076. doi: 10.1046/j.1365-2958.1998.00993.x. [DOI] [PubMed] [Google Scholar]
- 36.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reis O., Sousa S., Camejo A., Villiers V., Gouin E., Cossart P., Cabanes D. LapB, a novel Listeria monocytogenes LPXTG surface adhesin, required for entry into eukaryotic cells and virulence. J. Infect. Dis. 2010;202:551–562. doi: 10.1086/654880. [DOI] [PubMed] [Google Scholar]
- 38.Cotter P.D., Emerson N., Gahan C.G., Hill C. Identification and disruption of lisRK, a genetic locus encoding a two-component signal transduction system involved in stress tolerance and virulence in Listeria monocytogenes. J. Bacteriol. 1999;181:6840–6843. doi: 10.1128/jb.181.21.6840-6843.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kallipolitis B.H., Ingmer H. Listeria monocytogenes response regulators important for stress tolerance and pathogenesis. FEMS Microbiol. Lett. 2001;204:111–115. doi: 10.1111/j.1574-6968.2001.tb10872.x. [DOI] [PubMed] [Google Scholar]
- 40.Cotter P.D., Guinane C.M., Hill C. The LisRK signal transduction system determines the sensitivity of Listeria monocytogenes to nisin and cephalosporins. Antimicrob. Agents Chemother. 2002;46:2784–2790. doi: 10.1128/AAC.46.9.2784-2790.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kallipolitis B.H., Ingmer H., Gahan C.G., Hill C., Sogaard-Andersen L. CesRK, a two-component signal transduction system in Listeria monocytogenes, responds to the presence of cell wall-acting antibiotics and affects beta-lactam resistance. Antimicrob. Agents Chemother. 2003;47:3421–3429. doi: 10.1128/AAC.47.11.3421-3429.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nielsen P.K., Andersen A.Z., Mols M., van d.V., Abee T., Kallipolitis B.H. Genome-wide transcriptional profiling of the cell envelope stress response and the role of LisRK and CesRK in Listeria monocytogenes. Microbiology. 2012;158:963–974. doi: 10.1099/mic.0.055467-0. [DOI] [PubMed] [Google Scholar]
- 43.Gopel Y., Gorke B. Rewiring two-component signal transduction with small RNAs. Curr. Opin. Microbiol. 2012;15:132–139. doi: 10.1016/j.mib.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 44.Shimizu T., Yaguchi H., Ohtani K., Banu S., Hayashi H. Clostridial VirR/VirS regulon involves a regulatory RNA molecule for expression of toxins. Mol. Microbiol. 2002;43:257–265. doi: 10.1046/j.1365-2958.2002.02743.x. [DOI] [PubMed] [Google Scholar]
- 45.Koenig R.L., Ray J.L., Maleki S.J., Smeltzer M.S., Hurlburt B.K. Staphylococcus aureus AgrA binding to the RNAIII-agr regulatory region. J. Bacteriol. 2004;186:7549–7555. doi: 10.1128/JB.186.22.7549-7555.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Halfmann A., Kovacs M., Hakenbeck R., Bruckner R. Identification of the genes directly controlled by the response regulator CiaR in Streptococcus pneumoniae: five out of 15 promoters drive expression of small non-coding RNAs. Mol. Microbiol. 2007;66:110–126. doi: 10.1111/j.1365-2958.2007.05900.x. [DOI] [PubMed] [Google Scholar]
- 47.Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R., et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 48.Simons R.W., Kleckner N. Biological regulation by antisense RNA in prokaryotes. Annu. Rev. Genet. 1988;22:567–600. doi: 10.1146/annurev.ge.22.120188.003031. [DOI] [PubMed] [Google Scholar]
- 49.Eguchi Y., Itoh T., Tomizawa J. Antisense RNA. Annu. Rev. Biochem. 1991;60:631–652. doi: 10.1146/annurev.bi.60.070191.003215. [DOI] [PubMed] [Google Scholar]
- 50.Brunel C., Marquet R., Romby P., Ehresmann C. RNA loop-loop interactions as dynamic functional motifs. Biochimie. 2002;84:925–944. doi: 10.1016/s0300-9084(02)01401-3. [DOI] [PubMed] [Google Scholar]
- 51.Geissmann T., Chevalier C., Cros M.J., Boisset S., Fechter P., Noirot C., Schrenzel J., Francois P., Vandenesch F., Gaspin C., et al. A search for small noncoding RNAs in Staphylococcus aureus reveals a conserved sequence motif for regulation. Nucleic Acids Res. 2009;37:7239–7257. doi: 10.1093/nar/gkp668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guillier M., Gottesman S., Storz G. Modulating the outer membrane with small RNAs. Genes Dev. 2006;20:2338–2348. doi: 10.1101/gad.1457506. [DOI] [PubMed] [Google Scholar]
- 53.Jordan S., Hutchings M.I., Mascher T. Cell envelope stress response in Gram-positive bacteria. FEMS Microbiol. Rev. 2008;32:107–146. doi: 10.1111/j.1574-6976.2007.00091.x. [DOI] [PubMed] [Google Scholar]
- 54.Vogel J., Papenfort K. Small non-coding RNAs and the bacterial outer membrane. Curr. Opin. Microbiol. 2006;9:605–611. doi: 10.1016/j.mib.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 55.Valentin-Hansen P., Johansen J., Rasmussen A.A. Small RNAs controlling outer membrane porins. Curr. Opin. Microbiol. 2007;10:152–155. doi: 10.1016/j.mib.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 56.Sleator R.D., Hill C. A novel role for the LisRK two-component regulatory system in listerial osmotolerance. Clin. Microbiol. Infect. 2005;11:599–601. doi: 10.1111/j.1469-0691.2005.01176.x. [DOI] [PubMed] [Google Scholar]
- 57.Brencic A., McFarland K.A., McManus H.R., Castang S., Mogno I., Dove S.L., Lory S. The GacS/GacA signal transduction system of Pseudomonas aeruginosa acts exclusively through its control over the transcription of the RsmY and RsmZ regulatory small RNAs. Mol. Microbiol. 2009;73:434–445. doi: 10.1111/j.1365-2958.2009.06782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raengpradub S., Wiedmann M., Boor K.J. Comparative analysis of the sigma B-dependent stress responses in Listeria monocytogenes and Listeria innocua strains exposed to selected stress conditions. Appl. Environ. Microbiol. 2008;74:158–171. doi: 10.1128/AEM.00951-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Milohanic E., Glaser P., Coppee J.Y., Frangeul L., Vega Y., Vazquez-Boland J.A., Kunst F., Cossart P., Buchrieser C. Transcriptome analysis of Listeria monocytogenes identifies three groups of genes differently regulated by PrfA. Mol. Microbiol. 2003;47:1613–1625. doi: 10.1046/j.1365-2958.2003.03413.x. [DOI] [PubMed] [Google Scholar]
- 60.Quillin S.J., Schwartz K.T., Leber J.H. The novel Listeria monocytogenes bile sensor BrtA controls expression of the cholic acid efflux pump MdrT. Mol. Microbiol. 2011;81:129–142. doi: 10.1111/j.1365-2958.2011.07683.x. [DOI] [PubMed] [Google Scholar]
- 61.Toledo-Arana A., Repoila F., Cossart P. Small noncoding RNAs controlling pathogenesis. Curr. Opin. Microbiol. 2007;10:182–188. doi: 10.1016/j.mib.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 62.Loh E., Dussurget O., Gripenland J., Vaitkevicius K., Tiensuu T., Mandin P., Repoila F., Buchrieser C., Cossart P., Johansson J. A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell. 2009;139:770–779. doi: 10.1016/j.cell.2009.08.046. [DOI] [PubMed] [Google Scholar]
- 63.Chaudhuri S., Gantner B.N., Ye R.D., Cianciotto N.P., Freitag N.E. The Listeria monocytogenes ChiA chitinase enhances virulence through suppression of host innate immunity. MBio. 2013;4:e00617–12. doi: 10.1128/mBio.00617-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lenz D.H., Mok K.C., Lilley B.N., Kulkarni R.V., Wingreen N.S., Bassler B.L. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell. 2004;118:69–82. doi: 10.1016/j.cell.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 65.Tu K.C., Bassler B.L. Multiple small RNAs act additively to integrate sensory information and control quorum sensing in Vibrio harveyi. Genes Dev. 2007;21:221–233. doi: 10.1101/gad.1502407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shao Y., Feng L., Rutherford S.T., Papenfort K., Bassler B.L. Functional determinants of the quorum-sensing non-coding RNAs and their roles in target regulation. EMBO J. 2013;32:2158–2171. doi: 10.1038/emboj.2013.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reichenbach B., Maes A., Kalamorz F., Hajnsdorf E., Gorke B. The small RNA GlmY acts upstream of the sRNA GlmZ in the activation of glmS expression and is subject to regulation by polyadenylation in Escherichia coli. Nucleic Acids Res. 2008;36:2570–2580. doi: 10.1093/nar/gkn091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Urban J.H., Vogel J. Two seemingly homologous noncoding RNAs act hierarchically to activate glmS mRNA translation. PLoS. Biol. 2008;6:e64. doi: 10.1371/journal.pbio.0060064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schnorpfeil A., Kranz M., Kovacs M., Kirsch C., Gartmann J., Brunner I., Bittmann S., Bruckner R. Target evaluation of the non-coding csRNAs reveals a link of the two-component regulatory system CiaRH to competence control in Streptococcus pneumoniae R6. Mol. Microbiol. 2013;89:334–349. doi: 10.1111/mmi.12277. [DOI] [PubMed] [Google Scholar]
- 70.Bordi C., Lamy M.C., Ventre I., Termine E., Hachani A., Fillet S., Roche B., Bleves S., Mejean V., Lazdunski A., et al. Regulatory RNAs and the HptB/RetS signalling pathways fine-tune Pseudomonas aeruginosa pathogenesis. Mol. Microbiol. 2010;76:1427–1443. doi: 10.1111/j.1365-2958.2010.07146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Romeo T., Vakulskas C.A., Babitzke P. Post-transcriptional regulation on a global scale: form and function of Csr/Rsm systems. Environ. Microbiol. 2013;15:313–324. doi: 10.1111/j.1462-2920.2012.02794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu M.Y., Gui G., Wei B., Preston J.F., III, Oakford L., Yuksel U., Giedroc D.P., Romeo T. The RNA molecule CsrB binds to the global regulatory protein CsrA and antagonizes its activity in Escherichia coli. J. Biol. Chem. 1997;272:17502–17510. doi: 10.1074/jbc.272.28.17502. [DOI] [PubMed] [Google Scholar]
- 73.Moll S., Schneider D.J., Stodghill P., Myers C.R., Cartinhour S.W., Filiatrault M.J. Construction of an rsmX co-variance model and identification of five rsmX non-coding RNAs in Pseudomonas syringae pv. tomato DC3000. RNA. Biol. 2010;7:508–516. doi: 10.4161/rna.7.5.12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jorgensen M.G., Thomason M.K., Havelund J., Valentin-Hansen P., Storz G. Dual function of the McaS small RNA in controlling biofilm formation. Genes Dev. 2013;27:1132–1145. doi: 10.1101/gad.214734.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Geissmann T., Marzi S., Romby P. The role of mRNA structure in translational control in bacteria. RNA. Biol. 2009;6:153–160. doi: 10.4161/rna.6.2.8047. [DOI] [PubMed] [Google Scholar]
- 76.Boisset S., Geissmann T., Huntzinger E., Fechter P., Bendridi N., Possedko M., Chevalier C., Helfer A.C., Benito Y., Jacquier A., et al. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev. 2007;21:1353–1366. doi: 10.1101/gad.423507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chabelskaya S., Gaillot O., Felden B. A Staphylococcus aureus small RNA is required for bacterial virulence and regulates the expression of an immune-evasion molecule. PLoS. Pathog. 2010;6:e1000927. doi: 10.1371/journal.ppat.1000927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koraimann G., Teferle K., Markolin G., Woger W., Hogenauer G. The FinOP repressor system of plasmid R1: analysis of the antisense RNA control of traJ expression and conjugative DNA transfer. Mol. Microbiol. 1996;21:811–821. doi: 10.1046/j.1365-2958.1996.361401.x. [DOI] [PubMed] [Google Scholar]
- 79.Wagner E.G., Simons R.W. Antisense RNA control in bacteria, phages, and plasmids. Annu. Rev. Microbiol. 1994;48:713–742. doi: 10.1146/annurev.mi.48.100194.003433. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.