


Abstract
DNA topoisomerase II (topo II) changes DNA topology by cleavage/re-ligation cycle(s) and thus contributes to various nuclear DNA transactions. It is largely unknown how the enzyme is controlled in a nuclear context. Several studies have suggested that its C-terminal domain (CTD), which is dispensable for basal relaxation activity, has some regulatory influence. In this work, we examined the impact of nuclear localization on regulation of activity in nuclei. Specifically, human cells were transfected with wild-type and mutant topo IIβ tagged with EGFP. Activity attenuation experiments and nuclear localization data reveal that the endogenous activity of topo IIβ is correlated with its subnuclear distribution. The enzyme shuttles between an active form in the nucleoplasm and a quiescent form in the nucleolus in a dynamic equilibrium. Mechanistically, the process involves a tethering event with RNA. Isolated RNA inhibits the catalytic activity of topo IIβ in vitro through the interaction with a specific 50-residue region of the CTD (termed the CRD). Taken together, these results suggest that both the subnuclear distribution and activity regulation of topo IIβ are mediated by the interplay between cellular RNA and the CRD.
INTRODUCTION
Type II DNA topoisomerases (topos II) catalyze interconversion of DNA's topological states by swapping the spacial position of two segments of duplex DNA (1). This is achieved through a catalytic cycle composed of multiple steps that are highly concerted (2). Structural basis for the reaction comes from three mobile ‘gates’ that are formed between two identical subunits associated head-to-head orientation (3) (see Supplementary Figure S1). Two segments of DNA enter from the N-terminal gate (N-gate) whose closure and opening is controlled by binding and hydrolysis of adenosine triphosphate (ATP), respectively.
While invertebrates have only one form of topo II, vertebrates have two isoforms (α and β) encoded by distinct genes (4–6). Although the two isoforms possess very similar basic structure and mode of reaction, they are clearly different in several aspects. Topo IIα relaxes positively supercoiled DNA more readily than negative supercoils in vitro (7). Expression of topo IIα is restricted to proliferating cells and is regulated in a cell cycle-dependent manner (8,9). It plays a critical role in the decatenation of entangled sister chromatids before segregation, thus being essential for cell proliferation. In contrast, topo IIβ appears to play more specialized roles in cellular physiology. Topo IIβ is indispensable for gene regulation in the final stage of neuronal differentiation when the enzyme shows elevated expression (10–13). In immortalized cell lines, however, its expression is de-regulated to become constitutive and even dispensable for cell survival (8,14).
Amino acid sequence of topo II isoforms is highly homologous each other (80–90% similarity) except for ∼400 residues in the C-terminal domain (CTD). Whether the difference in CTD sequence can explain the differential behavior of topo II isoforms is an obvious question, which is not fully answered. Remarkably, CTD is dispensable for the basal activity of topo II (15). There has been little insight from structural studies since the domain is an intrinsically disordered region with little higher-order structure (16). Several reports have already addressed the functional ambiguity of CTD. Specifically, comparison between topo IIα wild-type (WT) and CTD-truncated mutant revealed that CTD confers preference to positive supercoils (7). It would be reasonable to assume that CTD has some regulatory role not only in the catalytic reaction per se, but also in living cells. The latter may be complex due to unknown interactive factors. The primary function of CTD would be to confine the enzyme to the nucleus as multiple nuclear localization signals (NLSs) have been located in CTD (17–19). Experiments where the CTDs were swapped between the two isoforms exhibited the importance of CTD in the isoform-specific functioning in vivo (20).
Most nuclear proteins localize to certain subnuclear regions because they are either in an operational mode or in a transient holding pattern. During M-phase, nucleoplasmic proteins redistribute to daughter cells by associating with chromosomes, while others diffuse out through the cytoplasm after the breakdown of nuclear membrane. Nuclear dynamics of topo II isoform distribution has been evaluated in a cellular context using photobleaching (21). The results showed clearly that in mitosis topo IIα migrates with chromosomes, whereas topo IIβ becomes cytoplasmic. In interphase, both are concentrated in nucleoli in a dynamic process, as attested by the rapid movement to nucleoplasm following treatment with topo II-specific poisons such as etoposide, which covalently traps the enzyme on genomic DNA.
Despite the recent progress in this area, regulation of topo II in the nuclear milieu remains unsettled. One issue in particular is how topo II operates catalytically in an environment replete with RNA, given that RNA is strongly inhibitory at least in vitro (22,23). We have shown recently that the RNA inhibition is neutralized by an nucleoplasmic RNA-binding protein hnRNP U/SAF-A/SP120 (23), thus, providing a rational built-in mechanism for activity regulation. In the present study, we examined the mechanism of topo IIβ activity regulation by analyzing the dynamics of subnuclear localization in response to activity changes. Results show that topo IIβ shuttles between nucleoplasm and nucleolus in a dynamic equilibrium that is determined by its residence time on nuclear DNA. We also investigated the roles of CTD in vitro using immobilized topo IIβ on beads and successfully identified a subdomain in CTD that is required for the susceptibility to RNA, which suggests that RNA is an innate regulator of topo IIβ.
MATERIALS AND METHODS
Plasmid construction
The plasmid pFlag-top2b encoding the full-length rat topo IIβ (1614 amino acids) with Flag sequence tagged at the N-terminus was constructed. The original topo IIβ cDNA clone (AB262979) was amplified with primers containing restriction overhangs (Not I/Sma I) that are listed in the supplementary file (Supplementary Table S1). A high-fidelity DNA polymerase, Phusion Hot Start II (Thermo Scientific), was used throughout. The polymerase chain reaction (PCR) product was inserted in frame between the Not I/Sma I sites of pFlag-CMV-2 expression vector (Sigma-Aldrich). To obtain Flag-fused C-terminal truncation mutants (ΔCTD, ΔCTD’), the pFlag-top2b plasmid was amplified with primers listed in Supplementary Table S1 and the products were inserted into pFlag-CMV-2 between Not I/Sma I sites. To construct pFlag-top2b (ΔCRD) expression plasmid, a C-terminal portion of topo IIβ (CTD’; #1251–1614) was PCR amplified from pFlag-top2b with primers containing restriction overhangs (Sma I/Sma I). The PCR product was then inserted at the Sma I site of the pFlag-top 2b (ΔCTD) expression plasmid and a transformed colony containing the insert in correct orientation was selected by colony PCR using forward primer (5′-GTCTCTATGGTCTCTTAC-3′) and reverse primer (5′-TCCCCCGGGCTCCTTTTTCTCCCTTTT-3′).
The plasmid pEGFP-top2b that encodes the full-length rat topo IIβ with enhanced green fluorescent protein (EGFP) sequence fused at the C-terminus was constructed. The pFLAG-top2b plasmid was PCR amplified with primers containing restriction overhangs (Xho I/Sma I) and the product was inserted in frame between the Xho I/Sma I sites of pEGFP-N1 expression vector (Clontech). Amino acid substitution mutants of topo IIβ (G173I, L178F and Y814S) were prepared using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent) with the pEGFP-Top2b plasmid as a template and with primers containing the desired mutation (underlined in Supplementary Table S1).
To express proteins with Flag-tag at the N-terminus and EGFP-tag at the C-terminus, pFlag-CMV-2-EGFP expression vector was constructed. We first obtained EGFP cDNA from pEGFP-N1 vector by PCR amplification with a primer pair (sense, 5′-GGGAATTCTCGAGTAGATCTGCCGGTCGCCACCATGGTG-3′; antisense, 5′-CGGGATCCCGCTTTACTTGTACAGCTC-3′) containing Eco RI or Bam HI overhangs (underlined). The EGFP cDNA was then inserted in frame between the Eco RI/Bgl II sites of the pFlag-CMV-2 vector. DNA fragments encoding the full-length or domain mutants of topo IIβ were PCR amplified with primers containing restriction overhangs (Not I/Sma I or Sma I/Sma I) from pFlag-top2b and subcloned into the pFlag-CMV-2-EGFP vector.
All the constructs used in this work were sequenced to confirm the absence of unintended mutations.
Cell culture and transfection
The human embryonal kidney cell line HEK293E (designated HEK hereafter), a rat fibroblast-like cell line (Rat-1) and a mouse neuroblastoma cell line (Neuro 2a) cells were grown at 37°C in a humidified atmosphere of 5% CO2 in a Dulbecco's modified Eagle's medium (DMEM) (Sigma-Aldrich) supplemented with 10% fetal calf serum (FCS) and 100 μg/ml kanamycin sulfate.
For ATP depletion studies, cells were washed with phosphate buffered saline (PBS), and incubated in glucose-free DMEM (catalog no. 11966–025; GIBCO BRL) containing 10 mM sodium azide, 6 mM 2-deoxy-d-glucose, 100 μg/ml kanamycin sulfate and 10% FCS (hereafter referred to as ‘ATP depletion medium’) for times indicated in the figure legends (24). Cellular ATP was quantified by a luciferin–luciferase-based ATP assay kit (Wako, Japan) according to the manufacturer's instructions.
For in situ digestion of cellular RNA or DNA, cells were permeabilized with 50 μg/ml digitonin in 20 mM HEPES-KOH (pH 7.3), 110 mM KOAc, 5 mM MgCl2 and 2 mM dithiothreitol for 5 min at 37°C, followed by digestion with 0.4 mg/ml RNase A (Qiagen) or 1.4 U/μl DNase I (InvitrogenTM) in 50 mM HEPES-NaOH (pH 7.4), 120 mM NaCl and 10 mM MgCl2 for 20 min at 37°C.
For cell transfection, HEK cells (2 × 105 cells/35-mm dish) were cultured for 24 h and then incubated with 3 μl of FuGENE 6 Transfection Reagent (Promega), and 1 μg of pFlag-CMV-2, pEGFP-N1 or pFlag-CMV-2-EGFP expression vectors harboring full-length or mutant topo IIβ cDNA. After 24 h, cells were subjected to analyses.
Immunocytochemistry
Cells were grown on 13-mm round coverslips immersed in DMEM with 10% FCS and 100 μg/ml kanamycin sulfate. Cells at subconfluency were fixed with 4% paraformaldehyde (PFA) in PBS kept at 37°C or on ice beforehand as indicated in figure legends, permeabilized then with 0.3% Triton X-100 in PBS and blocked with 10% goat serum in PBS containing 0.3% Triton X-100 (TPBS). After incubation with primary antibodies diluted in PBS supplemented with 1% goat serum, sections were washed with PBS, followed by incubation with fluorescent conjugates of goat secondary antibodies in PBS (9,10). Cells were finally stained for DNA with 0.25 μg/ml 4′, 6-diamidino-2-phenylindole (DAPI) in PBS.
Fluorescence microscopy
To capture images of living cells, HEK cells expressing EGFP-tagged recombinant proteins on 35-mm glass-bottomed dishes were examined under an inverted fluorescence microscope equipped with ApoTome device (Axiovert 200M; Carl Zeiss) and a cooled CCD camera (AxioCam MRm) using Axiovision 4.01 software (Zeiss).
For time-lapse imaging, the glass-bottomed dishes with HEK/topo IIβ-EGFP cells in 2 ml of growth medium were placed into an on-stage heating and cooling chamber (Temperable Insert P S1, Carl Zeiss) equipped with F25-ME Refrigerated/Heating Circulator (Carl Zeiss), and images were recorded at 1-min intervals for 60 min in the environmental chamber ensuring a constant change of temperature.
Fluorescence recovery after photobleaching (FRAP) analysis of living cells was performed at either 37 or 18°C with a confocal microscope (FV-1000; Olympus; operated by the built-in software version 2.1.1.4) equipped with a CO2-controlled on-stage heating chamber using a PLAPON 60XOSC NA 1.4 oil immersion lens. Images were taken before (10 images) and after (120 images) bleaching of a circular area of 1-μm diameter at 100% 488-nm laser transmission with four iterations. The imaging scans were acquired with a laser power attenuated to 0.1% of the bleached intensity using settings of 205 ms/frame; 2 μs/pixel; pinhole 300 μm (25). Binding kinetics was analyzed as described (26).
Protein expression and purification
To express Flag-tagged proteins for in vitro experiments, HEK cells grown to subconfluency on 100-mm dishes (1.2 × 106 cells/dish) were transfected with 3 μg of the construct plasmids using FuGENE 6 and cultured for 3 days. Cells were lysed on ice in 1 ml/dish of ice-cold high salt buffer (HSB) consisted of 50 mM HEPES-NaOH (pH7.4), 1 mM ethylenediaminetetraacetic acid (EDTA), 500 mM NaCl, 1 mM dithiothreitol, 0.1% NP-40 and Protease Inhibitor Cocktail (PIC, EDTA-free; Roche). Clear lysates were prepared by repeated centrifugation. Immunoprecipitation was performed at 4°C for 2 h with Dynabeads Protein G that had been pre-coated with anti-tag antibodies. After incubation, the beads were washed three times with HSB, suspended in 50 mM Tris–HCl (pH 8.0), 120 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol, 0.5 mM EDTA, PIC and 50% glycerol. The beads suspension was stored at −80°C until use.
To release Flag-tagged proteins from the Dynabeads, the protein-bound beads were incubated with 150 μg/ml 3× Flag peptide (Sigma) in 50 mM Tris–HCl (pH 8.0), 120 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol, 0.5 mM EDTA and PIC for 30 min on ice. The released proteins were frozen quickly in liquid N2 and stored at −80°C until use.
Protein concentrations of purified fractions were determined by densitometric scanning of SYPRO Ruby-stained sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) gel bands using bovine serum albumin (BSA) as a standard.
Procedures that involve western blotting
Western blotting was carried out as described (11). Cells grown on culture dishes were lysed directly in SDS-PAGE sample buffer (50 mM Tris–HCl: pH 6.8, 2% SDS, 1.25% 2-mercaptoethanol, 250 mM sucrose, 0.0025% bromophenol blue), subjected to 6.5% SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane. Protein-blotted membranes were incubated with anti-topo IIβ monoclonal antibody (clone 3B6, 1.5 μg/ml) (10) and then with horseradish peroxidase-conjugated second antibody (KPL) according to standard procedure. The peroxidase activity was detected by a chemiluminescence method using an ECL kit (GE Healthcare) and recorded on VersaDoc MP 5000 Imaging Systems (Bio-Rad). Protein bands were quantified by densitometry.
For band depletion assay, cells grown on 35 mm-culture dishes were incubated with 200 μM etoposide (VP-16) in DMEM for 30 min at 37°C in a humidified atmosphere of 5% CO2 to stabilize topo IIβ–DNA cleaved complex. The etoposide-treated cells were lysed directly in 100 μl of SDS-PAGE sample buffer and 10 μl-aliquots were subjected to western blot analyses.
Relaxation assay
Relaxation of supercoiled pUC18 (form I) DNA by immunopurified Flag-topo IIβ, either WT or a deletion mutant, was carried out for 30 min at 30°C in 10 μl of reaction mixture containing 5 or 50 ng DNA, 50 mM Tris–HCl (pH 8.0), 120 mM KCl, 10 mM MgCl2, 0.5 mM dithiothreitol, 0.5 mM EDTA, 0.5 mM ATP and 30 μg/ml BSA, unless stated otherwise (27). Reaction products were treated with 1% SDS and 100 μg/ml proteinase K at 55°C for 30 min before applying to 1% agarose gel electrophoresis. Topoisomer DNA bands were visualized by staining with 1/10 000x SYBR Green I (Takara Bio, Japan) or 0.5 μg/ml ethidium bromide. Fully supercoiled DNA substrates that remained unrelaxed after the reaction were quantified by densitometry.
For relaxation of supercoiled DNA in ‘on-bead assay’, the Flag-tagged enzyme protein immobilized on the Dynabeads through anti-Flag antibody was incubated with substrate DNA as described above. After reaction, DNA products were fractionated immediately into enzyme-bound and free in solution by magnetic separation and treated with 1% SDS and 100 μg/ml proteinase K prior to electrophoresis on 1% agarose gel.
RNA-binding assay
RNA was purified from HEK cells by using RNeasy Mini kit (Qiagen). This total RNA was separated into Poly(A)+ RNA and Poly(A)− RNA fractions using Dynabeads mRNA Purification Kit (InvitrogenTM). Purified RNAs were incubated at 30°C for 30 min with 80 fmol Flag-tagged proteins (WT topo IIβ or deletion mutants) immobilized on the Dynabeads in 10 μl of binding mixture containing 50 mM Tris–HCl (pH 8.0), 120 mM KCl, 10 mM MgCl2, 0.5 mM dithiothreitol, 0.5 mM EDTA, 30 μg/ml BSA and 4U RNase inhibitor from porcine liver (Takara Bio). Following magnetic separation, bead-bound RNA (bound to topo IIβ) and free RNA in solution were quantified directly using Quant-iTTM RiboGreen RNA Assay Kit (InvitrogenTM) and GENios multi-detection microplate reader (Tecan).
DNA-binding assay
Flag-tagged proteins (80 fmol of WT topo IIβ or deletion mutants) were immobilized on Dynabeads through anti-Flag antibody and incubated with 5 ng pUC18 plasmid DNA (supercoiled or linear) in 10 μl of the RNA-binding mixture stated above (but without RNase inhibitor). Following magnetic separation, bead-bound DNA (bound to topo IIβ) and free DNA in solution were treated with 1% SDS and 100 μg/ml proteinase K. DNAs were electrophoresed on 1% agarose gel, followed by visualization with SYBR Green I and quantification by densitometry.
RESULTS
Relocation of topo IIβ in interphase nuclei
The catalytic activity of topo IIβ in neuronal cells remains quite high during the terminal differentiation when the enzyme distributes in both nucleoplasm and nucleoli. As the cells develop to mature neurons, it shows decreased activity in vivo, which is measured by its cross-linking to genomic DNA following etoposide treatment. In concert with this change, the enzyme accumulates in nucleoli (9,10). These results suggest that a subpopulation of topo IIβ is engaged in catalytic action by interacting with DNA in the nucleoplasm and another located in nucleoli has little or no access to DNA. In cultured cells, the nuclear distribution of topo IIβ detected by immunostaining varies depending on cell types and preparation methods (17,21,28,29). Since topo IIβ in interphase nuclei of living cells is quite mobile (21), cellular conditions at the time of fixation can be a critical factor for the variable localization.
We used three different cell lines (HEK293, Rat-1, Neuro 2a) to assess the effects of temperature, topo II inhibition and cellular ATP content on distribution (Figure 1). Nucleoli were identified by double-immunostaining with an antibody against nucleolin (9). To our surprise, the nuclear distribution of topo IIβ differed dramatically depending on the temperature of fixation. When cells were fixed at 37°C, topo IIβ distributed throughout the nucleus with remarkable enhancement in nucleoli in all three cell types (Figure 1A). When cells were fixed on ice, however, the signal in nucleoli decreased substantially and the nucleoplasmic stain increased reciprocally, co-localizing with DAPI-staining signal (Figure 1B). A similar relocation of the enzyme was observed by treating the cells with ICRF-193 (Figure 1C), a catalytic inhibitor of type II topos that stabilizes the closed clamp conformation around target DNA during the catalytic reaction (30). In both low temperature and ICRF treatments, some topo IIβ signals in nucleoplasm co-localized with heterochromatic regions (chromocenters), which is most obvious in mouse Neuro 2a cells. Similar pattern of localization was observed by treating mouse cells with etoposide (VP-16), a topo II poison that stabilizes the enzyme–DNA covalent complexes (31). As topo IIβ requires ATP for its catalytic activity (32), cells were depleted of ATP to see whether its nuclear distribution changes. The treatment abolished nucleoplasmic topo IIβ and essentially all fluorescence was localized to the nucleolar region (Figure 1D). These results are in good agreement with the notion that the catalytically active topo IIβ is largely nucleoplasmic, while the nucleolar form is inactive.
Figure 1.
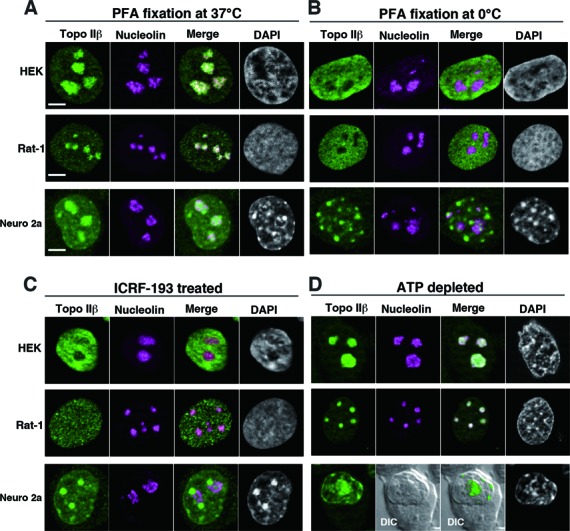
Subnuclear localization of topo IIβ under different conditions. Topo IIβ and nucleolin (as a nucleolus marker) were immunostained with cross-reactive antibodies in human (HEK293), rat (Rat-1) and mouse (Neuro 2a) cell lines under four different conditions. Topo IIβ (green) and nucleolin (magenta) are shown together with merged images and DNA (DAPI). Scale bars, 5 μm. (A) Cells were fixed with PFA at 37°C. (B) Cells were fixed with PFA at 0°C. (C) Cells were first treated with 30 μM ICRF-193 for 15 min and then fixed at 37°C. (D) Cells were first treated with 10 mM sodium azide and 6 mM 2-deoxy-d-glucose for 40 min and then fixed at 37°C.
Dynamic relocation of topo IIβ-EGFP between nucleoplasm and nucleoli in living cells
To further investigate the relationship between the enzyme activity and nuclear distribution in a cellular context, EGFP-fused topo IIβ was transiently expressed in HEK293 cells. As with endogenous enzyme (Figure 1A and C), EGFP signals in live cells were concentrated in nucleoli, and migrated to nucleoplasm upon ICRF-193 treatment (Figure 2B, leftmost column). When cellular ATP levels were reduced to <10% by sodium azide and 2-deoxyglucose for the times indicated (Figure 2A), nucleoplasmic EGFP signals were again strongly reduced. Concurrently, topo IIβ was clearly located in nucleolar regions (Figure 2B, upper panel). In the ATP-depleted cells, the nucleolar signal did not re-distribute to nucleoplasm upon ICRF-193 treatment (Figure 2B, lower panels), suggesting that inactive enzyme remains in the nucleoli. The process was rapid and completely reversible as subsequent recovery of cellular ATP level increased EGFP signals in nucleoplasm back to basal levels and restored the effect of ICRF-193 (Figure 2A and B, rightmost column).
Figure 2.
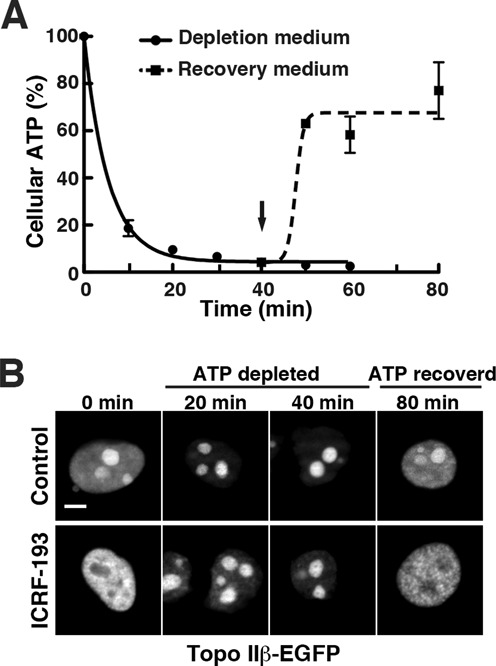
Topo IIβ shuttles between nucleoplasm and nucleoli depending on the cellular content of ATP. At time zero, culture medium was changed to depletion medium containing sodium azide and 2-deoxy-d-glucose. After 40 min, the medium was replaced with normal medium and continued to incubate at 37°C (indicated by arrow). (A) Time course of the relative cellular ATP levels after depletion/recovery treatments. Cells were removed at the time points indicated and cellular ATP was determined by fluorometry and plotted in the graph. Data points represent mean/SD (n = 3). Regression curves were drawn using a software, GraphPad Prism 5 (logistic curve fitting). (B) HEK cells were transfected with pEGFP-top2b and after 24 h, the ATP depletion/recovery procedure was done as in (A). Fluorescence micrographs of nuclei taken at the indicated times are shown in the upper row (control). Shown in the lower row are cells treated with 30 μM ICRF-193 for 15 min at the times indicated. Note that ICRF-induced relocation of topo IIβ does not occur under low ATP condition. Scale bar, 5 μm.
Molecular mechanism underlying the relocation of topo IIβ from nucleoli to nucleoplasm at lowered temperature (Figure 1B) was investigated by time-lapse fluorescence microscopy and FRAP. Medium temperature was lowered continuously from 37 to 15°C in 30 min, and then brought back to 37°C in the next 30 min. Nuclear distribution of topo IIβ-EGFP was recorded in real time and shown in the upper panel (Figure 3A). The nucleolar signal faded out at temperatures below 20°C (20-min incubation) and recovered by the time when the temperature reached 37°C (60-min incubation), indicating that the temperature-induced relocation process is not only rapid, but also reversible. We next compared the motility of nucleoplasmic and nucleolar topo IIβ-EGFP by FRAP under different temperatures. As shown in Figure 3B, the recovery of fluorescence in bleached areas was notably faster in nucleoplasm than in nucleoli at 37°C (binding time = 1.38 ± 0.64 and 3.75 ± 0.98 s, respectively), in good agreement with a previous report (21). At 18°C, however, the mobility of nucleoplasmic topo IIβ-EGFP dropped remarkably. In contrast, the nucleolar mobility change was relatively small (binding time = 7.61 ± 1.83 and 5.89 ± 2.53 s, respectively). Thus, the accumulation of nucleoplasmic topo IIβ at the lower temperature may be due to increased binding times or residence times in the nucleoplasm.
Figure 3.
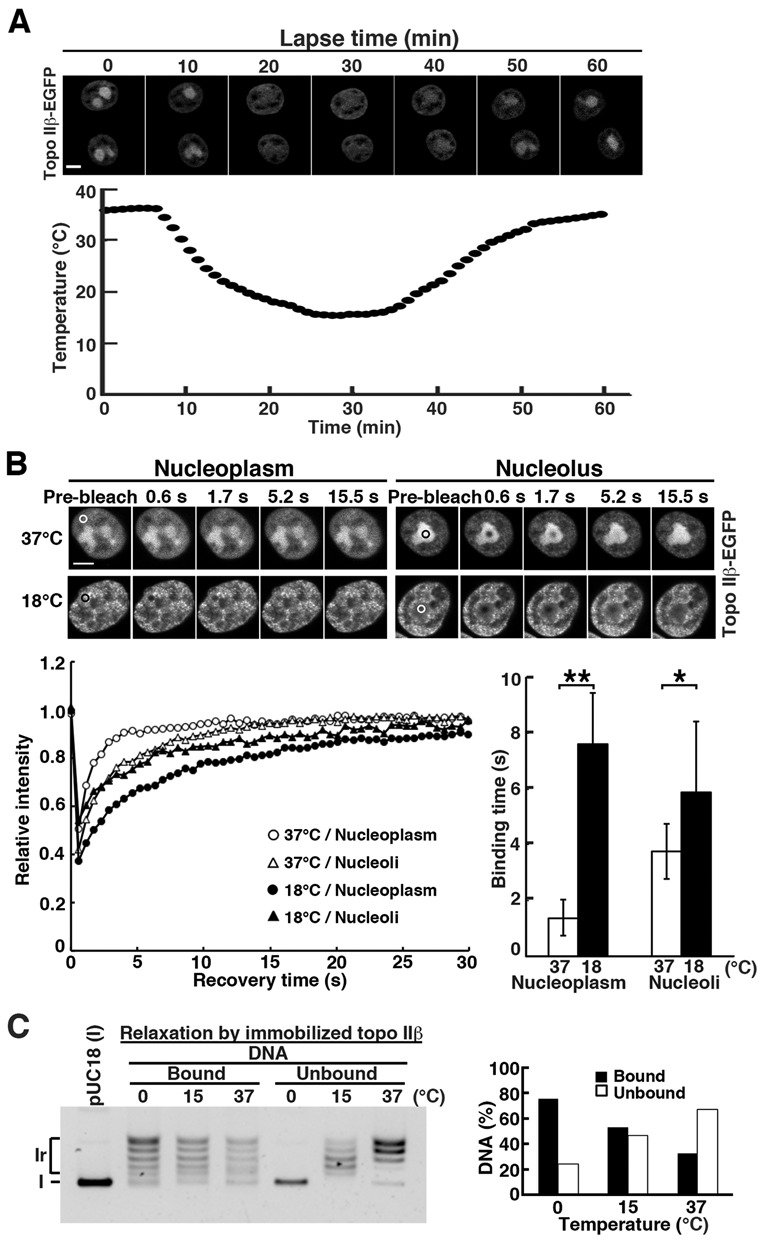
Cold-induced translocation of topo IIβ from nucleolus to nucleoplasm is accounted for by its lowered catalytic rate. (A) Simultaneous recordings of medium temperature and nuclear distribution of topo IIβ-EGFP expressed in HEK cells. Medium temperature was varied using an on-stage heating/cooling device and monitored by a thermocouple thermometer. Images were recorded at 1-min intervals for 60 min. Scale bar, 5 μm. (B) HEK cells transfected with topo IIβ-EGFP were subjected to FRAP analysis either at 37°C or at 18°C. Fluorescence images were recorded after bleaching the circled areas in nucleoplasm or nucleolus. Representative images and recovery curves (fluorescence relative to pre-bleach) are shown. Plotted in the bar graph are binding times in seconds that were calculated from kinetics data of 24 nuclei for each condition using ImageJ 1.4.6. Bars, mean/SD (n = 24); **P = 1.8 × 10−17, *P = 6 × 10−3 by Student's t-test. Scale bar, 5 μm. (C) Temperature dependency of relaxation products in on-bead assay: enzyme-bound versus released DNAs. DNA bands in each lane were quantified by densitometry and relative amounts at each temperature were graphed. Note that the DNA in unbound fraction is fully relaxed at 15°C, but remained supercoiled at 0°C.
To correlate nucleoplasmic-nucleolar dynamics with catalytic activity, we fractionated free and enzyme-bound DNA using immobilized topo IIβ on magnetic beads (on-bead assay). Topo IIβ protein, Flag-tagged at the N-terminus, was expressed in HEK cells and pulled down onto the beads coated with anti-Flag antibody. Washed beads were used for relaxation of supercoiled plasmid DNA. At 37°C, fully relaxed DNA was released from the enzyme and recovered in unbound fraction, whereas partially relaxed DNA remained on the beads (Figure 3C, left panel). The beads-bound fraction reflects the DNA directly bound to topo IIβ since control beads without enzyme or antibody do not bind DNA (data not shown). When the reaction temperature was lowered to 15°C, more plasmid DNA remained on the beads (Figure 3C, right panel). At 0°C, essentially all partially relaxed DNA was bound to the enzyme and unrelaxed substrate was recovered in unbound fraction. At 15°C, the DNA in unbound fraction is fully relaxed. The relative difference in the positions of topoisomer bands between 37 and 15°C is due to the temperature-dependent change in the writhe, which was confirmed by electrophoresis in the presence of ethidium bromide and by time course studies at each temperature (results not shown). These findings recapitulate our in vivo observations, specifically, that the catalytic cycle slows as temperature decreases and thus a higher proportion of topo IIβ would be retained on chromatin DNA in the nucleoplasm.
We further observed differential nuclear localization of topo IIβ mutants with single amino acid changes that cause functional defects (Figure 4A). The constructs were fused with EGFP to track their nuclear location and to unambiguously discriminate from the endogenous WT enzyme. Activity measurements using a band depletion assay with transfected cells confirmed expected properties of these mutants (Figure 4B). This assay is based on the fact that protein bands for catalytically active topo II on immunoblots are reduced after treating cells with a topo II poison such as VP-16. The reduction is due to formation of covalent topo II/DNA complexes (33).
Figure 4.
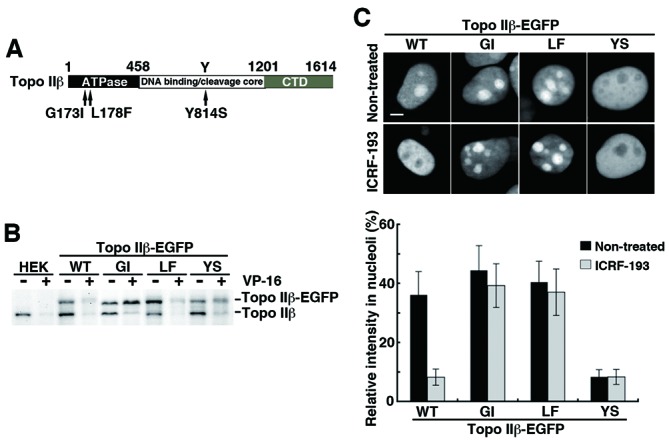
Migratory behavior of functionally defective topo IIβ mutants expressed in nucleus that are tagged with EGFP at C-terminus. (A) Summary of amino acid changes introduced to WT topo IIβ. (B) Assessment of in vivo topo IIβ activity by the band depletion assay. Activities for endogenous enzyme (labeled topo IIβ) and EGFP-fused exogenous one (topo IIβ-EGFP) can be discriminated by the size difference. EGFP-fused mutants are abbreviated: GI, G173I; LF, L178F; YS, Y814S. (C) Changes in the nuclear distribution of topo IIβ mutants after ICRF-193 treatment. Shown here are EGFP fluorescence images (upper panel; scale bar, 5 μm) and the ratio of nucleolar fluorescence to whole nuclear fluorescence (lower graph). The ratio was determined by using the ‘threshold’ and the ‘integrated density (IntDen)’ functions of ImageJ software. Bars, mean/SD (n = 50).
The Gly-164 to Ile mutation in human topo IIα disrupts the ATP binding and hydrolysis activities (34). Therefore, Gly-173 in rat topo IIβ, the counterpart residue of Gly-164 of human α, was altered to Ile (G173I). As shown in Figure 4B, the EGFP-fused G173I mutant (GI-EGFP) expressed in HEK cells did not possess enzymatic activity. The inactive mutant GI-EGFP remained in nucleolar region both in the absence and presence of ICRF-193 (44.5 ± 8.3 and 39.3 ± 7.4%, respectively), as compared to WT-EGFP (36.2 ± 7.9 versus 8.2 ± 2.8%, Figure 4C).
Mutation of Leu-178 to Phe in rat topo IIβ (L178F) converted the enzyme to an ICRF-193-resistant form just like the human topo IIα counterpart, L169F (35). Although LF-EGFP was enzymatically active (Figure 4B), it was resistant to ICRF-193 (Figure 4C), as it accumulated in nucleoli in the presence or absence of ICRF-193 (37.1 ± 7.9 and 40.4 ± 7.1%, respectively). The resistance of L178F/LF-EGFP toward ICRF-193 was also confirmed by in vitro activity assays (results not shown).
Tyr-814 of rat topo IIβ is an essential residue for the activity. The Tyr to Ser mutant (Y814S) was incompetent in the formation of a DNA cleavage complex (Figure 4B) as in the previously characterized human topo IIα mutant, Y805S (36). Figure 4C shows that YS-EGFP is largely non-nucleolar, even in the absence of ICRF-193 (8.3 ± 2.4% in nucleoli). Treatment of the cells with ICRF-193 did not change the distribution (8.3 ± 2.5% in nucleoli). Since the active site tyrosine mutant of human topo IIα (Y805S) was shown to be able to close the N-gate (36), nucleoplasmic YS-EGFP is likely due to trapping by the N-terminal clamp.
Association of topo IIβ with RNA in nucleoli
Although immobile topo IIβ molecules are virtually absent in living cells, the enzyme is not as freely diffusible as free EGFP (21). A simple model analysis for nuclear topo IIβ distribution showed that a higher proportion of the enzyme is ‘bound’ as compared to ‘free’ in nucleolus, as well as in nucleoplasm (37). In addition to this, except for the Y814S mutant, almost half of topo IIβ located in nucleoli in living cells (Figure 4C), and motility of the WT enzyme in nucleoli was even smaller than that in nucleoplasm at 37°C (Figure 3B). These findings strongly suggest that some components in the nucleolus associate with topo IIβ. We assume that RNA might be one of the major candidates. To test this, HEK cells transfected with WT-EGFP were permeabilized with digitonin and treated in situ with RNase A or DNase I. The specificity and completeness of digestion was confirmed by agarose gel electrophoresis (Supplementary Figure S2A). The nucleolar localization of WT-EGFP did not change by permeabilization or DNase treatment. However, RNase digestion resulted in relocation of EGFP signal from nucleoli to nucleoplasm (Supplementary Figure S2B), suggesting that nucleolar retention of topo IIβ is mediated primarily through RNA–topo IIβ interactions.
A novel domain in the C-terminal region of topo IIβ is involved in nucleolar retention
RNA interactions with a DNA-binding protein like topo II may be due to non-specific binding activity to a polyanion scaffold. The likelihood of a non-specific, non-physiological artifact is reduced if binding is selectively mediated by specific protein domains. To examine the domains responsible for topo IIβ–RNA interaction and nucleolar preference, we prepared topo IIβ deletion mutants and examined their nuclear localization together with RNA-binding abilities in vitro. The canonical CTD of human topo IIβ starts from amino acid residue #1202 and ends at the C-terminus #1621 (38) that corresponds to rat topo IIβ #1195–1614. In this study, the rat enzyme was simply divided into two portions, a C-terminal segment (CTD, #1201–1614) and its complementary segment (ΔCTD, #1–1199), which contains the ATPase domain, dimerization domain and DNA-binding/cleavage core. The CTD’ (#1251–1614), missing N-terminal 50 residues of CTD, is equivalent to the human topo IIβ #1258–1621 that contains three NLSs and is capable of transferring a marker protein (β-galactosidase) into nuclei (17). WT enzyme and the truncation mutants were dually tagged with Flag on N-terminus and EGFP on C-terminus, and expressed in HEK cells (Figure 5A).
Figure 5.
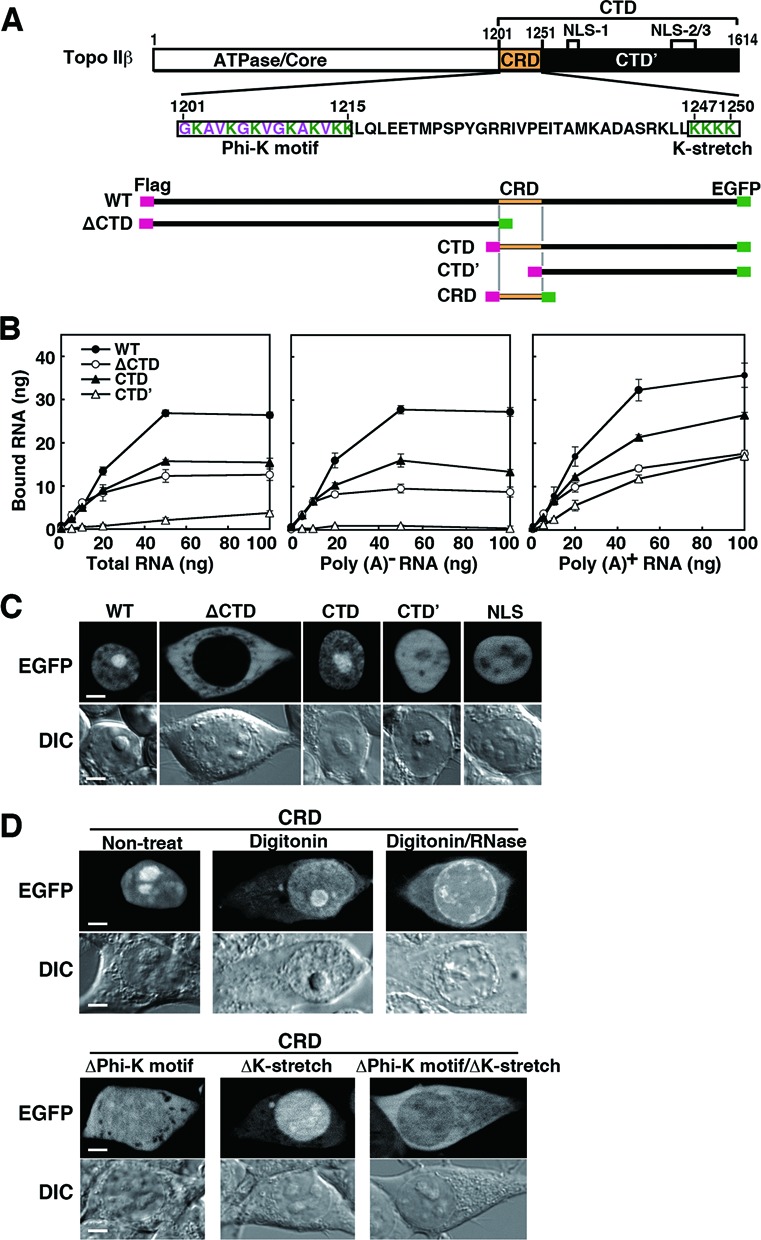
RNA-binding ability and cellular localization of topo IIβ domain-deletion mutants. (A) Domain diagram of deletion mutants used in this experiment that are dually tagged with Flag/EGFP. Amino acid sequence for CRD (C-terminal regulatory domain) and its subdomains (boxed) are given in the middle. (B) Binding of cellular RNA fractions with topo IIβ deletion mutants immobilized on magnetic beads. Plotted data are expressed in mean with SD (n = 4). (C) Cellular localization of topo IIβ deletion mutants. EGFP images are shown along with differential interference contrast (DIC) images. Images for the SV40 NLS (PKKKRKV) cloned in pEGFP-N1 are put on the right as a control. Scale bars, 5 μm. (D) Localization of CRD in intact, digitonin-treated and digitonin/RNase-treated cells (upper panel). The subdomains boxed in A were further deleted from CRD and their cellular localizations were examined (lower panel). EGFP images are shown along with DIC images. Scale bars, 5 μm.
Tagged proteins were pulled down with anti-Flag antibody-coated magnetic beads, followed by incubation either with total RNA, poly (A)− RNA, or poly (A)+ RNA isolated from HEK cells. The WT topo IIβ bound RNA regardless of poly (A) tails (Figure 5B). Both ΔCTD and CTD retained the RNA-binding activity, although bound RNA levels were lower than those of WT. As shown in Figure 5B (center panel), CTD’ did not bind poly (A)− RNA, whereas the mutant did bind poly (A)+ RNA (Figure 5B, right panel), indicating that the N-terminal 50 residues of CTD are required for the binding of poly (A)− RNA.
Correlations between RNA-binding activity and cellular localization of these mutants were analyzed. Although ΔCTD binds to both poly (A)− and poly (A)+ RNAs, this mutant was localized exclusively in cytoplasm most likely due to the absence of an NLS (Figure 5C). Just like the WT enzyme, the EGFP signal for CTD localized in nuclei with nucleolar enrichment. However, CTD’ did not accumulate in nucleoli, although the mutant exclusively localized in nuclei. This observation agrees with the previous report (17). The EGFP-tagged SV40 NLS showed similar localization pattern (Figure 5C). These data suggest that NLS alone is not sufficient for the nucleolar accumulation and the region (#1201–1250) is essential for the topo IIβ CTD to accumulate in nucleoli and for its binding to RNA.
Since this 50-residue domain appears to have a prime importance in the enzyme's transactions, we refer to this domain as the CRD (or ‘C-terminal regulatory domain’) and performed further analyses by expressing CRD in HEK cells as an EGFP-tagged protein (Figure 5D). The localization pattern of CRD was similar to that of WT and CTD, being exclusively in nuclei with nucleolar accumulation (Figure 5D, upper panel). Also like the WT-EGFP, the CRD-EGFP relocated from nucleoli to nucleoplasm upon in situ digestion with RNase in digitonin-permeabilized cells. Thus, nucleolar retention of CRD may be mediated by its binding to RNA in nucleoli.
CRD contains some characteristic sequence motifs within lysine clusters on both ends (Figure 5A). The #1201–1215 region is composed of regularly spaced repeats of lysine (K) and small hydrophobic residues: glycine (G), alanine (A) or valine (V), which is termed here Phi-K motif. The #1247–1250 region contains four consecutive lysine residues (K-stretch). When Phi-K motif was deleted from CRD (ΔPhi-K motif), the EGFP signal distributed diffusely throughout the cell (Figure 5D, lower panel). Deletion of K-stretch (ΔK-stretch) from CRD did not affect its nuclear localization, whereas its nucleolar accumulation was almost lost, suggesting that the K-stretch is indispensable for nucleolar localization of CRD. When both Phi-K motif and K-stretch were deleted from CRD, the mutant distributed in cytoplasm. These results clearly indicate that Phi-K motif is responsible for the nuclear retention and that Phi-K motif and K-stretch cooperate together for the accumulation of CRD in nucleoli.
CRD regulates enzymatic activity of topo IIβ through RNA binding
The function of CRD was further examined using topo IIβ deletion mutants illustrated in Figure 6A. The ΔCTD’ is a truncation mutant lacking CTD’. The WT and mutant enzymes were Flag-tagged on their N-termini, expressed in HEK cells and immunopurified with anti-Flag antibody. Purified proteins were subjected to relaxation assay with supercoiled plasmid DNA. Topo IIβ released from antibody beads showed similar relaxation activities regardless of the presence of deletion (Supplementary Figure S3A and B). We showed previously that relaxation activity of topo IIβ was inhibited by cellular RNA (23). As expected, total RNA purified from HEK cells inhibited the relaxation of Flag-tagged WT topo IIβ, whereas that of ΔCTD was not inhibited at all (Figure 6B). Remarkably, however, RNA did inhibit the reaction with ΔCTD’. Therefore, we examined whether topo IIβ lacking only CRD (ΔCRD) was resistant to RNA and found that this is indeed the case. To confirm the resistance of ΔCRD to RNA, enzyme inputs were varied in a dilution series keeping the RNA amount constant (Supplementary Figure S4A). These data show that relaxation activity was essentially independent of enzyme/RNA ratios.
Figure 6.
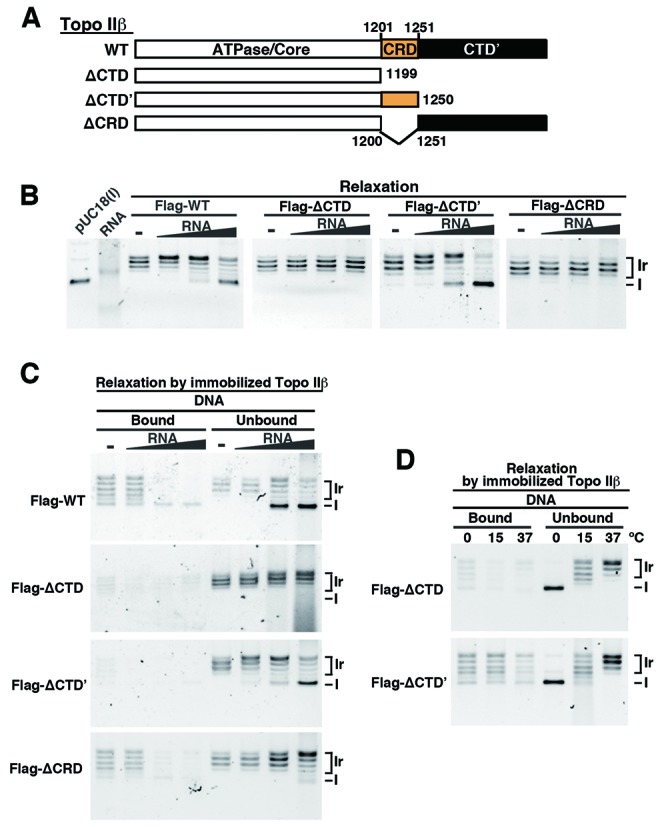
Effects of RNA on the relaxation activity of topo IIβ domain-deletion mutants. (A) Domain structure of deletion mutants used here that are Flag-tagged on N-terminus (not shown in the figure). (B) Inhibitory effects of total RNA on the relaxation activity. Tag-purified topo IIβ and the domain mutants (20 fmol) were incubated with 5 ng of supercoiled pUC18 DNA. Increasing amounts of RNA (5, 50, 500 ng) were added to the reaction. Positions of supercoiled form (I) and relaxed form (Ir) DNA are indicated on the right. (C) Fractionation of relaxation intermediates into enzyme-bound and released DNA by using 80 fmol immobilized topo IIβ domain mutants (on-bead assay). After incubation with 5 ng of supercoiled DNA as described in ‘Methods’ section, product DNA was fractionated into ‘bound’ and ‘unbound’ by magnetic separation. (D) Effects of reaction temperature on the retention of relaxation intermediates on the enzyme. DNA in unbound fraction indicates that the supercoiled substrate remained almost unreacted at 0°C, but no unreacted substrate remained at 15°C. Difference in the ladder position between 15 and 37°C is due to the temperature-dependent alteration of the writhe for relaxed form (Ir). Results for the WT enzyme are shown in Figure 3C.
When the inhibition experiments were repeated under the same conditions using RNA samples fractionated into poly (A)− and poly (A)+ RNAs, we obtained basically the same results with unfractionated total RNA. Thus, CRD is very likely responsible for the regulation of enzyme activity mediated by RNA regardless of poly (A) tailing.
CRD is involved, but not essential, in the processivity of topo IIβ
The use of a bead-bound topo IIβ assay allowed us to assess the topological status of substrate DNA that is free versus enzyme bound. Negatively supercoiled DNA was relaxed by the immobilized WT or mutant topo IIβ in the presence of increasing RNA inputs (Figure 6C). Without RNA, WT enzyme did not release the product until DNA was fully relaxed, reflecting the processive nature of WT enzyme. To see whether CRD contributes to the processivity, topo IIβ mutants were compared. When incubated at 30°C, it appeared that CRD has little contribution to the processive mode of reaction since ΔCTD’ that contains CRD did not hold partially relaxed DNA, while ΔCRD did (Figure 6C). However, when the reaction was performed at lower temperatures (to reduce catalytic rates), ΔCTD’ did retain the intermediate products (Figure 6D, lower panel). In contrast, ΔCTD did not bind partially relaxed DNA even at lower temperatures (Figure 6D, upper panel), or at decreased activity with less ΔCTD protein (Supplementary Figure S4B). The results indicate that without CTD, topo IIβ–DNA interaction is highly distributive, viz. non-processive.
Thus, CRD appears to be involved, but not essential, in the processive reaction and the CTD’ is more likely responsible for processivity of topo IIβ through retention of partially relaxed DNA. The binding or retention of these intermediate products is inhibited by increasing RNA inputs (Figure 6C).
CRD assists the RNA-mediated inhibition of supercoiled DNA binding to topo IIβ
Previous studies on topo II–DNA interactions demonstrated that the enzyme prefers to bind crossovers between duplex DNA segments, which occur frequently in supercoiled conformation (39,40). More recent studies suggested the involvement of CTD in capturing the G-segment that is strongly bent between the paired winged-helix domains (41,42). Little biochemical knowledge is available, however, concerning the DNA-binding properties of topo II relative to supercoils. We took advantage of the on-bead assay to investigate the binding of DNA and effects of RNA on the binding in the absence of ATP. Without ATP, the enzyme would behave like a simple DNA binder with constitutively open N-gate.
DNA-binding reaction was first optimized by incubating equimolar mixtures of negatively supercoiled (form I) and linear (form III) DNAs with constant amount of immobilized WT-topo IIβ (Supplementary Figure S5). The enzyme bound supercoiled DNA preferentially as total DNA amounts increase to a saturation level, but both forms were equally bound 100% at a lower dosage (5 ng each). Under the latter conditions, competitive binding assay was performed with WT and deletion mutants (Figure 7A). All mutants showed preference toward supercoiled DNA over linear DNA, although with reduced overall affinity. Reduction of DNA-binding ability was also reported with enzyme–oligonucleotide interactions when C-terminal region (#1264–1621) was deleted from human topo IIβ (43). The deleted region corresponds to #1257–1614 of rat topo IIβ. Deletion of CTD (ΔCTD) significantly decreased linear DNA binding. Adding back either CRD or CTD’ however restored binding to 50–60%. Without CTD’, CRD was essential for linear DNA binding.
Figure 7.
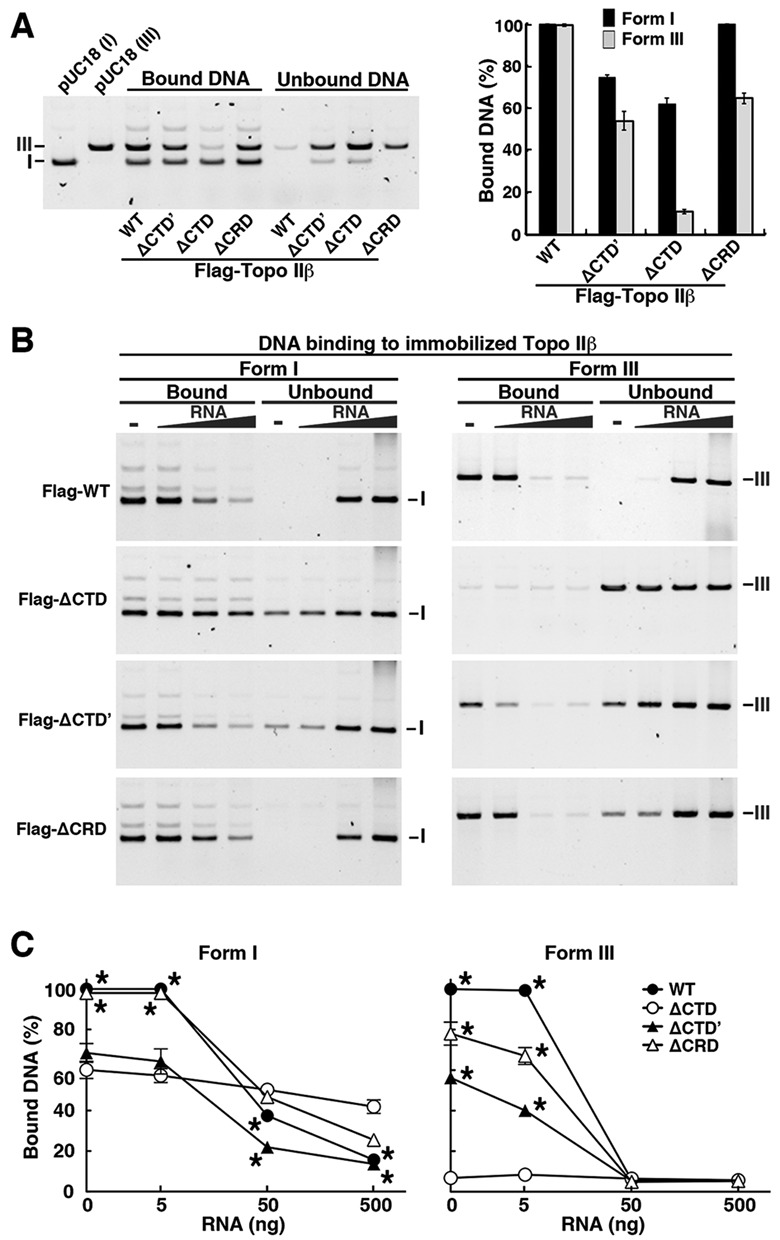
DNA-binding assays of Flag-tagged topo IIβ domain-deletion mutants in the absence of ATP. (A) Competitive binding of supercoiled (form I) and linear (form III) DNA to the enzyme (80 fmol) immobilized on beads. After incubating equimolar mixture (5 ng each) of supercoiled and linear DNA with the beads, bound and unbound DNA was fractionated by magnetic separation. DNA amounts in agarose gel bands were quantified by densitometry and plotted in the graph as percentages of input DNA (n = 3). (B) Effects of RNA on DNA binding. Five nanograms of supercoiled and linear DNAs were incubated separately with the enzyme (80 fmol). RNA was added as in Figure 6C. (C) Relative amounts of bound DNA as determined by densitometry of the gel images shown in (B). Supercoiled or linear DNA amounts bound to WT enzyme in the absence of RNA were set to 100%. Data points are mean with SD bar (n = 5). Asterisks indicate a significant deviation from ΔCTD (P < 0.005).
Similar results were obtained when supercoiled and linear DNAs were incubated separately (Figure 7B, leftmost lanes). The data clearly indicate that CTD, including CRD, is not essential for supercoiled DNA binding, but CTD’ assists the binding. In contrast, CTD is essential for linear DNA binding (Figure 7C, right panel) and mutants harboring either CRD or CTD’ retained linear DNA-binding activity with reduced levels, suggesting that CRD alone retains the ability to bind linear DNA.
In the presence of RNA, binding of supercoiled DNA to ΔCTD decreased slightly in a dose-dependent manner (Figure 7B and C), although the change was statistically insignificant (P > 0.10). In contrast, the RNA inhibition was significant (P < 0.01) when topo IIβ contains CRD (compare ΔCTD and ΔCTD’). Similarly, ΔCRD was less susceptible to RNA compared to WT (P < 0.01). Thus, CRD appears to be a principal element for the RNA interference on the association between topo IIβ and supercoiled DNA. Importantly, this parallels the effects of RNA on catalytic activity (Figure 6). As for linear DNA, RNA strongly inhibited both CRD- and CTD’–DNA interactions (Figure 7C, right panel).
These data suggest that supercoiled DNA is recognized by and bound to the N-terminal catalytic core (ΔCTD) that contains minimal topo IIβ activities of ATPase and DNA binding/cleavage. The consecutive short-segment, CRD, is not required for the supercoiled DNA binding, but it regulates the association between the enzyme core and substrate DNA by an RNA-mediated mechanism, probably through direct interaction with RNA. Taken together, CRD is a critical domain that controls topo IIβ activity by conferring its susceptibility to RNA and directionality toward nucleolus.
DISCUSSION
Subnuclear localization and catalytic activity of topo IIβ are linked
Topo IIβ in interphase nuclei was reported to be highly mobile and free to exchange between nuclear subcompartments (21). The enzyme relocates from nucleoli to nucleoplasm upon stabilizing the catalytic intermediates, indicating that it is most actively engaged in DNA catalysis in nucleoplasm. The notion is in good agreement with our previous observation that in differentiating neuronal cells, a large proportion of topo IIβ is involved in catalytic action and distributed in nucleoplasm, whereas in mature neuronal cells the enzyme is concentrated in nucleoli and has a limited access to chromatin DNA, although isolated enzyme is fully active on plasmid DNA in vitro. (9,10).
Using topo IIβ mutants, we have directly demonstrated in this study that enzymatically incompetent topo IIβ accumulates in the nucleolus. In ATP-depleted cells, both endogenous and EGFP-fused topo IIβ are exclusively nucleolar. The G173I mutant lacking ATP-binding activity was exclusively nucleolar when expressed in HEK cells. We also showed that nucleoplasmic topo IIβ is the catalytically active form. Topo IIβ clamped around target DNA by ICRF-193 treatment distributed predominantly in nucleoplasm. As expected, enzymatically incompetent topo IIβ (the G173I mutant and WT topo IIβ in ATP-depleted cells) did not change the nucleolar localization even after ICRF-193 treatment. The inactive tyrosine mutant, Y814S, was most likely immobilized on chromatin by the N-terminal clamp and thus heavily enriched in the nucleoplasm.
To our knowledge, the reversible translocation of topo IIβ induced by temperature has not been previously described. FRAP experiments and DNA relaxation with immobilized enzyme reveal that this effect is due to slower catalytic rates of topo IIβ at suboptimal temperatures, which results in prolonged residence time in nucleoplasm in living cells.
RNA is a major determinant of topo IIβ localization in nucleoli
The binding time of topo IIβ at 37°C estimated from FRAP was much longer in nucleoli than in nucleoplasm (Figure 3B), which is in good agreement with Christensen et al. (21). They further showed later using a mathematical distribution model with simple differential equations that nuclear topo IIβ can be distinguished between free and bound fractions, indicating a higher proportion of bound form in nucleoli than in nucleoplasm (37). Whereas the binding time for nucleoplasmic topo IIβ increased remarkably at lower temperatures because of its decreased turnover rate on DNA, the binding time in nucleoli did not increase significantly (Figure 3B), indicating that the enzyme is not interacting with DNA. Therefore, the putative binding partner(s) of topo IIβ in nucleoli is unlikely to be genomic DNA (presumably ribosomal DNA repeats).
Salt extraction experiments with whole cells indicated that the interaction between topo IIβ and cellular components is stable in 0.12 M NaCl, but disrupted in 0.42 M NaCl, suggesting the ionic nature of this interaction (data not shown). Furthermore, treatment of permeabilized cells with RNase, but not DNase, resulted in the relocation of topo IIβ-EGFP from nucleoli to nucleoplasm (Supplementary Figure S2B). We have also presented more direct evidence for the binding of cellular RNA with topo IIβ that is immobilized on magnetic beads (Figure 5B). These data suggest that RNA or an RNA-containing complex is a major holding element of topo IIβ in nucleoli that contributes to the prolonged residence of the enzyme in nucleoli.
Identification of a new domain in CTD that mediates topo IIβ–RNA interactions and nucleolar localization
EGFP-fused CTD expressed in HEK293 cells localized exclusively in nuclei with enhanced targeting to nucleoli. CTD’-EGFP, however, localized predominantly to nucleoplasmic regions (and was depleted from nucleoli). The human counterpart of CTD’ fragment that contains multiple NLSs also localizes in nucleoplasm but not in nucleoli (17). It appears, therefore, the N-terminal 50 residues of CTD (CRD) are indispensable for nucleolar accumulation of CTD.
The CRD is located in a region predicted to be intrinsically disordered (Supplementary Figure S6A). However, its sequence is highly conserved among rat, mouse and human topo IIβ, but diversified from topo IIα (Supplementary Figure S6B). EGFP-fused CRD is small enough to passively diffuse into nuclei. For nuclear retention of CRD, the ‘Phi-K motif’ composed of a 15-amino acid stretch with alternating Lys and hydrophobic amino acids seems to be essential (Figure 5D). The Phi-K motif, however, does not behave like a functional NLS since ΔCTD’-EGFP that contains the Phi-K motif but not a canonical NLS localized predominantly in cytoplasm (data not shown). CRD also contains a stretch of four Lys residues (termed K-stretch) that was necessary, but not sufficient for nucleolar accumulation of CRD, which required both Phi-K motif and K-stretch.
Experimental validation of nucleolar localization signals (NoLSs) revealed that NoLSs are mainly comprised of a stretch of basic residues (Arg or Lys) and are localized in regions predicted to be α-helices or coils (44). Secondary structure of CRD around Phi-K motif and K-stretch predicted by Jpred 3 (45) did not contradict this view. Thus, the Lys-rich regions in CRD (Phi-K motif and K-stretch) are likely to function together as a NoLS also in full-length topo IIβ.
Unlike the nucleus and other membrane-bound organelles, there is no membrane separating the nucleolus from the surrounding nucleoplasm; thus, any soluble molecule could diffuse in and out of the nucleolar compartment. Targeting of a specific molecule to the nucleolus requires direct or indirect interaction with one of the nucleolar building blocks composed of rDNA, its transcripts, small nucleolar RNAs and a number of ribosomal/non-ribosomal proteins (46). A short stretch of positively charged amino acids of human T-cell leukemia virus Rex proteins that localize in nucleoli contributes to the binding of Rex to its target RNA (47). An Arg/Lys-rich peptide derived from TRBP, a cellular protein that binds HIV-1 trans-activation responsive (TAR) RNA, was necessary and sufficient to ensure binding to the upper-stem/loop site of TAR that contains a double-stranded RNA (dsRNA). The Arg/Lys-rich peptide of TRBP is composed of 15 amino acid residues with alternations of Lys/Arg and hydrophobic amino acid stretches (48). The dsRNA elements can be formed by base pairing of complementary sequences within primary RNA transcripts. Based on these reports and our current results, we speculate that topo IIβ stays in nucleoli through interaction between CRD and nucleolar RNA.
CRD is an essential element for RNA-mediated regulation of topo IIβ activity
The CTD-truncated mutant (ΔCTD) was catalytically as active as full-length topo IIβ (15). However, CTD was essential for the RNA-mediated inhibition of topo IIβ catalytic activity. We have shown previously that relaxation of supercoiled DNA by full-length topo IIβ was inhibited by cellular RNA (23). In the present study, it became clear that the RNA inhibition is abolished by truncation of CTD. A responsive element of the RNA-mediated inhibition was more finely mapped to CRD, since relaxation of supercoiled DNA by CRD-deleted mutants (ΔCTD and ΔCRD) was insensitive to RNA, whereas the relaxation by the CRD containing mutant (ΔCTD’) was inhibited.
To understand the molecular mechanism of RNA-mediated regulation of topo IIβ through CRD, we compared DNA binding of immobilized topo IIβ with supercoiled (form I) and linear (form III) targets in the absence of ATP. The model shown in Figure 8A and B will be helpful for understanding the following discussions. Without ATP, N-gate of the enzyme is supposed to be open and supercoiled DNA will not be relaxed (49,50). Full-length topo IIβ can bind with both supercoiled and linear DNA with preference to supercoils (Figure 7A and Supplementary Figure S5), which is consistent with the notion that eukaryotic type II topos prefer supercoiled substrates because DNA crossings are more prevalent in superhelical DNA compared to unconstrained DNA (39,40,50,51). All the mutants we studied bound supercoiled DNA, although the ability decreased somewhat when CTD or CTD’ was deleted. To our surprise, in contrast to supercoiled DNA, linear DNA did not bind to the ΔCTD mutant (both CTD’ and CRD were deleted), although it retained binding activity to supercoiled DNA.
Figure 8.
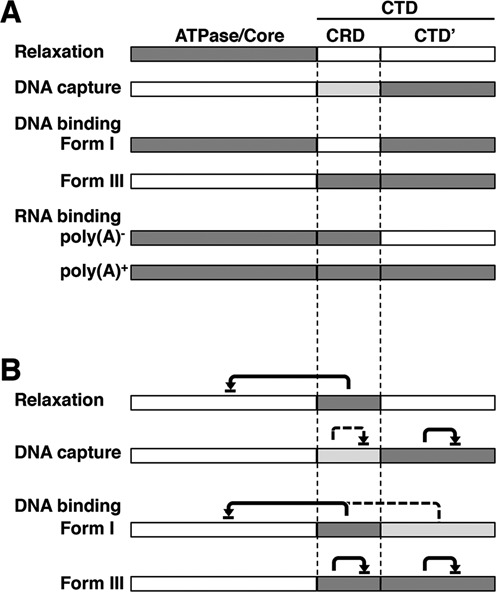
Summary for relative contributions of topo IIβ functional domains. (A) Domains involved in the activities shown on the left are shaded. Dark shade and light shade represent full and partial contribution, respectively. (B) RNA-mediated inhibition. Inhibitory domains are shaded depending on their relative contributions as in (A). Domains pointed by arrowhead are inhibited domains. Solid line and broken line designate the strong and weak inhibitions, respectively. Form I, supercoiled DNA; Form III, linear DNA.
These data suggest that supercoiled DNA binds to the core region of topo IIβ as an initial step and the CTD facilitates this binding. Inhibitory effects of RNA on supercoiled DNA binding to topo IIβ mutants resembled those on DNA relaxation activity of the mutants. This implies that the binding of RNA with CRD inhibits the interaction of supercoiled DNA substrate with the core region, resulting in the inhibition of relaxation (Figure 8B). The inhibition appears to be non-competitive in nature and CRD behaves like a built-in RNA-responsive element for the regulation of topo IIβ activity. Similarly, inhibitory effects of RNA on linear DNA binding to topo IIβ mutants resembled those on DNA capture by the enzyme during catalysis (Figure 8B). The CTD-truncated topo IIβ does not bind linear DNA and lacks the ability to hold partially relaxed DNA, suggesting that interaction between CTD and substrate DNA is required for the retention of G-segment during the relaxation in a processive mode. RNA may, thus, interfere with the DNA retention by binding competitively to CTD (especially to CTD’ portion).
Although crystallographic information on CTD is not available at present, it would be possible to extrapolate the position of CRD relative to the known structure because CRD resides right next to the C-terminus of the crystalized fragments (3,38,42,52). CTD’ may protrude away from the core region of topo IIβ and involve in holding the G-segment to drive the enzyme toward processive mode. However, CRD is probably placed close to the DNA gate and the active site tyrosine, thus regulating cleavage reaction more or less directly through interaction with RNA. Future studies on CRD structure would help clarify matters. The topo IIβ CRD and corresponding region of topo IIα are not highly homologous, although they share some features in common (Supplementary Figure S6B). It is not known whether this region in topo IIα serves as a functional counterpart of topo IIβ CRD.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We are grateful to Dr Mark T. Muller for his critical reading of the manuscript.
Footnotes
Present address:
Shinji Kawano, Department of Biochemistry, Faculty of Science, Okayama University of Science, 1-1 Ridai-cho, Kita-ku, Okayama 700-0005, Japan.
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
Grant-in-Aid for Scientific Research funding [23310133 to K. T.]; Science and Technology funding [to K.M.T.] from the Ministry of Education, Science, Sports and Culture of Japan. Source of Open Access funding: University infrastructure funding.
Conflict of interest statement. None declared.
REFERENCES
- 1.Wang J.C. DNA topoisomerases. Annu. Rev. Biochem. 1996;65:635–692. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- 2.Berger J.M. Type II DNA topoisomerases. Curr. Opin. Struct. Biol. 1998;8:26–32. doi: 10.1016/s0959-440x(98)80006-7. [DOI] [PubMed] [Google Scholar]
- 3.Berger J.M., Gamblin S.J., Harrison S.C., Wang J.C. Structure and mechanism of DNA topoisomerase II. Nature. 1996;379:225–232. doi: 10.1038/379225a0. [DOI] [PubMed] [Google Scholar]
- 4.Austin C.A., Fisher L.M. Isolation and characterization of a human cDNA clone encoding a novel DNA topoisomerase II homologue from HeLa cells. FEBS Lett. 1990;266:115–117. doi: 10.1016/0014-5793(90)81520-x. [DOI] [PubMed] [Google Scholar]
- 5.Jenkins J.R., Ayton P., Jones T., Davies S.L., Simmons D.L., Harris A.L., Sheer D., Hickson I.D. Isolation of cDNA clones encoding the beta isozyme of human DNA topoisomerase II and localisation of the gene to chromosome 3p24. Nucleic Acids Res. 1992;20:5587–5592. doi: 10.1093/nar/20.21.5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsutsui K., Tsutsui K., Okada S., Watanabe M., Shohmori T., Seki S., Inoue Y. Molecular cloning of partial cDNAs for rat DNA topoisomerase II isoforms and their differential expression in brain development. J. Biol. Chem. 1993;268:19 076–19 083. [PubMed] [Google Scholar]
- 7.McClendon A.K., Gentry A.C., Dickey J.S., Brinch M., Bendsen S., Andersen A.H., Osheroff N. Bimodal recognition of DNA geometry by human topoisomerase II alpha: preferential relaxation of positively supercoiled DNA requires elements in the C-terminal domain. Biochemistry. 2008;47:13 169–13 178. doi: 10.1021/bi800453h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woessner R.D., Mattern M.R., Mirabelli C.K., Johnson R.K., Drake F.H. Proliferation- and cell cycle-dependent differences in expression of the 170 kilodalton and 180 kilodalton forms of topoisomerase II in NIH-3T3 cells. Cell Growth Differ. 1991;2:209–214. [PubMed] [Google Scholar]
- 9.Tsutsui K., Tsutsui K., Hosoya O., Sano K., Tokunaga A. Immunohistochemical analyses of DNA topoisomerase II isoforms in developing rat cerebellum. J. Comp. Neurol. 2001;431:228–239. doi: 10.1002/1096-9861(20010305)431:2<228::aid-cne1067>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 10.Tsutsui K., Tsutsui K., Sano K., Kikuchi A., Tokunaga A. Involvement of DNA topoisomerase IIbeta in neuronal differentiation. J. Biol. Chem. 2001;276:5769–5778. doi: 10.1074/jbc.M008517200. [DOI] [PubMed] [Google Scholar]
- 11.Sano K., Miyaji-Yamaguchi M., Tsutsui K.M., Tsutsui K. Topoisomerase IIbeta activates a subset of neuronal genes that are repressed in AT-rich genomic environment. PLoS One. 2008;3:e4103. doi: 10.1371/journal.pone.0004103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyu Y.L., Lin C.P., Azarova A.M., Cai L., Wang J.C., Liu L.F. Role of topoisomerase IIbeta in the expression of developmentally regulated genes. Mol. Cell. Biol. 2006;26:7929–7941. doi: 10.1128/MCB.00617-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tiwari V.K., Burger L., Nikoletopoulou V., Deogracias R., Thakurela S., Wirbelauer C., Kaut J., Terranova R., Hoerner L., Mielke C., et al. Target genes of Topoisomerase IIbeta regulate neuronal survival and are defined by their chromatin state. Proc. Natl Acad. Sci. U.S.A. 2012;109:E934–E943. doi: 10.1073/pnas.1119798109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khelifa T., Casabianca-Pignede M.R., Rene B., Jacquemin-Sablon A. Expression of topoisomerases II alpha and beta in Chinese hamster lung cells resistant to topoisomerase II inhibitors. Mol. Pharmacol. 1994;46:323–328. [PubMed] [Google Scholar]
- 15.Crenshaw D.G., Hsieh T. Function of the hydrophilic carboxyl terminus of type II DNA topoisomerase from Drosophila melanogaster. I. In vitro studies. J. Biol. Chem. 1993;268:21 328–21 334. [PubMed] [Google Scholar]
- 16.Schoeffler A.J., Berger J.M. DNA topoisomerases: harnessing and constraining energy to govern chromosome topology. Q. Rev. Biophys. 2008;41:41–101. doi: 10.1017/S003358350800468X. [DOI] [PubMed] [Google Scholar]
- 17.Cowell I.G., Willmore E., Chalton D., Marsh K.L., Jazrawi E., Fisher L.M., Austin C.A. Nuclear distribution of human DNA topoisomerase IIbeta: a nuclear targeting signal resides in the 116-residue C-terminal tail. Exp. Cell Res. 1998;243:232–240. doi: 10.1006/excr.1998.4150. [DOI] [PubMed] [Google Scholar]
- 18.Mirski S.E., Gerlach J.H., Cummings H.J., Zirngibl R., Greer P.A., Cole S.P. Bipartite nuclear localization signals in the C terminus of human topoisomerase II alpha. Exp. Cell Res. 1997;237:452–455. doi: 10.1006/excr.1997.3805. [DOI] [PubMed] [Google Scholar]
- 19.Mirski S.E., Gerlach J.H., Cole S.P. Sequence determinants of nuclear localization in the alpha and beta isoforms of human topoisomerase II. Exp. Cell Res. 1999;251:329–339. doi: 10.1006/excr.1999.4587. [DOI] [PubMed] [Google Scholar]
- 20.Linka R.M., Porter A.C., Volkov A., Mielke C., Boege F., Christensen M.O. C-terminal regions of topoisomerase IIalpha and IIbeta determine isoform-specific functioning of the enzymes in vivo. Nucleic Acids Res. 2007;35:3810–3822. doi: 10.1093/nar/gkm102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christensen M.O., Larsen M.K., Barthelmes H.U., Hock R., Andersen C.L., Kjeldsen E., Knudsen B.R., Westergaard O., Boege F., Mielke C. Dynamics of human DNA topoisomerases IIalpha and IIbeta in living cells. J. Cell Biol. 2002;157:31–44. doi: 10.1083/jcb.200112023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park S.W., Parrott A.M., Fritz D.T., Park Y., Mathews M.B., Lee C.G. Regulation of the catalytic function of topoisomerase II alpha through association with RNA. Nucleic Acids Res. 2008;36:6080–6090. doi: 10.1093/nar/gkn614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawano S., Miyaji M., Ichiyasu S., Tsutsui K.M., Tsutsui K. Regulation of DNA Topoisomerase IIbeta through RNA-dependent association with heterogeneous nuclear ribonucleoprotein U (hnRNP U) J. Biol. Chem. 2010;285:26 451–26 460. doi: 10.1074/jbc.M110.112979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwoebel E.D., Ho T.H., Moore M.S. The mechanism of inhibition of Ran-dependent nuclear transport by cellular ATP depletion. J. Cell Biol. 2002;157:963–974. doi: 10.1083/jcb.200111077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hayashi-Takanaka Y., Yamagata K., Nozaki N., Kimura H. Visualizing histone modifications in living cells: spatiotemporal dynamics of H3 phosphorylation during interphase. J. Cell Biol. 2009;187:781–790. doi: 10.1083/jcb.200904137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kimura H., Cook P.R. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 2001;153:1341–1353. doi: 10.1083/jcb.153.7.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsutsui K., Tsutsui K., Sakurai H., Shohmori T., Oda T. Levels of topoisomerase II and DNA polymerase alpha are regulated independently in developing neuronal nuclei. Biochem. Biophys. Res. Commun. 1986;138:1116–1122. doi: 10.1016/s0006-291x(86)80398-9. [DOI] [PubMed] [Google Scholar]
- 28.Petrov P., Drake F.H., Loranger A., Huang W., Hancock R. Localization of DNA topoisomerase II in Chinese hamster fibroblasts by confocal and electron microscopy. Exp. Cell Res. 1993;204:73–81. doi: 10.1006/excr.1993.1010. [DOI] [PubMed] [Google Scholar]
- 29.Zini N., Santi S., Ognibene A., Bavelloni A., Neri L.M., Valmori A., Mariani E., Negri C., Astaldi-Ricotti G.C., Maraldi N.M. Discrete localization of different DNA topoisomerases in HeLa and K562 cell nuclei and subnuclear fractions. Exp. Cell Res. 1994;210:336–348. doi: 10.1006/excr.1994.1046. [DOI] [PubMed] [Google Scholar]
- 30.Roca J., Ishida R., Berger J.M., Andoh T., Wang J.C. Antitumor bisdioxopiperazines inhibit yeast DNA topoisomerase II by trapping the enzyme in the form of a closed protein clamp. Proc. Natl Acad. Sci. U.S.A. 1994;91:1781–1785. doi: 10.1073/pnas.91.5.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cowell I.G., Papageorgiou N., Padget K., Watters G.P., Austin C.A. Histone deacetylase inhibition redistributes topoisomerase IIbeta from heterochromatin to euchromatin. Nucleus. 2011;2:61–71. doi: 10.4161/nucl.2.1.14194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osheroff N., Shelton E.R., Brutlag D.L. DNA topoisomerase II from Drosophila melanogaster. Relaxation of supercoiled DNA. J. Biol. Chem. 1983;258:9536–9543. [PubMed] [Google Scholar]
- 33.Sorensen M., Sehested M., Jensen P.B. Effect of cellular ATP depletion on topoisomerase II poisons. Abrogation of cleavable-complex formation by etoposide but not by amsacrine. Mol. Pharmacol. 1999;55:424–431. [PubMed] [Google Scholar]
- 34.Skouboe C., Bjergbaek L., Oestergaard V.H., Larsen M.K., Knudsen B.R., Andersen A.H. A human topoisomerase II alpha heterodimer with only one ATP binding site can go through successive catalytic cycles. J. Biol. Chem. 2003;278:5768–5774. doi: 10.1074/jbc.M210332200. [DOI] [PubMed] [Google Scholar]
- 35.Patel S., Jazrawi E., Creighton A.M., Austin C.A., Fisher L.M. Probing the interaction of the cytotoxic bisdioxopiperazine ICRF-193 with the closed enzyme clamp of human topoisomerase IIalpha. Mol. Pharmacol. 2000;58:560–568. doi: 10.1124/mol.58.3.560. [DOI] [PubMed] [Google Scholar]
- 36.Oestergaard V.H., Knudsen B.R., Andersen A.H. Dissecting the cell-killing mechanism of the topoisomerase II-targeting drug ICRF-193. J. Biol. Chem. 2004;279:28 100–28 105. doi: 10.1074/jbc.M402119200. [DOI] [PubMed] [Google Scholar]
- 37.Athale C.A., Christensen M.O., Eils R., Boege F., Mielke C. Inferring a system model of subcellular topoisomerase II beta localization dynamics. OMICS. 2004;8:167–175. doi: 10.1089/1536231041388339. [DOI] [PubMed] [Google Scholar]
- 38.Wu C.C., Li T.K., Farh L., Lin L.Y., Lin T.S., Yu Y.J., Yen T.J., Chiang C.W., Chan N.L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science. 2011;333:459–462. doi: 10.1126/science.1204117. [DOI] [PubMed] [Google Scholar]
- 39.Zechiedrich E.L., Osheroff N. Eukaryotic topoisomerases recognize nucleic acid topology by preferentially interacting with DNA crossovers. EMBO J. 1990;9:4555–4562. doi: 10.1002/j.1460-2075.1990.tb07908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roca J., Berger J.M., Wang J.C. On the simultaneous binding of eukaryotic DNA topoisomerase II to a pair of double-stranded DNA helices. J. Biol. Chem. 1993;268:14 250–14 255. [PubMed] [Google Scholar]
- 41.Seol Y., Gentry A.C., Osheroff N., Neuman K.C. Chiral discrimination and writhe-dependent relaxation mechanism of human topoisomerase IIalpha. J. Biol. Chem. 2013;288:13 695–13 703. doi: 10.1074/jbc.M112.444745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dong K.C., Berger J.M. Structural basis for gate-DNA recognition and bending by type IIA topoisomerases. Nature. 2007;450:1201–1205. doi: 10.1038/nature06396. [DOI] [PubMed] [Google Scholar]
- 43.Gilroy K.L., Austin C.A. The impact of the C-terminal domain on the interaction of human DNA topoisomerase II alpha and beta with DNA. PLoS One. 2011;6:e14693. doi: 10.1371/journal.pone.0014693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scott M.S., Boisvert F.M., McDowall M.D., Lamond A.I., Barton G.J. Characterization and prediction of protein nucleolar localization sequences. Nucleic Acids Res. 2010;38:7388–7399. doi: 10.1093/nar/gkq653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cole C., Barber J.D., Barton G.J. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–W201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carmo-Fonseca M., Mendes-Soares L., Campos I. To be or not to be in the nucleolus. Nat. Cell Biol. 2000;2:E107–E112. doi: 10.1038/35014078. [DOI] [PubMed] [Google Scholar]
- 47.Grassmann R., Berchtold S., Aepinus C., Ballaun C., Boehnlein E., Fleckenstein B. In vitro binding of human T-cell leukemia virus rex proteins to the rex-response element of viral transcripts. J. Virol. 1991;65:3721–3727. doi: 10.1128/jvi.65.7.3721-3727.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Erard M., Barker D.G., Amalric F., Jeang K.T., Gatignol A. An Arg/Lys-rich core peptide mimics TRBP binding to the HIV-1 TAR RNA upper-stem/loop. J. Mol. Biol. 1998;279:1085–1099. doi: 10.1006/jmbi.1998.1831. [DOI] [PubMed] [Google Scholar]
- 49.Roca J. The path of the DNA along the dimer interface of topoisomerase II. J. Biol. Chem. 2004;279:25 783–25 788. doi: 10.1074/jbc.M402555200. [DOI] [PubMed] [Google Scholar]
- 50.Roca J., Wang J.C. DNA transport by a type II DNA topoisomerase: evidence in favor of a two-gate mechanism. Cell. 1994;77:609–616. doi: 10.1016/0092-8674(94)90222-4. [DOI] [PubMed] [Google Scholar]
- 51.Timsit Y., Duplantier B., Jannink G., Sikorav J.L. Symmetry and chirality in topoisomerase II-DNA crossover recognition. J. Mol. Biol. 1998;284:1289–1299. doi: 10.1006/jmbi.1998.2281. [DOI] [PubMed] [Google Scholar]
- 52.Wendorff T.J., Schmidt B.H., Heslop P., Austin C.A., Berger J.M. The structure of DNA-bound human topoisomerase II alpha: conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J. Mol. Biol. 2012;424:109–124. doi: 10.1016/j.jmb.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.