


Abstract
Global transcriptome investigations often result in the detection of an enormous number of transcripts composed of non-co-linear sequence fragments. Such ‘aberrant’ transcript products may arise from post-transcriptional events or genetic rearrangements, or may otherwise be false positives (sequencing/alignment errors or in vitro artifacts). Moreover, post-transcriptionally non-co-linear (‘PtNcl’) transcripts can arise from trans-splicing or back-splicing in cis (to generate so-called ‘circular RNA’). Here, we collected previously-predicted human non-co-linear RNA candidates, and designed a validation procedure integrating in silico filters with multiple experimental validation steps to examine their authenticity. We showed that >50% of the tested candidates were in vitro artifacts, even though some had been previously validated by RT-PCR. After excluding the possibility of genetic rearrangements, we distinguished between trans-spliced and circular RNAs, and confirmed that these two splicing forms can share the same non-co-linear junction. Importantly, the experimentally-confirmed PtNcl RNA events and their corresponding PtNcl splicing types (i.e. trans-splicing, circular RNA, or both sharing the same junction) were all expressed in rhesus macaque, and some were even expressed in mouse. Our study thus describes an essential procedure for confirming PtNcl transcripts, and provides further insight into the evolutionary role of PtNcl RNA events, opening up this important, but understudied, class of post-transcriptional events for comprehensive characterization.
INTRODUCTION
Of the post-transcriptional events, non-co-linear (designated as ‘PtNcl’) transcripts are a relatively less investigated class, which consist of sequence segments that are topologically inconsistent with their corresponding DNA sequences in the reference genome (1). PtNcl transcripts can arise from trans-splicing or back-splicing in cis. The former joins exons from two or more separate precursor mRNAs (pre-mRNAs) originating from the same gene (intragenic trans-splicing) or two or more different genes (intergenic trans-splicing) (2,3), whereas the latter joins exons within a single pre-mRNA, thereby forming circular RNAs in which the exon order is a circular permutation of that encoded by the corresponding genomic sequence (4,5). Trans-spliced RNAs have been shown to be associated with anti-apoptotic roles (1,6,7) and prostate cancer (1,8). We recently reported the important role of trans-splicing in pluripotency maintenance of human embryonic stem cells (ESCs) (9). On the other hand, circular RNAs have been reported to be ubiquitous throughout Eukaryotes (10–12). They may be important in the regulation of micro RNA activity, suggesting they play a role in neurological disorders and brain tumor development (13–15). Analysis of gene expression data suggested that over 1% of all human genes may be involved in the formation of non-co-linear RNAs (16). The ENCODE project further revealed that ∼65% of their tested genes may contribute to non-co-linear transcripts (1,17). The emergence of next-generation sequencing has enabled the development of several RNA-seq-based bioinformatics methods for global investigation of transcriptomes (18–24), which have resulted in the detection of hundreds to thousands of non-co-linear transcript candidates in various species (25–34).
Identification of PtNcl transcripts is often severely hampered by false positives arising from sequencing errors, alignment errors, genetic rearrangements and experimental artifacts (27,30,35,36). Almost no overlap was observed between non-co-linear RNA candidates obtained using different tools, suggesting that the majority of ab initio predicted non-co-linear RNAs are likely to be false positives (22,37–39). In particular, template switching events, which are experimental artifacts generated during reverse transcription (RT) and frequently emerge in cDNA products (40,41), have been reported to be the most significant challenge in the detection of PtNcl transcripts (9,27,41). For example, a prominent study using hybrid mRNAs (i.e. Drosophila melanogaster females versus Drosophila sechellia males) demonstrated that experimental artifacts are the predominant source of the observed non-co-linear RNA products (27). However, it would be impossible to apply such a system to humans. It should be noted that in vitro artifacts, such as those arising from template switching events, do not occur completely stochastically; as such, these artifacts cannot be easily eliminated by increasing the primer annealing temperature during RT (9,41) (which was earlier suggested to suppress the occurrence of template switching (40,42)) or controlling for the depth of supported RNA-seq reads (9,27). Furthermore, we previously demonstrated that preparation of two independent cDNA datasets by RT-PCR with the same RTase was also insufficient to filter out template switching events (9). Therefore, although a dramatic (and increasing) number of non-co-linear RNA products have been detected in human, only a few PtNcl transcripts have been verified or well-documented to date. It is thus imperative to develop an effective procedure to screen for false positives among nominated non-co-linear RNAs. Moreover, genetic rearrangements can also form non-co-linear RNAs (1,35,43), and these are not easily distinguished from PtNcl transcripts. If the detected non-co-linear transcript candidates are confirmed to be post-transcriptionally generated in vivo, a further challenge emerges in discriminating between trans-spliced and circular RNAs.
To address these obstacles, we collected non-co-linear RNA candidates from several well-known datasets, and designed a pipeline integrating in silico filters with multiple experimental validation steps to further eliminate false positives (including alignment errors and experimental artifacts), and to distinguish between trans-spliced RNAs and circular RNAs. We then asked the following questions: (1) What is the proportion of in vitro artifacts in the previously-nominated non-co-linear RNA candidates? (2) Which of the experimentally-verified PtNcl RNA events are spliced in trans (trans-spliced RNAs) and which in cis (circular RNAs)? (3) Are the PtNcl RNA events evolutionarily conserved? (4) Do trans-spliced and circular RNAs share the same non-co-linear junction site? (5) If so, are the PtNcl splicing forms (i.e. trans-splicing, circular RNA or both sharing the same junction) evolutionarily conserved? Our results revealed that over 50% of the experimentally-tested non-co-linear RNA candidates were RT artifacts, even though some of these had been previously reported to pass preliminarily experimental validations. After excluding the possibility of genetic rearrangements, we further distinguished between trans-spliced and circular RNAs, and found that these two splicing types can share the same non-co-linear junction sites. Through comparative analysis of multiple ESC lines of human, rhesus macaque and mouse, we confirmed that six PtNcl RNA events were conserved across primates and four were even conserved across mammals. Importantly, the corresponding PtNcl splicing types (trans-splicing RNA, circular RNA or both types sharing the same junction) of all of these events were also conserved across species, further supporting their potential biological significance.
MATERIALS AND METHODS
Data retrieval and availability
The non-co-linear RNA candidates were collected from four well-known datasets: that of Li et al. (44), ChimerDB 2.0 (25) (http://ercsb.ewha.ac.kr/fusiongene), ChiTaRS (43) (http://chitars.bioinfo.cnio.es/) and the PTES dataset (34). The human genomic sequences (hg19 or GRCh37) were downloaded from the UCSC Genome Browser at http://genome.ucsc.edu/. The BLAT package was downloaded from the Ensembl Genome Browser at http://www.ensembl.org/. The 40 non-co-linear RNA candidates extracted by our in silico filters (see Figure 1) are shown in Supplementary Dataset S1. The RT-PCR/qRT-PCR primers used in this study are listed in Supplementary Dataset S2. The exome sequence data generated by this study have been deposited into the National Center Biotechnology Information (NCBI) Sequence Read Archive, under accession number SRR1284284.
Figure 1.
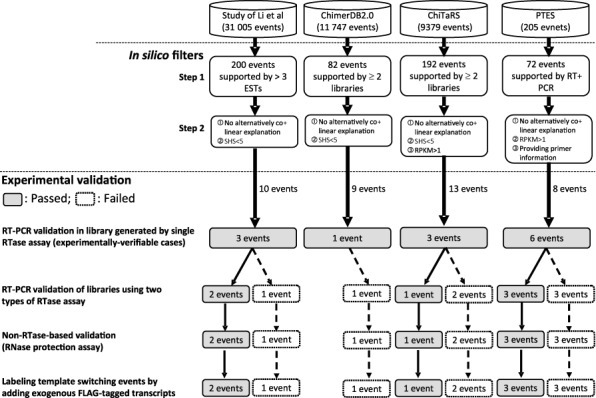
Flowchart of the designed pipeline, including in silico filters and multiple experimental validation steps, for removing in vitro artifacts from previously-annotated non-co-linear RNA candidates. SHS: short homologous sequences. RPKM: reads per kilobase per million mapped reads.
Removing possible co-linear RNA events from the collected non-co-linear RNA candidates
Different BLAT parameters may generate different alignment results; therefore, the use of BLAT-alignments with a single set of parameters is insufficient to detect all possible co-linear explanations of an expressed sequence (EST/mRNA/RNA-seq). As such, we aligned the extracted candidates (Figure 1) against the reference genome with two sets of BLAT parameters: (1) −stepSize = 5 and −repMatch = 2253 (default parameters of the Web-based version of BLAT); and (2) −stepSize = 11 and −repMatch = 1024 (default parameters of the stand-alone version of BLAT). A candidate was not considered if it exhibited an alternative co-linear explanation within a single gene in any one of the two BLAT alignments.
RNA extraction and RT-PCR
Total RNA was extracted using TRI reagent (Ambion) according to the manufacturer's instructions, and treated with DNase I to remove genomic DNA contamination. The cDNA libraries generated using MMLV-derived RTase (Superscript III, Invitrogen) and AMV-derived RTase (Promega) were primed with random hexamers, and reverse transcribed at 50°C for 2 h in buffers provided by the manufacturers. Random hexamer primers consist of a mixture of oligonucleotides, which represent all possible combinations of hexamer sequences; therefore, random hexamers can recognize all sequences, including any given region of PtNcl transcripts. PtNcl transcripts may arise from either trans-spliced isoforms with poly-A tails or cis-spliced circular isoforms without poly-A tails; random hexamer primers were used in order to simultaneously prime both types of PtNcl transcripts, without requiring prior knowledge of the sequences. polymerase chain reaction (PCR) amplification (32 cycles) of chimeric transcripts in cDNA libraries was performed using GoTag Green Master Mix (Promega). Primer sequences are listed in Supplementary Dataset 2.
Probe generation and RNase protection assay
In accordance with the instructions provided for the MAXIscript In Vitro Transcription Kit (Ambion), cDNA fragments containing chimeric junctions for probe synthesis were tagged with SP6- (antisense strand) and T7- (sense strand) targeting sequences by PCR. SP6 or T7 polymerase was added to generate 32P-UTP labeled antisense transcripts or non-labeled sense transcripts, respectively. The 32P-UTP labeled antisense transcripts were used as protective probes in RNase protection assay (RPA). RPA was performed using the RPA III kit (Ambion). In brief, 10 μg total RNA and 100 ng antisense probes were hybridized at 56°C for 16 h, and non-hybridized fragments were then digested by RNase A and T1. The protected fragments were separated on a 5% denaturing (8 M Urea) polyacrylamide gel, and signals were developed by exposure on X-OMAT blue films (Kodak).
Embryonic stem cell culture and normal human adult tissues
To generate feeder cells to support ESCs, mouse embryonic fibroblasts (MEFs) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS, Level), 1× non-essential amino acids (NEAA, Invitrogen), and 2 mM L-glutamine (Invitrogen), and treated with Mitomycin C. All primate ESCs (human: NTU1, H1, H9 (WiCell Bank); rhesus macaque: ORMES6, ORMES8) were grown on Mitomycin C-treated MEF feeders (2 × 104 cells/cm2) in DMEM/F12 media plus 20% Knockout Serum Replacement (Invitrogen) and 4 ng/ml bFGF (Sigma-Aldrich). Primate ESC colonies were manually expanded to new feeder cells every four days (9,45). Mouse ESC D3 and K1 derived from C57BL/6 × ICR embryos were cultured in DMEM containing 15% FBS (level), 1x NEAA, 2 mM L-glutamine (Invitrogen) and 103 units/ml of leukemia inhibitory factor (Millipore) on Mitomycin C-treated MEF feeders (5 × 104 cells/cm2). Mouse ESCs were expanded to new feeder cells with 0.1% trypsin treatment every three days. Total RNA samples from normal human adult tissues (brain, breast, colon, heart, kidney, liver, lung, muscle, pancreas and testis) were purchased from BioChain, and treated with DNase I to remove possible genomic contamination.
Tagging template switching events
To generate sequence-verified DNA templates, the 3′ partner genes of PtNcl transcripts were amplified from cDNA libraries and cloned into vector pCMV-Tag 4B, in which the FLAG sequence is fused to the 3′ terminus of the partner gene. The FLAG-tagged RNAs were then transcribed with T7 RNA polymerase. Since high copies of a single transcript may induce template switching, we diluted (1:1000) the in vitro transcripts (41) within human ESC total RNA, to (i) mimic the endogenous concentration of spliced transcripts and (ii) ensure that template switching was not induced by high concentrations of tagged RNA transcripts. Finally, reverse transcription was performed from the FLAG adaptor using AMV- and MMLV-derived RTases. The template switching experiments were repeated three times. Mixtures containing the same number of tagged mRNA transcripts were used for both the RT-dependent and RT-independent samples.
RNase R treatment
DNase-treated total RNA (2 μg) or purified mRNA (100 ng) were incubated for 30 min at 37°C with or without RNase R (3U/μg, Epicentre Biotechnologies). RNA was subsequently purified by phenol-chloroform extraction, reverse transcribed with the indicated RTases according to the manufacturer's protocol, and used as template in PCR and qRT-PCR.
Purification of mRNAs from total RNA
The mRNAs were purified using the Oligotex mRNA Min Kit (QIAGEN), according to the manufacturer's instructions. After DNase treatment, 20 μg of total RNA were heated at 70°C for 3 min to disrupt RNA secondary structure; the RNA was then incubated with Oligotex suspension at 30°C for 10 min. Oligotex-bound mRNAs were then centrifuged, washed and eluted with buffers provided in the kit.
RESULTS
Extraction of experimentally-verifiable non-co-linear RNA candidates
To determine the authenticity of previously-nominated non-co-linear RNA candidates in human, we first collected non-co-linear RNA candidates from four well-known datasets: that of Li et al. (44), ChimerDB 2.0 (25), ChiTaRS (43) and the post-transcriptional exon shuffling (PTES) dataset (34). These candidates were derived from varied types of expressed data. The PTES dataset was derived from RNA-seq reads, including long Roche 454 reads and short Illumina reads. The other three datasets were initially predicted based on EST/mRNA sequences, which were gathered from a wide variety of samples. The candidates within the ChimerDB 2.0 and ChiTaRS datasets were also inferred from RNA-seq reads. For accuracy, different in silico filters were applied to different datasets according to the available information. As shown in Figure 1, the candidates selected from these four datasets must be supported by >3 ESTs (for the Li dataset), both ESTs/mRNAs and RNA-seq data (for the ChimerDB 2.0 and ChiTaRS datasets) or RT-PCR experimental validation (for the PTES dataset). It should be noted that a non-co-linear RNA candidate may be misidentified due to gene duplications in the genome. To minimize such false positive events, we aligned the candidates against the reference genome with two different sets of BLAT parameters (‘Materials and Methods’ section). We eliminated all candidates with alternative co-linear explanations in one or both BLAT alignments. Next, we filtered out the non-co-linear RNA candidates containing short homologous sequences (SHSs) or gaps at non-co-linear junction sites, because such candidates were likely to be artifacts arising from temple switching (9,27). The ChiTaRS and PTES datasets provide information on expression levels and primer sequences, respectively; as such, we exploited this information to further increase the verifiability of the non-co-linear RNA candidates by selecting only those ChiTaRS candidates with a high expression level (RPKM > 1) (43) and PTES candidates with available primer information (34). After the multiple in silico screening steps, the number of candidates was dramatically reduced from >40 000 to 40 (Figure 1). Considering potential tissue-/sample-specificity, we performed RT-PCR with MMLV (Moloney Murine Leukemia Virus)- and AMV (Avian Myeloblastosis Virus)-derived RTase with primers against these 40 candidates in multiple tissues/cell lines, including 10 normal human tissues and one human ESC line, H9 (‘Materials and Methods’ section). Candidates detected by at least one of these two types of RTase-based experiments were defined as experimentally-verifiable PtNcl candidates. In this way, we extracted 13 experimentally-verifiable PtNcl candidates (Figure 1) and subjected them to the analyses and validations described below. As these 13 candidates were readily detected in multiple tissues/cell lines (Figure 2), they are highly unlikely to have arisen from genetic rearrangement events.
Figure 2.
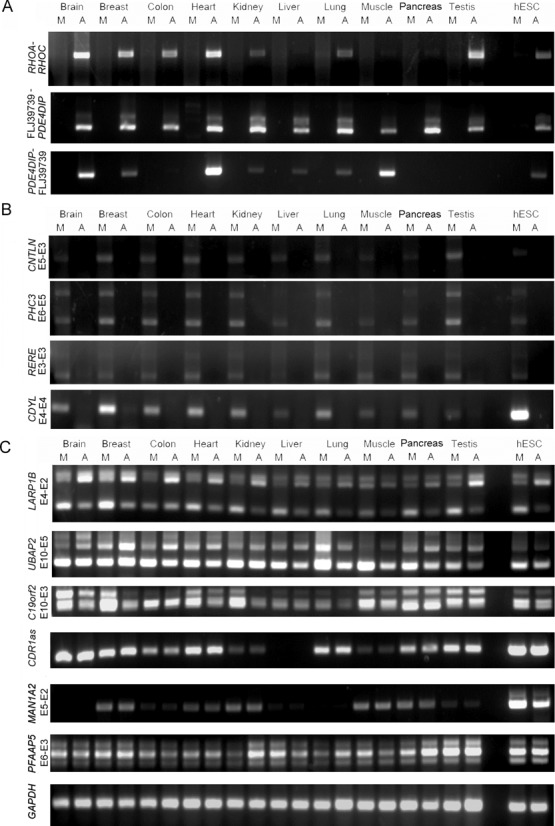
Comparisons of two different RTase products (AMV- and MMLV-derived) derived from 13 experimentally-verifiable non-co-linear RNA candidates (Figure 1) in 10 normal human tissues (brain, breast, colon, heart, kidney, liver, lung, muscle, pancreas and testis) and human ESCs, under identical conditions: (A) AMV-RTase-dependent, (B) MMLV-RTase-dependent and (C) RTase-independent candidates. Non-co-linear transcripts with shuffled exons are described from the 5′ to the 3′ terminus. All primers used in RT-PCR validation are listed in Supplementary Dataset 2. M: MMLV; A: AMV.
Over 50% of experimentally-verifiable candidates are in vitro artifacts
We proceeded to determine whether the 13 experimentally-verifiable PtNcl candidates were truly generated in vivo. According to the findings of the RTase-based experiments, the 13 PtNcl candidates can be classified into three groups: AMV-RTase-dependent candidates (i.e. RHOA–RHOC, FLJ39739-PDE4DIP and PDE4DIP-FLJ39739; Figure 2A), MMLV-RTase-dependent candidates (i.e. CNTNL E5-E3, PHC3 E6-E5, RERE E3-E3 and CDYL E4-E4; Figure 2B) and RTase-independent candidates (i.e. LARP1B E4-E2, UBAP2 E10-E5, C19orf2 E10-E3, CDR1as, MAN1A2 E5-E2 and PFAAP5 E6-E3; Figure 2C). RTase-dependent candidates were specifically detected in experiments using the indicated RTase, whereas RTase-independent candidates were detected in both types of RTase experiment. We suspected that the seven AMV- or MMLV-RTase-dependent candidates have been RT-based artifacts. To control for such artifacts, we subjected the 13 candidates to the RNase protection assay (RPA; ‘Materials and Methods’ section), a non-RTase-based assay (46), using total RNA from human H9 ESCs. RPA directly detects non-co-linear RNA products using complementary probes, as only the RNA duplex of true PtNcl-probe hybrids are protected from RNase A and RNase T1 degradation. In other words, RT-based artifact probes would be degraded in this assay, as no authentic PtNcl RNA products would form a RNA duplex with the probes. We observed that the probes of the seven RTase-dependent candidates were degraded (Supplementary Figures S1A and B), whereas those of the six RTase-independent ones were not (Supplementary Figure S1C). Sequence chromatograms of amplicons containing non-co-linear junctions and their corresponding flanking sequences for the six RTase-independent candidates are shown in Supplementary Figure S2 and Table S1. These experiments thus confirm that the RTase-dependent RNA products are indeed RT-based artifacts, indicating that the examined non-co-linear datasets exhibit a high false positive rate (53.8%). This result also confirms that comparing the products of different RTases is effective at confirming the authenticity of PtNcl RNA events.
It has been previously reported that spurious splicing frequently arises from template switching by RT (41). We therefore designed an experiment to determine whether the detected RT-based artifacts arose from template switching events (‘Materials and Methods’ section). As RT processes from the 3′ to 5′ terminus of RNA, we cloned the 3′ partner gene (with a FLAG tag at its 3′-most exon) into an expression vector (e.g. pre-mRNA-B′ in Figure 3A). The expression vector was then transcribed in vitro to generate an RNA product carrying a FLAG-tag (Figure 3A). The tagged transcript was mixed with total RNA from human ESCs at a low molar ratio (1:1000), to serve as templates for RT and prevent template switching induced by the presence of high copies of a single transcript (41). The mixture was then reverse transcribed from the FLAG sequence using both AMV- and MMLV-based RTases. FLAG-primed RT should only generate cDNA from our FLAG-tagged transcripts, and therefore, any PtNcl RNA candidates detected using FLAG-primed RT will be a consequence of template switching events (Figure 3A). Indeed, we observed that AMV- and MMLV-RTase-dependent transcripts were specifically induced by their corresponding RTase (Figure 3B), whereas the RTase-independent candidates were induced by neither RTase (Figure 3C). Crucially, we excluded the possibility that the FLAG sequence induced template switching by performing FLAG-primed RT-PCR in the absence of FLAG-tagged transcripts (Figure 3B). No PtNcl events were detected under such conditions, indicating that the FLAG sequence did not induce template switching events. For example, the AMV-RTase-dependent PtNcl event, AU126261 RHOA–RHOC, was only detected when using FLAG-tagged transcripts as template in AMV-based FLAG-primed RT-PCR. Taken together, we have demonstrated that the seven RTase-dependent candidates were RT artifacts arising from template switching events, while the six RTase-independent cases are genuine PtNcl RNA events.
Figure 3.
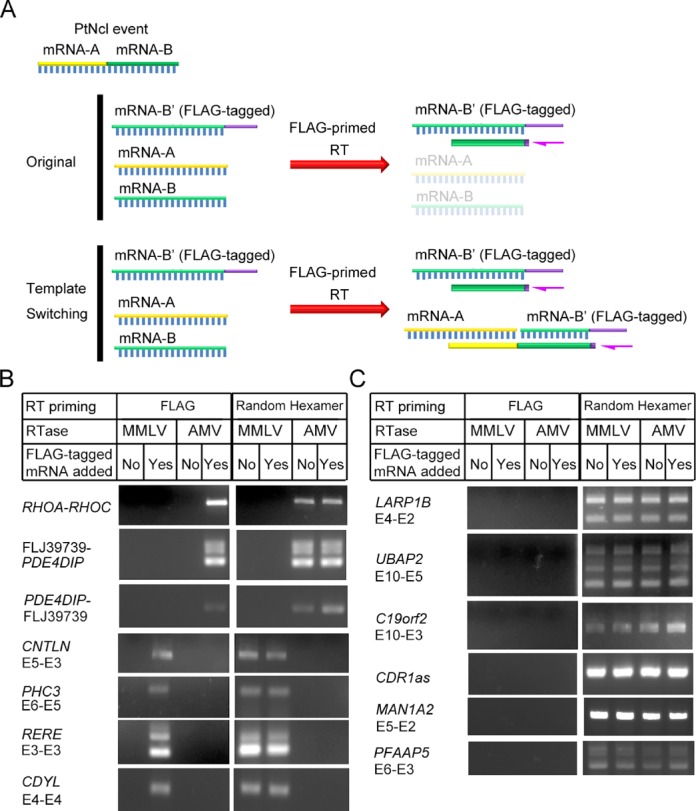
Detection of template switching events using tagged 3′ partner genes of PtNcl candidates in RT. (A) Procedure by which RT template switching events were identified in tagged transcripts. Total RNA from human ESCs were mixed with FLAG-tagged 3′ partner genes of the PtNcl candidates and reversely transcribed from the FLAG sequence (FLAG-primed RT). In the absence of a template switching event, FLAG-primed RT will generate a co-linear cDNA from the FLAG-tagged transcript. In contrast, FLAG-primed RT will generate a non-co-linear RT cDNA following a template switching event. (B-C) PCR detection of (B) MMLV-/AMV-RTase-dependent (seven cases) and (C) RTase-independent (six cases) non-co-linear RNA candidates following FLAG-primed RT.
Trans-spliced and circular RNAs can share the same non-co-linear junction sites
We subsequently investigated whether the observed PtNcl transcripts arose from trans-splicing or back-splicing in cis (circular RNAs); in addition, we asked whether it is possible for trans-spliced and circular RNAs to share the same non-co-linear junction sites. To answer these questions, we treated total RNA from human ESCs with RNase R, which degrades linear RNA with free terminal ends. Circular RNAs, which have no free terminal ends, are resistant to RNase R degradation (‘Materials and Methods’ section). Subsequent RT-PCR and qRT-PCR showed that C19orf2 E10-E3 and UBAP2 E10-E5 were degraded by RNase R treatment, while the other four transcripts (CDR1as, LARP1B E4-E2, MAN1A2 E5-E2 and PFAAP5 E6-E3) were unaffected (Figures 4A and 5B). These results indicate that the latter are circular RNAs, whereas C19orf2 E10-E3 and UBAP2 E10-E5 are not. However, it remains possible that these four circular RNA events share non-co-linear junction sites with trans-splicing events. We proceeded to perform qRT-PCR analyses of the six PtNcl RNA events using purified mRNAs with poly-A tails as template; we detected signals for all PtNcl RNA events except for CDR1as (Figure 4C). Potential contamination by circular RNAs upon purification was prevented, as purified mRNAs are degraded by RNase R treatment. These results indicate that CDR1as RNA exists in a circular, but not trans-spliced, form in human ESCs. On the other hand, LARP1B E4-E2, MAN1A2 E5-E2 and PFAAP5 E6-E3 can exist as both circular (Figure 4A and B) and poly-A tailed RNAs (Figure 4C), indicating that these two PtNcl splicing types (i.e. trans-spliced and circular RNAs) can share the same non-co-linear junction sites. Therefore, the observed PtNcl events may manifest as (1) trans-spliced RNAs only (i.e. C19orf2 E10-E3 and UBAP2 E10-E5); (2) both trans-spliced and circular RNAs (i.e. LARP1B E4-E2, MAN1A2 E5-E2 and PFAAP5 E6-E3); or (3) circular RNAs only (i.e. CDR1as). Of note, the splicing types of the six PtNcl RNA events were readily observed in multiple human ESC lines (H1, H9 and NTU1; Figure 4A–C), indicating that these PtNcl RNA events were not generated by chance. This also implies that these events may play a role in the biological processes associated with human pluripotent stem cells.
Figure 4.
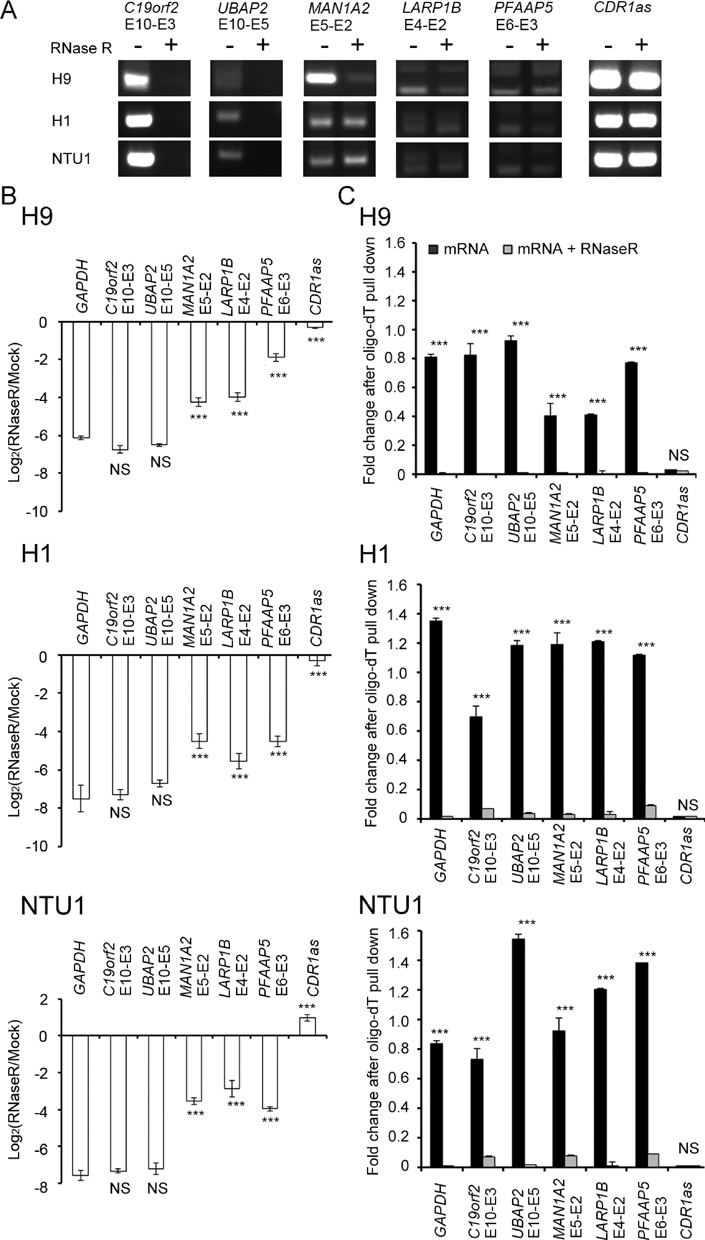
Discrimination between trans-spliced and circular RNAs for the six confirmed PtNcl RNA events. (A) RT-PCR results and (B) expression fold changes (as determined by qRT-PCR) for the six PtNcl RNA events in the indicated human ESC lines, before and after RNase R treatment. (C) Comparisons of the expression fold changes for the six PtNcl RNA events in poly-A tailed RNAs (purified mRNAs) and poly-A tailed RNAs treated with RNase R in the indicated human ESC lines. Error bars represent the mean ± standard deviation. P-values were estimated by the two-sample, two-tailed t-test. Significance: ***P < 0.001. NS: not significant.
Figure 5.
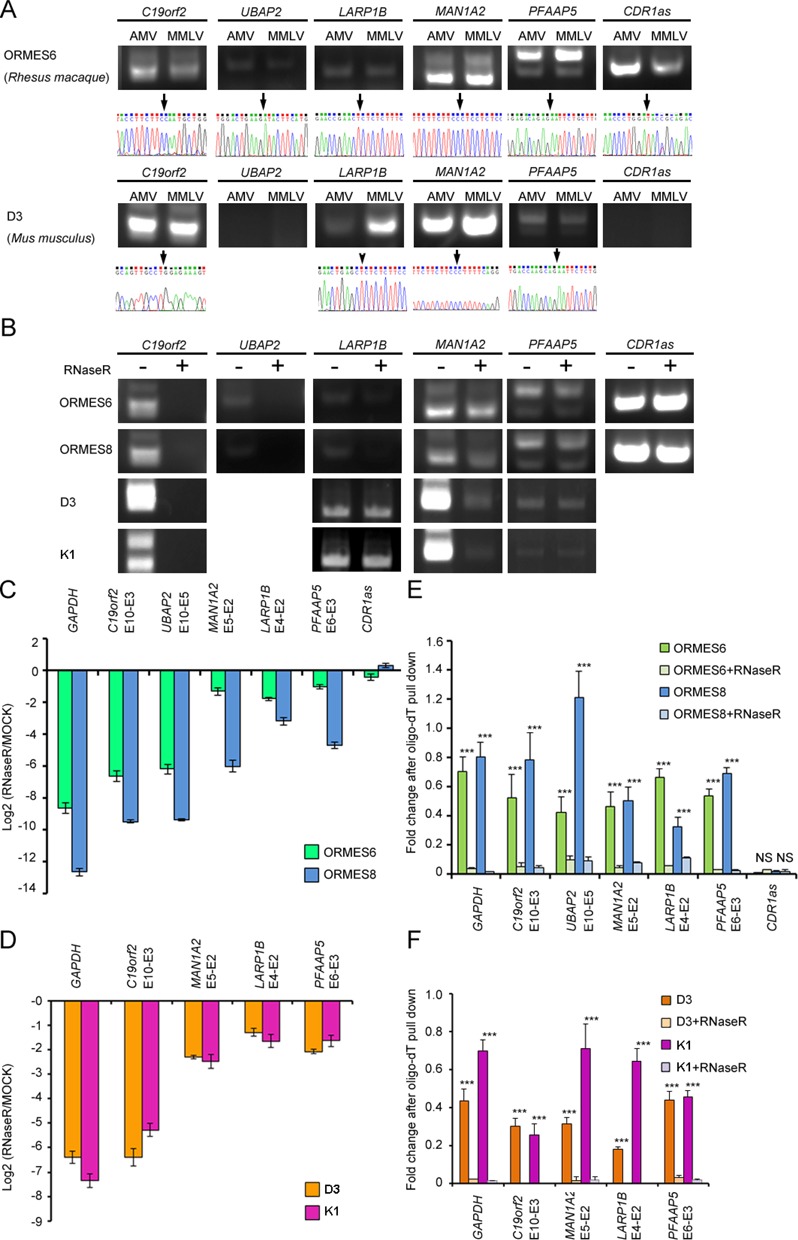
Examination of the evolutionary conservation of six human PtNcl RNA events and their corresponding splicing forms (i.e. trans-spliced and/or circular RNAs) in rhesus macaque and mouse. (A) Comparisons of AMV- and MMLV-derived-RTase products (top) and sequence chromatograms (bottom) for the six PtNcl RNA events in the indicated rhesus macaque and mouse ESCs. (B) RT-PCR results and (C-D) expression fold changes (as determined by qRT-PCR) for the six PtNcl RNA events in the indicated rhesus macaque and mouse ESCs lines, before and after RNase R treatment. (E-F) Comparisons of the expression fold changes for the six PtNcl RNA events in poly-A tailed RNAs (purified mRNAs) and poly-A tailed RNAs treated with RNase R in the indicated rhesus macaque and mouse ESC lines. Error bars represent the mean ± standard deviation. P-values were estimated by the two-sample, two-tailed t-test. Significance: ***P < 0.001. NS: not significant.
The PtNcl RNA events and their splicing types are evolutionarily conserved
As conservation of exonic structures across species is often believed to imply conservation of biological function, we examined whether the six confirmed PtNcl RNA events are also present in non-human mammals. Through comparative analysis of human, rhesus macaque and mouse ESCs, we observed that all six PtNcl RNA events were RTase-independent in rhesus macaque ESCs, and four of these events (LARP1B E4-E2, C19orf2 E10-E3, MAN1A2 E5-E2 and PFAAP5 E6-E3) were RTase-independent in mouse ESCs. The identity of the non-co-linear junction sites was validated by sequencing the RT-PCR amplicons (Figure 5A). These results demonstrate that the studied PtNcl RNA events are also present in non-human mammals. Interestingly, UBAP2 E10-E5 and CDR1as were observed in human and rhesus macaque but not in mouse, suggesting that they may be primate-specific PtNcl RNA events. Next, we examined the splicing types of the PtNcl RNA events (i.e. trans-splicing, circular RNA or both sharing the same junction) in rhesus macaque and mouse ESCs, using the same validation steps described above (i.e. RNase R treatment and mRNA purification; see also Figure 4). We observed that these PtNcl RNA events in both rhesus macaque and mouse ESCs exhibited the same PtNcl splicing types (Figure 5B–F) as in human ESCs (Figure 4A–C). Importantly, the splicing types of the four mammalian PtNcl RNA events and two primate-specific PtNcl RNA events were observed in multiple ESC lines of rhesus macaque (ORMES6 and ORMES8) and mouse (D3 and K1) (Figure 5B–F). The observed evolutionary conservation of these PtNcl RNA events and their corresponding splicing types in multiple ESC lines of human, rhesus macaque and mouse indicate that these events may play an important role in primate/mammalian ESCs.
DISCUSSION
Reverse transcriptases can be error prone, and frequently generate splicing artifacts in vitro (40,41,47). It remains a considerable challenge to systematically eliminate such RT-based artifacts while detecting non-co-linear RNAs. Although a considerable number of non-co-linear candidates have been identified in the human, mouse, fly and rice transcriptomes (25–31,33–35,44,48), control experiments to effectively eliminate potential in vitro artifacts were not performed. Several studies have made use of certain preliminary screens to eliminate apparent false positives from the identified non-co-linear RNA candidates. For example, an integrative approach based on the comparison of different types of expressed sequence data has been used to dramatically filter out false positives (>95% of the total candidates) (9,30). In addition, RT-PCR based on a single RTase has often been applied to validate the non-co-linear RNA events (30,33,34). However, we demonstrate here that even non-co-linear RNA candidates passing these criteria may still be false positives. By collecting non-co-linear RNA candidates previously identified in four well-known datasets (Figure 1), we extracted experimentally-verifiable candidates and designed a validation procedure to identify genuine non-co-linear RNA events. The validation results, which are summarized in Table 1, showed that over 50% (7/13) of the tested candidates were in vitro artifacts. Therefore, experimental artifacts comprised a considerable number of each of the four non-co-linear RNA datasets, even those that were supported by multiple types of expressed sequence data (Figure 1 and Table 1). Furthermore, a high percentage of candidates that had been previously validated by RT-PCR were still derived from in vitro artifacts (Figure 1); this casts considerable doubt on the authenticity of in silico predicted candidates that have not undergone any experimental validations. We thus emphasize the necessity of carefully confirming whether non-co-linear RNA candidates are genuine through control experiments to eliminate experimental artifacts.
Table 1. Validation of the experimentally-verifiable non-co-linear RNA candidates.
| PtNcl RNA event | Type of supported expressed data (dataset) | Canonical splicing site | Annotated exon-intron boundary | Fusion type | Experimental validation |
|---|---|---|---|---|---|
| PDE4DIP-FJL39739 | EST/mRNA, short Illumina RNA-seq read (ChiTaRS) | No | No | Intergenic | in vitro |
| FJL39739-PDE4DIP | EST/mRNA, short Illumina RNA-seq read (ChiTaRS) | No | No | Intergenic | in vitro |
| RHOA–RHOC | EST/mRNA, short Illumina RNA-seq read (ChimerDB 2.0) | Yes | Yes | Intergenic | in vitro |
| RERE E3-E3 | Long 454 and short Illumina RNA-seq reads, RT-PCR (PTES) | Yes | Yes | Intragenic | in vitro |
| CNTNL E5-E3 | Long 454 and short Illumina RNA-seq reads, RT-PCR (PTES) | Yes | Yes | Intragenic | in vitro |
| PHC3 E6-E5 | Long 454 and short Illumina RNA-seq reads, RT-PCR (PTES) | Yes | Yes | Intragenic | in vitro |
| CDYL E4-E4 | EST/mRNA (Li) | Yes | Yes | Intragenic | in vitro |
| C19orf2 E10-E3 | Long 454 and short Illumina RNA-seq reads, RT-PCR (PTES) | Yes | Yes | Intragenic | in vivo (trans-splicing event) |
| UBAP2 E10-E5 | Long 454 and short Illumina RNA-seq reads, RT-PCR (PTES) | Yes | Yes | Intragenic | in vivo (trans-splicing event) |
| MAN1A2 E5-E2 | EST/mRNA (Li) | Yes | Yes | Intragenic | in vivo (trans-splicing and circular RNA events) |
| LARP1B E4-E2 | Long 454 and short Illumina RNA-seq reads, RT-PCR (PTES) | Yes | Yes | Intragenic | in vivo (trans-splicing and circular RNA events) |
| PFAAP5 E6-E3 | EST/mRNA (Li) | Yes | Yes | Intragenic | in vivo (trans-splicing and circular RNA events) |
| CDR1as | EST/mRNA, short Illumina RNA-seq read (ChiTaRS) | Yes | No | Intragenic | in vivo (circular RNA event) |
Previously, the presence of (i) canonical splicing signals at non-co-linear junction sites and (ii) fusion boundaries that match the well-annotated exon boundaries was often regarded as measures of confidence for non-co-linear RNAs (25,34,40). However, we have demonstrated that these properties do not guarantee that a PtNcl RNA candidate is genuine. We found that >70% (5/7) of candidates derived from RT-based artifacts contain both canonical splicing signals at their non-co-linear junction sites and fusion boundaries matching the well-annotated exon boundaries (Table 1). On the other hand, although most of the experimentally-verified PtNcl RNA events possess the aforementioned properties, the fusion boundary of one genuine PtNcl (CDR1as) does not match the well-annotated exon boundary (Table 1). These results underscore the effectiveness of our validation pipeline, suggesting that the pipeline is an essential procedure for screening out in vitro artifacts when detecting PtNcl RNAs.
Another interesting finding is that a gene transcript can manifest as both trans-splicing and circular forms, and these can arise from the same non-co-linear junction site. Thus, all PtNcl transcripts can be divided into three categories: those that result from trans-splicing events alone; those that result from both trans-splicing and circular RNA events; and those that result from circular RNA events alone. It is worth noting that the splicing types of the PtNcl RNA events described here were not only conserved in multiple human ESC lines (Figure 4), but also in multiple rhesus macaque and mouse ESC lines (Figure 5). The observed conservation of PtNcl splicing forms across species suggests that the trans-splicing and circular forms of a gene may have differential roles in pluripotent stem cells.
Moreover, of the six experimentally-confirmed PtNcl RNA events, four are evolutionarily conserved across mammals (human, rhesus macaque and mouse) and two are present in primates only (human and rhesus macaque). Alignment of the six events against the entire NCBI EST database revealed that PFAAP5 E6-E3 and LARP1B E4-E2 are further supported by both sheep and dog ESTs (Supplementary Figure S3). These results suggest that PtNcl RNA events may play a lineage-specific role in primate/mammal transcriptome evolution. It has also been shown that non-co-linear forms may differ functionally from their corresponding co-linear forms, even if the latter is just a source for the former (9), suggesting that such a PtNcl splicing strategy is biologically advantageous. The PtNcl splicing strategy may provide an evolutionary advantage to complex organisms by introducing additional regulators of gene expression (e.g. CDR1as can act as a microRNA sponge (14,15)), so as to increase the plasticity of transcriptome. In addition, a striking example of trans-splicing, JAZF1-JJAZ1, was previously suggested to be a precondition for genetic rearrangement (1,49); this implies that PtNcl splicing may also enable the generation of variants at a relatively low evolutionary cost, as the source gene is retained in the wider population.
All six validated PtNcl RNA events were verified not only in multiple tissues/cell lines of human (including 10 types of human normal tissue and three types of human ESC line; see Figures 2 and 4), but also in rhesus macaque and mouse (multiple types of rhesus macaque and mouse ESC line; Figure 5). It seems unlikely that somatic recombination events would simultaneously occur in multiple biological samples and across different species (27), and we thus propose that these six non-co-linear transcripts are not the product of genomic rearrangements. To confirm this hypothesis, we captured and sequenced the exonic regions of human ESC line H9, and measured the read depths of the non-co-linear exon pairs of the PtNcl RNA events. CDR1as has been previously confirmed to be a circular RNA event (13–15); we therefore examined the possibility of genomic rearrangement origins for the five other PtNcl RNA events (i.e. C19orf2 E10-E3, UBAP2 E10-E5, MAN1A2 E5-E2, LARP1B E4-E2 and PFAAP5 E6-E3) by comparing read depths between their corresponding non-co-linear exon pairs. If an observed non-co-linear exon pair is subject to a genomic rearrangement event, the relative ratio of read-depth of 3′ end exon to that of 5′ end exon (e.g. the ratio of read-depth of Exon 3 to that of Exon 10 for C19orf2 E10-E3) should correspond to a 2-fold change. However, the relative ratios of the exon pairs for the five non-co-linear transcript events are all considerably <2 (Supplementary Table S2), further supporting that these events are not genomic rearrangements.
It is also important to note that C19orf2 E10-E3, which was confirmed to exist in only a trans-splicing form in human, rhesus macaque and mouse ESCs in this study (Figures 4 and 5), was predicted in silico to be a circular RNA in HEK293 cells in a previous study (14). Two possible scenarios may account for this discrepancy. First, the splicing type of C19orf2 E10-E3 may be cell type-dependent. Second, the in silico prediction may be incorrect, and the non-co-linear RNA product may indeed be derived from a trans-splicing event. We addressed this question by showing that C19orf2 E10-E3 RNA from HEK293 cells was degraded by RNase R treatment (Supplementary Figure S4), eliminating the possibility that C19orf2 E10-E3 exists in a circular form in the HEK293 line. In addition, C19orf2 E10-E3 has been previously confirmed to be polyadenlyated through sequencing from oligo-dT primed templates (34). We thus conclude that the second possibility is more likely. The above results also indicate that an observed intragenic PtNcl RNA product may arise from trans-splicing and/or back-splicing in cis, highlighting the importance of distinguishing between these two PtNcl splicing forms.
Although trans-splicing can occur within a single gene or between different genes (1,50), we did not experimentally verify any trans-spliced RNAs transcribed from multiple genes in this study (Table 1). This is consistent with the earlier reports that intragenic splicing is more common than intergenic splicing, and that the majority of observed intergenic trans-splicing candidates are, in fact, experimental artifacts (9,27). A possible explanation is that intragenic splicing within a single gene results in a higher local concentration of transcripts than intergenic splicing between different genes. Therefore, trans-splicing, which couples different pre-mRNAs, is more likely to occur within the same gene than between two or more separate genes. In this study, we focused purely on PtNcl transcripts arising from trans-splicing or back-splicing in cis. However, trans-splicing can also occur in a co-linear fashion, with trans-spliced RNAs being generated from different alleles or opposite strands of the same gene; the best-known examples of this type are lola and mod(mdg4) in Drosophila (51,52). Therefore, there are four possible types of intragenic splicing: (1) cis-splicing in a co-linear form; (2) cis-splicing in a non-co-linear form; (3) trans-splicing in a co-linear form and (4) trans-splicing in a non-co-linear form (examples are provided in Figure 6; Cases 1–4, respectively). Although the co-linearly cis-splicing form (Case 1) is generally regarded as the main path of RNA processing from primary to mature RNA molecules (1), the other three types of splicing (Cases 2–4) provide a higher level of flexibility for segmentation of genomic information in an RNA context. These varied types of splicing thus provide an alternative way to increase the complexity of transcriptomes (or even proteomes (53–57)) and processing machinery, and appear to be more complicated than previously anticipated.
Figure 6.
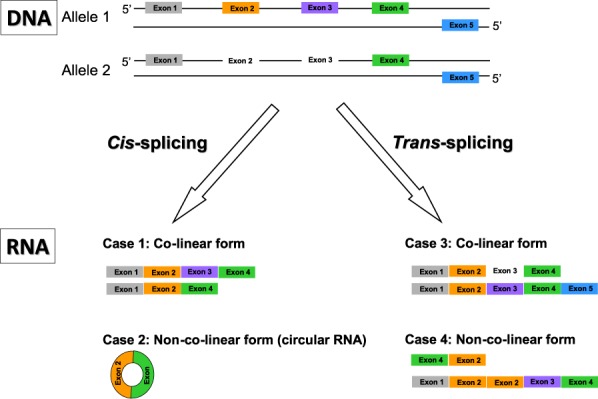
Four types of intragenic splicing: co-linear cis-splicing (Case 1), non-co-linear cis-splicing (i.e. circular RNAs; Case 2), co-linear trans-splicing (i.e. the formation of trans-spliced RNAs from different alleles or opposite strands of the same gene; Case 3) and non-co-linear trans-splicing (i.e. the formation of trans-spliced RNAs from the same gene by exon rearrangement; Case 4).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online including [1–2].
FUNDING
Genomics Research Center and the Institute of Cellular and Organismic Biology, Academia Sinica, Taiwan; National Science Council (Ministry of Science and Technology), Taiwan (under contracts NSC102-2621-B-001-003-, MOST 103-2628-B-001 -001-MY4 (to T.J.C.), NSC 102-2321-B-001-012 and NHRI-Ex103-10320SI- (to H.C.K.)). Funding for open access charge: Genomics Research Center and the Institute of Cellular and Organismic Biology, Academia Sinica, Taiwan; National Science Council, Taiwan (under contracts NSC102-2621-B-001-003- (to T.J.C.), NSC 102-2321-B-001-012 and NHRI-Ex103-10320SI- (to H.C.K.)).
Conflict of interest statement. None declared.
REFERENCES
- 1.Gingeras T.R. Implications of chimaeric non-co-linear transcripts. Nature. 2009;461:206–211. doi: 10.1038/nature08452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Konarska M.M., Padgett R.A., Sharp P.A. Trans splicing of mRNA precursors in vitro. Cell. 1985;42:165–171. doi: 10.1016/s0092-8674(85)80112-4. [DOI] [PubMed] [Google Scholar]
- 3.Solnick D. Trans splicing of mRNA precursors. Cell. 1985;42:157–164. doi: 10.1016/s0092-8674(85)80111-2. [DOI] [PubMed] [Google Scholar]
- 4.Hsu M.T., Coca-Prados M. Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature. 1979;280:339–340. doi: 10.1038/280339a0. [DOI] [PubMed] [Google Scholar]
- 5.Nigro J.M., Cho K.R., Fearon E.R., Kern S.E., Ruppert J.M., Oliner J.D., Kinzler K.W., Vogelstein B. Scrambled exons. Cell. 1991;64:607–613. doi: 10.1016/0092-8674(91)90244-s. [DOI] [PubMed] [Google Scholar]
- 6.Li H., Wang J., Mor G., Sklar J. A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science. 2008;321:1357–1361. doi: 10.1126/science.1156725. [DOI] [PubMed] [Google Scholar]
- 7.Schoenfelder S., Clay I., Fraser P. The transcriptional interactome: gene expression in 3D. Curr. Opin. Genet. Dev. 2010;20:127–133. doi: 10.1016/j.gde.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 8.Rickman D.S., Pflueger D., Moss B., VanDoren V.E., Chen C.X., de la Taille A., Kuefer R., Tewari A.K., Setlur S.R., Demichelis F., et al. SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res. 2009;69:2734–2738. doi: 10.1158/0008-5472.CAN-08-4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu C.S., Yu C.Y., Chuang C.Y., Hsiao M., Kao C.F., Kuo H.C., Chuang T.J. Integrative transcriptome sequencing identifies trans-splicing events with important roles in human embryonic stem cell pluripotency. Genome Res. 2014;24:25–36. doi: 10.1101/gr.159483.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salzman J., Gawad C., Wang P.L., Lacayo N., Brown P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 2012;7:e30733. doi: 10.1371/journal.pone.0030733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jeck W.R., Sorrentino J.A., Wang K., Slevin M.K., Burd C.E., Liu J., Marzluff W.F., Sharpless N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19:141–157. doi: 10.1261/rna.035667.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang P.L., Bao Y., Yee M.C., Barrett S.P., Hogan G.J., Olsen M.N., Dinneny J.R., Brown P.O., Salzman J. Circular RNA is expressed across the eukaryotic tree of life. PLoS One. 2014;9:e90859. doi: 10.1371/journal.pone.0090859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hansen T.B., Wiklund E.D., Bramsen J.B., Villadsen S.B., Statham A.L., Clark S.J., Kjems J. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J. 2011;30:4414–4422. doi: 10.1038/emboj.2011.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., Maier L., Mackowiak S.D., Gregersen L.H., Munschauer M., et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 15.Hansen T.B., Jensen T.I., Clausen B.H., Bramsen J.B., Finsen B., Damgaard C.K., Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 16.Dixon R.J., Eperon I.C., Hall L., Samani N.J. A genome-wide survey demonstrates widespread non-linear mRNA in expressed sequences from multiple species. Nucleic Acids Res. 2005;33:5904–5913. doi: 10.1093/nar/gki893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Birney E., Stamatoyannopoulos J.A., Dutta A., Guigo R., Gingeras T.R., Margulies E.H., Weng Z., Snyder M., Dermitzakis E.T., Thurman R.E., et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim D., Salzberg S.L. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Francis R.W., Thompson-Wicking K., Carter K.W., Anderson D., Kees U.R., Beesley A.H. FusionFinder: a software tool to identify expressed gene fusion candidates from RNA-Seq data. PLoS One. 2012;7:e39987. doi: 10.1371/journal.pone.0039987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McPherson A., Hormozdiari F., Zayed A., Giuliany R., Ha G., Sun M.G., Griffith M., Heravi Moussavi A., Senz J., Melnyk N., et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput. Biol. 2011;7:e1001138. doi: 10.1371/journal.pcbi.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakarya O., Breu H., Radovich M., Chen Y., Wang Y.N., Barbacioru C., Utiramerur S., Whitley P.P., Brockman J.P., Vatta P., et al. RNA-Seq mapping and detection of gene fusions with a suffix array algorithm. PLoS Comput Biol. 2012;8:e1002464. doi: 10.1371/journal.pcbi.1002464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abate F., Acquaviva A., Paciello G., Foti C., Ficarra E., Ferrarini A., Delledonne M., Iacobucci I., Soverini S., Martinelli G., et al. Bellerophontes: an RNA-Seq data analysis framework for chimeric transcripts discovery based on accurate fusion model. Bioinformatics. 2012;28:2114–2121. doi: 10.1093/bioinformatics/bts334. [DOI] [PubMed] [Google Scholar]
- 23.Piazza R., Pirola A., Spinelli R., Valletta S., Redaelli S., Magistroni V., Gambacorti-Passerini C. FusionAnalyser: a new graphical, event-driven tool for fusion rearrangements discovery. Nucleic Acids Res. 2012;40:e123. doi: 10.1093/nar/gks394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoffmann S., Otto C., Doose G., Tanzer A., Langenberger D., Christ S., Kunz M., Holdt L., Teupser D., Hackermueller J., et al. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing, and fusion detection. Genome Biol. 2014;15:R34. doi: 10.1186/gb-2014-15-2-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim P., Yoon S., Kim N., Lee S., Ko M., Lee H., Kang H., Kim J. ChimerDB 2.0–a knowledgebase for fusion genes updated. Nucleic Acids Res. 2010;38:D81–D85. doi: 10.1093/nar/gkp982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ha K.C., Lalonde E., Li L., Cavallone L., Natrajan R., Lambros M.B., Mitsopoulos C., Hakas J., Kozarewa I., Fenwick K., et al. Identification of gene fusion transcripts by transcriptome sequencing in BRCA1-mutated breast cancers and cell lines. BMC Med. Genomics. 2011;4:75. doi: 10.1186/1755-8794-4-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McManus C.J., Duff M.O., Eipper-Mains J., Graveley B.R. Global analysis of trans-splicing in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 2010;107:12975–12979. doi: 10.1073/pnas.1007586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang G., Guo G., Hu X., Zhang Y., Li Q., Li R., Zhuang R., Lu Z., He Z., Fang X., et al. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010;20:646–654. doi: 10.1101/gr.100677.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Q., Caballero O.L., Levy S., Stevenson B.J., Iseli C., de Souza S.J., Galante P.A., Busam D., Leversha M.A., Chadalavada K., et al. Transcriptome-guided characterization of genomic rearrangements in a breast cancer cell line. Proc. Natl. Acad. Sci. U.S.A. 2009;106:1886–1891. doi: 10.1073/pnas.0812945106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maher C.A., Kumar-Sinha C., Cao X., Kalyana-Sundaram S., Han B., Jing X., Sam L., Barrette T., Palanisamy N., Chinnaiyan A.M. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maher C.A., Palanisamy N., Brenner J.C., Cao X., Kalyana-Sundaram S., Luo S., Khrebtukova I., Barrette T.R., Grasso C., Yu J., et al. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc. Natl. Acad. Sci. U.S.A. 2009;106:12353–12358. doi: 10.1073/pnas.0904720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma L., Yang S., Zhao W., Tang Z., Zhang T., Li K. Identification and analysis of pig chimeric mRNAs using RNA sequencing data. BMC Genomics. 2012;13:429. doi: 10.1186/1471-2164-13-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inaki K., Hillmer A.M., Ukil L., Yao F., Woo X.Y., Vardy L.A., Zawack K.F., Lee C.W., Ariyaratne P.N., Chan Y.S., et al. Transcriptional consequences of genomic structural aberrations in breast cancer. Genome Res. 2011;21:676–687. doi: 10.1101/gr.113225.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Al-Balool H.H., Weber D., Liu Y., Wade M., Guleria K., Nam P.L., Clayton J., Rowe W., Coxhead J., Irving J., et al. Post-transcriptional exon shuffling events in humans can be evolutionarily conserved and abundant. Genome Res. 2011;21:1788–1799. doi: 10.1101/gr.116442.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shao X., Shepelev V., Fedorov A. Bioinformatic analysis of exon repetition, exon scrambling and trans-splicing in humans. Bioinformatics. 2006;22:692–698. doi: 10.1093/bioinformatics/bti795. [DOI] [PubMed] [Google Scholar]
- 36.Ozsolak F., Milos P.M. RNA sequencing: advances, challenges and opportunities. Nat. Rev. Genet. 2011;12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nacu S., Yuan W., Kan Z., Bhatt D., Rivers C.S., Stinson J., Peters B.A., Modrusan Z., Jung K., Seshagiri S., et al. Deep RNA sequencing analysis of readthrough gene fusions in human prostate adenocarcinoma and reference samples. BMC Med. Genomics. 2011;4:11. doi: 10.1186/1755-8794-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carrara M., Beccuti M., Cavallo F., Donatelli S., Lazzarato F., Cordero F., Calogero R.A. State of art fusion-finder algorithms are suitable to detect transcription-induced chimeras in normal tissues. BMC Bioinformatics. 2013;14(Suppl. 7):11. doi: 10.1186/1471-2105-14-S7-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carrara M., Beccuti M., Lazzarato F., Cavallo F., Cordero F., Donatelli S., Calogero R.A. State-of-the-art fusion-finder algorithms sensitivity and specificity. Biomed. Res. Int. 2013:340620. doi: 10.1155/2013/340620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cocquet J., Chong A., Zhang G., Veitia R.A. Reverse transcriptase template switching and false alternative transcripts. Genomics. 2006;88:127–131. doi: 10.1016/j.ygeno.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 41.Houseley J., Tollervey D. Apparent non-canonical trans-splicing is generated by reverse transcriptase in vitro. PLoS One. 2010;5:e12271. doi: 10.1371/journal.pone.0012271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ouhammouch M., Brody E.N. Temperature-dependent template switching during in vitro cDNA synthesis by the AMV-reverse transcriptase. Nucleic Acids Res. 1992;20:5443–5450. doi: 10.1093/nar/20.20.5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frenkel-Morgenstern M., Gorohovski A., Lacroix V., Rogers M., Ibanez K., Boullosa C., Andres Leon E., Ben-Hur A., Valencia A. ChiTaRS: a database of human, mouse and fruit fly chimeric transcripts and RNA-sequencing data. Nucleic Acids Res. 2013;41:D142–D151. doi: 10.1093/nar/gks1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li X., Zhao L., Jiang H., Wang W. Short homologous sequences are strongly associated with the generation of chimeric RNAs in eukaryotes. J. Mol. Evol. 2009;68:56–65. doi: 10.1007/s00239-008-9187-0. [DOI] [PubMed] [Google Scholar]
- 45.Mitalipov S., Kuo H.C., Byrne J., Clepper L., Meisner L., Johnson J., Zeier R., Wolf D. Isolation and characterization of novel rhesus monkey embryonic stem cell lines. Stem Cells. 2006;24:2177–2186. doi: 10.1634/stemcells.2006-0125. [DOI] [PubMed] [Google Scholar]
- 46.Djebali S., Lagarde J., Kapranov P., Lacroix V., Borel C., Mudge J.M., Howald C., Foissac S., Ucla C., Chrast J., et al. Evidence for transcript networks composed of chimeric RNAs in human cells. PLoS One. 2012;7:e28213. doi: 10.1371/journal.pone.0028213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo G.X., Taylor J. Template switching by reverse transcriptase during DNA synthesis. J. Virol. 1990;64:4321–4328. doi: 10.1128/jvi.64.9.4321-4328.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herai R.H., Yamagishi M.E. Detection of human interchromosomal trans-splicing in sequence databanks. Brief. Bioinform. 2010;11:198–209. doi: 10.1093/bib/bbp041. [DOI] [PubMed] [Google Scholar]
- 49.Eychene A., Rocques N., Pouponnot C. A new MAFia in cancer. Nat. Rev. Cancer. 2008;8:683–693. doi: 10.1038/nrc2460. [DOI] [PubMed] [Google Scholar]
- 50.Horiuchi T., Aigaki T. Alternative trans-splicing: a novel mode of pre-mRNA processing. Biol. Cell. 2006;98:135–140. doi: 10.1042/BC20050002. [DOI] [PubMed] [Google Scholar]
- 51.Dorn R., Krauss V. The modifier of mdg4 locus in Drosophila: functional complexity is resolved by trans splicing. Genetica. 2003;117:165–177. doi: 10.1023/a:1022983810016. [DOI] [PubMed] [Google Scholar]
- 52.Goeke S., Greene E.A., Grant P.K., Gates M.A., Crowner D., Aigaki T., Giniger E. Alternative splicing of lola generates 19 transcription factors controlling axon guidance in Drosophila. Nat. Neurosci. 2003;6:917–924. doi: 10.1038/nn1105. [DOI] [PubMed] [Google Scholar]
- 53.Parra G., Reymond A., Dabbouseh N., Dermitzakis E.T., Castelo R., Thomson T.M., Antonarakis S.E., Guigo R. Tandem chimerism as a means to increase protein complexity in the human genome. Genome Res. 2006;16:37–44. doi: 10.1101/gr.4145906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Akiva P., Toporik A., Edelheit S., Peretz Y., Diber A., Shemesh R., Novik A., Sorek R. Transcription-mediated gene fusion in the human genome. Genome Res. 2006;16:30–36. doi: 10.1101/gr.4137606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frenkel-Morgenstern M., Valencia A. Novel domain combinations in proteins encoded by chimeric transcripts. Bioinformatics. 2012;28:i67–i74. doi: 10.1093/bioinformatics/bts216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frenkel-Morgenstern M., Lacroix V., Ezkurdia I., Levin Y., Gabashvili A., Prilusky J., Del Pozo A., Tress M., Johnson R., Guigo R., et al. Chimeras taking shape: potential functions of proteins encoded by chimeric RNA transcripts. Genome Res. 2012;22:1231–1242. doi: 10.1101/gr.130062.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harrow J., Nagy A., Reymond A., Alioto T., Patthy L., Antonarakis S.E., Guigo R. Identifying protein-coding genes in genomic sequences. Genome Biol. 2009;10:201. doi: 10.1186/gb-2009-10-1-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.