


Abstract
Eukaryotic mRNAs with premature translation-termination codons (PTCs) are recognized and eliminated by nonsense-mediated mRNA decay (NMD). NMD substrates can be degraded by different routes that all require phosphorylated UPF1 (P-UPF1) as a starting point. The endonuclease SMG6, which cleaves mRNA near the PTC, is one of the three known NMD factors thought to be recruited to nonsense mRNAs via an interaction with P-UPF1, leading to eventual mRNA degradation. By artificial tethering of SMG6 and mutants thereof to a reporter mRNA combined with knockdowns of various NMD factors, we demonstrate that besides its endonucleolytic activity, SMG6 also requires UPF1 and SMG1 to reduce reporter mRNA levels. Using in vivo and in vitro approaches, we further document that SMG6 and the unique stalk region of the UPF1 helicase domain, along with a contribution from the SQ domain, form a novel interaction and we also show that this region of the UPF1 helicase domain is critical for SMG6 function and NMD. Our results show that this interaction is required for NMD and for the capability of tethered SMG6 to degrade its bound RNA, suggesting that it contributes to the intricate regulation of UPF1 and SMG6 enzymatic activities.
INTRODUCTION
In order to guarantee the accuracy of gene expression, eukaryotic cells have evolved numerous intricate quality control mechanisms. One of the best studied of these mechanisms is the nonsense-mediated mRNA decay pathway (NMD) that was archetypically known as a pathway acting to selectively identify and degrade mRNAs containing a premature translation-termination codon (PTC), and hence reduces the accumulation of potentially toxic truncated proteins. However, NMD also targets various physiological mRNAs, signifying a role for NMD in post-transcriptional gene expression regulation in eukaryotes (1–3). Therefore, NMD probably controls a large and diverse inventory of transcripts which reflects the important influence of NMD on the metabolism of the cell and consequently in many human diseases (4,5). In order to develop pharmacological reagents and to better understand the influence of NMD on disease, it is essential to unravel the molecular mechanisms that underpin NMD.
A plausible current model of NMD in human cells postulates that the decision of whether the pathway is to be initiated relies upon competition between up-frame shift 1 (UPF1), a core NMD factor that exhibits 5′-3′ helicase and nucleic acid-dependent adenosine triphosphatase (ATPase) activities (6), and cytoplasmic poly-A binding protein for binding to eukaryotic release factor 3 (eRF3) on the terminating ribosome (7–11). Suppressor with morphogenetic effect on genitalia protein 1 (SMG1), which is a phosphatidylinositol 3-kinase-related protein kinase (PIKK) (12), is also recruited by ribosomes terminating translation prematurely through interactions with the eRF1/3 and this complex of UPF1, SMG1 and the eRF1/3 is termed the SURF complex (13). In the presence of UPF2 and UPF3, most likely bound to downstream exon junction complexes (EJCs) on the mRNA, SMG1 phosphorylates UPF1 (13–15) creating an N-terminal binding platform for SMG6 and a C-terminal binding site for the SMG5–SMG7 complex, the latter of which has been reported to recruit mRNA decay factors (16,17) and these interactions at the N and C-termini of UPF1 are essential for NMD (18). SMG5, SMG6 and SMG7 each contain a 14-3-3-like domain, which in the case of SMG6 and SMG7 has been experimentally confirmed to bind phosphorylated residues of UPF1 (18,19). SMG6 can also associate with the mRNA surveillance complex through its ability to directly bind the EJC via conserved motifs called EJC binding motifs (EBMs) (20). SMG5 and SMG6 both contain a C-terminal PIN (PilT N-terminus) domain adopting a similar overall fold related to ribonucleases of the RNase H family but only SMG6 harbors the canonical triad of aspartic acid residues crucial for nuclease activity (21–23). Thereafter, SMG6 was revealed to be the endonuclease in human and Drosophila melanogaster cells that cleaves nonsense mRNAs in the vicinity of the PTC (24,25).
However, less is known about the actual mRNA degradation aspect of NMD but an emerging consensus is that phosphorylated UPF1 (P-UPF1) is the common starting point for all of the multiple decay routes that have been reported to be possible in NMD (26). SMG6 is one of the several proteins that are able to interact with P-UPF1 to ultimately induce RNA decay.
So far, it is not known if and how the endonuclease activity of SMG6 is regulated so that it is used only when and where it is needed and how this regulation would be orchestrated. Similarly, it is not clear exactly how SMG6 achieves target specificity; how exactly it is recruited to target mRNAs. In this study, we have investigated what is required for SMG6-mediated endonucleolytic cleavage of mRNA. Through in vivo functional assays, protein–protein interaction studies and in vitro experiments, we found that SMG6 activity specifically requires SMG1 and UPF1. However, this requirement is not dependent on the previously documented interaction between SMG6 and UPF1 phosphorylated at threonine 28 (18) but rather due to a newly identified phosphorylation-independent interaction between SMG6 and the unique stalk region in the UPF1 helicase domain, along with a contribution from the proximal portion of the SQ domain. We confirm that this interaction is critical for NMD and give insight into how this novel interaction may contribute to the regulation of UPF1 and SMG6.
MATERIALS AND METHODS
Plasmids
A comprehensive description of the plasmids used and made in this study can be found in the Supplementary Data section.
Tethered function and NMD assays
Cells were seeded into 6-well plates and DNA co-transfections were carried out using Lipofectamine 2000 (Life Technologies) according to the manufacturer's guidelines. In all tethered function assays (TFAs), 100-ng pEGFP-C3, 100-ng pcβ-globin6MS2 and 2-μg pCMV-SMG6-MS2-HA or 1-μg pCMV-LacZ-MS2-HA or 1-μg pCMV-HA-MS2-LacZ, along with either 400-ng pSUPuro scrambled plasmid (control) or 400-ng pSUPuro plasmid expressing an shRNA targeting specific NMD factors, was used. In Figure 1D, 2-μg pCMV-SMG6-MS2-HA, 1-μg pCMV-SMG6m14-3-3-MS2-HA and pCMV-SMG6mEBM-MS2-HA and 4 μg of pCMV-SMG6mPIN-MS2-HA were used. In Figure 2B–D, 400 ng of pSUPuro plasmids expressing shRNAs against SMG1, SMG5, SMG7, UPF1 or UPF2 were co-transfected. In Figure 3A–C, 600 ng of RNAi-resistant (RNAiR) pcDNA3-Flag-SMG1 or 550-ng RNAiR pcDNA3-HA-UPF1 plasmids were also co-transfected and the amount of DNA in each co-transformation mixture was made equal by the addition of the corresponding empty plasmid. In the NMD assays, 100 ng of each plasmid producing mini-μ or β-globin reporters containing a PTC as well as the wild-type counterpart, 100-ng pmCMV-rGPx1-TGC, along with either 400-ng pSUPuro control plasmid or 400 ng of plasmids expressing two shRNA targets against UPF1, were co-transfected. In Figure 7A, 550-ng RNAiR pcDNA3-HA-UPF1 wild-type, 600-ng RNAiR pcDNA3-HA-UPF1ΔStalk and 500-ng of RNAiR pcDNA3-HA-UPF1Δ1C were included in the co-transformation mixtures of both the NMD rescue experiments and the TFA rescue experiments. The shRNA target sequence for SMG5 was defined in (27) and the rest have been described elsewhere (28). The remainder of the pSUPuro-based knockdown procedure, cell harvesting for both protein and RNA samples (which were taken from the same sample), total RNA extraction and measurement of relative mRNA levels by quantitative reverse transcription polymerase chain reaction (RT-qPCR) was done as previously explained (29). qPCR assays have been described elsewhere (27,28), except for the TaqMan assay to measure eukaryotic green fluorescent protein (eGFP) mRNA levels that is comprised of 5′-TGAGCAAAGACCCCAACGA-3′, 5′-GGCGGCGGTCACGAA-3′ and 5′-FAM-GCGGATCACATGGTCCTGCTGG-BHQ1 -3′. For the assays in Figures 3 and 6, total RNA was extracted using the Total RNA Miniprep Kit (Sigma-Aldrich). To examine protein levels from the assays, generally 35 000 whole cell equivalents were separated using 6–12% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and the proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Westran Clear Signal, GE Healthcare) and probed with the indicated primary antibodies and appropriate fluorophore-coupled secondary antibodies. Fluorescent signals were captured using the Odyssey Infrared Imaging System (LI-COR Biosciences). A full inventory of all the antibodies used in this study can be found in the Supplementary data.
Figure 1.
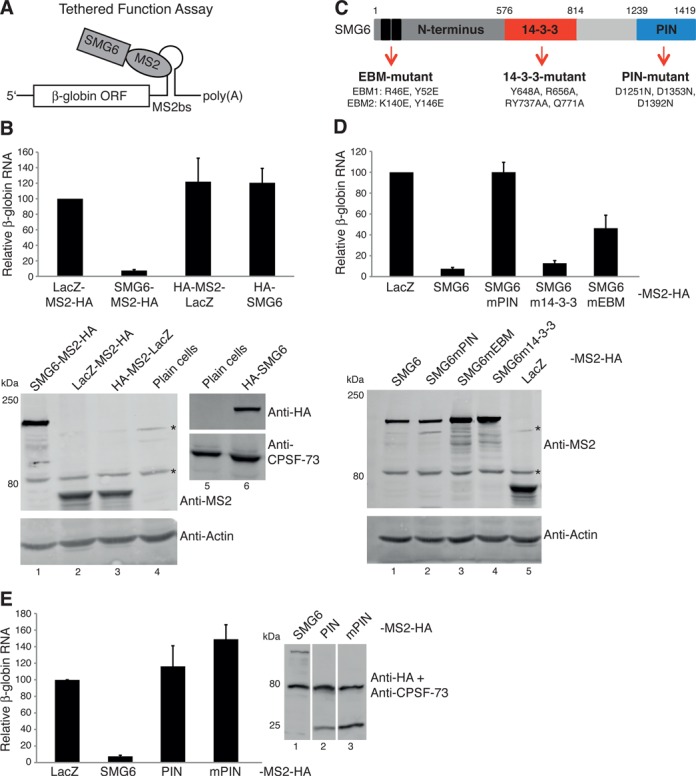
The SMG6 PIN domain is necessary but not sufficient for the degradation of reporter mRNA. (A) Schematic of the SMG6-tethered function assay (TFA). (B) SMG6-TFA. HeLa cells were transfected with pcβ-globin-6-MS2bs, pEGFP (serving as a co-transfection control) and with plasmids expressing each of the indicated factors fused to an MS2-HA moiety or HA-SMG6 that lacks the MS2 moiety. RT-qPCR analysis was used to evaluate the β-globin mRNA levels, normalized to GFP mRNA levels and with mRNA levels in cells expressing the LacZ-MS2-HA set as 100. The bars represent the mean of >6 independent experiments. Error bars indicate standard deviations (SD) and P-values are displayed in Supplementary Figure S1A. Western blots showing the expression of the MS2-HA fusion proteins (below left blot) and that of HA-SMG6 protein (below right blot). The antibody used is indicated at the right of each blot. * indicates unspecific bands detected using the anti-MS2 antibody. (C) Illustration of the human SMG6 protein annotated with the mutants used in this study and the specification of each mutant. (D) As in (B), except performed using plasmids expressing SMG6-MS2-HA and the indicated mutants thereof. Western blot displaying the expression of the specified SMG6-MS2-HA fusion proteins. (E) As in (B), except performed with plasmids expressing the designated MS2-HA fusion proteins, where the SMG6 PIN domain (AA 1239-1419 of SMG6) fused to the MS2 coat protein is simply denoted as PIN and its endonucleolytically inactive counterpart as mPIN. Western blot documenting the expression of the indicated MS2-HA fusion proteins.
Figure 2.
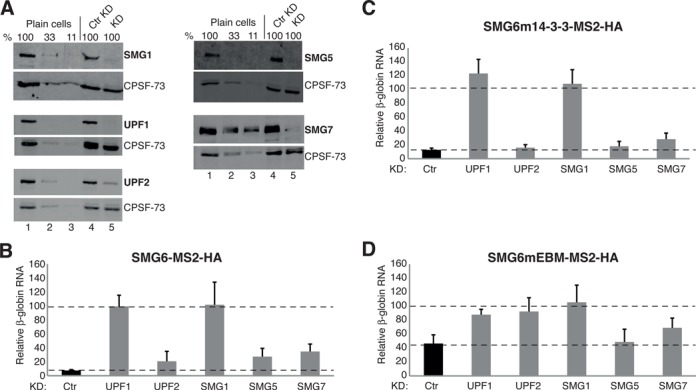
SMG6-mediated mRNA decay requires SMG1 and UPF1. (A) Western blots examining the knockdown efficiencies of various NMD factors from samples corresponding to the experiments shown in (B–D). Lanes 1–3 represent a serial dilution (100, 33 and 11%) of cell lysate from untransfected HeLa cells (plain cells). Lanes 4 and 5 show the protein levels from cells expressing a control shRNA (Ctr KD) and the shRNA targeting the designated NMD factor (KD), respectively. The detected protein is denoted at the right of each blot. (B–D) SMG-TFA as in Figure 1B, using plasmids expressing SMG6-MS2-HA in (B), SMG6m14-3-3-MS2-HA in (C) and SMG6mEBM-MS2-HA in (D), except performed in cells depleted of various NMD factors, whereby a plasmid expressing either a control shRNA or an shRNA targeting one of the indicated NMD factors was also included in the co-transfection mixture. β-globin reporter mRNA levels have been normalized to GFP mRNA levels and with mRNA levels in cells expressing LacZ-MS2-HA set as 100 (not shown for clarity). The bar charts depict the relative mRNA levels under control knockdown conditions (black bars) and the designated knockdown conditions (named below each gray bar). The bars represent the mean of >6 independent experiments. Error bars indicate SD and P-values are displayed in Supplementary Figure S1B.
Figure 3.
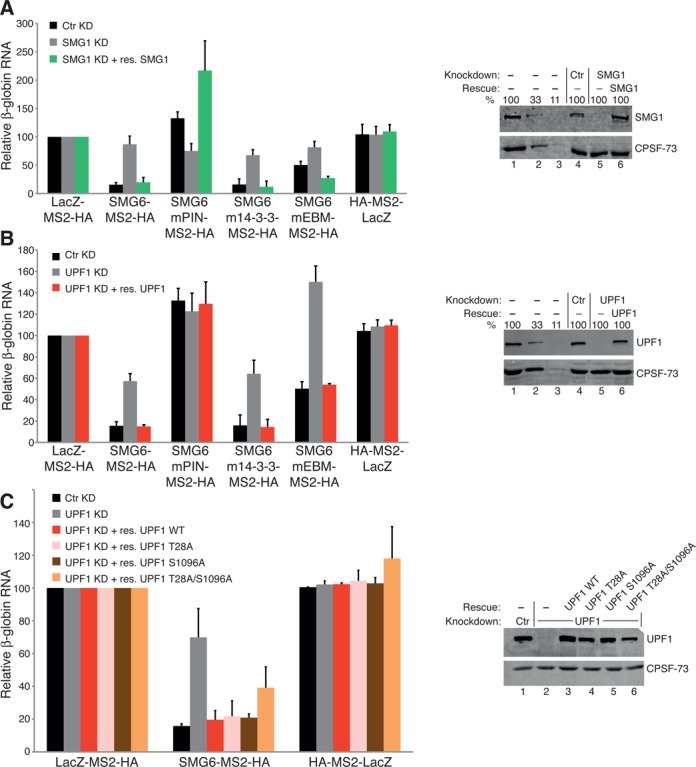
UPF1 phospho-site mutants still rescue SMG6 activity in UPF1-depleted cells. (A) SMG6 TFA as in Figure 1B using the labeled SMG-MS2-HA and the designated LacZ control plasmids, except a plasmid either expressing a control shRNA (black bars), an shRNA targeting SMG1 (gray bars) or a plasmid expressing an shRNA targeting SMG1 and a plasmid expressing an RNAi resistant (RNAiR) SMG1 (green bars) have also been transfected. β-globin mRNA levels, normalized to GFP mRNA levels and with mRNA levels in cells expressing LacZ-MS2-HA under no knockdown, SMG1 knockdown and SMG1 rescue conditions set as 100. The bars represent the mean of three independent experiments. Error bars indicate SD and P-values are presented in Supplementary Figure S1C. Western blot showing SMG1 levels. Lanes 1–3 represent a serial dilution (100, 33 and 11%) of cell lysate from untransfected HeLa cells. Lane 4 represents control shRNA (Ctr) and lane 5 represents the samples expressing the shRNA targeting SMG1. Lane 6 represents the samples expressing the shRNA targeting SMG1 and the exogenous expression of RNAiR full-length SMG1. The detected protein is indicated on the right of the blot. (B) SMG6-TFA performed exactly as in (A), except endogenous UPF1 levels were knocked down via expression of two shRNAs targeting UPF1 and complemented using exogenously expressed RNAiR UPF1 wild-type (red bars). (C) SMG6-TFA executed as in (B), except plasmids exogenously expressing RNAiR UPF1-T28A (pink bars), -S1096A (brown bars) and -T28A/S1096A (orange bars) mutants were also included and their expression levels are shown in lanes 4–6 of the corresponding western blot.
Figure 7.
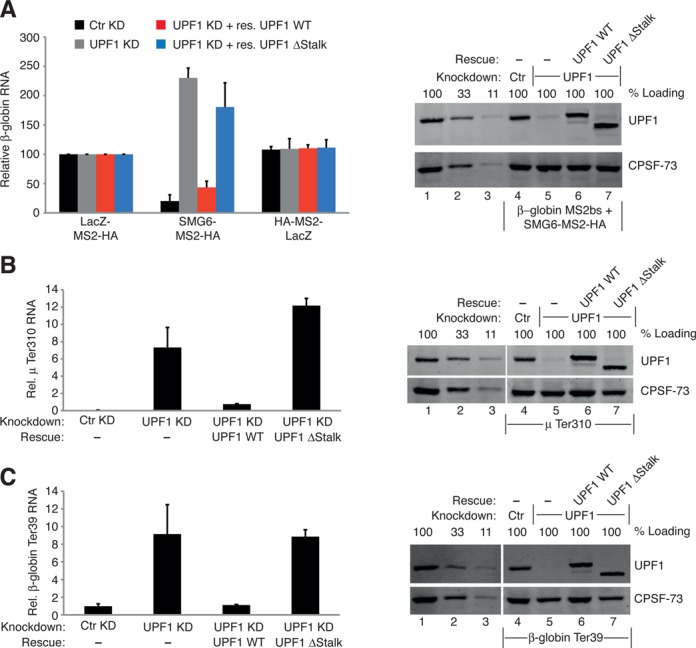
Functional assays confirm that the phospho-independent interaction between UPF1 and SMG6 is crucial for NMD. (A) SMG6-TFA completed as in Figure 3B, except the plasmid expressing RNAiR UPF1 ΔStalk (blue bars) was also included. The knockdown was extended by 1 day in these experiments and the bars represent the mean of three independent experiments. Error bars are SD and P-values are show in Supplementary Figure S1D. Western blot showing UPF1 levels. Lanes 1–3 represent a serial dilution (100, 33 and 11%) of cell lysate from untransfected HeLa cells. Lane 4 represents control shRNA (Ctr) and lane 5 represents the samples expressing two shRNAs targeting UPF1. Lanes 6 and 7 represent the samples expressing the shRNAs targeting UPF1 and the exogenous expression of RNAiR UPF1 WT and UPF1 ΔStalk, respectively. The detected protein is indicated on the right of the blot. (B) NMD rescue assays. HeLa cells were transfected with plasmids expressing mini-μ mRNA with a PTC at position 310 (μ Ter310), pmCMV-rGPx1-TGC, serving as a co-transfection control and with plasmids expressing either a control shRNA or shRNAs targeting UPF1 and where indicated plasmids expressing RNAiR UPF1 WT or RNAiR UPF1 ΔStalk were also co-transfected. RT-qPCR analysis was used to evaluate the relative μ mRNA levels, normalized to GPx1 mRNA levels, and the levels of normalized μ WT mRNA in control knockdown cells were set to 100% (not shown for clarity). The bars represent the mean of three independent experiments. Error bars are SD and P-values are show in Supplementary Figure S1D. Right panel: western blots from μ Ter310 expressing cells are as in (A). (C) Performed and analyzed as in (B), except plasmids expressing β-globin mRNA with a PTC at position 39 (β-globin Ter39) were co-transfected.
Figure 6.
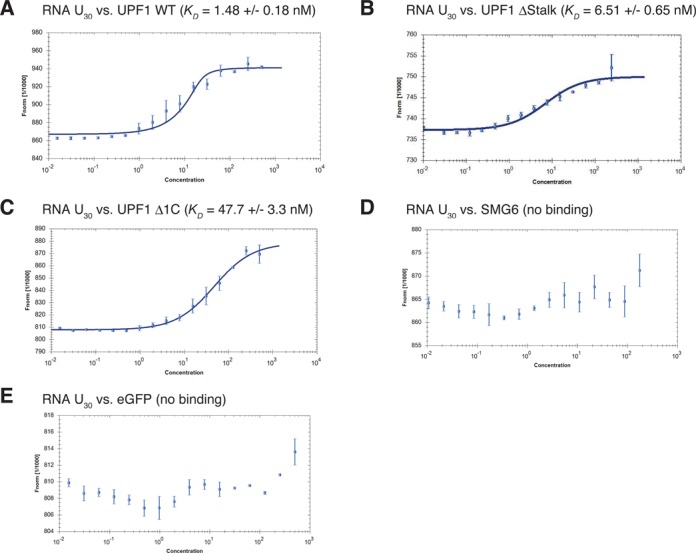
Purified UPF1 ΔStalk still interacts with RNA. (A–E) RNA binding studies using MicroScale Thermophoresis (MST). By fitting the change in thermophoretic depletion upon titration of (A) UPF1 WT, (B) UPF1 ΔStalk, (C) UPF1 Δ1C, (D) SMG6 and (E) eGFP to a constant amount (20 nM) of Cy5-labeled U30 RNA to the quadratic solution of the mass action law, the binding constants in nM (KD) were determined. The normalized fluorescence (Fnorm) is plotted on a linear y-axis in per thousand (‰) against the total concentration of the titrated protein (nM) on a log10 x-axis. At least two independent measurements were made for each assay and error bars on the individual data points represent the SD between these repetitions.
Yeast two-hybrid β-galactosidase plate assays
Two hundred and fifty nanograms of the designated pGADT7 and pGBKT7 plasmids were co-transformed into Mav99 cells (31) according to the high-efficiency LiOAc/single-stranded carrier DNA (Clontech)/polyethylene glycol ((Clontech)/PEG) method of transformation (32). Four co-transformants from each co-transformation reaction were spread in equal concentrated patches on fresh selective plates and incubated at 30°C for 3 days. Thereafter, non-lethal X-gal/β-galactosidase plate assays were performed (33). Briefly, the plates were then flooded with chloroform, completely immersing the colonies. After 5 min, the chloroform was decanted and the plates were inverted and dried for 5 min. Each plate was then overlaid with 1% low melting agarose (Promega, V2111) containing 1-mg ml−1 X-Gal and 100-mM KPO4, pH 7.0 cooled to 42°C. Once the agarose was hardened, the plates were inverted and incubated at 30°C for 24 h and then at 4°C for 24 h before being photographed.
Recombinant protein production
The recombinant proteins were produced in HEK293T cells by transfecting pEFh_SBP-eGFP, pEF_Flag-HA-SBP-SMG6, pSR_myc-SBP-UPF1, pEFh_SBP-UPF1 (codon optimized) and derivatives thereof and following a purification procedure formerly described (34), except that the proteins were eluted in 1x HBS-EP+ buffer (GE Healthcare BR-1008-26).
In vitro protein-binding experiments
For in vitro protein-binding assays, 3 × 107 of 293T cell extracts transfected with pEF_Flag-HA-SBP-SMG6 or pEF_Flag-HA-maltose-binding protein (MBP) were lysed in NF buffer [20-mM Tris HCl (pH 7.5), 150-mM NaCl, 0.25-M sucrose, 1% Triton X-100, 0.5% Tween 20, 1-mM dithiothreitol (DTT), 1 x protease inhibitor (Nacalai), 1 x phosphatase inhibitor (Nacalai)] containing 50-μg/ml RNase A and incubated with 0.5 mg of anti-HA-tag mAb-Magnetic beads (TANA2, MBL) for 2 h at 4°C. The beads were washed by NF buffer and separated into 5–6 tubes. The separated beads were incubated with 1000 nM of SBP-eGFP, or SBP-UPF1 and derivatives in T buffer [20-mM HEPES-KOH (pH 7.5), 150-mM NaCl, 2.5-mM MgCl2 and 0.05% Tween 20] complemented with 1-mM MgCl2, 1-mM MnCl2, 1-μM ZnCl2, 1-mM DTT and 0.01% bovine serum albumin. After an incubation of 4 h, the beads were washed four times with T buffer containing 1-mM DTT. The bound proteins were eluted with SDS sample buffer lacking reducing agents. After addition of reducing agents and heat denaturation, the eluted proteins were separated by SDS-PAGE, transferred to a membrane and probed using antibodies stipulated in the Supplementary data section. The bands were detected using either an ECL western blotting detection kit (GE Biotech) or Luminata forte (Millipore) using a Lumino-Imager, LAS-4000 and Science Lab 2001 Image Gauge software (FujiPhoto Film).
In vitro RNA–protein interaction studies using MicroScale Thermophoresis
The MicroScale Thermophoresis (MST) assays were set up, performed and analyzed as previously outlined (35–38). Specifically, the 5′-labeled Cy5 RNA U30 oligonucleotide was purchased from Microsynth.ch and diluted to 40 nM using 1 x HBS-EP+ buffer. The solution of each designated unlabeled recombinant protein (see recombinant protein production) was serially diluted from a concentration of 500 nM down to 0.015 nM (with the exception of SMG6 that was diluted from 350 nM to 0.01 nM) in the presence of constant labeled RNA (20 nM). The RNA–protein mixtures were analyzed in hydrophilic capillaries (NanoTemper Technologies ref: K004) using the Monolith NT.115T MST instrument (NanoTemper Technologies) at room temperature (RT), where the light-emitting diode (LED) was set at 40% (except in Figure 6E where the LED was set to 70%), the IR-laser power was set at 20% and laser on and off times were fixed at 30 s and 5 s, respectively. The Nanotemper Technologies Analysis software, version 1.5.41, was used to obtain normalized fluorescence versus concentration curves and to determine the corresponding KD using the law of mass action.
RESULTS
Tethering of the SMG6 PIN domain alone is not sufficient to reduce reporter RNA levels
To build upon what is known about SMG6 and further delineate the molecular mechanisms surrounding SMG6 activity, we established an SMG6 TFA, a comprehensive description of which can be found elsewhere (29). Full-length SMG6, mutants thereof and a fragment of LacZ serving as a control were fused to the MS2 coat protein, which in turn can be artificially tethered to a β-globin reporter mRNA containing six MS2 binding sites in its 3′-untranslated region (3′UTR) (β-globin-6-MBS; Figure 1A). SMG6-MS2 fusion proteins were co-expressed with the β-globin-6-MBS reporter and a GFP expressing plasmid in HeLa cells, and the steady-state levels of the reporter mRNA were quantified and normalized to the levels of GFP mRNA. Expression of SMG6-MS2-HA reduced the steady-state levels of the reporter mRNA to below 10% of the levels detected in cells expressing the LacZ-MS2-HA or the HA-MS2-LacZ controls, which encode a fragment of the β-galactosidase fused to a C-terminally located MS2-HA or an N-terminally located HA-MS2 moiety, respectively. To control for potential pleiotropic effects caused by over-expression of proteins, SMG6 without the MS2-moiety was also tested (Figure 1B). Western blotting confirmed that the MS2 fusion proteins and the SMG6 lacking the MS2 moiety were all expressed (Figure 1B, lower panels).
To address what aspect of SMG6 was important for inducing reporter level reduction when tethered to the mRNA, we expressed various SMG6 mutants fused to MS2-HA (Figure 1C). In the SMG6-PIN mutant (SMG6mPIN), the endonuclease activity was abolished by mutating the three crucial aspartic acids in the catalytic center to asparagines (24). In the SMG6-14-3-3 mutant (SMG6m14-3-3), four highly conserved amino acids in the 14-3-3-like domain were changed (19) with the expectation that this should prevent SMG6 from binding to UPF1 phosphorylated at T28, as previously shown with such an SMG6 mutant (18). Finally, in the SMG6 EBM mutant (SMG6mEBM), point mutations were introduced in the EBMs 1 and 2 in the very N-terminus of SMG6 (20). Transfection conditions were adapted to achieve a similar expression of all of the SMG6-MS2-HA mutants as displayed in the western blot in the lower part of Figure 1D. When tethered to the β-globin reporter mRNA, we observed that the SMG6mPIN-MS2-HA did not reduce the reporter RNA levels at all (Figure 1D). By contrast, the SMG6m14-3-3-MS2-HA behaved like its wild-type counterpart and was able to reduce the reporter RNA levels to ∼12% of the levels detected in cells expressing the LacZ-MS2-HA control. Finally, the ability of the tethered SMG6mEBM-MS2-HA in reducing the reporter RNA levels was significantly compromised compared to tethering wild-type SMG6 (P-value 3.9 × 10−5; see Supplementary Figure S1A) yet significantly different from the LacZ-MS2-HA control (P-value 1.5 × 10−11). The same results were reached when the β-globin RNA levels were analyzed by northern blotting and also when the same SMG6 fusion proteins were tethered to a Renilla luciferase reporter mRNA containing six MS2 binding sites in its 3′UTR (Supplementary Figure S2). Next, we wondered if tethering of the PIN domain alone was enough to elicit decreased β-globin reporter mRNA levels. As shown in Figure 1E, tethering the SMG6 PIN domain alone was not sufficient to bring about the reduction in reporter mRNA levels. Expression of the PIN-MS2-HA and mPIN-MS2-HA are documented in lanes 2 and 3 of the accompanying western blot.
In summary, mutations affecting the N-terminal EBM motifs compromise the ability of SMG6 to reduce reporter mRNA levels despite the fact that their proposed role of recruiting SMG6 to the RNA (20) is bypassed in these assays, begging the question as to what function these motifs contribute to the endonuclease activity of SMG6. The SMG6 14-3-3 mutant that should no longer be able to interact with P-UPF1 induced reporter mRNA reduction as well as its wild-type SMG6, consistent with the view that normally SMG6 is recruited to NMD-targeted messenger ribonucleoprotein particles (mRNPs) by this interaction and our tethering assay bypasses this requirement. Importantly, the endonucleolytic activity of SMG6 is required but not sufficient by itself for inducing reporter mRNA level reduction, demonstrating that additional parts of the SMG6 protein are necessary for endowing the PIN domain with endonuclease activity, presumably by interacting with additional proteins.
The SMG6-mediated reporter mRNA level reduction requires SMG1 and UPF1
To test whether additional NMD factors are required to stimulate the endonuclease activity of SMG6, we performed TFAs with the SMG6-MS2-HA constructs described in Figure 1 in HeLa cells depleted of various NMD factors. UPF1, UPF2, SMG1, SMG5 or SMG7 was knocked down by expressing corresponding shRNAs in parallel with a control pSUPuro plasmid expressing a scrambled sequence with no predicted specific targets in human cells. A fraction of the cell lysates were used to extract RNA and determine the relative β-globin reporter mRNA levels (see below), and western blots were performed with the remainder of each lysate to assess the knockdown efficiencies of the stipulated NMD factors (Figure 2A). The effectiveness of the stipulated RNAi-mediated knockdowns was also documented at the mRNA level (Supplementary Figure S3A–E). The β-globin reporter mRNA level for each experimental condition is depicted relative to the level in cells expressing LacZ-MS2-HA and normalized to GFP mRNA encoded on a co-transfected expression plasmid to account for differences in transfection efficiencies among the samples (Figure 2B–D and Supplementary Figure S1B). We found that SMG6-MS2-HA-induced reporter mRNA level reduction significantly requires UPF1 and SMG1 but is only marginally compromised by the knockdown of UPF2, SMG5 or SMG7 (Figure 2B). Essentially the same result was obtained when SMG6m14-3-3-MS2-HA was tethered (Figure 2C). The already compromised destabilizing activity of the SMG6mEBM was also lost under UPF1 and SMG1 knockdown conditions (Figure 2D). Intriguingly, the modest reduction of reporter RNA induced by tethering of SMG6mEBM was further lost when UPF2 was knocked down, in contrast to tethered SMG6 wild-type, which was barely affected by UPF2. Knockdown of UPF3B gave a similar result; it barely affected the strong drop in reporter mRNA levels induced by tethering wild-type SMG6, but it significantly abrogated the modest decrease of reporter levels induced by tethering of SMG6mEBM (Supplementary Figures S3F and G and S1E). This suggests that UPF2 and UPF3B are also involved in activation of the SMG6 endonuclease under physiological conditions but possibly their contribution is weaker than those of UPF1 and SMG1, and hence it can only be detected under conditions that reduce the otherwise dominant contributions, such as in the case of the EBM mutant. In summary, the data identify UPF1 and SMG1 as necessary co-factors for the endonuclease activity of SMG6 and also indicate a minor involvement of UPF2 and UPF3B. In contrast, the roles of SMG5 and SMG7 seem marginal at best in our SMG6 TFA.
Rescue experiments confirm that SMG6 activity requires UPF1 and SMG1
To ascertain that the loss of the SMG6 induced reporter mRNA level reduction was specifically caused by the depletion of SMG1 and UPF1 and not by an off-target effect of the knockdown, we tried to rescue the observed SMG6-mediated effect on the mRNA reporter by expression of exogenous RNAiR versions of these proteins. Figure 3A depicts the SMG1 rescue experiment where the specified SMG6-MS2-HA constructs, or the controls LacZ-MS2-HA and HA-MS2-LacZ, were tethered in HeLa cells expressing a control shRNA (black bars), cells depleted of SMG1 (gray bars) or cells depleted of SMG1 and expressing exogenous RNAiR SMG1 (green bars). Our data show that expression of RNAiR SMG1 significantly and specifically rescues the steady-state reporter mRNA levels back to those observed under no knockdown conditions (see Supplementary Figure S1C for P-values). Western blotting confirmed that the RNAi mediated SMG1 knockdown was effective and that the expression of the exogenous RNAiR SMG1 replenished the pool of intracellular SMG1 to a similar level as without or with a control knockdown. The UPF1 rescue experiment was analogously performed and gave a similar result (Figure 3B and Supplementary Figure S1C). It showed that exogenously expressed RNAiR UPF1 was able to significantly rescue the mRNA reporter levels under endogenous UPF1 knockdown conditions back to those seen in HeLa cells with a control knockdown. As for SMG1, western blotting confirmed that the UPF1 knockdown was effective and that the expression of the RNAiR UPF1 in cells depleted of UPF1 yielded UPF1 protein levels similar to those observed in cells without or with a control knockdown. These results confirm that tethered SMG6-mediated reporter mRNA level reduction definitively requires the presence of both UPF1 and SMG1.
UPF1 phosphorylation mutants still support SMG6-facilitated RNA reporter level reduction, regardless of the need for SMG1 and UPF1
To decipher the domains or functions of UPF1 and SMG1 required for the activity of SMG6, RNAiR UPF1 constructs harboring various previously described mutations were expressed in SMG6 tethering rescue experiments. We tested three different UPF1 phospho-site (P-site) mutants: T28A, S1096A and a double mutant comprising both of these point mutations (18). As before, the UPF1 knockdown effectiveness and the expression levels of the RNAiR UPF1 constructs were monitored by western blotting (Figure 3C, right panel). The efficient UPF1 knockdown was determined (lane 2) and the levels of all RNAiR UPF1 mutants (lanes 4–6) were comparable to the endogenous UPF1 levels in control knockdown cells (lane 1). As previously observed (Figure 3B), wild-type RNAiR UPF1 rescued the increased mRNA reporter levels resulting from the loss of UPF1 back to the levels observed when the assay was performed without knockdown (Figure 3C, red bars). Interestingly, RNAiR UPF1-T28A (pink bars) and UPF1-S1096A (brown bars) rescued the reduced reporter levels induced by SMG6 as efficiently as wild-type UPF1 (all P-values >0.1 when compared to the effect induced by SMG6 under control knockdown conditions; Supplementary S1C) and even the double mutant T28A/S1096A (orange bars) was almost as effective as the wild-type counterpart in these SMG6 tethering rescue experiments (the P-values when compared to the other P-site mutant rescues are >0.1 but the P-value is 0.01 compared to the control knockdown condition).
This result was unexpected in light of the previously reported dependence of the UPF1–SMG6 interaction on phosphorylated T28 in UPF1 (18). Therefore, we wanted to confirm that the mutants designed to disrupt the reported interaction between P-UPF1 and SMG6 were successfully abolishing such an interaction in our assays. Hence, we co-expressed HA-UPF1 with our various SMG6 mutants fused to MS2-HA and performed immunoprecipitations (IPs) with an anti-MS2 antibody (MS2-IP) to test the SMG6-MS2-HA proteins for their ability to co-IP P-UPF1 (Supplementary Figure S4A). Since only a very small fraction of UPF1 is phosphorylated at steady state (39), we co-expressed HA-UPF1 and added okadaic acid (a potent inhibitor of serine/threonine phosphatases 1, 2A and 2B) prior to cell lysis to increase the abundance of intracellular P-UPF1 (40). Intriguingly, all of the described SMG6-MS2-HA proteins co-immunoprecipitated P-UPF1 (lanes 6, 9, 12, 15 and 18) as determined by using a phospho-(Ser/Thr) ATM/ATR substrate antibody that specifically recognizes P-UPF1 at the correct molecular weight, albeit, we observed that the SMG6mEBM-MS2-HA and the SMG6m14-3-3-MS2-HA versions co-precipitated much less P-UPF1 than the SMG6-MS2-HA wild-type and SMG6mPIN-MS2-HA constructs (compare lanes 12 and 15 with 6 and 9, respectively). Furthermore, we detected a small and similar fraction of UPF1 in the precipitates of all SMG6-MS2-HA constructs using an anti-UPF1 antibody, even when the lysate was treated with RNase A (lane 18). In contrast, the LacZ-MS2-HA control did not co-IP any P-UPF1 (lane 3). In a vice-versa approach, we expressed our MS2-UPF1 P-site mutants together with HA-SMG6, performed MS2-IPs and examined the extent of SMG6 co-precipitation (Supplementary Figure S4B). Noteworthy, the MS2-UPF1-T28A still co-precipitated a small fraction of SMG6 (lane 9). In fact, all of the UPF1 P-site mutants still weakly co-precipitated SMG6 (lanes 12 and 15), to a similar or slightly lesser extent than the wild-type UPF1 protein and regardless of RNase A treatment (lanes 6 and 18, respectively). HA-MS2-LacZ did not co-IP any SMG6 which confirmed the specificity of the IPs (lane 3).
Given that the UPF1 P-site mutants still co-immunoprecipitated with SMG6, we wondered whether the kinase activity of SMG1 was required for SMG6-mediated RNA decay in the TFA or whether perhaps another function of SMG1 supported SMG6 activity. Therefore, RNAiR SMG1 D2331A where the kinase activity was abolished by mutating a crucial aspartic acid in the PIKK catalytic domain to an alanine (41,42) was expressed in the SMG6 TFA. As previously observed (Figure 3A), wild-type RNAiR SMG1 rescued the increased mRNA reporter levels resulting from the loss of SMG1 back to the levels observed when the assay was performed without knockdown (Supplementary Figure S5A, green bars and Supplementary Figure S1F for P-values). In contrast, the RNAiR SMG1 D2331 (purple bars) did not rescue the SMG6-induced reduced reporter RNA levels. The efficient SMG1 knockdown was determined (Supplementary Figure S5B, lane 5) and the levels of RNAiR SMG1 WT and the D2331A mutant (lanes 6 and 7) were comparable to the endogenous SMG1 levels in untreated cells and in the control knockdown cells (lanes 1 and 4, respectively). This result indicates that SMG1 kinase activity is required for SMG6 activity in the TFA. Since tethered SMG6 needs UPF1 and kinase-competent SMG1 for decreasing the reporter mRNA levels, suggesting a requirement for P-UPF1, while on the other hand the phospho-epitope binding SMG6 14-3-3 mutant still induced strong reporter mRNA level loss and the UPF1 T28A and S1096A mutants rescued SMG6-mediated effects on the reporter mRNA in UPF1 knockdown cells as efficiently wild-type UPF1, we conclude that these SMG1-mediated phosphorylation events needed for SMG6 activity occur either in other proteins or in UPF1 at P-sites other than T28 and S1096.
Moreover, we discovered that more of the SMG6 protein than just the PIN or 14-3-3 domain was needed for its activity and we always observed a minor but constant interaction between UPF1 and SMG6, regardless of the mutations made in either protein. Based on all of this, we postulated that at least a part of the SMG6–UPF1 interaction may occur in a phosphorylation-independent manner as previously hinted to but never explored (18,20). We anticipated that such a phospho-independent interaction might contribute a regulatory aspect to each enzyme and would be important for the actual mRNA degradation induced by SMG6.
Discovery of a phosphorylation-independent interaction between SMG6 and the helicase domain of UPF1
To identify such a phospho-independent interaction between UPF1 and SMG6, we utilized the yeast two-hybrid system (43). First, we tested several truncations of UPF1 (Figure 4A) fused to the GAL4-DNA-binding domain (BD) for an interaction with full-length SMG6 fused to the GAL4-activation domain (AD) using a LacZ reporter gene, which allows detection of colonies expressing β-galactosidase by addition of the chromogenic substrate X-gal to the plates. Of each co-transformation, four different colonies (A–D) were analyzed. GAL4-DNA-BD-eRF3a and GAL4-AD-eRF1 served as positive control since the interaction between eRF1 and eRF3 is well documented (44,45) (Figure 4B, row 15). Additional controls ruled out self-activation of any of the GAL4-DNA-BD fusion proteins in the absence of the GAL4-AD-SMG6 (Figure 4B, rows 8–14) while the control shown in row 7 confirmed that GAL4-AD-SMG6 is also not able to self-activate the reporter gene. Co-expression of BD-UPF1 and AD-SMG6 resulted in blue colonies, indicating an interaction between the UPF1 and SMG6 full-length proteins (row 1). While we did not observe any interaction between SMG6 and the CH or SQ domains of UPF1 (rows 2 and 4), we scored an interaction between the UPF1 HD domain and SMG6 (row 3), which was abolished by the presence of the CH domain (row 5). On the other hand, we noted that the interaction between the UPF1 HD and SMG6 appeared consistently stronger when the HD still had its SQ domain present (row 6). These results indicate that the presence of the CH domain inhibits and the presence of the SQ domain enhances the interaction of SMG6 with the UPF1 HD. The expression levels of the utilized UPF1 constructs and of the SMG6 full-length construct in yeast cells were confirmed by western blotting using an anti-c-Myc antibody recognizing the in-frame c-Myc tag located between the DNA-BD and the start of the UPF1 ORF and an anti-HA antibody identifying the in-frame HA-epitope tag located between the AD and the start of the SMG6 ORF (Supplementary Figure S6A and B, respectively).
Figure 4.
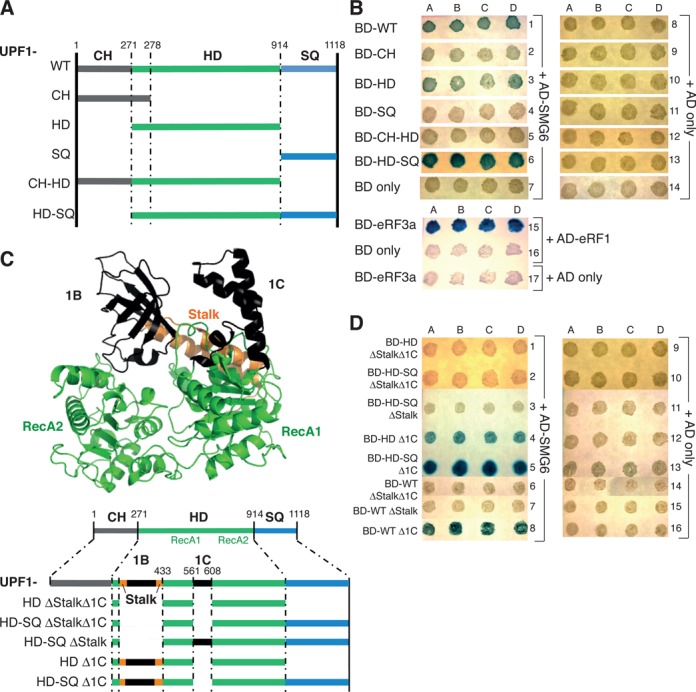
SMG6 interacts with UPF1 HD in a phospho-independent manner. (A) Diagram of the UPF1 aspect of various GAL4-DNA-BD-UPF1 constructs tested. (B) Yeast two-hybrid data. In rows 1–6, plasmids expressing GAL4-DNA-binding domain UPF1 fusion protein (BD-UPF1) and derivatives thereof were co-transformed with a plasmid expressing GAL4-activation domain SMG6 fusion protein (AD-SMG6) into Mav99 cells. The designated co-transformations in rows 7–14 were performed as negative controls, where BD only represents transformation of a plasmid expressing GAL4-DNA-BD-empty and AD only represents transformation of a plasmid expressing GAL4-AD-empty. In row 15, plasmids expressing GAL4-DNA-BD-eRF3a co-transformed with a plasmid expressing GAL4-AD-eRF1 served as a positive control, along with the necessary controls in rows 16–17. Four colonies (denoted as A–D) from each co-transformation were selected for the β-galactosidase plate assay. An interaction between the BD and the AD fusion proteins activates β-galactosidase expression, which produces a blue color by hydrolyzing X-gal. (C) Top: PyMOL view of the structure of human UPF1 HD based on pdb file 2xzp (30) showing the two RecA domains (in green), the additional protruding sub-domains 1B and 1C (both in black) along with the stalk helices (in orange). Bottom: schematic of the UPF1 part of the additional GAL4-DNA-BD-UPF1 constructs tested. Note the same deletions were created and tested in full-length UPF1. (D) As in (B), except the co-transformations were performed using plasmids expressing the stipulated GAL4-DNA-BD-UPF1 proteins (BD-UPF1) with the plasmid expressing GAL4-DNA-AD-SMG6 (AD-SMG6) shown in rows 1–8. Rows 9–16 represent the corresponding negative controls expressing empty GAL-AD (AD only).
As illustrated in the crystal structure of the UPF1 HD (30) in Figure 4C, it consists of two domains shown in green, each of which comprises RecA-like α/β domains that are responsible for nucleotide and RNA binding and have been referred to as RecA1 (or 1A) and RecA2 (or 2A), respectively (30,46). The adenosine triphosphate (ATP) binding site is located in a deep cleft separating these two RecA-like domains. On the first domain, two further sub-domains termed 1B and 1C exist (shown in black). 1B comprises amino acids (AA) 325-414 and takes the form of a β-barrel consisting of six anti-parallel strands located above the boundary between the two RecA-like domains (6,46–47). 1B is connected to RecA1 by two α-helices that are not preserved in any other known helicase structures. These two α-helices were termed the stalk (colored in orange) because they protrude from RecA1 (30). 1C comprises AA 556-609 and is made up of three helices that pack against the outer surface of Rec1A and makes a few contacts to sub-domain 1B. Since the 1B and 1C insertions are unique for UPF1, form structural entities above the RecA-like domains and seem to be important for NMD, at least in yeast (30,46), we wondered if SMG6 interacted with these structures, which would confer specificity in the UPF1–SMG6 interaction. To test this, we generated full-length UPF1, UPF1 HD or UPF1 HD-SQ fused to the GAL4-DNA-BD that lacks the stalk plus the intervening 1B insertion (ΔStalk, AA 271-433), the 1C sub-domain (Δ1C, AA 561-608) or both (ΔStalkΔ1C) as illustrated in Figure 4C below the crystal structure. When tested in the yeast two-hybrid assay, we found that even when the 1C region is removed from the UPF1 HD, it still interacted with SMG6 (Figure 4E, rows 4, 5 and 8). Notably, the enhancing effect of the presence of the SQ domain was again clearly seen in these tests (compare rows 4 and 5). However, when the stalk region was removed, the interaction was lost (Figure 4D, rows 1–3 and 6–7). This dependence on the stalk region strongly suggested that the interaction point of UPF1 with SMG6 lies somewhere within AA 271-433. Again, the negative controls ruled out self-activation of the various protein fusions alone (rows 9–16).
Given that this result is based upon a loss of interaction, it was essential to confirm by western blotting that the constructs depicted in Figure 4C and assayed in Figure 4D were all made into proteins in the yeast cells (Supplementary Figure S6C). We could not detect expression of the BD-UPF1 HD ΔStalk construct (not shown), but we could detect expression of the same mutant incorporated into BD-UPF1-HD-SQ (left panel, lane 3) and in full-length UPF1 (right panel, lane 8).
Concurrently with mapping the region in UPF1 required for this novel interaction between UPF1 and SMG6, we also tried to identify the interaction point to UPF1 in SMG6 using the yeast two-hybrid approach. We constructed several plasmids encoding the different SMG6 portions fused to the GAL4-AD but we were not able to conclusively map the interaction point in SMG6 because many of the SMG6 fusion proteins involving the unstructured N-terminus were not well expressed in the yeast cells (data not shown). However, SMG6 PIN domain (AA 1239-1419), 14-3-3 domain (AA 576–814) and SMG6 AA 814-1419 fused to the GAL4-AD all expressed well in the yeast cells but we did not observe an interaction of any of these SMG6 fusion proteins with UPF1 (Supplementary Figure S6D and E). The interaction between UPF1 and SMG6 14-3-3 domain is probably reliant upon phosphorylation and the yeast cells lack SMG1 to phosphorylate UPF1 for this to occur (12). Therefore, it is clear that the interaction between SMG6 and UPF1 probably involves the N-terminal part of SMG6 and is not mediated by the PIN domain of SMG6.
UPF1 Stalk and SQ regions are involved in binding to SMG6 in vitro
We next analyzed in vitro the binding between SMG6 and UPF1 to confirm the interaction identified using the yeast two-hybrid assay by testing if purified SMG6 can pull-down purified UPF1 and variants thereof depicted in Figure 5A. Specifically, Flag-HA-SBP-SMG6 and Flag-HA-SBP-MBP, serving as a control were expressed in HEK293T cells and captured on anti-HA magnetic beads (Figure 5B) and streptavidin-binding protein (SBP) tagged UPF1 WT, UPF1 ΔStalk, UPF1 Δ1C and eGFP were expressed in HEK293T cells. Such cells were treated with 7-mM caffeine for 4 h to abolish UPF1 phosphorylation and affinity purified using streptavidin sepharose (Figure 5C). Aliquots of immobilized Flag-HA-SBP-SMG6 and Flag-HA-SBP-MBP were mixed with equimolar amounts of the purified UPF1 protein variants or with eGFP as a control and the retained proteins were detected by western blot using an anti-UPF1 or anti-GFP antibody, respectively (Figure 5D). All three UPF1 protein variants co-precipitated specifically with Flag-HA-SBP-SMG6 but the amount of SMG6 associated with UPF1 ΔStalk was reduced compared with UPF1 WT and UPF1 Δ1C (Figure 5D, compare lane 11 with lanes 10 and 12, respectively). This result corroborates our yeast two-hybrid data by confirming that the stalk region is an important part of UPF1 for its interaction with SMG6. Since we consistently observed in our yeast two-hybrid analyses that the SQ domain of UPF1 enhanced the binding between UPF1 and SMG6 (Figure 4) and removal of the stalk region did not completely abolish the interaction between SMG6 and UPF1, we also tested if the SQ domain of UPF1 contributes to the interaction. Thus, two additional SBP-tagged C-terminally truncated UPF1 ΔStalk constructs lacking the last 99 amino acids (UPF1 ΔStalkΔSQ1; see Figure 5A) or the entire SQ domain (ΔStalkΔSQ2) were also expressed in HEK293T cells as before (Figure 5E). Again, the UPF1 WT protein co-precipitated specifically with Flag-HA-SBP-SMG6 (Figure 6F, lane 11) and the amounts of SMG6-associated UPF1 ΔStalk and ΔStalkΔSQ1 were reduced to similar extents compared with UPF1 WT (Figure 5F, compare lane 11 with lanes 12 and 13, respectively). Strikingly, almost no UPF1 ΔStalkΔSQ2 was pulled down by SMG6 (lane 14). Therefore, the UPF1 Stalk region of the HD and the proximal 105 amino acids of the UPF1 SQ domain are involved in binding to SMG6. This result is in line with the fact that Chakrabarti et al. also simultaneously detected a contribution of the SQ domain to this newly identified interaction between UPF1 and SMG6 (48). Since yeast cells do not appear to have an SMG1 ortholog (12) and because the UPF1 used in our in vitro assays was hypo-phosphorylated, this identified interaction between UPF1 and SMG6 likely occurs independently of phosphorylation. Further to this, there are no annotated P-sites existing within the UPF1 region spanning AA 271-433 and the two annotated P-sites in the proximal SQ domain at Y946 and Y958 are not well conserved and have not been site-specifically analyzed so far (49,50).
Figure 5.
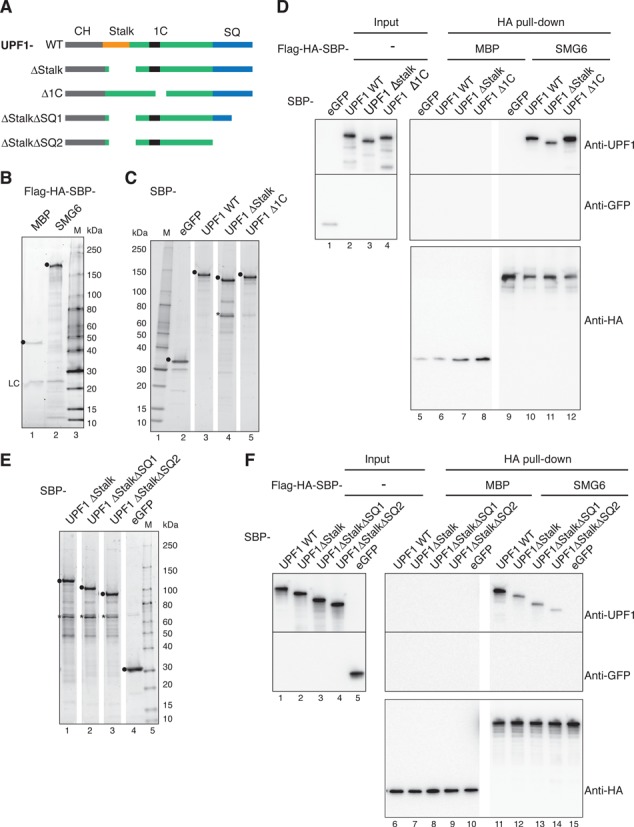
UPF1 Stalk and SQ regions are involved in binding to SMG6 in vitro. (A) Schematic of the tested UPF1 constructs. UPF1 WT, ΔStalk and Δ1C were defined in Figure 4C. UPF1 ΔStalk ΔSQ1 and UPF1 ΔStalk ΔSQ2 lack the last 99 or all 204 AA of the UPF1 SQ domain (in blue), respectively. (B) Twenty percent of Flag-HA-SBP-MBP (as control) or Flag-HA-SBP-SMG6 bound to anti-HA magnetic beads were eluted in SDS sample buffer, subjected to SDS-PAGE and stained and visualized with ORIOLE fluorescent gel stain. The positions of the full-length proteins and the lgG light chain are marked by black dots and LC, respectively. (C, E) Hundred nanograms of the indicated proteins, expressed in 293T cells and purified using SBP-tag affinity chromatography, were separated on an SDS-PAGE and as in (B). Black dots (•) and asterisk (*) indicate the positions of purified proteins and a degradation product of UPF1 ΔStalk, respectively. (D, F) Pull-down assays testing the interaction of SMG6 with UPF1 mutants. Flag-HA-SBP-MBP (as control) or Flag-HA-SBP-SMG6 immobilized on anti-HA magnetic beads were incubated with the indicated SBP-tagged purified proteins (1000 nM each). Seventeen percent of each sample was set aside as input and subjected to SDS-PAGE (lanes 1–4 in (D) and lanes 1–5 in (F)) and the remaining material was precipitated and subjected to SDS-PAGE (lanes 5–12 in (D) and lanes 6–15 in (F)). UPF1, eGFP and HA-tagged MBP and SMG6 were detected by immunoblotting using the indicated antibodies.
UPF1 ΔStalk can still bind RNA while UPF1 Δ1C has a greatly reduced affinity for RNA in vitro
The activity of the UPF1 HD is regulated by its neighboring N-terminal histidine-rich (CH) domain and C-terminal SQ domain. The CH domain of UPF1 makes intramolecular connections to HD which seems to hide its enzymatic activity and this repression is only alleviated when the UPF2/UPF3 complex binds to the CH domain, which induces a large conformational change causing UPF1 to relax its grip on the RNA and leading to the activation of UPF1 ATPase/helicase activity (30,51,52). Similarly, the C-terminal region of human UPF1 also appears to interact directly with the HD to suppress UPF1 enzymatic activities. Again, this repression is only diminished when a currently unknown factor binds to the area of the SQ proximal to the HD, causing a rearrangement allowing for enzymatic activation (53). Ultimately these regulatory effects are a consequence of tempering with the extent of RNA binding and the sub-domains 1B and 1C of the HD have been implicated to be important in controlling RNA binding (30,46).
Given the postulated roles of 1B and 1C in UPF1's RNA binding behavior, it was vital to examine how deletions of the stalk region (encompassing sub-domain 1B) and the sub-domain 1C in the HD affect the ability of UPF1 to bind single-stranded RNA. We wanted to know whether the reduced interaction between UPF1 ΔStalk and SMG6 was due to the loss of an interaction point or due to a damaging effect on the ability of UPF1 to bind RNA. Moreover, we also wanted to test if SMG6 could bind to RNA itself. Therefore, interactions of our recombinant proteins with RNA were examined in solution using MST (38). We used fluorescently labeled U30 RNA at a fixed concentration (20 nM) and titrated the unlabeled designated protein partner in a range starting at concentrations above the expected dissociation constant (KD) and ending with sub-stoichiometric concentrations with respect to the labeled RNA. Under our experimental conditions, UPF1 exhibited a high affinity for RNA with a calculated KD of 1.48 nM (Figure 6A). Deletion of the stalk/1B structure led to a moderate weakening of the interaction between UPF1 and RNA (KD of 6.51 nM) (Figure 6B). In contrast, deletion of the 1C region dramatically reduced UPF1's binding affinity for RNA by 32.2-fold compared to UPF1 WT (Figure 6, compare A and C). Contrary to another study (54), we found no evidence for binding of SMG6 to RNA in our assay (Figure 6D). Finally, we did not observe eGFP binding to RNA, which served as a control in these assays (Figure 6E). Thus, our result indicates that SMG6 has to be recruited to its target mRNAs by a protein–protein interaction, for example by UPF1.
Furthermore, we validated the functionality of the recombinant full-length SMG6 protein in vitro by performing endocleavage assays. Wild-type SMG6 protein degraded a radioactively labeled U30 RNA while its endonucleolytically inactive counterpart could not degrade the RNA. We also demonstrated that this ribonuclease activity requires Mn2+ (Supplementary Figure S7) as was shown by Glavan et al. using bacterially produced SMG6 PIN domain (21). SMG6 activity in these in vitro assays was poor and we ambitiously tried to increase the activity of SMG6 by adding UPF1. However, we did not conclusively find UPF1 to promote SMG6 activity in these assays, most likely because we were still lacking other regulatory factors that are essential for optimal SMG6 activity (data not shown).
In conclusion, we have shown that only the deletion of 1C abrogated the ability of UPF1 to effectively bind RNA, which is in line with earlier results using filter binding assays (46), while the UPF1 ΔStalk protein is able to bind RNA. This strongly indicates that our observed loss of interaction between UPF1 ΔStalk and SMG6 observed in the yeast two-hybrid assays and in the in vitro pull-down assays is not because UPF1 can no longer bind RNA but rather due to the fact that the stalk region of the UPF1 HD (AA 271-433) encompasses a phosphorylation-independent interaction surface for SMG6.
It had not escaped our attention that the stalk region includes the area of difference between human UPF1 isoform 1 (UniProtKB: Q92900-1) and isoform 2 (Q92900-2). Compared to hUPF1 isoform 1, isoform 2 lacks AA 353-363 (Supplementary Figure S8A) due to the usage of an alternative 5′ splice site in exon 7. Both isoforms have been used in publications but possible functional differences between them have never been examined. So far, we had only worked with UPF1 isoform 2 and thus we wanted to examine if the extra 11 amino acids in isoform 1 altered the affinity of UPF1 for SMG6. HeLa cells produce mRNAs for both isoforms, with isoform 2 being the more abundant isoform (Supplementary Figure S8B). Similar to results gained using UPF1 isoform 2 in Figure 4B, we found that small amounts of SMG6 also co-immunoprecipiated with MS2-UPF1 iso1 (Supplementary Figure S8C) and RNAiR UPF1 iso1 was able to rescue the ability of SMG6 to induce reporter mRNA level reduction as efficiently as RNAiR UPF1 iso2 (Supplementary Figure S8D). In addition, both isoforms were able to restore NMD of different PTC-containing Ig-μ and TCRβ reporter mRNAs in cells with reduced endogenous UPF1 isoform 1 and 2 protein levels (data not shown). These data suggest that both isoforms of UPF1 can support the mapped interaction with SMG6 in human cells.
The interaction between the UPF1 Stalk region and SMG6 is crucial for SMG6 activity in the TFA and for NMD
To determine the functional significance of the interaction between UPF1 HD and SMG6, we first examined how important the stalk region of the HD of UPF1 was to the induction of SMG6-mediated mRNA reporter level reduction. The SMG6 TFA was performed as before, along with an RNAi-mediated depletion of endogenous UPF1 and expression of RNAiR UPF1ΔStalk. As shown in Figure 7A and Supplementary Figure S1D, the RNAiR UPF1 ΔStalk (blue bars) was not able to rescue the steady-state mRNA reporter levels under UPF1 knockdown conditions back to those seen in HeLa cells with a control knockdown. Notably, the UPF1 knockdown in this experiment was prolonged by 1 day, which accounts for the stronger effects observed in the UPF1 knockdown conditions compared those documented in Figure 3C. As shown in the western blot, the RNAiR UPF1 versions were detected in cells lacking endogenous UPF1 and were each expressed to a similar level as their wild-type counterpart (Figure 7A, lanes 6 and 7). This result confirmed a functional necessity for the UPF1 stalk region in SMG6-mediated degradation of reporter mRNA levels.
In addition, we also performed classical NMD assays using two well-characterized NMD reporter genes, one based on an immunoglobulin μ mini-gene (55) and the other on the β-globin gene (56). We enforced UPF1 knockdowns and rescued the loss of NMD using RNAiR UPF1 WT as well as RNAiR UPF1ΔStalk. As anticipated, the levels of the μ Ter 310 and β-globin Ter 39 reporter mRNAs were reduced to less than 1% of their corresponding wild-type counterparts due to NMD (Figure 7B and C, respectively, and Supplementary Figure S1D). Depletion of UPF1 and hence abolishment of NMD caused the μ Ter 310 and β-globin Ter 39 reporter mRNA levels to increase by over 10-fold. When RNAiR wild-type UPF1 was expressed, the μ Ter 310 and β-globin Ter 39 reporter mRNA levels were rescued almost back to that observed in the control-treated cells. In contrast, no rescue at all was observed when RNAiR UPF1ΔStalk was expressed. Western blotting confirmed that the UPF1 knockdown was efficient (Figure 7B, lane 5) and that the expression of the RNAiR UPF1 versions in cells depleted of endogenous UPF1 yielded UPF1 protein levels similar to those observed in cells without or with a control knockdown (compare lanes 6 and 7 with 1 or 4, respectively). Similarly, the accompanying western blot of Figure 7C also confirmed that the UPF1 knockdown was effective (Figure 7C, lane 5) and that the exogenously expressed RNAiR UPF1 proteins were detected (lanes 6 and 7) in the cells expressing β-globin Ter 39. These results establish that the region of UPF1 HD that we have identified as being involved in a phosphorylation-independent interaction with SMG6 is essential for NMD in human cells.
DISCUSSION
SMG6 is an essential component of the NMD apparatus in mammalian cells and we have sought to further delineate the molecular mechanisms surrounding the regulation of its endonucleolytic activity in the NMD pathway. To examine what is required for SMG6 to induce degradation uncoupled from what is needed for its recruitment to the mRNA, we established an SMG6-TFA in HeLa cells. The combination of the TFA with knockdowns of other NMD factors allowed us to identify NMD factors required for SMG6-mediated decay in NMD.
Contrary to a previous study (57) but in keeping with a recent study (17), we found that tethered SMG6 resulted in strongly reduced reporter mRNA levels, bypassing the need for a PTC (Figure 1B and Supplementary Figure S2). We also examined the SMG6 14-3-3-like domain and the EBMs, both of which have been implicated in the recruitment of SMG6 to mRNPs. We reasoned that if these parts of SMG6 are solely involved in the function of recruiting SMG6 to NMD-targeted mRNPs, then their function should be bypassed by the MS2-mediated tethering and thus no longer critical for SMG6 activity in the TFA. This is what we observed with the SMG6-14-3-3 mutant, which was fully active in the TFA albeit its association with P-UPF1 was strongly diminished (Figure 1D and Supplementary Figure S4A). On the contrary, mutations made to the EBMs in the N-terminus of SMG6 compromised the activity of SMG6 in the TFA (Figure 1D), suggesting that in addition to its role in recruitment of SMG6 to the mRNP, the EBM also seems to play a role in activating SMG6 activity. It was proposed that SMG6 gained its target specificity by binding to the EJC via these conserved motifs, yet they are lacking from D. melanogaster SMG6 (20,58), where it has been demonstrated that the components of the EJC are not essential for NMD and that PTC definition occurs independently of exon boundaries (59). Furthermore, the EBMs most likely do not contribute to the SMG6 recruitment to mRNPs in the EJC-independent mode of NMD in mammalian cells (27) but the involvement of non-canonical EJC (60,61) in the EJC-independent NMD mode cannot be formally excluded. Since our data also show that SMG6 cannot bind RNA itself (Figure 6D), it seems highly plausible that P-UPF1 recruits SMG6 via its 14-3-3-like domain to the mRNP.
When brought to the target RNA, SMG6 is known to induce endonucleolytic cleavage (24,25) and in line with this, we were able to show that the PIN domain of SMG6 is absolutely essential for the ability of tethered SMG6 to induce reduction of reporter mRNA levels, yet we also discovered that the PIN domain on its own is not sufficient in human cells (Figure 1E). This is contrary to what has been reported for SMG6 in D. melanogaster, where experiments performed in S2 cells showed that tethering of the SMG6 PIN domain could degrade reporter mRNA as well if not even slightly better than full-length SMG6 (21). Nonetheless, we deduced that in human cells an intact SMG6 protein is required for its activity in vivo, which already strongly hinted at other parts of SMG6 being important and most likely other factors being essential for SMG6-mediated decay. This is in line with a recent study that showed in SMG6 lacking cells that all parts of SMG6 were required to rescue NMD of classical reporter mRNAs (TCR-β and β-globin +/− PTCs) and that the N-terminus of SMG6 can co-IP with most other NMD factors (20). Such a conclusion might also explain why we (Supplementary Figure S7) and other studies observe such poor SMG6 endonuclease activity in vitro where other factors are simply lacking for efficient enzymatic activities (21,24,25).
To identify which other NMD factors are needed for SMG6 function, we looked to see if UPF1, UPF2, SMG1, SMG5 and SMG7 were needed for SMG6 activity in our assays. Indeed, we could specifically show that the presence of both SMG1 and UPF1 was needed for efficient SMG6 activity in the TFAs, even in the case of the SMG6-4-3-3 and EBM mutants (Figure 2 and Supplementary Figure S3 A, B, D, F and H). Intriguingly, the SMG6 EBM mutant also had no activity in cells lacking UPF2 or UPF3B (Figure 2D and Supplementary Figure S3C, F, G and H). Thus, it seems that UPF2 and UPF3B become essential for SMG6 activation when SMG6 cannot associate with the EJC, consistent with our previous observation that the EJC-independent NMD mode was more sensitive to depletion of UPF2 and UPF3B than EJC-enhanced NMD (27). Additionally, we found that while the EBM mutant retained its weak interaction to UPF1, it strongly reduced its interaction with P-UPF1 (Supplementary Figure S4A), which may signify that SMG6 connects to the EJC while it is being recruited by P-UPF1, meaning that it is recruited by two means; one by anchoring on to the EJC and the other by P-UPF1, adding extra specificity to the SMG6 recruitment process. However, collectively our results suggest that there is more to the role of the EBM motifs in SMG6 than simply a means to recruit SMG6 to target mRNPs and future work should try to determine the contribution these motifs play to SMG6 activity in NMD.
The observation that SMG6 decay function requires UPF1 and kinase-competent SMG1 immediately insinuated a requirement for UPF1 phosphorylation (Figure 2B and Supplementary Figure S5). However, and in contrast to recent work (17), the UPF1 T28A mutant rescued SMG6 tethering mediated reporter mRNA reduction in cells depleted of endogenous UPF1 as efficiently as UPF1 WT (Figure 3C), arguing that UPF1 could activate SMG6 through a contact different from the previously characterized interaction between the UPF1 P-T28 and the SMG6 14-3-3-like domain. Our results advocate that the well-documented P-UPF1-SMG6 interaction probably represents the means to get SMG6 to the target mRNP, a step bypassed in the TFA, while the subsequent activation of endonuclease activity of SMG6 depends on a phosphorylation-independent interaction with UPF1. With regards to SMG1, it may be that its kinase activity is needed to phosphorylate sites on another NMD factor. For example, it is known that human and yeast UPF2 can be phosphorylated but the function of P-UPF2 in NMD is not yet known (62,63). It is also very likely that SMG1 can phosphorylate UPF1 at various P-sites and by doing so dictates the function of UPF1 in many aspects of NMD such as RNA binding, enzymatic activity and recruitment of various factors beyond SMG6 and SMG5-7. Recently, it was documented in yeast that two newly identified P-sites in UPF1 act to promote ATP hydrolysis, NMD efficiency and translation termination fidelity (50). Further work is needed to delineate the roles of each individual UPF1 P-site and their contribution to UPF1 function.
Our hypothesis of a phosphorylation-independent interaction between UPF1 and SMG6 was corroborated by finding that both proteins consistently co-immunoprecipitated each other, regardless of the mutations made in either protein (Supplementary Figure S4). This has also been observed in other studies but never further investigated (18,20). Using yeast two-hybrid assays, we discovered a phosphorylation-independent interaction between the HD of UPF1 and SMG6 (Figure 4B) and narrowed down the interaction point within UPF1 HD to the stalk structure comprising AA 271-433 (Figure 4D). We were initially concerned that the observed loss of interaction between the UPF1 ΔStalk and SMG6 in our interaction studies was actually to do with a detrimental effect on UPF1 RNA binding but we demonstrated that the deletion of the stalk and hence sub-domain 1B did not abolish RNA binding (Figure 6B). Furthermore, UPF1 and SMG6 could still co-IP even in the presence of RNase A (Supplementary Figure S4), and UPF1 lacking 1C, which hindered RNA binding (Figure 6C), still interacted with SMG6 (Figures 4D and 5D), further indicating that the stalk/1B structure of the UPF1 HD directly interacts with SMG6.
In mapping AA 271-433 of UPF1 isoform 2 to bind to SMG6, we realized that this fragment covers the only alteration between human UPF1 isoform 2 and isoform 1: at amino acid position 353, the less abundant isoform 1 has an insertion of 11 amino acids. Since the alternative splicing event generating the two isoforms appears to be highly conserved and because we could detect the mRNA of both isoforms in our cells, we examined the tantalizing possibility that UPF1 isoform 1 might have a different affinity for binding to SMG6. However, we could not detect any difference between the two human UPF1 isoforms with regard to binding SMG6, being needed for SMG6 activity (Supplementary Figure S8), or even for its involvement in NMD. However, it still does beg the question as to why the cell produces two UPF1 isoforms, differing only by 11 amino acids.
Finally, UPF1 ΔStalk could not rescue tethered SMG6-mediated RNA degradation in cells lacking endogenous UPF1, validating that this region of UPF1 is essential for SMG6 activity (Figure 7A). In addition, we could demonstrate that the UPF1 stalk structure is required for NMD (Figure 7B and C).
With regard to which parts of SMG6 interact with UPF1, our results hint toward the N-terminal part of SMG6 (AA 1-576), because neither the SMG6 814-1419 construct nor the C-terminal PIN domain or the 14-3-3-like domain interacted with UPF1 in yeast two-hybrid assays (Supplementary Figure S6D and E). This is in line with a previous study showing that removal of the SMG6 N-terminal 576 AA abolished co-IPs between SMG6 and UPF1 (20). Unfortunately, our constructs containing the unstructured N-terminal region of SMG6 did not express in yeast, which prevented further mapping of this region. Notably, it has been suggested that the interacting complexes in NMD contain disordered protein components and that their flexibility plays a crucial role in the formation of long-reaching protein–protein interactions. Within the disordered N-terminus of SMG6, eight protein–protein interaction sites have been predicted, one of which maps to the EBM1 at the very N-terminus (64). From the seven remaining sites, one site spanning AA 401-424 is a conserved site that could potentially be the site needed for an interaction with the UPF1 HD.
In further assessing the yeast two-hybrid data, we consistently observed that the presence of the CH domain weakened the interaction of UPF1 with SMG6 in the yeast two-hybrid assays, while the interaction between the UPF1 HD and SMG6 appeared consistently stronger when the HD still had its SQ domain present (Figure 4). Despite the data being predominately qualitative, these observations indicated that the presence of the CH domain inhibits and the presence of the SQ domain enhances the interaction of SMG6 with the UPF1 HD. Therefore, when we sought to confirm the interaction between UPF1 stalk region and SMG6 in vitro by pull-down experiments, we also tested truncations of the SQ domain. While deletion of the stalk region reduced UPF1 binding to SMG6 only partially, additional removal of the proximal part of the SQ domain almost completely abolished the interaction between UPF1 and SMG6 (Figure 5D). Therefore, we can show along with Chakrabarti et al. that the SQ domain makes a significant contribution to the interaction between UPF1 stalk and SMG6 (48). The CH and SQ domains of UPF1 engage in intramolecular interactions with the HD to suppress its enzymatic activities and only when UPF2 binds to the CH domain and another unidentified factor binds to the SQ domain, this repression is lifted and UPF1 enzymatic activities are induced (30,53). It was further shown that when UPF2 binds to UPF1, triggering ATPase and helicase activities, this is accompanied by a switch from an RNA clamping mode to a more relaxed UPF1 grip on the RNA (30) and this is keeping with the fact that we observed in vitro that UPF1 F192E, which inhibits the allosteric inhibition from the CH domain to RecA2 (30,65), and P-UPF1 both had reduced affinity for RNA compared to wild-type UPF1 (data not shown). Based on our observations, we speculated that SMG6 may also be involved in this intricate regulation of UPF1 enzymatic activities and/or RNA binding affinities. The observed inhibitory effect of the CH domain on the interaction between SMG6 and UPF1 HD in the yeast two-hybrid assays could be explained by the absence of UPF2 in the yeast nucleus, which would result in the UPF1 conformation in which the CH domain is repressing the HD domain. In NMD, UPF2 binding probably precedes the interaction between UPF1 and SMG6. Yet, the interaction between UPF1 and SMG6 also involves the proximal part of the SQ domain. It is very plausible that the unidentified factor that specifically binds to the SQ domain to abolish the intramolecular interactions repressing the HD activities may be SMG6. This possibility is in line with the fact that SMG6 is involved in the UPF1 dissociation from mRNA (18), which already suggested that SMG6 may have a role to play in ATP binding, ATPase and/or RNA binding activity of UPF1. Moreover, UPF1 ATPase activity is required for UPF1 dissociation from mRNA but there has been no analysis performed to date that would implicate the involvement of SMG6. Structural studies and further biochemical studies beyond the scope of this investigation will have to address this exciting possibility.
UPF1, SMG1 and SMG6 are the only enzymes specific to the NMD pathway and recent studies have started to give insight into how the enzymatic activities of UPF1 (30,46,51–53) and SMG1 (14,34,66) are managed in the NMD pathway. Here, we have been able to show that SMG6 activity is also under regulation by additional proteins. The future of trying to understand the complex regulatory dynamic between the three enzymes may not only be important in trying to understand the role of these proteins in NMD but also in other cellular processes where these three enzymes have been implicated to also function, such as in DNA replication, telomere metabolism and genome maintenance pathways (67,68).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We are very grateful to Jens Lykke-Andersen (UCSD, USA) and to Shigeo Ohno (Yokohama City University, Japan) for kindly sharing various antibodies and plasmids, to François-Xavier Ogi (NanoTemper Technologies GmbH) for his assistance with the MST measurements, to Berndt Muller (University of Aberdeen, UK) for generously sending the pGBKT7 and pGADT7 plasmids, to Ian Stansfield (University of Aberdeen, UK) for kindly giving us various yeast strains, to Alain Hauser for his statistical support (University of Berne), to Kyoko Aoyagi, Matthias Dreier, Valentin Flury and Reto Michel for their contributions to the cloning of various pEFh_SBP-UPF1 derivatives, MS2-IPs, constructing pCK-Flag-RNAiR-SMG1 and to the isoform 1 related experiments, respectively, and to Christoph Schweingruber for his helpful comments concerning this study.
FUNDING
European Research Council [ERC-StG 207419]; Swiss National Science Foundation [31003A-127614, -143717]; Novartis Foundation for Biomedical Research and the Canton Bern [to O.M.]; Japan Society for the Promotion of Science KAKENHI [23687025, 23112718, 21115004]; Takeda Science Foundation [to A.Y.]. Funding for open access charge: University of Bern.
Conflict of interest statement. None declared.
REFERENCES
- 1.Mendell J.T., Sharifi N.A., Meyers J.L., Martinez-Murillo F., Dietz H.C. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet. 2004;36:1073–1078. doi: 10.1038/ng1429. [DOI] [PubMed] [Google Scholar]
- 2.He F., Li X., Spatrick P., Casillo R., Dong S., Jacobson A. Genome-wide analysis of mRNAs regulated by the nonsense-mediated and 5’ to 3’ mRNA decay pathways in yeast. Mol. Cell. 2003;12:1439–1452. doi: 10.1016/s1097-2765(03)00446-5. [DOI] [PubMed] [Google Scholar]
- 3.Rehwinkel J., Letunic I., Raes J., Bork P., Izaurralde E. Nonsense-mediated mRNA decay factors act in concert to regulate common mRNA targets. RNA. 2005;11:1530–1544. doi: 10.1261/rna.2160905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frischmeyer P.A., Dietz H.C. Nonsense-mediated mRNA decay in health and disease. Hum. Mol. Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- 5.Keeling K.M., Bedwell D.M. Suppression of nonsense mutations as a therapeutic approach to treat genetic diseases. Wiley Interdiscip. Rev. RNA. 2011;2:837–852. doi: 10.1002/wrna.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhattacharya A., Czaplinski K., Trifillis P., He F., Jacobson A., Peltz S.W. Characterization of the biochemical properties of the human Upf1 gene product that is involved in nonsense-mediated mRNA decay. RNA. 2000;6:1226–1235. doi: 10.1017/s1355838200000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Czaplinski K., Ruiz-Echevarria M.J., Paushkin S.V., Han X., Weng Y., Perlick H.A., Dietz H.C., Ter Avanesyan M.D., Peltz S.W. The surveillance complex interacts with the translation release factors to enhance termination and degrade aberrant mRNAs. Genes Dev. 1998;12:1665–1677. doi: 10.1101/gad.12.11.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eberle A.B., Stalder L., Mathys H., Orozco R.Z., Muhlemann O. Posttranscriptional gene regulation by spatial rearrangement of the 3’ untranslated region. PLoS Biol. 2008;6:e92. doi: 10.1371/journal.pbio.0060092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ivanov P.V., Gehring N.H., Kunz J.B., Hentze M.W., Kulozik A.E. Interactions between UPF1, eRFs, PABP and the exon junction complex suggest an integrated model for mammalian NMD pathways. EMBO J. 2008;27:736–747. doi: 10.1038/emboj.2008.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silva A.L., Ribeiro P., Inacio A., Liebhaber S.A., Romao L. Proximity of the poly(A)-binding protein to a premature termination codon inhibits mammalian nonsense-mediated mRNA decay. RNA. 2008;14:563–576. doi: 10.1261/rna.815108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh G., Rebbapragada I., Lykke-Andersen J. A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense-mediated mRNA decay. PLoS Biol. 2008;6:e111. doi: 10.1371/journal.pbio.0060111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamashita A. Role of SMG-1-mediated Upf1 phosphorylation in mammalian nonsense-mediated mRNA decay. Genes Cells. 2013;18:161–175. doi: 10.1111/gtc.12033. [DOI] [PubMed] [Google Scholar]
- 13.Kashima I., Yamashita A., Izumi N., Kataoka N., Morishita R., Hoshino S., Ohno M., Dreyfuss G., Ohno S. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 2006;20:355–367. doi: 10.1101/gad.1389006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamashita A., Izumi N., Kashima I., Ohnishi T., Saari B., Katsuhata Y., Muramatsu R., Morita T., Iwamatsu A., Hachiya T., et al. SMG-8 and SMG-9, two novel subunits of the SMG-1 complex, regulate remodeling of the mRNA surveillance complex during nonsense-mediated mRNA decay. Genes Dev. 2009;23:1091–1105. doi: 10.1101/gad.1767209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Izumi N., Yamashita A., Iwamatsu A., Kurata R., Nakamura H., Saari B., Hirano H., Anderson P., Ohno S. AAA+ proteins RUVBL1 and RUVBL2 coordinate PIKK activity and function in nonsense-mediated mRNA decay. Sci. Signal. 2010;3:1–13. doi: 10.1126/scisignal.2000468. [DOI] [PubMed] [Google Scholar]
- 16.Loh B., Jonas S., Izaurralde E. The SMG5–SMG7 heterodimer directly recruits the CCR4–NOT deadenylase complex to mRNAs containing nonsense codons via interaction with POP2. Genes Dev. 2013;27:2125–2138. doi: 10.1101/gad.226951.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho H., Han S., Choe J., Park S.G., Choi S.S., Kim Y.K. SMG5–PNRC2 is functionally dominant compared with SMG5–SMG7 in mammalian nonsense-mediated mRNA decay. Nucleic Acids Res. 2013;41:1319–1328. doi: 10.1093/nar/gks1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okada-Katsuhata Y., Yamashita A., Kutsuzawa K., Izumi N., Hirahara F., Ohno S. N- and C-terminal Upf1 phosphorylations create binding platforms for SMG-6 and SMG-5:SMG-7 during NMD. Nucleic Acids Res. 2012;40:1251–1266. doi: 10.1093/nar/gkr791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fukuhara N., Ebert J., Unterholzner L., Lindner D., Izaurralde E., Conti E. SMG7 Is a 14-3-3-like adaptor in the nonsense-mediated mRNA decay pathway. Mol. Cell. 2005;17:537–547. doi: 10.1016/j.molcel.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 20.Kashima I., Jonas S., Jayachandran U., Buchwald G., Conti E., Lupas A.N., Izaurralde E. SMG6 interacts with the exon junction complex via two conserved EJC-binding motifs (EBMs) required for nonsense-mediated mRNA decay. Genes Dev. 2010;24:2440–2450. doi: 10.1101/gad.604610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glavan F., Behm-Ansmant I., Izaurralde E., Conti E. Structures of the PIN domains of SMG6 and SMG5 reveal a nuclease within the mRNA surveillance complex. EMBO J. 2006;25:5117–5125. doi: 10.1038/sj.emboj.7601377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takeshita D., Zenno S., Lee W.C., Saigo K., Tanokura M. Crystallization and preliminary X-ray analysis of the PIN domain of human EST1A. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006;62:656–658. doi: 10.1107/S1744309106020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takeshita D., Zenno S., Lee W.C., Saigo K., Tanokura M. Crystal structure of the PIN domain of human telomerase-associated protein EST1A. Proteins. 2007;68:980–989. doi: 10.1002/prot.21351. [DOI] [PubMed] [Google Scholar]
- 24.Eberle A.B., Lykke-Andersen S., Muhlemann O., Jensen T.H. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat. Struct. Mol. Biol. 2009;16:49–55. doi: 10.1038/nsmb.1530. [DOI] [PubMed] [Google Scholar]
- 25.Huntzinger E., Kashima I., Fauser M., Sauliere J., Izaurralde E. SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA. 2008;14:2609–2617. doi: 10.1261/rna.1386208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicholson P., Mühlemann O. Cutting the nonsense: the degradation of PTC-containing mRNAs. Biochem. Soc. Trans. 2010;38:1615–1620. doi: 10.1042/BST0381615. [DOI] [PubMed] [Google Scholar]
- 27.Metze S., Herzog V.A., Ruepp M.-D., Mühlemann O. Comparison of EJC-enhanced and EJC-independent NMD in human cells reveals two partially redundant degradation pathways. RNA. 2013;19:1432–1448. doi: 10.1261/rna.038893.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yepiskoposyan H., Aeschimann F., Nilsson D., Okoniewski M., Mühlemann O. Autoregulation of the nonsense-mediated mRNA decay pathway in human cells. RNA. 2011;17:2108–2118. doi: 10.1261/rna.030247.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicholson P., Joncourt R., Mühlemann O. Analysis of nonsense-mediated mRNA decay in mammalian cells. Curr. Protoc. Cell Biol. 2012:1–61. doi: 10.1002/0471143030.cb2704s55. Unit 27.4. [DOI] [PubMed] [Google Scholar]
- 30.Chakrabarti S., Jayachandran U., Bonneau F., Fiorini F., Basquin C., Domcke S., Le Hir H., Conti E. Molecular mechanisms for the RNA-dependent ATPase activity of Upf1 and its regulation by Upf2. Mol. Cell. 2011;41:693–703. doi: 10.1016/j.molcel.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 31.Vidal M., Brachmann R.K., Fattaey A., Harlow E., Boeke J.D. Reverse two-hybrid and one-hybrid systems to detect dissociation of protein-protein and DNA-protein interactions. Proc. Natl Acad. Sci. U.S.A. 1996;93:10315–10320. doi: 10.1073/pnas.93.19.10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gietz D., St Jean A., Woods R.A., Schiestl R.H. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duttweiler H.M. A highly sensitive and non-lethal beta-galactosidase plate assay for yeast. Trends Genet. 1996;12:340–341. doi: 10.1016/s0168-9525(96)80008-4. [DOI] [PubMed] [Google Scholar]
- 34.Arias-Palomo E., Yamashita A., Fernández I.S., Núñez-Ramírez R., Bamba Y., Izumi N., Ohno S., Llorca O. The nonsense-mediated mRNA decay SMG-1 kinase is regulated by large-scale conformational changes controlled by SMG-8. Genes Dev. 2011;25:153–164. doi: 10.1101/gad.606911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zillner K., Jerabek-Willemsen M., Duhr S., Braun D., Längst G., Baaske P. Part V protein analysis II: functionalcharacterization. In: Kaufmann M, Klinger C, editors. Microscale Thermophoresis as a Sensitive Methodto Quantify Protein: Nucleic Acid Interactions in Solution. Vol. 815. New York, NY: Springer; 2012. pp. 241–252. [DOI] [PubMed] [Google Scholar]
- 36.Seidel S.A.I., Dijkman P.M., Lea W.A., van den Bogaart G., Jerabek-Willemsen M., Lazic A., Joseph J.S., Srinivasan P., Baaske P., Simeonov A., et al. Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods. 2013;59:301–315. doi: 10.1016/j.ymeth.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jerabek-Willemsen M., Wienken C.J., Braun D., Baaske P., Duhr S. Molecular interaction studies using microscale thermophoresis. Assay Drug Dev. Technol. 2011;9:342–353. doi: 10.1089/adt.2011.0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wienken C.J., Baaske P., Duhr S., Braun D. Thermophoretic melting curves quantify the conformation and stability of RNA and DNA. Nucleic Acids Res. 2011;39:e52. doi: 10.1093/nar/gkr035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Page M.F., Carr B., Anders K.R., Grimson A., Anderson P. SMG-2 is a phosphorylated protein required for mRNA surveillance in Caenorhabditis elegans and related to Upf1p of yeast. Mol. Cell. Biol. 1999;19:5943–5951. doi: 10.1128/mcb.19.9.5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohnishi T., Yamashita A., Kashima I., Schell T., Anders K.R., Grimson A., Hachiya T., Hentze M.W., Anderson P., Ohno S. Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG-5 and hSMG-7. Mol. Cell. 2003;12:1187–1200. doi: 10.1016/s1097-2765(03)00443-x. [DOI] [PubMed] [Google Scholar]
- 41.Yamashita A., Ohnishi T., Kashima I., Taya Y., Ohno S. Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev. 2001;15:2215–2228. doi: 10.1101/gad.913001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morita T., Yamashita A., Kashima I., Ogata K., Ishiura S., Ohno S. Distant N- and C-terminal domains are required for intrinsic kinase activity of SMG-1, a critical component of nonsense-mediated mRNA decay. J. Biol. Chem. 2007;282:7799–7808. doi: 10.1074/jbc.M610159200. [DOI] [PubMed] [Google Scholar]
- 43.Fields S., Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 44.Stansfield I., Jones K.M., Kushnirov V.V., Dagkesamanskaya A.R., Poznyakovski A.I., Paushkin S.V., Nierras C.R., Cox B.S., Ter-Avanesyan M.D., Tuite M.F. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J. 1995;14:4365–4373. doi: 10.1002/j.1460-2075.1995.tb00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhouravleva G., Frolova L., Le Goff X., Le Guellec R., Inge-Vechtomov S., Kisselev L., Philippe M. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J. 1995;14:4065–4072. doi: 10.1002/j.1460-2075.1995.tb00078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng Z., Muhlrad D., Lim M.K., Parker R., Song H. Structural and functional insights into the human Upf1 helicase core. EMBO J. 2007;26:253–264. doi: 10.1038/sj.emboj.7601464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Applequist S.E., Selg M., Raman C., Jack H.M. Cloning and characterization of HUPF1, a human homolog of the Saccharomyces cerevisiae nonsense mRNA-reducing UPF1 protein. Nucleic Acids Res. 1997;25:814–821. doi: 10.1093/nar/25.4.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chakrabarti S., Bonneau F., Schüssler S., Eppinger E., Conti E. Phospho-dependent and phospho-independent interactions of the helicase UPF1 with the NMD factors SMG5–SMG7 and SMG6. Nucleic Acid Res. 2014 doi: 10.1093/nar/gku578. doi:10.1093/nar/gku562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hornbeck P.V., Kornhauser J.M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2011;40:D261–D270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lasalde C., Rivera A.V., León A.J., González-Feliciano J.A., Estrella L.A., Rodríguez-Cruz E.N., Correa M.E., Cajigas I.J., Bracho D.P., Vega I.E., et al. Identification and functional analysis of novel phosphorylation sites in the RNA surveillance protein Upf1. Nucleic Acids Res. 2013;42:1916–1929. doi: 10.1093/nar/gkt1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clerici M., Mourao A., Gutsche I., Gehring N.H., Hentze M.W., Kulozik A., Kadlec J., Sattler M., Cusack S. Unusual bipartite mode of interaction between the nonsense-mediated decay factors, UPF1 and UPF2. EMBO J. 2009;28:2293–2306. doi: 10.1038/emboj.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chamieh H., Ballut L., Bonneau F., Le Hir H. NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat. Struct. Mol. Biol. 2008;15:85–93. doi: 10.1038/nsmb1330. [DOI] [PubMed] [Google Scholar]
- 53.Fiorini F., Boudvillain M., Le Hir H. Tight intramolecular regulation of the human Upf1 helicase by its N- and C-terminal domains. Nucleic Acids Res. 2013;41:2404–2415. doi: 10.1093/nar/gks1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Redon S., Reichenbach P., Lingner J. Protein RNA and protein protein interactions mediate association of human EST1A/SMG6 with telomerase. Nucleic Acids Res. 2007;35:7011–7022. doi: 10.1093/nar/gkm724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buhler M., Paillusson A., Muhlemann O. Efficient downregulation of immunoglobulin mu mRNA with premature translation-termination codons requires the 5’-half of the VDJ exon. Nucleic Acids Res. 2004;32:3304–3315. doi: 10.1093/nar/gkh651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thermann R., Neu-Yilik G., Deters A., Frede U., Wehr K., Hagemeier C., Hentze M.W., Kulozik A.E. Binary specification of nonsense codons by splicing and cytoplasmic translation. EMBO J. 1998;17:3484–3494. doi: 10.1093/emboj/17.12.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Unterholzner L., Izaurralde E. SMG7 acts as a molecular link between mRNA surveillance and mRNA decay. Mol. Cell. 2004;16:587–596. doi: 10.1016/j.molcel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 58.Schoenberg D.R. Mechanisms of endonuclease-mediated mRNA decay. Wiley Interdiscip. Rev. RNA. 2011;2:582–600. doi: 10.1002/wrna.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gatfield D., Unterholzner L., Ciccarelli F.D., Bork P., Izaurralde E. Nonsense-mediated mRNA decay in Drosophila: at the intersection of the yeast and mammalian pathways. EMBO J. 2003;22:3960–3970. doi: 10.1093/emboj/cdg371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singh G., Kucukural A., Cenik C., Leszyk J.D., Shaffer S.A., Weng Z., Moore M.J. The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell. 2012;151:750–764. doi: 10.1016/j.cell.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sauliere J., Murigneux V., Wang Z., Marquenet E., Barbosa I., Le Tonqueze O., Audic Y., Paillard L., Roest Crollius H., Le Hir H. CLIP-seq of eIF4AIII reveals transcriptome-wide mapping of the human exon junction complex. Nat. Struct. Mol. Biol. 2012;19:1124–1131. doi: 10.1038/nsmb.2420. [DOI] [PubMed] [Google Scholar]
- 62.Chiu S.Y., Serin G., Ohara O., Maquat L.E. Characterization of human Smg5/7a: a protein with similarities to Caenorhabditis elegans SMG5 and SMG7 that functions in the dephosphorylation of Upf1. RNA. 2003;9:77–87. doi: 10.1261/rna.2137903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang W., Cajigas I.J., Peltz S.W., Wilkinson M.F., Gonzalez C.I. Role for Upf2p phosphorylation in Saccharomyces cerevisiae nonsense-mediated mRNA decay. Mol. Cell. Biol. 2006;26:3390–3400. doi: 10.1128/MCB.26.9.3390-3400.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kalmar L., Acs V., Silhavy D., Tompa P. Long-range interactions in nonsense-mediated mRNA decay are mediated by intrinsically disordered protein regions. J. Mol. Biol. 2012;424:125–131. doi: 10.1016/j.jmb.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 65.Kadlec J., Guilligay D., Ravelli R.B., Cusack S. Crystal structure of the UPF2-interacting domain of nonsense-mediated mRNA decay factor UPF1. RNA. 2006;12:1817–1824. doi: 10.1261/rna.177606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fernández I.S., Yamashita A., Arias-Palomo E., Bamba Y., Bartolomé R.A., Canales M.A., Teixidó J., Ohno S., Llorca O. Characterization of SMG-9, an essential component of the nonsense-mediated mRNA decay SMG1C complex. Nucleic Acids Res. 2011;39:347–358. doi: 10.1093/nar/gkq749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Azzalin C.M., Lingner J. The double life of UPF1 in RNA and DNA stability pathways. Cell Cycle. 2006;5:1496–1498. doi: 10.4161/cc.5.14.3093. [DOI] [PubMed] [Google Scholar]
- 68.Chawla R., Azzalin C.M. The telomeric transcriptome and SMG proteins at the crossroads. Cytogenet. Genome Res. 2008;122:194–201. doi: 10.1159/000167804. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.