


Abstract
The possible effect of transfer ribonucleic acid (tRNA) concentrations on codons decoding time is a fundamental biomedical research question; however, due to a large number of variables affecting this process and the non-direct relation between them, a conclusive answer to this question has eluded so far researchers in the field. In this study, we perform a novel analysis of the ribosome profiling data of four organisms which enables ranking the decoding times of different codons while filtering translational phenomena such as experimental biases, extreme ribosomal pauses and ribosome traffic jams. Based on this filtering, we show for the first time that there is a significant correlation between tRNA concentrations and the codons estimated decoding time both in prokaryotes and in eukaryotes in natural conditions (−0.38 to −0.66, all P values <0.006); in addition, we show that when considering tRNA concentrations, codons decoding times are not correlated with aminoacyl-tRNA levels. The reported results support the conjecture that translation efficiency is directly influenced by the tRNA levels in the cell. Thus, they should help to understand the evolution of synonymous aspects of coding sequences via the adaptation of their codons to the tRNA pool.
INTRODUCTION
The way in which intracellular transfer ribonucleic acid (tRNA) levels affect messenger RNA (mRNA) decoding times is still debatable, due to the complexity of quantifying these effects (1–11). First, gene expression is affected by a large number of factors; for example, gene translation efficiency is determined by various features of the transcript (e.g. mRNA folding (2,12), context of the start codon (13,14), length of the different parts of the transcripts (15), charge of the amino acids (16,17), intracellular concentrations of mRNA molecules (18), ribosomes (19), tRNA molecules (20–23), aminoacyl-tRNA synthetases (aaRS) (24) and the intracellular concentrations of dozens of initiation and elongation factors (24,25)); thus it is impossible to completely control for non-causal relations between these two variables (1–3,6,7). Second, heterologous gene expression, which is often used to study such relations, may not reflect the decoding time of endogenous transcripts since they tend to violate the natural intracellular regimes (1,5,7). Third, although most large-scale measurements of the different stages of gene expression do not directly measure translation elongation rates, they are nevertheless used as a proxy of this variable (e.g. protein levels are often used as a proxy for translation rates (1–3,5,8,26), neglecting additional levels of control that govern the synthesis and degradation of mRNAs and proteins).
The current cutting-edge methodology for studying mRNA translation is ribosome profiling (Ribo-seq), which is based on deep sequencing of ribosome-protected mRNA fragments and produces a detailed account of ribosome occupancy on specific mRNAs under endogenous conditions (27). Recently, several studies (5,10,11) using ribosome profiling data found insignificant correlations between tRNA levels and codons decoding times, inconsistent with previous studies based on other methodologies and data sources (1,4,8,9,28–30).
In this study, we develop a novel statistical approach specifically tailored for analyzing ribosome profiling data of both prokaryotes and eukaryotes. The new approach enables a better understanding of the different variables that contribute to the codon decoding time. We show for the first time that when filtering out rare events such as long pauses in translation elongation, the correlation between codon decoding times and tRNA levels is significant for endogenous transcripts in all analyzed organisms. This relationship is not only fundamental for human health (31–33) but also affects biotechnology (7,8) and disciplines such as molecular evolution (1,5,30,34,35) and functional genomics (6,9,26,28,29,36).
MATERIALS AND METHODS
Reconstruction of the Open Reading Frame (ORFs) ribosomal profiles of the analyzed organisms
Saccharomyces cerevisiae ribosomal profiles were reconstructed using the data published in the GEO database, accession number GSE13750 (GSM346111, GSM346114) (27). Caenorhabditis elegans ribosomal profiles of genes expressed in the L4 larval stage were built from Illumina sequencing results (NCBI Sequence Read Archive, accession number SRR52883) (37). Escherichia coli and Bacillus subtilis profiles were built from the published Illumina sequencing results (GEO database, accession number GSE35641) (11). Full details regarding the alignment method appear in the Supplementary text.
Calculating the normalized footprint count (NFC) - data normalization
To avoid analyzing ribosomal profiles of genes with many missing read counts (RCs) that may result in a non-reliable estimation of the local ribosome density, only genes with a median RC above 1 were included in the analysis. Previous studies indicated an increase of RCs at the beginning of the ORF (10,38) and for some organisms at the end of ORF (11), therefore the first and last 20 codons were excluded when determining these thresholds or when calculating the average RCs per ORF. The exact number of genes included in the analysis after applying this filter is depicted in Supplementary Table S1.
To enable comparison and analysis of RCs of codons of the same type originating from different genes, RCs of each codon were normalized by the average RCs of each gene; this normalization controls for possible different mRNA levels and initiation rates of different genes and has been performed in previous studies (5,11). To prevent biasing the average with codons containing less than one RC, those were excluded from the analysis (a similar procedure has been performed in a previous study (11)). Therefore this normalization enables measuring the relative time a ribosome spends translating each codon in a specific gene relative to other codons in it, while considering the total number of codons in the gene, resulting in its normalized footprint count (NFC):
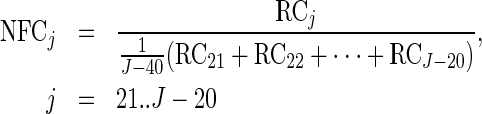 |
where J is the number of codons in the gene and j is the index of a codon; see also Figure 1B.
Figure 1.
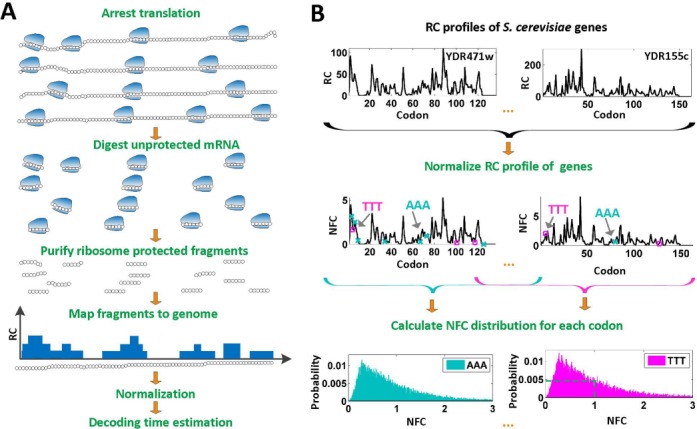
Schematic description of the ribosome profiling method and generating the NFC distributions. (A) Translation of codons on mRNAs (black circles) by ribosomes (blue shapes) is arrested, then exposed mRNA is digested. Protected mRNA footprints are then sequenced and mapped to the genome, creating for each gene its read count (RC) profile. (B). Illustration of the codons NFC distributions inference from a set of RC profiles: to control for different mRNA levels and translation initiation rates, RC profiles of S. cerevisiae (here only YDR471w and YDR155c RC profiles are illustrated) are normalized per gene by their mean RC value, resulting in NFC profiles. Then, NFC values of each specific codon type (NFC values of codons of type ‘AAA’ are marked by teal ‘x’ while NFC values of codons of type ‘TTT’ are marked by magenta squares) are collected from all analyzed genes and presented using a histogram, where the x-axis represents the NFC values and the y-axis represents the fraction of the time (probability) each NFC value appears in the analyzed genes, thus creating the NFC distribution of a codon. For example, the codon ‘TTT’ appears with an NFC value that equals 1 in the analyzed genes in 0.48% of the times (probability of 0.0048; dashed green lines in the ‘TTT’ histogram).
Calculating the NFC distributions
To study the translation time properties of different codons, for each codon type we generated a vector consisting of NFC values originating from all analyzed genes. These vectors were used to generate for each codon type a histogram reflecting the probability of observing each NFC value in the expressed genes (the number of times each NFC value occurs in the data normalized by the total number of times the codon appears in the data) which was named the ‘NFC distribution’ of the codon (see also Figure 1B). The black histograms in Supplementary Figures S1–S4 depict the NFC distributions of each type of codon, for all analyzed organisms. As summarized in Supplementary Table S2 the mean variance of NFC values of the codons differs between different organisms, such that prokaryotes have a higher NFC standard deviation range, which is associated with more prominent translational pauses (11).
The skewness of a distribution
Skewness measures the lack of symmetry of a distribution. Given a data set of samples 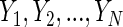 their skewness is defined as
their skewness is defined as
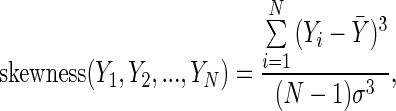 |
where 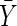 symbolizes the mean,
symbolizes the mean, 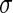 the standard deviation and
the standard deviation and 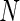 the number of points.
the number of points.
Inferring the two components of the NFC distributions
Based on the characteristics of the NFC distributions, we suggest that their topology could result from a superposition of two distributions/components: the first one describes the ‘typical’ decoding time of the ribosomes, which was modeled by a normal distribution characterized by its mean 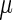 and variance
and variance 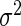 with a probability density function
with a probability density function 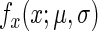 (for a random variable X) of
(for a random variable X) of
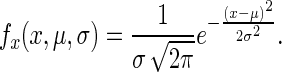 |
The second component describes relatively rare translational pauses and ribosomal interactions such as traffic jams due to the codons’ different translation efficiency and was modeled by a random variable with an exponentially distribution, characterized by one parameter λ with a probability density function, 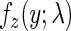 , (for a random variable Y) of
, (for a random variable Y) of
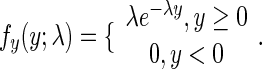 |
It is known that the distribution of a random variable w(t) that is a sum of two independent random variables 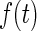 and
and 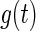 (i.e. w(t) =
(i.e. w(t) =
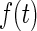 +
+ 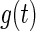 ) is calculated as convolution between the two distributions (39):
) is calculated as convolution between the two distributions (39):
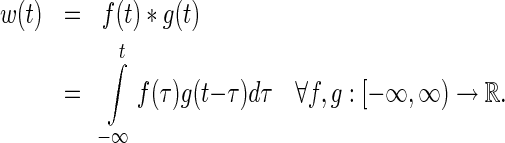 |
Thus, the summation of two independent normal and exponential random variables corresponding to the distributions mentioned above results in a distribution that is named ‘exponentially modified Gaussian’ (EMG) and is a convolution of a normal and exponential distribution; formally, the EMG distribution function, 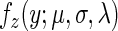 , of a random variable Z (where
, of a random variable Z (where 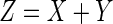 )(40) is
)(40) is
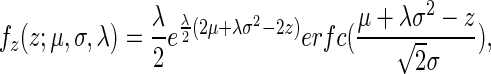 |
where
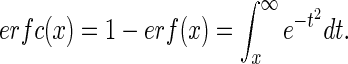 |
The parameters 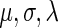 were estimated by fitting the measured NFC distributions to the EMG distribution, under the log-likelihood criterion. Estimated
were estimated by fitting the measured NFC distributions to the EMG distribution, under the log-likelihood criterion. Estimated 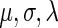 values appear in Supplementary Table S11. It should be mentioned that the EMG distribution does not necessarily define an equal weight for the Gaussian and exponential distributions. Rather, the optimization of the
values appear in Supplementary Table S11. It should be mentioned that the EMG distribution does not necessarily define an equal weight for the Gaussian and exponential distributions. Rather, the optimization of the 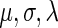 parameters defines their optimal weight. For additional insights regarding the expected relation between the
parameters defines their optimal weight. For additional insights regarding the expected relation between the 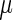 and
and 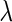 parameters see the Supplementary section.
parameters see the Supplementary section.
In addition, we fit the NFC data using an EMG model with an identical exponential distribution parameter for all codons; in this model, the λ parameter was optimized for all codons under the log-likelihood criterion. Moreover, the NFC distribution was fit also using the exponential and Gaussian distributions separately. The goodness of these fittings was compared and analyzed by using the Akaike information criterion (AIC) score (more details below).
Evaluating the goodness of models using the AIC score
Comparison between the different fitting models was evaluated using the AIC (41). This measure considers both the number of parameters in the model and the fit (log-likelihood) of the data to the model (i.e. it penalizes more complicated models). Lower AIC score is related to a model that is better fitted to the data.
Calculating codons adaptiveness value to tRNA levels 
The tRNA adaptation index (tAI) (42) uses the adaptiveness of the codons of a gene to the tRNA pool. Let us mark the adaptiveness value of codon of type i with 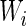 . Let
. Let 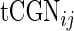 be the copy number of the j-th anti-codon that recognizes the i-th codon and let
be the copy number of the j-th anti-codon that recognizes the i-th codon and let 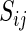 be the selective constraint of the codon–anti-codon coupling efficiency. The
be the selective constraint of the codon–anti-codon coupling efficiency. The 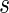 vector (42,43)
vector (42,43) 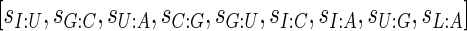 was defined for prokaryotes as [0, 0, 0, 0, 0.41, 0.63, 0.9749, 0.68, 0.95] and for eukaryotes as [0, 0, 0, 0, 0.561, 0.28, 0.9999, 0.68, 0.89]. Then, the absolute adaptiveness value of a codon is defined by
was defined for prokaryotes as [0, 0, 0, 0, 0.41, 0.63, 0.9749, 0.68, 0.95] and for eukaryotes as [0, 0, 0, 0, 0.561, 0.28, 0.9999, 0.68, 0.89]. Then, the absolute adaptiveness value of a codon is defined by
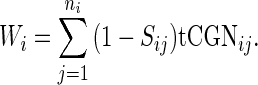 |
Let us mark the relative adaptiveness value of codon  with
with 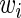 by normalizing each
by normalizing each 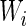 with the maximal
with the maximal 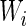 value among the 61
value among the 61 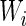 values. A codon typical decoding time
values. A codon typical decoding time 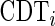 used for the Totally Asymmetric Simple Exclusion Process (TASEP) simulations was defined as
used for the Totally Asymmetric Simple Exclusion Process (TASEP) simulations was defined as
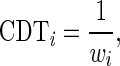 |
where codons with low adaptiveness values to the tRNA pool will be more slowly translated.
Simulating ribosome density profiles using the TASEP model
Ribosome density profiles were simulated using the TASEP biophysical translation model, previously used in different studies (44,45). In this model, the mRNA was simulated using a lattice of N sites, where N represents the number of codons of the ORF. Each ribosome was defined to cover 11 codons and its A site was located at the sixth codon. During translation, any codon could be covered at a time by a single ribosome at most. In each step of the simulation, a single ribosome was allowed to attach itself to the lattice/advance to the next codon if the first/next six codons were not occupied. The time between initiation attempts was set to be exponentially distributed with a constant rate 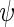 . Similarly, the time between jump attempts from site
. Similarly, the time between jump attempts from site 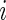 to site
to site 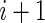 was assumed to be exponentially distributed with rate
was assumed to be exponentially distributed with rate 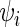 .
.
The time between events (initiation or jumping between sites) is therefore exponentially distributed (minimum of exponentially distributed random variables) with rate
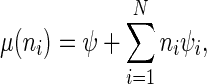 |
where 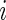 describes the site (codon) number on the lattice and
describes the site (codon) number on the lattice and 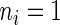 if codon
if codon 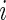 is being translated, otherwise
is being translated, otherwise 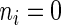 . Therefore the initiation probability is given by
. Therefore the initiation probability is given by 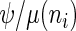 and the probability of a ribosome to jump from site
and the probability of a ribosome to jump from site 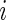 to site
to site 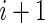 is given by
is given by 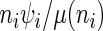 .
.
In this study S. cerevisiae genes that contained a sufficient number of RCs (see the criterion in Supplementary Table S1) and with a least 50 codons were simulated. Codons’ decoding rate 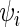 was set according to their adaptiveness to the tRNA pool (specific codon decoding times (CDT) values used in the simulation are presented in Supplementary Table S3), where
was set according to their adaptiveness to the tRNA pool (specific codon decoding times (CDT) values used in the simulation are presented in Supplementary Table S3), where
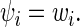 |
The initiation rate 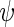 was set to be as the decoding rate of the fastest codon. To reach a steady-state distribution on the mRNA, the simulation of each mRNA copy was run until 200 ribosomes finished translating the gene. At this point of the simulation, codons covered by ribosomes were referred as ribosomal protected fragments. For each simulated gene the TASEP model was run for
was set to be as the decoding rate of the fastest codon. To reach a steady-state distribution on the mRNA, the simulation of each mRNA copy was run until 200 ribosomes finished translating the gene. At this point of the simulation, codons covered by ribosomes were referred as ribosomal protected fragments. For each simulated gene the TASEP model was run for 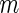 times, where
times, where 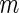 represents the mRNA level of the gene. This parameter was set to be the maximal measured RC value of each gene. The protected fragments of each one of the m simulations of a gene were used to create its ribosomal profile as previously described for S. cerevisiae.
represents the mRNA level of the gene. This parameter was set to be the maximal measured RC value of each gene. The protected fragments of each one of the m simulations of a gene were used to create its ribosomal profile as previously described for S. cerevisiae.
To simulate the effect of translational pauses on the NFC distributions, codons of S. cerevisiae with NFC higher than 3-fold of the mean NFC of the ORF (based on the real measurements, when excluding codons with zero RCs (11), i.e. codons with NFC higher than 3) were defined as locations of translational pauses. The decoding time of these codons was set to be proportional to the NFC of the measured pauses in the real ribosomal profiles. Specifically, we defined the simulated translational pauses translation time to be proportional to the mean simulated mean RCs, as in the real data, i.e.
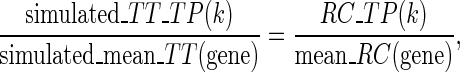 |
where 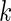 is the index of the codon;
is the index of the codon; 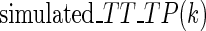 is the simulated translational time of codon
is the simulated translational time of codon 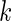 ;
; 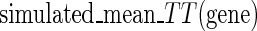 is the mean simulated translational time of all codons in the gene not defined as translational pauses;
is the mean simulated translational time of all codons in the gene not defined as translational pauses; 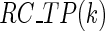 is the real RC of the translational pause of the gene at codon
is the real RC of the translational pause of the gene at codon 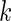 ; and
; and 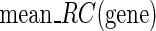 is the real measured mean RC of all codons in the gene not defined as translational pauses.
is the real measured mean RC of all codons in the gene not defined as translational pauses.
Protein abundance and mRNA level measurements of S. cerevisiae and E. coli
S. cerevisiae protein abundance (PA) measures were averaged from four quantitative large-scale measurements: two large-scale measurements in two conditions (46) and a large-scale PA measurement from two sources (47,48). mRNA levels were determined by averaging large-scale measurements of mRNA levels (27,49). E. coli PA measurements were downloaded from the PRIDE database (50) and mRNA level measurements were taken from another source (51). Aminoacyl-tRNA synthetase protein levels and quantities of S. cerevisiae (52) and E. coli (53) were used from (52) and (53), respectively.
Inferring the weighted typical decoding times and tRNA levels per amino acid
To enable comparison between aaRS levels, tRNA levels and typical decoding times, we defined the representative tRNA level of each amino acid as the sum of tRNA levels corresponding to the amino acid weighted by the frequency of codons in the ORFs that translate it. Let fi denote the frequency of codon i and 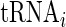 its corresponding tRNA level. Thus, the weighted tRNA level of an amino acid equals
its corresponding tRNA level. Thus, the weighted tRNA level of an amino acid equals
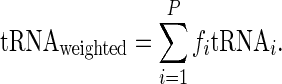 |
Similarly, the typical amino acid decoding time 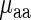 also took into consideration the codon frequencies of the codons coding it, thus was defined as
also took into consideration the codon frequencies of the codons coding it, thus was defined as
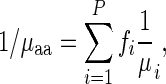 |
where P depicts the number of codons coding to a specific amino acid and 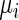 depicts the typical codon decoding time.
depicts the typical codon decoding time.
Estimating the typical amino acid decoding time using a linear regressor of the weighted tRNA levels and aaRS levels
To estimate to what extent the aaRS levels improve the prediction of the amino acid decoding time (relative to tRNA levels only), we have modeled the relationship between amino acid decoding times and the two aforementioned possible explanatory variables by using a linear regressor (54)
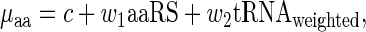 |
where the coefficients c, w1 and w2 are estimated as to minimize the mean square error difference between the prediction vector 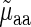 and the real measurements
and the real measurements 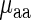 . Absolute Spearman correlation between
. Absolute Spearman correlation between 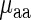 and
and 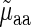 was compared to Spearman correlation between
was compared to Spearman correlation between 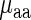 and
and 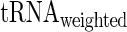 to determine if the linear regressor based on aaRS levels improves the correlation. The correlation coefficient between
to determine if the linear regressor based on aaRS levels improves the correlation. The correlation coefficient between 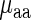 and
and 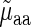 was adjusted due to an increased number of explanatory variables using the correction equation (55)
was adjusted due to an increased number of explanatory variables using the correction equation (55)
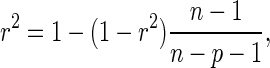 |
where r depicts the Spearman correlation coefficient, n depicts the sample size (in this case n = 20) and p depicts the total number of regressors (in this case p = 2). For each of the coefficients c, w1 and w2 their confidence interval (56) was calculated to determine the reliability of the estimates (at a 95% confidence interval). A coefficient's confidence interval that does not contain zeros implies that it significantly contributes to the regression, i.e. the coefficient is not zero (56).
RESULTS
Estimating the codons relative decoding times from ribosome profiling data
We began our analysis by reconstructing ribosome profiles for E. coli (11), B. subtilis (11), C. elegans (37) and S. cerevisiae (27) expressed genes. The ribosome profiling method produces ribosome footprint counts that are proportional to the time spent in decoding each codon of all translated transcripts in a genome, at single nucleotide resolution (see Figure 1A and Supplementary Methods). To enable a comparison between footprint counts of codons from different genes we applied to them a conventional normalization (11) (see the Materials and Methods section; Figure 1B) and named this value the Normalized Footprint Count (NFC). To gain an initial understanding of the NFC values in natural conditions, we calculated a histogram reflecting the distribution of the NFC values of each codon in the expressed genes for each codon in each organism; we call this histogram the ‘NFC distribution’ of the codon. Each NFC distribution (see Figure 1B and Supplementary Figures S1–S4) describes the probability (y-axis) of observing each of the codon's NFC values (x-axis) in the ORFs of the analyzed organism.
The mean codons decoding time is extremely sensitive to extreme values caused by translational pauses
The intuitive way for representing the decoding time of a codon is by calculating the average of all NFC values for each codon (we name this estimation mean NFC), which was implemented in several previous studies (5,11) with some variation. However, as can be seen in Figure 2A the NFC histograms show a right-skewed distribution with ‘right tails’, reflecting possible interactions between ribosomes such as ribosomal jams, rare but extreme events such as translational pauses (10,11) and/or experimental biases (10,57,58). Therefore, straightforward mean NFC estimations (see also Supplementary Tables S4 and S5) may have been biased by these factors, thus did not accurately represent the typical codon decoding time.
Figure 2.
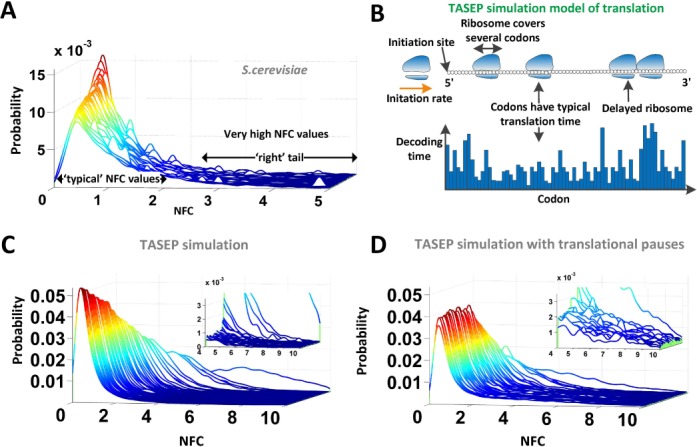
Visualization of the NFC distributions in real and simulated data. (A) NFC distributions (e.g. S. cerevisiae) consist of two components: one that is centered around the most frequent (typical) NFC values in the distribution and another that consists mainly of high NFC values, associated with extremely slow translation events, e.g. translation pauses. The graph shows the NFC distributions of all S. cerevisiae codons. (B) Description of the TASEP simulation model: each gene has its own specific initiation rate; ribosomes cover multiple codons and can continue translating if the adjacent downstream codons are not covered by ribosomes. Each codon is associated with its typical decoding time. (C) NFC distributions of all codons based on the S. cerevisiae genes simulated with the TASEP model without translation pauses. In the absence of translational pauses the ‘right tail’ region is negligible. (D) NFC distributions of all codons based on the simulated S. cerevisiae genes when 4% of the codons are altered to cause translation pauses. The additional translational pauses are reflected as increased peaks in the ‘right tail’ region of the distribution.
To illustrate the statistical problem and evaluate the suggested approach in a controlled environment where the codons decoding times are known, we started our analysis by first creating different TASEP simulations of the ribosome profiling experiment (see the Materials and Methods section and Figure 2B). In the first simulation, all codons decoding times were set to be exponentially distributed with the same parameter 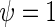 and with a low initiation rate of 0.3 (thus generally no ribosomal jamming was created; no translational pauses were added). The resulting NFC distribution of the codons (Supplementary Figure S05) shows that when no interactions between codons/biases/pauses are present, the expected NFC distribution is similar to a Normal distribution (comparison of codon NFC distribution to Normal distribution under Kolmogorov–Smirnov (KS) test: mean P-value = 0.27).
and with a low initiation rate of 0.3 (thus generally no ribosomal jamming was created; no translational pauses were added). The resulting NFC distribution of the codons (Supplementary Figure S05) shows that when no interactions between codons/biases/pauses are present, the expected NFC distribution is similar to a Normal distribution (comparison of codon NFC distribution to Normal distribution under Kolmogorov–Smirnov (KS) test: mean P-value = 0.27).
However, deviations from the theoretic Normal distribution are expected due to ribosomal jamming caused by different codons decoding efficiency, translational pauses and experimental biases such as a low number of mRNA copies per gene. To demonstrate this, we created an additional TASEP simulation of the ribosome profiling experiment where each codon was assigned a unique decoding time. According to the TASEP model, a queue of ribosomes (or ribosomal jamming) is expected upstream of a slow codon. These jams can result from slower codons (reflected by high RCs) or from translational pauses (reflected by very high RCs). Therefore the effective decoding time of a codon 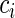 can be higher than its nominal decoding time if a slower codon (or a translational pause) appears downstream of it, causing a traffic jam that eventually affects codon
can be higher than its nominal decoding time if a slower codon (or a translational pause) appears downstream of it, causing a traffic jam that eventually affects codon 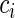 . As a result, the NFC distribution of a codon type can be skewed from the expected symmetric ‘Normal like’ distribution in the TASEP simulation where no pauses are present and all codons have equal translation decoding times. Thus, overall it is expected that codons with faster translation time will be more influenced by ribosomal jams and translational pauses, leading to an NFC distribution with a thicker ‘right tail’ and higher ‘skewness’ (i.e. asymmetricity; see the Materials and Methods section). As expected, in the presence of ribosomal jamming, the simulated NFC distributions deviated from expected Normal distribution, creating a right-skew/right tail (see Figure 2C and Supplementary Figure S06). Specifically, the mean/median/max skewness was 0.078/0.086/0.13, respectively. However, even when the codons decoding time differed, still a highly significant correlation was observed between the mean NFC values and simulated decoding times (Spearman correlation: r = 0.99; P = 3.5 × 10−52; Figure 3B).
. As a result, the NFC distribution of a codon type can be skewed from the expected symmetric ‘Normal like’ distribution in the TASEP simulation where no pauses are present and all codons have equal translation decoding times. Thus, overall it is expected that codons with faster translation time will be more influenced by ribosomal jams and translational pauses, leading to an NFC distribution with a thicker ‘right tail’ and higher ‘skewness’ (i.e. asymmetricity; see the Materials and Methods section). As expected, in the presence of ribosomal jamming, the simulated NFC distributions deviated from expected Normal distribution, creating a right-skew/right tail (see Figure 2C and Supplementary Figure S06). Specifically, the mean/median/max skewness was 0.078/0.086/0.13, respectively. However, even when the codons decoding time differed, still a highly significant correlation was observed between the mean NFC values and simulated decoding times (Spearman correlation: r = 0.99; P = 3.5 × 10−52; Figure 3B).
Figure 3.
tRNA levels are correlated with the estimated μ in real and simulated data. (A) NFC distribution of codon AAA of S. cerevisiae is depicted as ahistogram. The combined normal/exponential model fitting is plotted as a curve. The position of the mean NFC value is presented with a vertical line. The NFC distribution can be decomposed into a normal (which includes the parameter μ) and an exponential component using a log-likelihood fitting. (B) Estimated mean NFC/μ values of simulative ribosome profiles versus simulated decoding times in the absence and presence of translation pauses. (C) Estimated μ values of the real ribosome profiles plotted against tRNA levels (first row), tRNA copy numbers (second row) and tAI values (third row) for the various organisms analyzed. Fitting lines are also shown in all cases.
To demonstrate that the mean codon decoding time is very sensitive to rare and extreme pauses, we added to the previous TASEP simulation translational pauses in only 4% of the positions of the ORFs (see details in the Materials and Methods section). In the presence of translational pauses, the skewness of the NFC distributions (see Figure 2D and Supplementary Figure S07) increased (mean/median/max of skewness = 0.18/0.19/0.23, respectively) and dramatically reduced the correlation between mean NFC and simulated decoding times (Spearman correlation: r = −0.11; P = 0.39; Figure 3B). Therefore, this result suggests that the mean estimator is very sensitive to phenomena such as extreme values caused by translational pauses.
The estimated typical codon decoding times are significantly correlated with tRNA levels
To estimate the typical decoding time of each codon based on NFC distributions, we developed a novel statistical model, which takes into consideration the skewed nature of the NFC distribution that includes three parameters. In this model, the typical codon decoding time was described by a Normal distribution with two parameters: mean (μ) and standard deviation; the μ parameter represents the location of the mean of the theoretical Gaussian component that should be obtained if there are no phenomena such as pauses/biases/ribosomal traffic jams (see Supplementary Figure S5); σ represents the width of the Gaussian component, while λ represents the skewness of the NFC distribution. Rare and extreme translation pauses and translational features such as ribosomal jamming caused by codons with different decoding times were described by an exponential single-parameter distribution (see illustration in Figure 3A and more details in the Materials and Methods section). We used the conventional maximum likelihood criterion to estimate these three parameters for each codon and organism by fitting the suggested model to the NFC distribution (see the Materials and Methods section and Supplementary Figures S1–S4). When employing this approach on the simulated data generated in the presence of translation pauses as mentioned above, we obtained an extremely high correlation between the simulated decoding times and the estimated decoding time values (μ) over the entire set of codons (Spearman correlation: r = 0.99; P = 6.9 × 10−48; Figure 3B); this result demonstrates the high accuracy of the new approach.
Next, we applied our approach on real ribosome profiling data. First, we fit the NFC data to the EMG distribution. For comparison purposes the AIC score (see the Materials and Methods section) was calculated for this model and we compared the goodness of fitting when using only a Gaussian or an exponential fitting (see Supplementary Table S6). The results indicate that in all organisms the EMG exhibits a lower AIC score, supporting the conjecture that EMG distribution better describes the NFC distribution than either the normal or the exponential distributions alone.
In addition, we also considered an EMG model with an identical exponential distribution parameter for all codons (see the Materials and Methods section) and show that it also decreases the data fit (see Supplementary Table S7). This further showed that the EMG model with three free parameters better fits the NFC data.
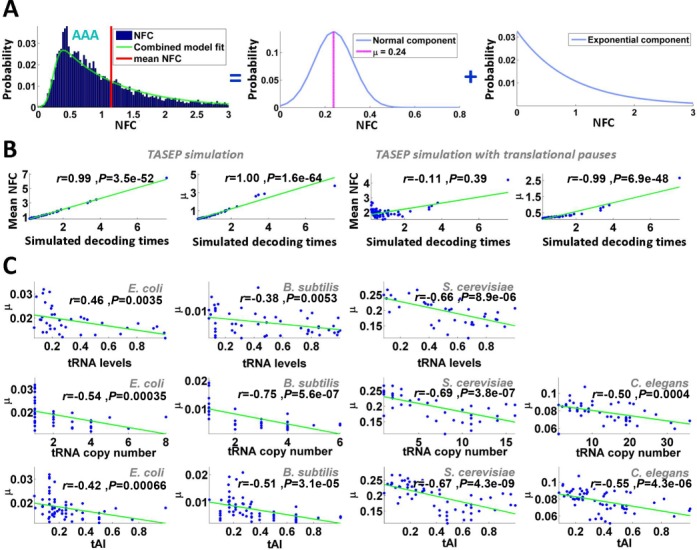
If tRNA levels affect codons translation efficiency, we would expect them to correlate with the estimated typical codon decoding times (μ). Indeed, we found such a significant correlation between μ and tRNA levels in all organisms with available tRNA level measurements (E. coli (59), B. subtilis (60) and S. cerevisiae (28); Spearman correlation was between −0.38 and −0.66, all P-values <0.006; see Figure 3C). Significant Spearman correlations were also obtained when tRNA copy numbers were used as a proxy for absolute tRNA levels (28,60) (r = −0.5 to −0.75, all P-values <0.0004; Figure 3C). A similar observation was made when using the tAI (2,42) of the different codons (see the Materials and Methods section), which considers not only codon–anti-codon interactions but also wobble interactions with other tRNA molecules (r = −0.42 to r = −0.67; all P-values <0.0007; Figure 3C). To control for possible biases resulting from codons which are more abundant and thus for example are known to be recognized by more abundant tRNA species (20,21,23,45,61), the analysis was repeated when calculating the NFC distributions based on an equal amount of RCs for all codons sampled from the real ribosomal profiling data; the results in this case were similar (Supplementary Figure S8). This indicates that our conclusions are not dependent on the amount of RCs per codon type, therefore eliminating a possible bias caused by this factor. Finally, we obtained similar significant correlation also when we considered an EMG model with an identical exponential distribution parameter for all codons (Supplementary Table S8).
The estimated typical codon decoding times are not correlated with aaRS levels given the tRNA levels
In the previous section, we showed that the estimated codon decoding times are significantly correlated with tRNA levels. However, the decoding time of codons could be affected by additional factors such as aaRS concentrations. In this section, we estimate the effect of aaRS concentrations on the typical decoding times and compare it to the effect of tRNA levels on the typical decoding times.
Each amino acid (or its precursor) has only one corresponding aaRS which catalyzes its esterification to one of all its compatible cognate tRNAs; thus in this section, we analyze the effect of aaRS PA and mean tRNA levels on the typical amino acid decoding times 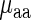 (see details in the Materials and Methods section). Specifically, the amino acid decoding times
(see details in the Materials and Methods section). Specifically, the amino acid decoding times 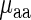 and their tRNA levels
and their tRNA levels 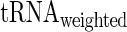 were computed via a weighted average that considers the relative frequencies of all codons coding for each specific amino acid (we analyzed two organisms with reliable aaRS protein levels, E. coli and S. cerevisiae).
were computed via a weighted average that considers the relative frequencies of all codons coding for each specific amino acid (we analyzed two organisms with reliable aaRS protein levels, E. coli and S. cerevisiae).
As expected, in both organisms aaRS protein levels negatively correlate with amino acid decoding times, i.e. higher protein levels correspond to shorter decoding times; only in S. cerevisiae this correlation was significant (r = −0.32, −0.66, P = 0.16/0.0018 for E. coli and S. cerevisiae, respectively). However, since tRNA levels and aaRS concentrations are expected to undergo co-evolution to optimize translation cost, they are expected to positively correlate, i.e. amino acids with higher aaRS protein levels tend to have higher levels of tRNA corresponding to their codons (r = 0.49/0.66; P = 0.031/0.0016/ for E. coli and S. cerevisiae, respectively) and thus are expected to also affect codon decoding time in the same direction.
To study the distinct statistical contribution of tRNA levels/aaRS protein levels to the measured 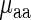 we calculated partial Spearman correlation between
we calculated partial Spearman correlation between 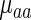 and
and 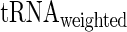 given the aaRS protein levels and also partial Spearman correlation between
given the aaRS protein levels and also partial Spearman correlation between 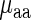 aaRS protein levels given
aaRS protein levels given 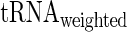 . We found that the partial correlation corr(
. We found that the partial correlation corr(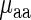 ,
, 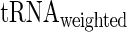 | aaRS protein levels) is very high and significant (r = −0.67/−0.63; P = 0.00017/0.0036 for E. coli and S. cerevisiae, respectively) while the partial correlation corr(
| aaRS protein levels) is very high and significant (r = −0.67/−0.63; P = 0.00017/0.0036 for E. coli and S. cerevisiae, respectively) while the partial correlation corr(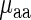 , aaRS protein levels |
, aaRS protein levels | 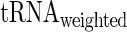 ) is not significant (r = 0.04/−0.20; P = 0.88/0.42 for E. coli and S. cerevisiae, respectively); these results support the conjecture that in the analyzed organisms weighted tRNA levels rather than aaRS levels explain the variance in
) is not significant (r = 0.04/−0.20; P = 0.88/0.42 for E. coli and S. cerevisiae, respectively); these results support the conjecture that in the analyzed organisms weighted tRNA levels rather than aaRS levels explain the variance in 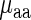 indicating that
indicating that 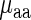 is more likely to be determined by the tRNA levels in the cell.
is more likely to be determined by the tRNA levels in the cell.
Similar results were obtained based on a linear regression analysis: when considering the number of explaining variables in the regressor (i.e. adjusting the correlation to the number of variables in the regression; see the Materials and Methods section), in both analyzed organisms the correlation between 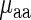 and its regressor based on aaRS protein levels and the weighted tRNA levels per amino acid (see the Materials and Methods section) was not higher than the absolute value of the correlation between tRNA levels and
and its regressor based on aaRS protein levels and the weighted tRNA levels per amino acid (see the Materials and Methods section) was not higher than the absolute value of the correlation between tRNA levels and 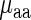 (r = 0.58/0.77; P = 0.0029/0.0001 versus r = −0.71/−0.81; P = 0.00063/1.8 × 10-5 for E. coli and S. cerevisiae, respectively); in addition, in both analyzed organisms the confidence interval of the regressor's aaRS protein levels coefficient was not significantly different than zero (see the Materials and Methods section), suggesting the insignificant contribution of aaRS protein levels to the prediction of
(r = 0.58/0.77; P = 0.0029/0.0001 versus r = −0.71/−0.81; P = 0.00063/1.8 × 10-5 for E. coli and S. cerevisiae, respectively); in addition, in both analyzed organisms the confidence interval of the regressor's aaRS protein levels coefficient was not significantly different than zero (see the Materials and Methods section), suggesting the insignificant contribution of aaRS protein levels to the prediction of 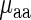 .
.
These results indicate that in the analyzed organisms aaRS protein levels do not significantly improve the explained variance of the typical codon decoding times given the tRNA levels; thus, our analyses support the conjecture that typically tRNA levels and not aaRS protein levels are a rate limiting factor during translation elongation.
The mean-estimated typical codons decoding times of genes significantly correlate with their number of proteins per mRNA molecules
To evaluate the influence of the estimated typical decoding times on the genes’ translation efficiency (i.e. the protein levels of a gene when controlling its mRNA levels or the number of proteins per mRNA molecule), we defined for each gene a translation efficiency measure. This measure was defined as the geometric Mean of the Typical Decoding Rates (1/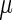 ) of its codons (MTDR), in a similar manner that previous related codon bias usage indexes have been defined (2,62).
) of its codons (MTDR), in a similar manner that previous related codon bias usage indexes have been defined (2,62).
Two organisms (E. coli and S. cerevisiae) with available large-scale measurements of protein and mRNA levels were analyzed. To prevent overfitting the results, the following procedure was performed: for each organism, codons decoding times were estimated based on only 60% of the genes (train set) with sufficient RCs (see details in the Materials and Methods section); then, for the rest of the genes (test set) with existing mRNA and protein abundance levels (not participating in the learning phase) we computed the MTDR based on the learned 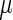 values.
values.
Finally, partial Spearman correlation between PA levels and MTDR when controlling mRNA levels corr(PA, MTDR | mRNA) was computed for the test set; the procedure was repeated 10 times (see also a flow diagram in Figure 4A), obtaining similar results: the correlation coefficient for E. coli was between 0.49 and 0.58 (all P < 3.1 × 10−16) and for S. cerevisiae the obtained correlation coefficient was between 0.5 and 0.57 (all P < 7.9 × 10−76). Results of a single representative run of the procedure are presented in Figure 4B–E. When controlling an equal amount of RCs for all codons, the partial correlation obtained for E. coli was between 0.44 and 0.61 (all P < 1.2 × 10−10) and for S. cerevisiae was between 0.56 and 0.58 (all P < 2.6 × 10−109).
Figure 4.
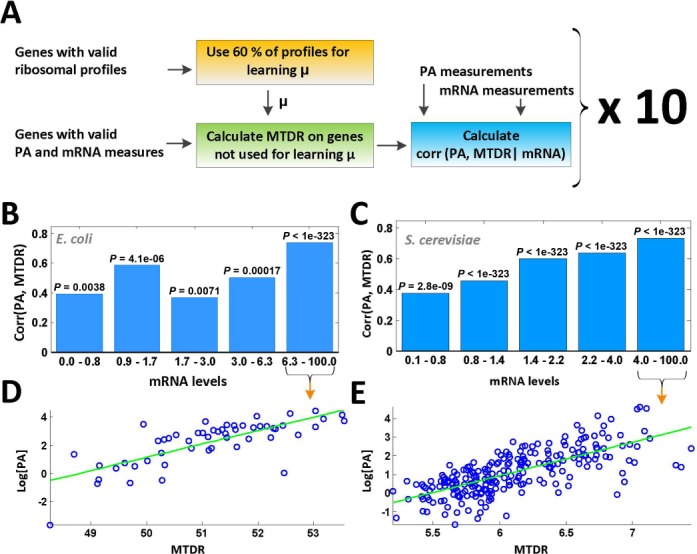
Translation efficiency index based on the estimated typical codons decoding time significantly explains the translation efficiency in E. coli and S. cerevisiae. (A) Diagram showing the process of the calculating Spearman partial correlation between protein abundance levels of S. cerevisiae and genes translation efficiency index (MDTR) when controlling mRNA levels (corr(PA, MTDR | mRNA)). (B) Calculating the correlation between protein abundance and MDTR measure (corr(PA, MTDR)) on E. coli genes for five equal-sized bins with similar mRNA level values, for a single representative run (Spearman partial correlation is 0.53, P < 1.2 × 10−20). The specific P-value calculated for each bin apart appears above it. mRNA level values for this presentation were normalized to range [0, 100]. (C) The same presentation as in (B) calculated for S. cerevisiae. The partial Spearman correlation for this run is 0.54 (P < 1.26 × 10−91). (D) Dot-plot of the log protein abundance (PA) values (normalized to range [0, 100]) versus MTDR values for genes in the bin obtaining the highest correlation presented in (B), marked with an arrow. (E) The same presentation as in (D) for S. cerevisiae.
Significant results were also obtained when computing the correlations between MTDR and PA/mRNA (the number of proteins per mRNA molecule) on the test set: correlation coefficient for E. coli was between 0.41 and 0.52 (all P < 3.1 × 10−11) and for S. cerevisiae the obtained correlation coefficient was between 0.25 and 0.29 (all P < 9.1 × 10−19). Similarly, when constraining an equal amount of RCs for all codons, the correlation obtained for E. coli was between 0.41 and 0.55 (all P < 1.3 × 10−8) and for S. cerevisiae between 0.29 and 0.31 (all P < 1 × 10−323). These results indicate that the translation efficiency of genes can be partially explained by the codons typical decoding times estimated using the newly suggested method, which in turn are significantly influenced by the abundance of tRNA molecules in the cell. Finally, a similar analysis based on the mean NFC estimator yields no significant correlation or very low correlation to protein abundance: the correlation for S. cerevisiae was between 0.10 and 0.2 (P < 0.0008) and for E. coli the correlation was between 0.02 and 0.11 (P < 0.77).
It should be mentioned that the mean NFC estimator did not result in a significant correlation with PA as it is very sensitive to extreme translational pauses and to other phenomena such as traffic jams and experimental biases which are non-codon specific and blur the actual effect of the nominal codon decoding time. The MDTR based on our approach estimates the typical/nominal translation speed of each codon, therefore considers the direct contribution of the codons to the elongation speed. Thus a correlation between PA and MDTR was observed, but not with the geometrical mean (over the codons in the ORF) of the mean codon NFC values.
It is important to explain that the fact that there is correlation between MTDR and PA per mRNA does not contradict the fact that initiation usually tends to be more rate limiting than elongation. Many previous studies have demonstrated that even though initiation has higher correlation with protein per mRNA (or other measures of translation rate), elongation can also significantly explain non-negligible portion of the variance in translation rate (2,3,8,20,21,23,63).
As translation efficiency is influenced not only by the elongation rate but also by initiation rates and other unknown additional factors, our correlation is, of course, significant but not perfect, i.e. r = 0.5–0.6. Thus, MTDR can explain around 25–36% of the variance in PA per mRNA levels. Therefore additional major factors (such as initiation) are needed to explain the variance in translation efficiency.
DISCUSSION
The analyses performed in this study support the conjecture that both in eukaryotes and prokaryotes tRNA concentrations affect codons decoding times. The decoding time of codons recognized by tRNA species with lower intracellular concentrations tends to be longer. It was suggested before that there is a relation between the adaptation of an ORF to the intracellular tRNA concentrations and its translation rate (1–4,6,7,20,22,23,26,28,29,31,34,36,47,64); however, this is the first time that a direct relation between these two variables is shown. We believe that due to noise and biases in the data (e.g. possibility to accurately determining the exact P-site position related to the read) the reported results are only lower bounds of the actual relations. Nevertheless our findings do not contradict the possible effect of other factors on translation elongation speed (2,11,45,65); for example, it was recently shown that the anti-Shine–Dalgarno-like sequences (11), charge of amino acids (45,65) and folding strength of the mRNA molecule (45,66) affect elongation rate and translation efficiency. Several different factors can affect this process and it is not always trivial to isolate the effect of each variable on it; furthermore in different organisms, conditions and/or genes, some factors may be more dominant than others. Thus, the results reported in this study emphasize the complexity of the translation elongation process and the way its efficiency is encoded in the ORF. Naturally, the reported relation between codon decoding times and tRNA concentrations has various fundamental ramifications.
First, we show that translation elongation speed is affected by the adaptation of codons to the tRNA pool, explaining a mechanism by which the codon content of the ORF can affect the organismal fitness. Thus, the reported results should help to understand the evolution of synonymous aspects of coding sequences via the adaptation of their codons to the tRNA pool (1,7,28,30,33–35,66).
Second, it was shown that the folding of proteins partially occurs during translation elongation (67–71). In addition, several studies also suggested that the speed of translation elongation affects co-translational folding of the nascent peptide chain (34,70,72–75); specifically, it was suggested that codon bias usage can affect the folding of proteins, among others due to the two points above. The results reported here suggest that the link between codons bias usage and protein co-translational folding is partially related to the effect of the adaptation of codons to the tRNA pool on their decoding speed. Thus, our results support previous studies that have suggested that codons adaptation to the tRNA pool may affect protein folding/function (2,4,26,29,34,36) and thus even cause human disease (31,32,65).
Third, various studies have suggested the existence of co-evolution between transcriptomic codon frequencies and the intra-cellular levels of the tRNA molecules recognizing them (5,61,76,77): codons with higher frequencies are recognized by tRNA molecules with higher intra-cellular levels; evolutionary changes that affect tRNA levels corresponding to a certain codon are followed by evolutionary changes that affect the codon frequency in transcripts in the same direction and vice versa. The results reported in this study suggest that this co-evolutionary process may at least partially be mediated by the effect of tRNA species on codon decoding times; specifically, increasing the frequency of codons recognized by highly concentrated tRNA species should improve elongation speed and thus the organismal fitness.
It was also suggested that since eventually there is a balance between demand (codon frequency) and supply (tRNA levels), the codon decoding times of all codon should be identical (5). Our study shows that this co-evolutionary process does not produce a ‘perfect’ balance as there is still a high correlation between codon decoding times and intra-cellular levels of tRNA levels (the decoding time of codon recognized by more abundant tRNA molecules is shorter). This scenario may be possible due to the fact that there may be a delay between evolutionary changes in the intracellular tRNA repertoire and the time it takes for evolutionary forces to shape codon frequencies to match this repertoire. It is also possible that there is a selection for non-prefect balance between tRNA levels and codon frequency; the possible functional advantage of non-perfect balance may be related to the fact that different codon decoding time may regulate translation and protein folding in various ways as mentioned above (7,26,28,29,34,36,70,72–75).
Finally, here we studied the effect of tRNA and aaRS levels on the typical codons decoding time; however, it should be mentioned that the typical codon decoding time is affected by several additional factors that the ribosome profiling method cannot directly and/or separately measure. Among others, these factors include the amino acid–tRNA accommodation time, peptidyl transfer time and the time for the whole EF-G-driven translocation process. It is not clear if the effect of these factors on elongation rate differs among different codons and amino acids; we also do not know to what extent these factors can explain the non-prefect correlation between codon decoding time and tRNA levels. The understanding of the contribution of these factors to codon decoding time and specific answers to these questions are deferred to future studies.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We would like to thank Orna Elroy-Stein, Martin Kupiec and Ranen Aviner for their helpful comments.
FUNDING
GIF (I-2327-1131.13/2012); Minerva ARCHES.
Conflict of interest statement. None declared.
REFERENCES
- 1.Kudla G., Murray A.W., Tollervey D., Plotkin J.B. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009;324:255–258. doi: 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tuller T., Waldman Y.Y., Kupiec M., Ruppin E. Translation efficiency is determined by both codon bias and folding energy. Proc. Natl Acad. Sci. U.S.A. 2010;107:3645–3650. doi: 10.1073/pnas.0909910107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Supek F., Smuc T. On relevance of codon usage to expression of synthetic and natural genes in Escherichia coli. Genetics. 2010;185:1129–1134. doi: 10.1534/genetics.110.115477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lithwick G., Margalit H. Hierarchy of sequence-dependent features associated with prokaryotic translation. Genome Res. 2003;13:2665–2673. doi: 10.1101/gr.1485203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qian W., Yang J.R., Pearson N.M., Maclean C., Zhang J. Balanced codon usage optimizes eukaryotic translational efficiency. PLoS Genet. 2012;8:e1002603. doi: 10.1371/journal.pgen.1002603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Novoa E.M., Ribas de Pouplana L. Speeding with control: codon usage, tRNAs, and ribosomes. Trends Genet. 2012;28:574–581. doi: 10.1016/j.tig.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Plotkin J.B., Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 2011;12:32–42. doi: 10.1038/nrg2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gustafsson C., Govindarajan S., Minshull J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004;22:346–353. doi: 10.1016/j.tibtech.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 9.Cannarozzi G., Schraudolph N.N., Faty M., von Rohr P., Friberg M.T., Roth A.C., Gonnet P., Gonnet G., Barral Y. A role for codon order in translation dynamics. Cell. 2010;141:355–367. doi: 10.1016/j.cell.2010.02.036. [DOI] [PubMed] [Google Scholar]
- 10.Ingolia N.T., Lareau L.F., Weissman J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li G.W., Oh E., Weissman J.S. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 2012;484:538–541. doi: 10.1038/nature10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zur H., Tuller T. Strong association between mRNA folding strength and protein abundance in S. cerevisiae. EMBO Rep. 2012;13:272–277. doi: 10.1038/embor.2011.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kozak M. Point mutations close to the AUG initiator codon affect the efficiency of translation of rat preproinsulin in vivo. Nature. 1984;308:241–246. doi: 10.1038/308241a0. [DOI] [PubMed] [Google Scholar]
- 14.Zur H., Tuller T. New universal rules of eukaryotic translation initiation fidelity. PLoS Comput. Biol. 2013;9:e1003136. doi: 10.1371/journal.pcbi.1003136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanguay R.L., Gallie D.R. Translational efficiency is regulated by the length of the 3’ untranslated region. Mol. Cell. Biol. 1996;16:146–156. doi: 10.1128/mcb.16.1.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trylska J., Konecny R., Tama F., Brooks C.L., III, McCammon J.A. Ribosome motions modulate electrostatic properties. Biopolymers. 2004;74:423–431. doi: 10.1002/bip.20093. [DOI] [PubMed] [Google Scholar]
- 17.Lu J., Deutsch C. Electrostatics in the ribosomal tunnel modulate chain elongation rates. J. Mol. Biol. 2008;384:73–86. doi: 10.1016/j.jmb.2008.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwanhausser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 19.Arava Y., Wang Y., Storey J.D., Liu C.L., Brown P.O., Herschlag D. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. U.S.A. 2003;100:3889–3894. doi: 10.1073/pnas.0635171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J. Mol. Biol. 1981;146:1–21. doi: 10.1016/0022-2836(81)90363-6. [DOI] [PubMed] [Google Scholar]
- 21.Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985;2:13–34. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- 22.Dittmar K.A., Goodenbour J.M., Pan T. Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2006;2:e221. doi: 10.1371/journal.pgen.0020221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duret L. tRNA gene number and codon usage in the C. elegans genome are co-adapted for optimal translation of highly expressed genes. Trends Genet. 2000;16:287–289. doi: 10.1016/s0168-9525(00)02041-2. [DOI] [PubMed] [Google Scholar]
- 24.Alberts B., Johnson A., Lewis J., Raff M., Roberts K., Walter P. Molecular Biology of the Cell. New York: Garland Science; 2002. [Google Scholar]
- 25.Jackson R.J., Hellen C.U., Pestova T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu Y., Ma P., Shah P., Rokas A., Liu Y., Johnson C.H. Non-optimal codon usage is a mechanism to achieve circadian clock conditionality. Nature. 2013;495:116–120. doi: 10.1038/nature11942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tuller T., Carmi A., Vestsigian K., Navon S., Dorfan Y., Zaborske J., Pan T., Dahan O., Furman I., Pilpel Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010;141:344–354. doi: 10.1016/j.cell.2010.03.031. [DOI] [PubMed] [Google Scholar]
- 29.Frenkel-Morgenstern M., Danon T., Christian T., Igarashi T., Cohen L., Hou Y.M., Jensen L.J. Genes adopt non-optimal codon usage to generate cell cycle-dependent oscillations in protein levels. Mol. Syst. Biol. 2012;8:572. doi: 10.1038/msb.2012.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Novoa E.M., Pavon-Eternod M., Pan T., Ribas de Pouplana L. A role for tRNA modifications in genome structure and codon usage. Cell. 2012;149:202–213. doi: 10.1016/j.cell.2012.01.050. [DOI] [PubMed] [Google Scholar]
- 31.Kimchi-Sarfaty C., Oh J.M., Kim I.W., Sauna Z.E., Calcagno A.M., Ambudkar S.V., Gottesman M.M. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315:525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 32.Sauna Z.E., Kimchi-Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet. 2011;12:683–691. doi: 10.1038/nrg3051. [DOI] [PubMed] [Google Scholar]
- 33.Chamary J.V., Parmley J.L., Hurst L.D. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet. 2006;7:98–108. doi: 10.1038/nrg1770. [DOI] [PubMed] [Google Scholar]
- 34.Pechmann S., Frydman J. Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat. Struct. Mol. Biol. 2013;20:237–243. doi: 10.1038/nsmb.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drummond D.A., Wilke C.O. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134:341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou M., Guo J., Cha J., Chae M., Chen S., Barral J.M., Sachs M.S., Liu Y. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature. 2013;495:111–115. doi: 10.1038/nature11833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stadler M., Artiles K., Pak J., Fire A. Contributions of mRNA abundance, ribosome loading, and post- or peri-translational effects to temporal repression of C. elegans heterochronic miRNA targets. Genome Res. 2012;22:2418–2426. doi: 10.1101/gr.136515.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ingolia N.T. Genome-wide translational profiling by ribosome footprinting. Methods Enzymol. 2010;470:119–142. doi: 10.1016/S0076-6879(10)70006-9. [DOI] [PubMed] [Google Scholar]
- 39.Damelin S.B., Miller W., Jr . The Mathematics of Signal Processing. New York: Cambridge University Press; 2011. [Google Scholar]
- 40.Grushka E. Characterization of exponentially modified Gaussian peaks in chromatography. Anal. Chem. 1972;44:1733–1738. doi: 10.1021/ac60319a011. [DOI] [PubMed] [Google Scholar]
- 41.Akaike H. A new look at the statistical model identification. Automatic Control, IEEE Trans. 1974;19:716–723. [Google Scholar]
- 42.dos Reis M., Savva R., Wernisch L. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res. 2004;32:5036–5044. doi: 10.1093/nar/gkh834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sabi R., Tuller T. Modeling the efficiency of codon-tRNA interactions based on codon usage bias. DNA Res. 2014 doi: 10.1093/dnares/dsu017. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shaw L.B., Zia R.K., Lee K.H. Totally asymmetric exclusion process with extended objects: a model for protein synthesis. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2003;68:021910. doi: 10.1103/PhysRevE.68.021910. [DOI] [PubMed] [Google Scholar]
- 45.Tuller T., Veksler-Lublinsky I., Gazit N., Kupiec M., Ruppin E., Ziv-Ukelson M. Composite effects of gene determinants on the translation speed and density of ribosomes. Genome Biol. 2011;12:R110. doi: 10.1186/gb-2011-12-11-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Newman J.R., Ghaemmaghami S., Ihmels J., Breslow D.K., Noble M., DeRisi J.L., Weissman J.S. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature. 2006;441:840–846. doi: 10.1038/nature04785. [DOI] [PubMed] [Google Scholar]
- 47.Ghaemmaghami S., Huh W.K., Bower K., Howson R.W., Belle A., Dephoure N., O'Shea E.K., Weissman J.S. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 48.Lee M.V., Topper S.E., Hubler S.L., Hose J., Wenger C.D., Coon J.J., Gasch A.P. A dynamic model of proteome changes reveals new roles for transcript alteration in yeast. Mol. Syst. Biol. 2011;7:514. doi: 10.1038/msb.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y., Liu C.L., Storey J.D., Tibshirani R.J., Herschlag D., Brown P.O. Precision and functional specificity in mRNA decay. Proc. Natl Acad. Sci. U.S.A. 2002;99:5860–5865. doi: 10.1073/pnas.092538799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vizcaino J.A., Cote R.G., Csordas A., Dianes J.A., Fabregat A., Foster J.M., Griss J., Alpi E., Birim M., Contell J., et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013;41:D1063–D1069. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taniguchi Y., Choi P.J., Li G.W., Chen H., Babu M., Hearn J., Emili A., Xie X.S. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010;329:533–538. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Craig R., Cortens J., Fenyo D., Beavis R. Using annotated peptide mass spectrum libraries for protein identification. J. Proteome Res. 2006;5:1843–1849. doi: 10.1021/pr0602085. [DOI] [PubMed] [Google Scholar]
- 53.Jakubowski H., Goldman E. Quantities of individual aminoacyl-tRNA families and their turnover in Escherichia coli. J. Bacteriol. 1984;158:769–776. doi: 10.1128/jb.158.3.769-776.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seber G.A., Lee A.J. Linear Regression Analysis. Hoboken, New Jersey: John Wiley & Sons; 2012. [Google Scholar]
- 55.Glantz S.A., Slinker B.K. Primer of Applied Regression and Analysis of Variance. New York, NY: McGraw-Hill; 1990. [Google Scholar]
- 56.Kendall M., Stuart A. Inference and Relationship. 4th edn. Vol. 2. London: Griffin; 1979. The advanced theory of statistics; p. 1. [Google Scholar]
- 57.Dana A., Tuller T. Determinants of translation elongation speed and ribosomal profiling biases in mouse embryonic stem cells. PLoS Comput. Biol. 2012;8:e1002755. doi: 10.1371/journal.pcbi.1002755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O'Connor P.B., Li G.W., Weissman J.S., Atkins J.F., Baranov P.V. rRNA:mRNA pairing alters the length and the symmetry of mRNA-protected fragments in ribosome profiling experiments. Bioinformatics. 2013;29:1488–1491. doi: 10.1093/bioinformatics/btt184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dong H., Nilsson L., Kurland C.G. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J. Mol. Biol. 1996;260:649–663. doi: 10.1006/jmbi.1996.0428. [DOI] [PubMed] [Google Scholar]
- 60.Kanaya S., Yamada Y., Kudo Y., Ikemura T. Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene. 1999;238:143–155. doi: 10.1016/s0378-1119(99)00225-5. [DOI] [PubMed] [Google Scholar]
- 61.Rocha E.P. Codon usage bias from tRNA's point of view: redundancy, specialization, and efficient decoding for translation optimization. Genome Res. 2004;14:2279–2286. doi: 10.1101/gr.2896904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharp P.M., Li W.H. The codon Adaptation Index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chu D., Kazana E., Bellanger N., Singh T., Tuite M.F., Haar T. Translation elongation can control translation initiation on eukaryotic mRNAs. EMBO J. 2014;33:21–34. doi: 10.1002/embj.201385651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gustafsson A., Baverud V., Gunnarsson A., Pringle J., Franklin A. Study of faecal shedding of Clostridium difficile in horses treated with penicillin. Equine Vet. J. 2004;36:180–182. doi: 10.2746/0425164044868657. [DOI] [PubMed] [Google Scholar]
- 65.Charneski C.A., Hurst L.D. Positively charged residues are the major determinants of ribosomal velocity. PLoS Biol. 2013;11:e1001508. doi: 10.1371/journal.pbio.1001508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shabalina S.A., Spiridonov N.A., Kashina A. Sounds of silence: synonymous nucleotides as a key to biological regulation and complexity. Nucleic Acids Res. 2013;41:2073–2094. doi: 10.1093/nar/gks1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fedorov A.N., Baldwin T.O. Cotranslational protein folding. J. Biol. Chem. 1997;272:32715–32718. doi: 10.1074/jbc.272.52.32715. [DOI] [PubMed] [Google Scholar]
- 68.Kramer G., Boehringer D., Ban N., Bukau B. The ribosome as a platform for co-translational processing, folding and targeting of newly synthesized proteins. Nat. Struct. Mol. Biol. 2009;16:589–597. doi: 10.1038/nsmb.1614. [DOI] [PubMed] [Google Scholar]
- 69.Komar A.A. A pause for thought along the co-translational folding pathway. Trends Biochem. Sci. 2009;34:16–24. doi: 10.1016/j.tibs.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 70.Zhang G., Ignatova Z. Folding at the birth of the nascent chain: coordinating translation with co-translational folding. Curr. Opin. Struct. Biol. 2011;21:25–31. doi: 10.1016/j.sbi.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 71.Hoffmann A., Becker A.H., Zachmann-Brand B., Deuerling E., Bukau B., Kramer G. Concerted action of the ribosome and the associated chaperone trigger factor confines nascent polypeptide folding. Mol. Cell. 2012;48:63–74. doi: 10.1016/j.molcel.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 72.Komar A.A., Jaenicke R. Kinetics of translation of gamma B crystallin and its circularly permutated variant in an in vitro cell-free system: possible relations to codon distribution and protein folding. FEBS Lett. 1995;376:195–198. doi: 10.1016/0014-5793(95)01275-0. [DOI] [PubMed] [Google Scholar]
- 73.Zhang G., Hubalewska M., Ignatova Z. Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat. Struct. Mol. Biol. 2009;16:274–280. doi: 10.1038/nsmb.1554. [DOI] [PubMed] [Google Scholar]
- 74.O'Brien E.P., Vendruscolo M., Dobson C.M. Prediction of variable translation rate effects on cotranslational protein folding. Nat. Commun. 2012;3:868. doi: 10.1038/ncomms1850. [DOI] [PubMed] [Google Scholar]
- 75.O'Brien E.P., Vendruscolo M., Dobson C.M. Kinetic modelling indicates that fast-translating codons can coordinate cotranslational protein folding by avoiding misfolded intermediates. Nat. Commun. 2014;5:2988. doi: 10.1038/ncomms3988. [DOI] [PubMed] [Google Scholar]
- 76.Ikemura T. Correlation between the abundance of yeast transfer RNAs and the occurrence of the respective codons in protein genes. Differences in synonymous codon choice patterns of yeast and Escherichia coli with reference to the abundance of isoaccepting transfer RNAs. J. Mol. Biol. 1982;158:573–597. doi: 10.1016/0022-2836(82)90250-9. [DOI] [PubMed] [Google Scholar]
- 77.Bulmer M. Coevolution of codon usage and transfer RNA abundance. Nature. 1987;325:728–730. doi: 10.1038/325728a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.