


Abstract
Trithorax and polycomb group proteins are generally thought to antagonize one another. The trithorax family member MLL (myeloid/lymphoid or mixed-lineage leukemia) is presumed to activate Hox expression, counteracting polycomb-mediated repression. PC4 and SF2 interacting protein 1 (PSIP1)/p75, also known as LEDGF, whose PWWP domain binds to H3K36me3, interacts with MLL and tethers MLL fusion proteins to HOXA9 in leukaemias. Here we show, unexpectedly, that Psip1/p75 regulates homeotic genes by recruiting not only MLL complexes, but also the polycomb group protein Bmi1. In Psip1−/− cells binding of Mll1/2, Bmi1 and the co-repressor Ctbp1 at Hox loci are all abrogated and Hoxa and Hoxd mRNA expression increased. Our data not only reveal a potential mechanism of action for Psip1 in the regulation of Hox genes but also suggest an unexpected interplay between proteins usually considered as transcriptional activators and repressors.
INTRODUCTION
The Homeotic (Hox) gene family encodes transcription factors essential for patterning the anterior–posterior body axis. The developmental pattern of Hox gene expression is thought to be maintained by two groups of proteins: Polycomb repressive complexes (PRCs) maintain Hox genes in a silent state (1). The PRC2 complex contains the Ezh2/Ezh1 histone methyltransferases (HMTs) that mediate H3K27 trimethylation (H3K27me3) (2) and PRC1 contains the Bmi1 or Mel18/Ring1a/b heterodimer which can ubiquitinate H2AK119 and can compact chromatin (3). Trithorax (Trx) proteins (MLL proteins in mammals) of the COMPASS-like family have histone H3 lysine 4 (H3K4) methyl transferase activity and are generally thought to maintain the active expression level of Hox genes (4,5), and may function as anti-repressors to prevent the repressive function of polycomb (6–8).
Six mammalian COMPASS-like complexes have been identified, each with a SET domain-containing HMT subunit including; Set1A/KMT2F, Set1B/KMT2G and four MLL-family proteins—MLL1/KMT2A, Mll2/KMT2B, MLL3/KMT2C, MLL4/KMT2D (9,10). Each of these complexes associates with proteins that can modulate target site selection and enzymatic activity. Set1A/B are associated with Wdr82 (11,12), Mll1 and Mll2 are associated with Menin (5), Mll3 and Mll4 with PTIP (13).
Among the MLL genes, MLL1 has been the most extensively studied as it is frequently involved in leukemia-associated chromosomal translocations, where its fusion to a variety of proteins is accompanied by dysregulated Hox expression in haematopoiesis (14). Mice mutant for Mll1, or for its Set domain, have homeotic transformations of the axial skeleton and aberrant Hox gene expression (15,16). In contrast to SET1A/B, Mll1 and Mll2 have few target genes, but these include Hox genes (17,18). However, the relationship between Mlls and H3K4 methylation is complex. Mll1 is dispensable for most of the H4K4me3 at Hox genes in fibroblasts (5) and in mouse embryonic stem cells (mESCs) (18). But H3K4me3 at some Hox promoters in mESCs requires Mll1, at others it requires Mll2, and at some it requires both. Many promoters do not require either Mll1 or Mll2 indicating that a third enzyme is responsible for H3K4 trimethylation on Hox genes (18). Indeed, recent in vitro evidence indicates that Trx, Mll1 and Mll2 catalyse H3K4 monomethylation rather than H3K4me3 (19).
In flies Trx complexes bind to specific response DNA elements (TREs). Mammalian TREs have not been identified and the mechanism of MLL recruitment to Hox genes is not clear. Two lncRNAs expressed from the Hoxa cluster, and linked to Hox gene activation, have been suggested to function through recruitment of the MLL1 complex. Mistral, located between Hoxa6 and Hoxa7, has been reported to recruit WDR5 and the MLL1 complex to activate Hoxa6 and Hoxa7 transcription (20). HOTTIP is transcribed in an antisense direction from the 5′ end of Hoxa13, and is reported to be important for targeting Wdr5 and MLL across HOXA and for maintaining 5′ Hoxa expression and H3K4me3 in distal tissues (21).
The menin tumor suppressor protein is a common component of MLL1 and 2 complexes, and has been reported to be important for MLL recruitment to target genes and for the regulation of Hox expression (22). Menin functions as an adaptor molecule, binding to MLL1while also interacting with the protein Psip1/p75 at a distinct surface. Neither menin nor MLL1 alone can interact with Psip1 (23–25).
PC4 and SF2 interacting protein 1 (Psip1), previously known as LEDGF, is a chromatin protein implicated in; transcriptional regulation—including of Hox genes (26), mRNA splicing (27,28), DNA repair (29) and HIV integration (30,31). Psip1 encodes two isoforms (p52 and p75) which share a common N-terminal PWWP domain that binds to H3K36me3 (28) and is required for MLL1-mediated leukemic transformation (32) (Figure 1A). Psip1 p52, but not p75, interacts with splicing factors and can modulate alternative splicing of weak exons (28). It is the C-terminal domain of p75, absent in p52, which interacts with MLL1 (32) and a variety of other proteins (33–35) (Figure 1A). The mechanism by which Psip1/p75 regulates transcription is not known.
Figure 1.
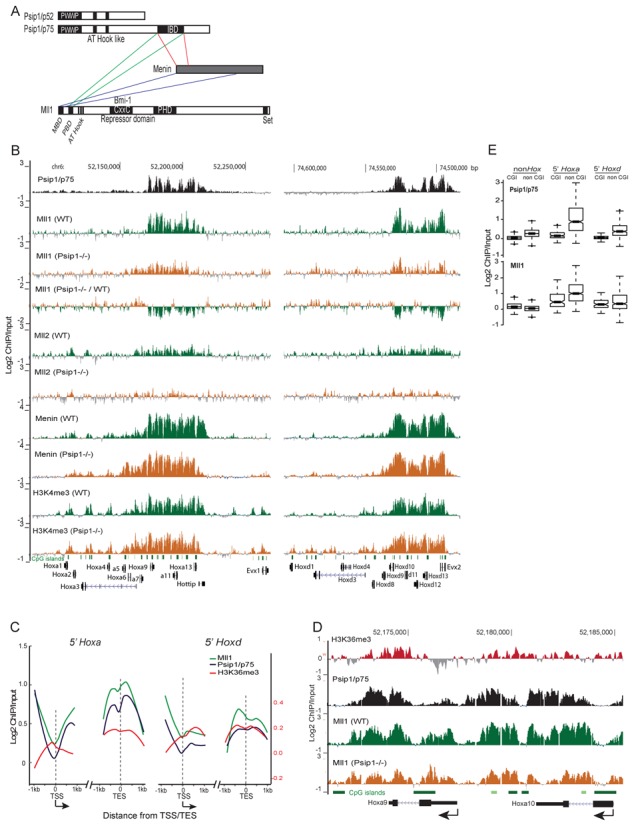
Psip1/p75, Mll1 and menin localization in wild-type and Psip1−/− cells. (A) Cartoon of Psip1 p52 and p75 isoforms showing the localization of the PWWP domain, AT hooks and also the integrase binding domain (IBD) domain at the C-terminus of p75 that interacts with menin and Mll1. The menin binding domain (MBD), Psip1 binding domain (PDB), CxxC domain and plant homeodomain (PHD) of Mll are also shown. (B) Mean Log2 ChIP/input for Psip1/p75 Mll1, Mll2, menin and H3K4me3 in WT and Psip1−/− MEFs over genomic regions encompassing HoxA (left) and HoxD loci (right). For Mll1, a difference plot for Mll1 ChIP in Psip1−/− versus WT cells is also shown. (n = 2 biological replicates.) Below, the positions of CpG islands and genes are shown. Genome co-ordinates (bp) from the mm9 version of the mouse genome assembly. (C) Averaged Log2 ChIP/input ratios for p75 (black lines), Mll1(green) and H3K36me3 (red) in 2 kb windows surrounding the transcription start site (TSS) or transcription end site (TES) of expressed genes from the 5′ portions of Hoxa and Hoxd. Arrow under TSS indicates the direction of transcription. (D) As in B, but zoomed in view of Psip1/p75, Mll1 and H3K36me3 distributions over Hoxa9 and Hoxa10 in WT MEFS. The Mll1 distribution in Psip1−/− MEFs is also shown. (E) Box plots showing the distribution of Log2 ChIP/input ratios for Psip1/p75 (top) and Mll1 (bottom) over the CpGisland (CGI) and non-CGI portions of expressed non-Hox genes and expressed genes from 5′ HoxA and HoxD.
Here, we show that Psip1/p75, rather than p52, is important in regulating Hoxa and Hoxd gene expression. Psip1/p75 interacts with Mll1, and in Psip1−/− mouse embryonic fibroblasts (MEFs) Mll1 and Mll2, but not H3K4me3, levels are reduced on expressed Hoxa and Hoxd genes. Unexpectedly, however, given the assumed association of Mll1 with Hox activation, Hox genes are up-regulated in the absence of Psip1 and the accompanying loss of Mll1, suggesting that p75, while recruiting Mll1—a supposed activator, acts to repress gene expression. Furthermore, we show that Psip1/p75 is also required to recruit the polycomb group protein Bmi1, and the co-repressor Ctbp1 to expressed Hox genes.
This study reveals a potential mechanism through which the p75 isoform of Psip1 regulates the expression of Hox genes and it highlights the unexpectedly complex relationship between the polycomb and trithorax machinery. It is clear that these systems cannot simply be considered as opposing repression versus activating protein complexes.
MATERIALS AND METHODS
Cell culture
Psip1−/− and corresponding WT immortalized MEFs (30) were a gift of Prof. Alan Engelman. Primary MEFs were derived from 13-day-old (E13) Psip1gt/gt embryos and their WT littermates as described previously (28). Psip1−/− MEFs were transduced with retroviral vectors containing p52 and p75 cDNAs -pLPX-p52HA and pLPX-p75HA (30) and packaged in PLAT-E cells according to a standard protocol (Clonetech). Transduced cells were selected with 2.5 μg/ml puromycin and stably expressed HA-tagged Psip1 isoforms were detected by immunoblotting with Psip1 antibodies (Bethyl lab. A300–847).
Lentiviral knockdown
Lentiviral micro RNA (Gift from Dr. Gijsbers, KU Leuven) specifically targeting p75, or both p52 and p75, isoforms of Psip1 were transduced into wild-type (WT) MEFs. Stably transduced MEFs were selected using blasticidin (10 μg/ml). The efficiency of knockdown was validated by immunoblotting with antibodies recognizing Psip1 p52 or p75 (28). Psip1 p75 was also depleted in 10.5 dpc mouse distal posterior limb cells (36) using lenti-viral shRNAs (Sigma Aldrich, TRCN0000012116 and TRCN0000012113) and stably transduced cells were selected using puromycin (3 μg/ml).
Chromatin immunoprecipitation (ChIP)
ChIP was performed as previously described (28), using antibodies for Psip1/p75 (Bethyl laboratory A300–848), Psip1/p52 (Bethyl laboratory A300–847) Mll1 (Active Motif 61295), BMI1 (Millipore 05–637), Ring1B (MBL D139–3), H3K4me3 (Millipore 07–473), menin (Abcam, ab4452–50), RNA PolII Ser2p (Millipore 04–1571, Clone 3E10), Ctbp1 (Santa Cruz SC-55502), CBX4 (Abcam ab139815) and Mll2 (Abcam ab15962). ChIPed DNA was amplified with WGA2 using the manufacturer's protocol (Sigma Aldrich) and hybridized to custom Hox arrays (28). All ChIP on chip experiments were done with at least two biological replicates (GEO accession number GSE 49182 for platform GPL13276).
Normalization and analysis of microarray data was as described previously (28). For CpG analysis, CpG islands (CGIs) were identified by finding probes with a minimum of 25 bp overlap with CGI found ±1 kb within genes using Galaxy software. CGI positions were taken from the University of California Santa Cruz (UCSC) table browser.
Enrichment analysis for ChIP and run-on data was performed for probes ±1 kb from transcription start sites (TSS) or transcription end site (TES). The smoothed conditional mean plots were generated using the R package ggplot2 and the geom_smooth function.
The following mm9 coordinates were used for quantification of ChIP enrichment; non-expressed 3′ Hoax(Hoxa1 to Hoxa7) genes chr6:52,101,011–52,172,728, expressed 5′ Hoxa genes (Hoxa9-Hoxa13) chr6:52,171,296–52,211,033, 3′ non-expressed Hoxd (Hoxd1 to Hoxd9 genes) chr2:74,534,258–74,606,421, expressed 5′ Hoxd genes (Hoxd9-Hoxd13) chr2:74,484,916–74,537,448. To test the significance of differential ChIP enrichment at genomic regions a Wilcoxon rank-sum test was performed with a correction for multiple testing (Holm method) using the R statistical program.
For sequential ChIP (SeqChIP), antibodies were covalently coupled to Dynabeads with antibody coupling kit, (Invitrogen Cat. 14311D), using the manufacture's protocol. The first ChIP was eluted with 10 mM DTT and the elute was diluted 30 times with Radio-Immunoprecipitation Assay (RIPA) (50 mMTris, pH 7.5, 150 mMNaCl, 1% IGEPAL CA-630, 0.5% deoxycholate) buffer before continuing with the second ChIP. Primers used for ChIP qPCR are given in Supplementary Table S1.
Expression analysis
Expression microarray was performed with four biological replicates of WT and, Psip1−/− MEFs, as described previously (37). Gene Ontology (GO; Biological Process) enrichment analysis was performed using the GO enrichment analysis and visualization web tool (GORilla). A False Discovery Rate (FDR) q-value cut-off of 0.01 was used to select significantly enriched GO terms.
For reverse transcriptase-polymerase chain reaction (RT-qPCR), cDNAs were prepared with Superscript II (Invitrogen) reverse transcriptase using random primers. The list of specific primers used is given in Supplementary Table S1. RT-qPCR was done with three biological replicates of WT and Psip1−/− MEFs and Psip1−/−MEFs rescued with p52-HA or p75-HA cDNA on a LightCycler 480 (Roche Diagnostics). Data were normalized to Gapdh and the error bars indicate standard error of mean (s.e.m.) from three biological replicates. Similarly, RT-qPCR for WT MEFs depleted for Psip1 isoforms was done for three biological replicates.
Run-on transcription
Approximately 107 MEFs were resuspended in hypotonic buffer (20 mM Hepes-KOH pH 7.9, 10 mMKCl, 1 mM MgCl2, 0.5% NP40, 20% Glycerol) and dounced 25 times on ice. The run-on transcription assay was performed as described previously (38). Run-on RNA was reverse transcribed using whole transcriptome amplification (WTA2) kit according to the manufacturer (Sigma Aldrich), and the resultant cDNA was labeled with Cy3 or Cy5 and hybridized to the same custom tiling arrays used for hybridizing ChIP DNA.
Nuclear extracts and immuno-precipitations
Cells from 14-cm dishes were trypsinized and pelleted, resuspended in 5 ml of ice-cold swelling buffer (10 mM Hepes, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, and protease inhibitors) for 5 min, and broken open to release nuclei using a pre-chilled Dounce homogenizer (20 strokes with a tight pestle). Nuclei were pelleted by centrifugation at 228 g for 5 min at 4°C and resuspended in 1 ml of RIPA buffer and protease inhibitors + Benzonase (Novagen; final concentration, 1.25 U/μl) and incubated for 30 min on ice. Extracts were cleared by centrifugation at 15500 g for 10 min at 4°C.
Protein A Dynabeads (Invitrogen), were incubated with 5 μg of α HA antibodies (for HA pulldown) or control immunoglobulin G (IgG) antibodies for 1 h in phosphate buffered saline. Equivalent nuclear protein amounts were incubated with the beads coupled to antibodies for 1 h. After 4 wash steps in RIPA buffer, bound proteins were eluted using 2× sodium dodecyl sulphate (SDS) loading buffer, and separated on a NuPAGE gel, blotted to Polyvinylidene fluoride (PVDF) membrane, and immunoblotted with antibodies recognizing; Mll1 (Active Motif 61295, 1:1000), BMI1 (Millipore 05–637, 1:1000), Ring1B (MBL D139–3, 1:2000), EZH2 (BD Biosciences 1:3000) and HA tag (Sigma H6908, 1:1000). For the GFP trap experiment, WT MEFs were stably transduced with green fluorescent protein-p75 (GFP-p75) (39), GFP-p75 complex was purified according to the manufacturer's protocol (ChromoTek).
RESULTS
Psip1/p75 localizes to expressed Hox genes
Surviving Psip1 gene-trap mutant mice show homeotic skeletal transformation phenotypes, similar to those of Hoxa4, 5 and 6 mutant animals (26). Moreover, knockdown of PSIP1 in human cells showed that mRNAs of 5′HOXA genes, but not genes of the other HOX loci, are among the most up-regulated (26,40). This suggests that the HoxA cluster may be a specific target for regulation by Psip1.
To identify Psip1/p75 occupancy on Hox loci we performed ChIP for endogenous p75 from WT immortalized MEFs with an antibody (A300–848), whose specificity for Psip1/p75 and inability to recognize the p52 Psip1 isoform we have confirmed previously (28). ChIP'd DNA was hybridized to custom arrays covering all four Hox loci and several other developmental genes (28). Psip1/p75 was enriched over HoxA and HoxD (Figure 1B) but was not detected at HoxB or HoxC loci in these cells (Supplementary Figure S1A). The preference for p75 occupancy over Hox A and D clusters does not reflect an intrinsic property of the DNA sequence there: ChIP in independent primary MEFs derived in our laboratory (28) showed specific occupancy of p75 over Hoxa, b and c genes which are expressed in those cells (data not shown), suggesting that Psip1/p75 generally binds to expressed Hox genes.
We found that the distribution of Mll1 and menin across HoxA and HoxD was highly similar to that of p75 (Spearman's correlation between p75 and Mll1 ρ = 0.82, P < 0.01) (Figure 1B–D) and, like p75, these proteins were also not detected at HoxB and HoxC clusters in these MEFs (Supplementary Figure S1A). Psip1 p75, Mll1, menin and H3K4me3 were abundant at the 5′ ends of HoxA and HoxD (Hoxa9 to a13, Hoxd10 to d13) but not at the 3′ end (Figure 1B). Mll1 and p75 were also not enriched at the silent non-Hox polycomb-target Shh gene (Supplementary Figure S1B and C).
At Myc, p75 is present at the 3′ end of this active gene, as has been seen more globally for Psip1/p52 (Supplementary Figure S1B and C), consistent with targeting to H3K36me3 via the PWWP domain common to both Psip1 isoforms (28) (Figure 1C and D). Mll1 and Menin have a broad distribution across Myc, suggesting Psip1 independent binding at this locus (Supplementary Figure S1C).
At 5′Hoxa and 5′Hoxd genes we found that, like p75, Mll1 is partially excluded from CGIs (Figure 1D and E) but is enriched ∼1 kb downstream from the TSSs, over gene bodies particularly near TESs and beyond (Figure 1C and D). This is a similar profile to that of H3K36me3 (Figure 1C and D) (28) and is consistent with previous analysis of Mll1 distribution over late HOXA genes in a human lymphoma cell line (41). This suggests that Mll1 recruitment to chromatin may be dependent on its interaction with Psip1 and not binding of its CXXC domain to unmethylated CGIs.
Psip1 is required for Mll1 recruitment to expressed HoxA and HoxD
The co-occurrence of Mll1 and Psip1/p75 are consistent with Psip1 being required to tether Mll1 at target genes (32). To test this, we performed ChIP for Mll1, Menin and H3K4me3 in Psip1 homozygous null MEFs (Psip1−/−) derived from mutant embryos that were littermates to the WT controls (28,30). There were not extensive changes in Mll1 or menin binding at non Hox genes in the mutant MEFs (Figure 2). However, loss of Psip1 significantly (P < 0.01) reduced Mll1 binding across 5′ Hoxa (a9-a13) - and Hoxd (d9-d13) genes (Figures 1B and 2A). Menin levels were not reduced (Figure 2C). Surprisingly, H3K4me3 levels were also not significantly changed at either 5′ HoxD or HoxA (Figures 1B and 2D) in the absence of Psip1, suggesting that Mll1 is dispensable for this histone modification at these sites.
Figure 2.
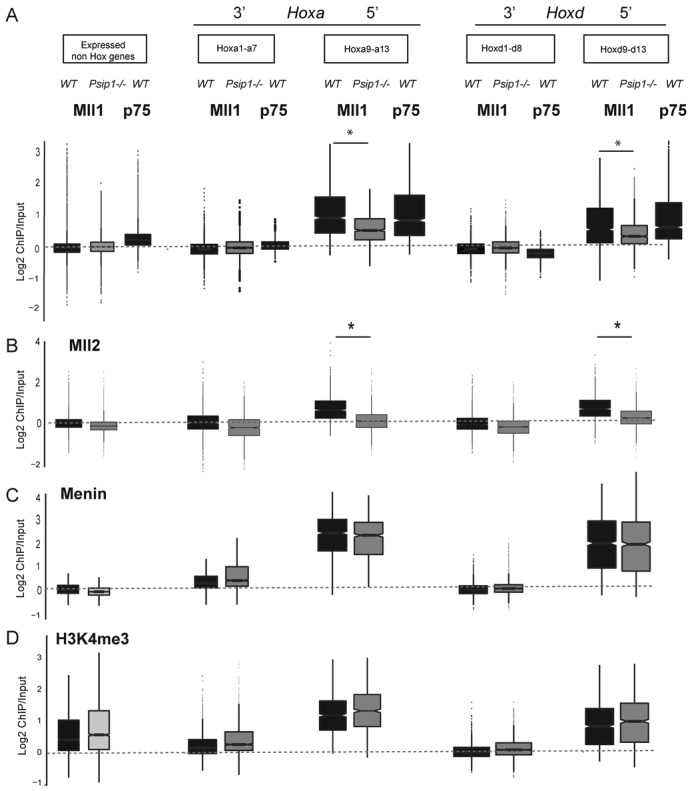
Psip1 loss results in reduced Mll1 and Mll2 at Hox loci. Box plots showing the distribution of; (A) Mll1, Psip1/p75, (B) Mll2 (C) menin and (D) H3K4me3 in WT (black boxes) and Psip1−/− (gray boxes) cells at expressed non-Hox genes, 3′ Hoxa genes (Hoxa1-a7), 5′ Hoxa genes (Hoxa9-a13), 3′ Hoxd genes (Hoxd1-d9) and Hoxd10-d13. Regions with a statistically significant (P < 0.01) difference in binding between WT and Psip1−/− cells as assessed with a Wilcoxon rank-sum test are indicated with an asterisk (*). Box plots showing the distribution of Psip p75 are also shown in (A).
To confirm that loss of Mll1 in Psip1−/−MEFs is not due to differences in cell types, we repeated the Mll1 ChIP in limb mesenchyme cells (36) specifically depleted for Psip1/p75 with a short-hairpin (Sh) RNA. Mll1 was significantly reduced over Hox genes in the p75 knockdown cells compared to controls. The p52 isoform of Psip1 was not ablated by the p75 ShRNA (Supplementary Figure S1D), confirming that it is indeed Psip1/p75 that is required for Mll1 targeting.
Both Mll1 and Mll2 are implicated in regulating Hox genes (5) and although a direct interaction between p75 and Mll2 has not been demonstrated, a recent report (42) and our GFP-p75 trap data, see below, indicate the presence of Mll2 with p75 complexes, possibly through the common interactor Menin (25). Indeed, we detected Mll2 over both HoxA and D loci in WT MEFs (Figure 1B) and Mll2 levels were significantly reduced over 5′Hoxa and Hoxd genes in Psip1−/− MEFs (Figure 2B).
Absence of Psip1/p75 leads to mis-expression of Hox genes
Microarray analysis shows elevated levels of mRNAs from several 5′ Hoxa and Hoxd genes in Psip1−/− MEFs compared to WT cells (Figure 3A) and, surprisingly, given the usual association of Trx proteins with gene activation, this included 5′ Hoxa genes (a9, a10 and a11), that lose Mll1 binding in the mutant cells. This was confirmed by quantitative RT-PCR (Figure 3B). GO analysis of genes differentially expressed (FDR q < 0.01) between WT and Psip1−/−show enrichment of terms associated with anatomical structure morphogenesis and developmental process (Supplementary Figure S2A), consistent with the craniofacial and skeletal abnormalities of surviving Psip1 mutant mice (26).
Figure 3.
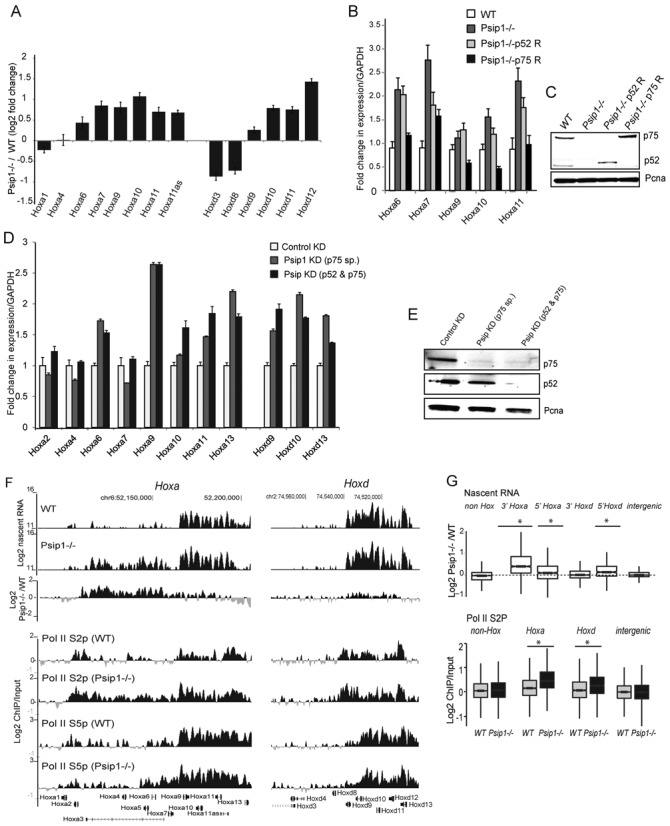
Psip1/p75 represses the expression of Hox genes. (A) Log2 fold change in mean (+/- s.e.m) microarray expression levels of Hoxa and Hoxd genes in Psip1−/− versus WT (n = 4 biological replicates each). (B) Mean (+/- s.e.m.) expression of Hoxa genes, normalized to Gapdh, assayed by RT-qPCR, in WT (white) and Psip1−/− (dark gray) cells, and in Psip1 mutant cells rescued with p52 (light gray) or p75 (black bars) Psip1 isoforms (n = 3 biologial replicates). (C) Immunoblot to detect p75 and p52 Psip1 isoforms in WT, Psip1−/−, p52 rescue, and p75 rescue cells. Pcna served as loading control. (D) Mean (+/- s.e.m.) Hoxa and Hoxd gene expression, normalized to Gapdh, in WT MEFs after specific knockdown of p75 isoform (gray bars), or both p52 and p75 isoforms of Psip1 (black bars) and a control scrambled micro-RNA (white bars). (E) Immunoblot to detect p75, p52 and Pcna in cells transfected with control, p75-specific, and p52 + p75 isoform specific lentiviral micro-RNAs. (F) Top: Mean log2 signal of run-on transcribed RNA from WT and Psip1−/− MEFs over HoxA (left) and HoxD (right), established by hybridization of cDNA from run-on transcripts to custom tiling arrays. (n = 3, 2 biological and 1 technical replicates). A difference plot for Psip1−/−versus WT cells is also shown. Bottom: Mean Log2 ChIP/input for Ser2 (S2p) and Ser5 (S5p) phosphorylated PolII in WT and Psip1−/− MEFs (n = 2 biological replicates). (G) Box plots show: Top—Log2 ratio of Psip1−/−/WT run-on transcribed RNA (Nascent RNA) over non Hox genes, genes from the 3′ and 5′ ends of HoxA and HoxdD, and intergenic regions. Bottom—Log2 ChIP/input for Ser2 phosporylated RNA Polymerase II (Pol II (S2p) in WT and Psip1−/− MEFs over non-Hox genes, Hoxagenes, Hoxd genes and intergenic regions (n = 2 biological replicates). Regions with a statistically significant difference (P < 0.05) in S2p between WT and Psip1−/− cells as assessed by the Wilcoxon rank-sum test are indicated with an asterisk (*).
To test whether mis-regulation is due to loss of the p52 or p75 Psip1 isoforms, we rescued each isoform by retroviral transduction of appropriate cDNAs into Psip1−/− MEFs (Figure 3C). Psip1 p75 was able to reverse the elevated mRNA levels of many of the up-regulated Hoxa genes, and p52 had modest or no effects on expression of the tested genes, with the exception of Hoxa7 (Figure 3B).
We further confirmed a direct effect of p75 loss on Hoxa mRNAs by knocking down Psip1/p75 protein levels in WT MEFs using two different miRNAs, one specific for the p75 isoform and that has no effect on Psip1/p52, and the other targeting the N-terminal domain common to both isoforms (Figure 3E). This resulted in increased expression of Hoxa9-a13, and Hoxd9-d13 mRNAs (Figure 3D). Steady-state levels of mRNAs from 3′ Hox (a2-a4) were unaltered, compatible with data from Psip1−/− cells (Figure 3A and B).
Elevated 5′ Hoxa mRNA levels, was also validated in primary MEFs derived from E13.5 Psip1gt/gt embryos compared to their wild-type littermates (26,28) (Supplementary Figure S2B). Together, these data suggest that Psip1 p75 has a specific role in regulating the expression level of Hox genes.
Given our previous demonstration of a role for Psip1 p52 in mRNA processing (28) we determined whether Psip p75 affected the level of transcription per se. Run-on transcription in WT MEFs detected high nascent RNA synthesis from the 5′ ends of the HoxA and HoxD clusters, and not from the 3′ regions (Figure 3F). In Psip1−/− cells, run-on RNA levels were elevated at 5′ Hoxa and Hoxd genes, but not at non-Hox genes or intergenic regions, or at the 3′ end of HoxD (Figure 3F and G, Supplementary Figure S2C). Intriguingly, elevated nascent RNA was also detected over part of the 3′ end of HoxA, even though there is no Psip1/p75 bound at this region and no change in levels of mature mRNA from Hoxa4 is detected in Psip1 mutant cells (Figure 3A and D). However, we note that the extent of this transcription corresponds to the large annotated Hoxa3 transcript whose TSS is located toward the 5’ of Hoxa, between Hoxa6 and Hoxa7. This signal may also arise from unstable, unprocessed RNA. We also noted slightly elevated levels of Menin and H3K4me3 at 3′HoxA in the mutant cells though this increase was not statistically significant (Figures 1B and 2C and D).
Our data are consistent with Psip1 playing some role in regulating transcription of Hox genes. Furthermore, the enrichment of p75 on gene bodies and toward the TES of transcribed Hoxa genes (Figure 1B–D), together with direct binding to H3K36me3 (28), suggests that Psip1 could function to regulate transcription elongation. To investigate this we examined the levels of the elongating serine 2 phosphorylated form of RNA polymerase II (PolII S2p) (43,44) by ChIP in WT and Psip1 mutant cells. The levels of PolII S2p were significantly elevated across HoxA and HoxD in Psip1−/− cells (Figure 3F and G). Levels of the serine 5 phosphorylated initiating form of PolII (PolII S5p) were not changed. These data suggests that Psip1/p75 functions to restrain the elongation of transcription from paused/poised Pol II at Hox loci and that this may operate through Mll1 retention—at least for 5′ genes of the cluster.
Absence of Psip1 leads to loss of Bmi1 and Ctbp1
To further investigate the mechanism through which Psip1 and Mll1 restrict expression from Hox genes, we performed a GFP trap experiment using stably expressed GFP-p75 in WT MEFs. Consistent with previous studies (8,32) and with our ChIP analyses, Mll1 and Mll2 were detected interacting with Psip1. Given the known role of the PRCs in repressing Hox gene expression via paused polII (44) and in antagonizing trithorax, we looked for members of the PRC1 and PRC2 complexes in the proteins pulled down with GFP-p75 (Figure 4A). We detected no Ezh2, the HMTase from PRC2, nor did we detect Mel18 from PRC1 or Rybp—a member of a non-canonical PRC1 complex (45–47). However, we did detect the PRC1 components Bmi1 and Cbx4. Interestingly, Ctbp1 a transcriptional co-repressor was also detected in the GFP-p75 trap. Interactions amongst Mll1, Bmi1 and Psip/75, but not p52, were confirmed by pulldown of HA-tagged Psip1 isoforms (Supplementary Figure S3A). Not all PRC1 members interact with Psip1/Mll1 complex—Ring1B, a core member of PRC1 was not detected (Supplementary Figure S3A).
Figure 4.
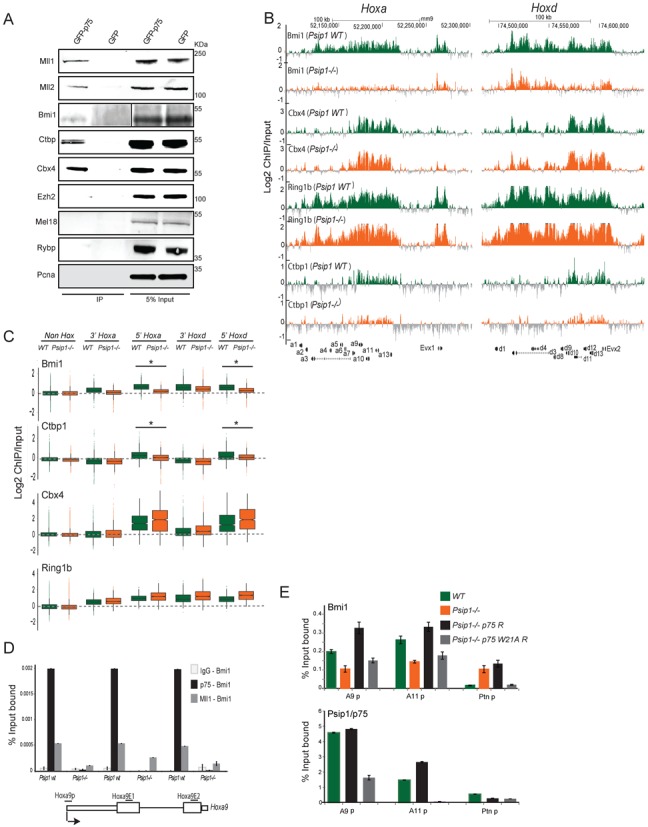
Loss of Psip1 leads to reduced Bmi1 and Ctbp1 on target genes. (A) Psip1/p75 complexes purified from stably transduced GFP-p75, and GFP (control) cells, and immunoblotted using antibodies against Mll1, Mll2, Bmi1, Ctbp1, Cbx4, Ezh2, Mel18, Rybp and Pcna. Note that 5% input extract were also loaded. (B) Mean Log2 ChIP/input for Bmi1, Cbx4, Ring1B and Ctbp1 in WT and Psip1−/− MEFs over HoxA (left) and HoxD clusters (right) using custom tiling arrays as in Figure 1. (C) Box plots showing Log2 ChIP/input distributions for Bmi1, Ctbp1, Cbx4 and Ring1B in WT and Psip1−/− MEFs over 5′ and 3′ regions of HoxA and HoxD clusters, and non-Hox genes (n = 2 biological replicates). Asterisk (*) indicates a significant difference in ChIP/input signal between WTand Psip1−/−(P < 0.01, Wilcoxon rank-sum test). (D) Sequential-ChIP qPCR over promoter (Hoxa9p), exon1 (Hoxa9E1) and exon 2 of Hoxa9 (Hoxa9E2) in Psip1 WT and Psip1−/− MEFs. First, ChIP was performed with covalently coupled IgG (IgG-Bmi1), p75 (p75-Bmi1) and Mll1 (Mll1-Bmi1), followed by Bmi1 antibodies for second ChIP. Schematic below shows the Hoxa9 gene and primers used for PCR. (E) ChIP qPCR for Bmi1 and p75 in WT (green) and Psip1−/− (orange) MEFS, and inPsip1−/− rescued with WT p75 (Psip1−/−p75 R; black) or p75 with W21A PWWP point mutation (Psip1−/−p75 W21A R; gray). Ptn promoter primers (Ptnp) were used as a control. Mean (+/- s.e.m.) percent (%) input bound (n = 3) are plotted.
Bmi1, Ctbp1 and Cbx4 have been previously shown to interact directly with the CxxC domain of Mll1 (48). These data prompted us to investigate the occupancy of these, and other polycomb-associated proteins, in WT and Psip1−/− MEFs. Bmi1, and Ring1B were detected at HoxA and D clusters, even over transcribed genes in WT MEFs (Figure 4B). Like Psip1 and Mll1, Cbx4 and Ctbp1 were enriched at the expressed 5′ Hoxa and d genes and not the 3′ regions of these clusters. The absence of Psip1 in mutant MEFs had no significant effects on Bmi1, Cbx4 and Ring1B levels at non-expressed HoxB and C (Supplementary Figure S3B), or at non-Hox genes (Figure 4C). However, Psip1 absence led to significant loss of Bmi1 and Ctbp1 over 5′ HoxA and D (Figure 4B and C). We also note that there is some reduction of Bmi1 detected at 3′ HoxA and D in Psip1 mutant cells but this does not reach statistical significance. Ctbp1 is not present at these regions even in wild-type cells.
To verify co-occupancy of Psip1/Mll1 and Bmi1 over target genes, we performed sequential ChIP, first using covalently coupled IgG, p75 and Mll1 antibodies and then the second ChIPs done with antibodies recognizing Bmi1. In WT MEFs, we detected high co-occupancy of p75 with Bmi1, as well as Mll with Bmi1, over the promoter, exon1 and exon2 of Hoxa9 (Figure 4D). In Psip1−/− MEFs, p75-Bmi co-occupancy was eliminated and Mll/Bmi1 occupancy greatly reduced (by 50–80%).
To confirm a direct role for Psip1 in Bmi1 retention at 5′ HoxA we performed ChIP for Bmi1 and Psip1/p75 in WT and Psip1−/− MEFs, and in Psip1−/− cells rescued with the p75 isoform. qPCR showed restoration of Bmi1 levels over tested Hoxa genes after p75 rescue (Figure 4E). A version of p75 in which a critical residue in the PWWP aromatic cage involved in H3K36me3 binding is mutated (W21A) (49) failed to restore Bmi1 binding (Figure 4E) and indeed W21A p75 failed to bind to Hox loci, consistent with a role for H3K36me3 recognition in p75 targeting (28).
DISCUSSION
It is generally assumed that members of polycomb complexes are involved in gene repression and that trithorax group members counteract this and help to maintain an active state (6–8). Here we have demonstrated that Psip1/p75 is required to retain Mll1/2 and Bmi1/Ctbp1 at HoxA and HoxD loci (Figures 1 and 2), with the net result of dampened gene expression. Contrary to the assumed role of Mll1 in maintaining gene activity, the loss of Mll1 (and Mll2) that occurs in the absence of Psip1 results in the upregulation of Hoxa and Hoxd gene transcription and mRNA levels. These data that we obtained in mouse embryonic fibroblasts are consistent with the upregulation of Hox genes that was reported after knockdown of PSIP1 in human 293 cells (26,40).
Psip1, Mll and menin
Menin has been reported to be important for MLL1/2 recruitment to target genes and for Hox gene regulation (22). Menin was suggested to bind to MLL1 and to interact with Psip1/p75 using distinct interaction surfaces (23–25) (32). Levels of Menin were not significantly changed in Psip1−/− cells suggesting that Psip1 is not required for menin targeting (Figure 2).
In MEFs we found similar genomic binding profiles for Psip1/p75, Mll1 and Mll2 (Figure 1) and in the absence of Psip1, Mll1/2 levels were reduced over expressed (5′) genes of the HoxA and HoxD clusters. However, we found no corresponding loss of H3K4 trimethylation suggesting that Mll1/2 are dispensable for H3K4me3 at these loci. Our data are consistent with persistence of H3K4me3 over some Hox genes in both Mll1−/− and Mll2−/− ES cells (18) and in Mll1−/− MEFs (5) and the suggestion that multiple H3K4 HMTs, including SET1, can be found co-bound at the same active genes (18).
Psip1 and Hoxa gene expression
PSIP1, and particularly its PWWP domain, is known to be required for MLL1-mediated leukemogenesis, and for targeting MLL1 fusion partners leading to uncontrolled expression of HOXA9 in leukemia (32). Similarly, Psip1 gene-trap mutant (Psip1gt/gt) mice have posterior skeletal transformations (26), similar to mice with mutation of Hoxa genes (50–52).
Here, we have demonstrated specific up-regulation of mRNA expression from 5′ genes of the HoxA and HoxD clusters (Figure 3) in the absence of Psip1 and have shown that this is dependent on the long p75 isoform of Psip1 and not p52—which we have previously demonstrated interacts with splicing factors to modulate alternative splicing (28). Consistent with recognition of H3K36me3 by the PWWP domain of Psip1 (28,49), p75 is distributed away from CGIs and toward the 3′ end of Hox genes (Figure 1). We show that the Psip1 PWWP domain is required for Bmi1 recruitment to 5′ HoxA and HoxD (Figure 4).
Psip1 and the interplay between polycomb and trithorax regulation
Our results suggest that Psip1/p75 functions to recruit Mll, and yet to restrain expression, from specific Hox loci. Clues to the mechanism underlying this may arise from our observation that Psip1 is also needed to recruit the repressors Bmi1 and Ctbp1 (Figure 4). We therefore suggest that at HoxA and HoxD, Psip1 tunes gene expression through its ability to recruit both Mll1 and Bmi1/Ctbp1.
Mll1 is a large multidomain protein that, by binding different proteins, can act either as a transcriptional activator or a repressor. Bmi1 has been reported to bind to the repressive (CxxC) domain of Mll1 (48) and we have confirmed that Bmi1 is present in chromatin that also contains Mll1 (Figure 4). Supporting a functional link between Psip1 and Bmi1, the up-regulation of 5′ Hoxa genes in Psip1−/− cells is similar to that seen in Bmi1−/− MEFs (53) and Psip1 gene-trap mutant (Psip1gt/gt) mice have posterior skeletal transformations (26) similar to mice with mutation of Bmi1 (54).
The HMTase activity of Mll1 is known to be dispensable for its essential functions (16). Since we show that H3K4me3 levels are not altered over 5′Hoxa genes in the absence of Psip1 and loss of Mll1 binding (Figures 1 and 2), we suggest that the Psip1-Mll1 interaction serves to recruit repressors, including the Bmi1 component of PRC1 and the co-repressor Ctbp1. Ctbp1 has been reported to colocalize with Bmi1 in the nucleus (55) and mutation of Ctbp1 in flies leads to loss of polycomb group protein recruitment to polycomb response elements (PREs) (56). Similarly, the increased expression of Hoxd genes reported in Mll1−/− MEFs (5) could be due to loss of repressors, such as Bmi1 and Ctbp1. Psip1 has been shown to promote homologous recombination (HR) by interacting with CtIP (29), CtIP interacts with CtBP1 and both proteins are components of the RBP-Jκ/SHARP corepressor complex (57). Recently, a role for H3K36me3 and Psip1 in promoting HR at transcriptionally active loci has been demonstrated (58), which is consistent with the recruitment of Psip1 to expressed gene bodies marked by H3K36me3 (28).
We have identified a new Psip1-dependent pathway of control of Hox loci that involves both repressors (Bmi1, Ctbp1) and proteins traditionally thought to be involved in maintaining gene activation (Mll1/2). This appears to operate, at least partially, at the level of transcription elongation (Figure 3), but we do not exclude that there may be effects at other levels of RNA processing as well. The polycomb and trithorax systems are usually considered to be antagonistic repressive and activating protein complexes. However, the colocalization of both Trx and polycomb at specific sites on Drosophila polytene chromosomes hints at an interaction between the two systems (59). Other studies have also indicated the presence of repressor complexes at active loci and their role in ‘fine-tuning’ of gene activation (60). Occurrence of proteins known to be implicated with gene repression—e.g. Bmi1, Ctbp1, CBX4 and Ring1B over the expressed Hox genes (Figure 4), and up-regulation of mRNAs from Hox genes with the loss of some of these proteins, suggests a more nuanced role of these proteins in fine tuning gene expression.
Finally, our data suggests a new pathway of gene control that may be important for the dysregulation of HOXA genes in leukemia (61). MLL is required to maintain HOXA9 expression in haematopoietic progenitor cells, but then is also required to later repress HOXA9 during the later stages of differentiation (62). Moreover, acute and chronic myeloid leukemias and myelodysplastic syndrome have been associated with fusion of PSIP1 to NUP-98 (63–65).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Prof. Alan Engelman (Harvard Medical School) for Psip1−/− MEFs and Psip1/p52 and Psip1/p75 retroviral rescue plasmids, Dr. Rik Gijsbers (KU Leuven) for Psip1 knockdown and GFP-p75 lentiviral vectors and Prof. Jay Hess (The University of Michigan) for Mll1−/− MEFs.
Footnotes
Present address:
Heidi Sutherland, Queensland University of Technology, Kelvin Grove, Queensland 4059, Australia.
FUNDING
Medical Research Council UK and Wellcome Trust [WT085767]. Source of open access funding: MRC University Unit programme grant (University of Edinburgh).
Conflict of interest statement. None declared.
REFERENCES
- 1.Soshnikova N., Duboule D. Epigenetic temporal control of mouse Hox genes in vivo. Science. 2009;324:1320–1323. doi: 10.1126/science.1171468. [DOI] [PubMed] [Google Scholar]
- 2.Shen X., Liu Y., Hsu Y.-J., Fujiwara Y., Kim J., Mao X., Yuan G.-C., Orkin S.H. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol. Cell. 2008;32:491–502. doi: 10.1016/j.molcel.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eskeland R., Leeb M., Grimes G.R., Kress C., Boyle S., Sproul D., Gilbert N., Fan Y., Skoultchi A.I., Wutz A., et al. Ring1B compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol. Cell. 2010;38:452–464. doi: 10.1016/j.molcel.2010.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Milne T.A., Briggs S.D., Brock H.W., Martin M.E., Gibbs D., Allis C.D., Hess J.L. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 5.Wang P., Lin C., Smith E.R., Guo H., Sanderson B.W., Wu M., Gogol M., Alexander T., Seidel C., Wiedemann L.M., et al. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol. Cell. Biol. 2009;29:6074–6085. doi: 10.1128/MCB.00924-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klymenko T., Müller J. The histone methyltransferases Trithorax and Ash1 prevent transcriptional silencing by Polycomb group proteins. EMBO Rep. 2004;5:373–377. doi: 10.1038/sj.embor.7400111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poux S., Horard B., Sigrist C.J., Pirrotta V. The Drosophila trithorax protein is a coactivator required to prevent re-establishment of polycomb silencing. Development. 2002;129:2483–2493. doi: 10.1242/dev.129.10.2483. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka Y., Kawahashi K., Katagiri Z.-I., Nakayama Y., Mahajan M., Kioussis D. Dual function of histone H3 lysine 36 methyltransferase ASH1 in regulation of Hox gene expression. PLoS One. 2011;6:e28171. doi: 10.1371/journal.pone.0028171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schuettengruber B., Martinez A.-M., Iovino N., Cavalli G. Trithorax group proteins: switching genes on and keeping them active. Nat. Rev. Mol. Cell Biol. 2011;12:799–814. doi: 10.1038/nrm3230. [DOI] [PubMed] [Google Scholar]
- 10.Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee J.-H., Skalnik D.G. CpG-binding protein (CXXC finger protein 1) is a component of the mammalian Set1 histone H3-Lys4 methyltransferase complex, the analogue of the yeast Set1/COMPASS complex. J. Biol. Chem. 2005;280:41725–41731. doi: 10.1074/jbc.M508312200. [DOI] [PubMed] [Google Scholar]
- 12.Wu M., Wang P.F., Lee J.S., Martin-Brown S., Florens L., Washburn M., Shilatifard A. Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol. Cell. Biol. 2008;28:7337–7344. doi: 10.1128/MCB.00976-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho Y.-W., Hong T., Hong S., Guo H., Yu H., Kim D., Guszczynski T., Dressler G.R., Copeland T.D., Kalkum M., et al. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 2007;282:20395–20406. doi: 10.1074/jbc.M701574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dharmarajan V., Cosgrove M.S. Antica Mariastefania., editor. Biochemistry of the mixed lineage leukemia 1 (MLL1) protein and targeted therapies for associated leukemia. Acute Leuk. - The Scientist's Perspective and Challenge In Tech. 2011 [Google Scholar]
- 15.Yu D.B., Hess J.L., Horning S.E., Brown G.A., Korsmeyer S.J. Altered Hox expression and segmental identity in Mll-mutant mice. Nature. 1995;378:505–508. doi: 10.1038/378505a0. [DOI] [PubMed] [Google Scholar]
- 16.Terranova R., Agherbi H., Boned A., Meresse S., Djabali M. Histone and DNA methylation defects at Hox genes in mice expressing a SET domain-truncated form of Mll. Proc. Natl. Acad. Sci. U.S.A. 2006;103:6629–6634. doi: 10.1073/pnas.0507425103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu D., Garruss A.S., Gao X., Morgan M.A., Cook M., Smith E.R., Shilatifard A. The Mll2 branch of the COMPASS family regulates bivalent promoters in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 2013;20:1093–1097. doi: 10.1038/nsmb.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Denissov S., Hofemeister H., Marks H., Kranz A., Ciotta G., Singh S., Anastassiadis K., Stunnenberg H.G., Stewart A.F. Mll2 is required for H3K4 trimethylation on bivalent promoters in embryonic stem cells, whereas Mll1 is redundant. Development. 2014;141:526–537. doi: 10.1242/dev.102681. [DOI] [PubMed] [Google Scholar]
- 19.Tie F., Banerjee R., Saiakhova A.R., Howard B., Monteith K.E., Scacheri P.C., Cosgrove M.S., Harte P.J. Trithorax monomethylates histone H3K4 and interacts directly with CBP to promote H3K27 acetylation and antagonize Polycomb silencing. Development. 2014;141:1129–1139. doi: 10.1242/dev.102392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bertani S., Sauer S., Bolotin E., Sauer F. The noncoding RNA Mistral activates Hoxa6 and Hoxa7 expression and stem cell differentiation by recruiting MLL1 to chromatin. Mol. Cell. 2011;43:1040–1046. doi: 10.1016/j.molcel.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Wang K.C., Yang Y.W., Liu B., Sanyal A., Corces-Zimmerman R., Chen Y., Lajoie B.R., Protacio A., Flynn R.A., Gupta R.A., et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–124. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milne T.A., Hughes C.M., Lloyd R., Yang Z., Rozenblatt-Rosen O., Dou Y., Schnepp R.W., Krankel C., Livolsi V. A, Gibbs D., et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2005;102:749–754. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yokoyama A., Wang Z., Wysocka J., Sanyal M., Aufiero D.J., Kitabayashi I., Herr W., Cleary M.L. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol. Cell. Biol. 2004;24:5639–5649. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yokoyama A., Somervaille T.C.P., Smith K.S., Rozenblatt-Rosen O., Meyerson M., Cleary M.L. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123:207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 25.Huang J., Gurung B., Wan B., Matkar S., Veniaminova N.A., Wan K., Merchant J.L., Hua X., Lei M. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature. 2012;482:542–546. doi: 10.1038/nature10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sutherland H.G., Newton K., Brownstein D.G., Holmes M.C., Kress C., Semple C.A., Bickmore W.A. Disruption of Ledgf/Psip1 results in perinatal mortality and homeotic skeletal transformations. Mol. Cell. Biol. 2006;26:7201–7210. doi: 10.1128/MCB.00459-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ge H., Si Y., Wolffe A P. A novel transcriptional coactivator, p52, functionally interacts with the essential splicing factor ASF/SF2. Mol. Cell. 1998;2:751–759. doi: 10.1016/s1097-2765(00)80290-7. [DOI] [PubMed] [Google Scholar]
- 28.Pradeepa M.M., Sutherland H.G., Ule J., Grimes G.R., Bickmore W.A. Psip1/Ledgf p52 Binds Methylated Histone H3K36 and Splicing Factors and Contributes to the Regulation of Alternative Splicing. PLoS Genet. 2012;8:e1002717. doi: 10.1371/journal.pgen.1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daugaard M., Baude A., Fugger K., Povlsen L.K., Beck H., Sørensen C.S., Petersen N.H.T., Sorensen P.H.B., Lukas C., Bartek J., et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat. Struct. Mol. Biol. 2012;19:803–810. doi: 10.1038/nsmb.2314. [DOI] [PubMed] [Google Scholar]
- 30.Shun M.-C., Raghavendra N.K., Vandegraaff N., Daigle J.E., Hughes S., Kellam P., Cherepanov P., Engelman A. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 2007;21:1767–1778. doi: 10.1101/gad.1565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marshall H.M., Ronen K., Berry C., Llano M., Sutherland H., Saenz D., Bickmore W., Poeschla E., Bushman F.D. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS One. 2007;2:e1340. doi: 10.1371/journal.pone.0001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yokoyama A., Cleary M.L. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell. 2008;14:36–46. doi: 10.1016/j.ccr.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maertens G.N., Cherepanov P., Engelman A. Transcriptional co-activator p75 binds and tethers the Myc-interacting protein JPO2 to chromatin. J. Cell Sci. 2006;119:2563–2571. doi: 10.1242/jcs.02995. [DOI] [PubMed] [Google Scholar]
- 34.Bartholomeeusen K., Christ F., Hendrix J., Rain J.-C., Emiliani S., Benarous R., Debyser Z., Gijsbers R., De Rijck J. Lens epithelium-derived growth factor/p75 interacts with the transposase-derived DDE domain of PogZ. J. Biol. Chem. 2009;284:11467–11477. doi: 10.1074/jbc.M807781200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cherepanov P., Maertens G., Proost P., Devreese B., Van Beeumen J., Engelborghs Y., De Clercq E., Debyser Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 2003;278:372–381. doi: 10.1074/jbc.M209278200. [DOI] [PubMed] [Google Scholar]
- 36.Williamson I., Eskeland R., Lettice L.a., Hill A.E., Boyle S., Grimes G.R., Hill R.E., Bickmore W.A. Anterior-posterior differences in HoxD chromatin topology in limb development. Development. 2012;139:3157–3167. doi: 10.1242/dev.081174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taylor G.C.A., Eskeland R., Hekimoglu-Balkan B., Pradeepa M., Bickmore W.A. H4K16 acetylation marks active genes and enhancers of embryonic stem cells, but does not alter chromatin compaction. Genome Res. 2013;23:2053–2065. doi: 10.1101/gr.155028.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clouaire T., Webb S., Skene P., Illingworth R., Kerr A., Andrews R., Lee J.-H., Skalnik D., Bird A. Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev. 2012;26:1714–1728. doi: 10.1101/gad.194209.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hendrix J., Gijsbers R., De Rijck J., Voet A., Hotta J., McNeely M., Hofkens J., Debyser Z., Engelborghs Y. The transcriptional co-activator LEDGF/p75 displays a dynamic scan-and-lock mechanism for chromatin tethering. Nucleic Acids Res. 2011;39:1310–1325. doi: 10.1093/nar/gkq933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ciuffi A., Llano M., Poeschla E., Hoffmann C., Leipzig J., Shinn P., Ecker J.R., Bushman F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005;11:1287–1289. doi: 10.1038/nm1329. [DOI] [PubMed] [Google Scholar]
- 41.Guenther M.G., Jenner R.G., Chevalier B., Nakamura T., Croce C.M., Canaani E., Young R.A. Global and Hox-specific roles for the MLL1 methyltransferase. Proc. Natl. Acad. Sci. U.S.A. 2005;102:8603–8608. doi: 10.1073/pnas.0503072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Nuland R., Smits A.H., Pallaki P., Jansen P.W.T.C., Vermeulen M., Timmers H.T.M. Quantitative dissection and stoichiometry determination of the human SET1/MLL histone methyltransferase complexes. Mol. Cell. Biol. 2013;33:2067–2077. doi: 10.1128/MCB.01742-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsin J.-P., Manley J.L. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 2012;26:2119–2137. doi: 10.1101/gad.200303.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stock J.K., Giadrossi S., Casanova M., Brookes E., Vidal M., Koseki H., Brockdorff N., Fisher A.G., Pombo A. Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat. Cell Biol. 2007;9:1428–1435. doi: 10.1038/ncb1663. [DOI] [PubMed] [Google Scholar]
- 45.Tavares L., Dimitrova E., Oxley D., Webster J., Poot R., Demmers J., Bezstarosti K., Taylor S., Ura H., Koide H., et al. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell. 2012;148:664–678. doi: 10.1016/j.cell.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao Z., Zhang J., Bonasio R., Strino F., Sawai A., Parisi F., Kluger Y., Reinberg D. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell. 2012;45:344–356. doi: 10.1016/j.molcel.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morey L., Aloia L., Cozzuto L., Benitah S.A., Di Croce L. RYBP and Cbx7 define specific biological functions of polycomb complexes in mouse embryonic stem cells. Cell Rep. 2013;3:60–69. doi: 10.1016/j.celrep.2012.11.026. [DOI] [PubMed] [Google Scholar]
- 48.Xia Z., Anderson M., Diaz M.O., Zeleznik-le N.J. MLL repression domain interacts with histone deacetylases, the polycomb group proteins HPC2 and BMI-1, and the corepressor C-terminal-binding protein. Proc. Natl. Acad. Sci. U.S.A. 2003;100:2–7. doi: 10.1073/pnas.1436338100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Nuland R., van Schaik F.M., Simonis M., van Heesch S., Cuppen E., Boelens R., Timmers H.M., van Ingen H. Nucleosomal DNA binding drives the recognition of H3K36-methylated nucleosomes by the PSIP1-PWWP domain. Epigenet. Chromatin. 2013;6:12. doi: 10.1186/1756-8935-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horan G.S., Wu K., Wolgemuth D.J., Behringer R.R. Homeotic transformation of cervical vertebrae in Hoxa-4 mutant mice. Proc. Natl. Acad. Sci. U.S.A. 1994;91:12644–12648. doi: 10.1073/pnas.91.26.12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jeannotte L., Lemieux M., Charron J., Poirier F., Robertson E.J. Specification of axial identity in the mouse: role of the Hoxa-5 (Hox1.3) gene. Genes Dev. 1993;7:2085–2096. doi: 10.1101/gad.7.11.2085. [DOI] [PubMed] [Google Scholar]
- 52.Kostic D., Capecchi M.R. Targeted disruptions of the murine Hoxa4 and Hoxa6 genes result in homeotic transformations of components of the vertebral column. Mech. Dev. 1994;46:231–247. doi: 10.1016/0925-4773(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 53.Cao R., Tsukada Y., Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol. Cell. 2005;20:845–854. doi: 10.1016/j.molcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 54.Van der Lugt N.M., Domen J., Linders K., van Roon M., Robanus-Maandag E., te Riele H., van der Valk M., Deschamps J., Sofroniew M., van Lohuizen M. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 1994;8:757–769. doi: 10.1101/gad.8.7.757. [DOI] [PubMed] [Google Scholar]
- 55.Palijan A., Fernandes I., Verway M., Kourelis M., Bastien Y., Tavera-Mendoza L.E., Sacheli A., Bourdeau V., Mader S., White J.H. Ligand-dependent corepressor LCoR is an attenuator of progesterone-regulated gene expression. J. Biol. Chem. 2009;284:30275–30287. doi: 10.1074/jbc.M109.051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Srinivasan L., Atchison M.L. YY1 DNA binding and PcG recruitment requires CtBP. Genes Dev. 2004;18:2596–2601. doi: 10.1101/gad.1228204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oswald F., Winkler M., Cao Y., Astrahantseff K., Bourteele S., Knochel W., Borggrefe T. RBP-J kappa / SHARP Recruits CtIP / CtBP Corepressors To Silence Notch Target Genes. Mol. Cell Biol. 2005;25:10379–10390. doi: 10.1128/MCB.25.23.10379-10390.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aymard F., Bugler B., Schmidt C.K., Guillou E., Caron P., Briois S., Iacovoni J.S., Daburon V., Miller K.M., Jackson S.P., et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014;21:366–374. doi: 10.1038/nsmb.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chinwalla V., Jane E.P., Harte P.J. The Drosophila trithorax protein binds to specific chromosomal sites and is co-localized with Polycomb at many sites. EMBO J. 1995;14:2056–2065. doi: 10.1002/j.1460-2075.1995.tb07197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reynolds N., O'Shaughnessy A., Hendrich B. Transcriptional repressors: multifaceted regulators of gene expression. Development. 2013;140:505–512. doi: 10.1242/dev.083105. [DOI] [PubMed] [Google Scholar]
- 61.Ferrando A.A., Armstrong S.A., Neuberg D.S., Sallan S.E., Silverman L.B., Korsmeyer S.J., Look A.T. Gene expression signatures in MLL-rearranged T-lineage and B-precursor acute leukemias: dominance of HOX dysregulation. Blood. 2003;102:262–268. doi: 10.1182/blood-2002-10-3221. [DOI] [PubMed] [Google Scholar]
- 62.Slany R.K. The molecular biology of mixed lineage leukemia. Haematologica. 2009;94:984–993. doi: 10.3324/haematol.2008.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grand F.H., Koduru P., Cross N.C.P., Allen S.L. NUP98-LEDGF fusion and t(9;11) in transformed chronic myeloid leukemia. Leuk. Res. 2005;29:1469–1472. doi: 10.1016/j.leukres.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 64.Hussey D.J., Moore S., Nicola M., Dobrovic A. Fusion of the NUP98 gene with the LEDGF/p52 gene defines a recurrent acute myeloid leukemia translocation. BMC Genet. 2001;2:20. doi: 10.1186/1471-2156-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamamoto K., Nakamachi Y., Yakushijin K., Funakoshi Y., Okamura A., Kawano S., Matsuoka H., Minami H. Expression of the novel NUP98/PSIP1 fusion transcripts in myelodysplastic syndrome with t(9;11)(p22;p15) Eur. J. Haematol. 2012;88:244–248. doi: 10.1111/j.1600-0609.2011.01736.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.