


Abstract
Biomolecular interactions have important cellular implications, however, a simple method for the sensing of such proximal events is lacking in the current molecular toolbox. We designed a dynamic DNA circuit capable of recognizing targets in close proximity to initiate a pre-programmed signal transduction process resulting in localized signal amplification. The entire circuit was engineered to be self-contained, i.e. it can self-assemble onto individual target molecules autonomously and form localized signal with minimal cross-talk. α-thrombin was used as a model protein to evaluate the performance of the individual modules and the overall circuit for proximity interaction under physiologically relevant buffer condition. The circuit achieved good selectivity in presence of non-specific protein and interfering serum matrix and successfully detected for physiologically relevant α-thrombin concentration (50 nM–5 μM) in a single mixing step without any further washing. The formation of localized signal at the interaction site can be enhanced kinetically through the control of temperature and probe concentration. This work provides a basic general framework from which other circuit modules can be adapted for the sensing of other biomolecular or cellular interaction of interest.
INTRODUCTION
Interactions between biomolecules, such as nucleic acid and proteins, are important for understanding cellular processes and have functional implications in, for example, disease progression and inflammatory responses. While a myriad of tools are available for the end-point detection of various biomolecules (1–3), there is limited toolbox available to probe for their interaction activities. Conventional assays, including immunostaining, pull-down assay and enzyme-linked immunosorbent assay, capture only the relative bulk expression levels under static condition and fail to track the interaction in real time. Another commonly used technique of fluorescence resonance energy transfer allows for the dynamic monitoring of protein interaction for interaction distance shorter than 10 nm (4). Many important cellular proximal interactions take place at distances greater than 10 nm, such as the interaction between growth factors and receptors (5,6) and for intracellular trafficking (7). Recently developed technique of proximity ligation assay allows for long-range interrogation but suffers from the disadvantages of having numerous experimental steps and the need for enzymatic manipulation (8).
This motivates the design of a simple molecular tool that can sense for biomolecular interactions without the need for complicated enzymatic manipulation or tedious washing steps. Recent advances in dynamic DNA nanotechnology offer an interesting design tool that allows for the modular-based programming of specific stimulus-triggered response, enabling the assembly of functional DNA nanomachines in a ‘bottom-up’ approach (9,10). One key application of the DNA circuit is in molecular detection where it recognizes specific target inputs and translates the recognition events into a series of DNA strand displacement processes (11) for the subsequent development of readout signals, which is often amplified by catalytic DNA designs (12,13).
Leveraging upon the basic circuit design concepts available for end-point biomolecule detection, we expanded the scope of the usual detection schemes to pick up signal from biomolecules in close proximity. Similar concept of binding-induced DNA assembly was recently reported by Le's group where the co-binding of two reporter molecules on a single protein molecule increased the local probe concentration and drastically enhanced the kinetic rate of strand exchange (14,15). While their proposed format works well for homogeneous assay (16,17), the fluorescence signal developed is freely moving in the bulk solution, and render it difficult to extend the design to pinpoint and visualize the site of interaction. Therefore, we aim to engineer a self-contained DNA circuit on the binding site itself where the localized readout signal is developed and amplified. The concept of ‘self-contained’ refers to the physical confinement of the DNA circuit onto the binding site where all circuit operations take place. We believe that this can broaden the utility of existing homogeneous assays beyond that of end-point detection and provide an additional dimension of spatial information to facilitate upstream biology studies, for example, pathogen–host interaction, stem cell homing and cancer metastasis (18).
The general idea here is to design a DNA circuit that is dynamically triggered by biomolecules at close proximity to self-assemble into localized signal indicative of the interaction event on individual target molecules (Figure 1). α-Thrombin was selected as a model protein in this proof-of-concept study as it has two well-characterized aptamer sequences known to bind to the fibrinogen- and heparin-recognition exosites located at opposite ends of the molecule (19). Aptamer was chosen as the functional moiety for target recognition as its structure-switching capability between complex secondary structure and duplex form facilitates target-responsive triggering of the circuit. We mimicked a bi-molecular recognition event by designing two initiator strands capable of binding to the two exosites. Both initiators contained a ‘key’ region designed to be locked up at equilibrium so that no signal developed under static condition. Every step in the dynamic assembly process required physical contact between the target-bound DNA components to proceed in a self-contained format, thereby reducing signal cross-talk by freely moving DNA strands and facilitated the development of localized signal.
Figure 1.
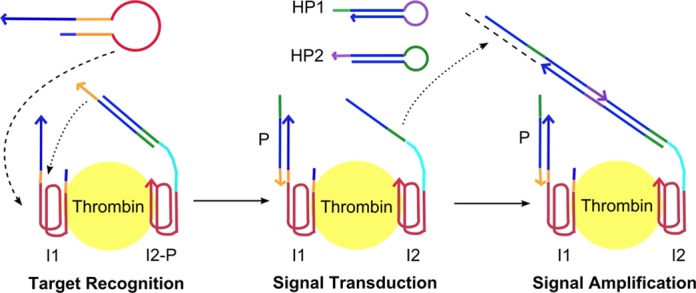
Schematic outline of the dynamic assembly process of the proposed self-contained DNA circuit for proximity-based biomolecule sensing. α-Thrombin was used as a model protein in this study. The binding of initiator 1 (I1) to α-thrombin opened the aptamer molecular switch and exposed the ‘key’ (orange) which unlocked the protector (P) strand to trigger signal transduction. During the signal transduction process, the initiator for HCR (green) was exposed which triggered the cascaded opening of two kinetically locked hairpins (HP1 and HP2) in a signal amplification process. All DNA components were designed to be bound onto the α-thrombin molecule during their participation in the self-assembly process.
MATERIALS AND METHODS
Materials
All DNA oligonucleotides used in this study were purchased from Integrated DNA (IDT), and high pressure liquid chromatography purified by IDT. The sequences are provided in Supplementary Table S1. The lyophilized DNA was reconstituted in 1× Tris-ethylenediaminetetraacetic acid (EDTA) buffer (1× TE, pH 8.0) to give 100 μM stock and stored at 4°C. The following chemicals were used as received: Human α−thrombin (8.9 mg/ml in 50% (v/v) glycerol/H2O) was purchased from Haematologic Technologies Inc. Sodium chloride (NaCl, ≥99.5%) and magnesium chloride (MgCl2, ≥98%) were purchased from Sigma Aldrich. Potassium chloride (KCl, ≥99.5%) was purchased from Applichem. SYBR gold nucleic acid stain (10 000× in dimethyl sulfoxide (DMSO)) was purchased from Invitrogen. Agarose (molecular biology grade) and 10× Tris-borate-EDTA (TBE, pH 8.3) were purchased from Vivantis. 1× TE (pH 8.0) was purchased from 1st BASE. Phosphate buffer (pH 7.4) was prepared using sodium phosphate dibasic (Na2HPO, anhydrous enzyme grade) from Fischer Scientific and sodium dihydrogen phosphate dihydrate (H6NaO6P) from Kanto Chemical Co. Inc. Mili-Q water with resistance >18.2 MΩ/cm was used throughout the experiment.
DNA design, reaction and analysis
All DNA sequences were designed using Nupack web server (20) and are listed in Supplementary Table S2. Stock DNA (100 μM in 1× TE, pH 8.0) was diluted to 5 μM working concentration in DNA binding buffer (10 mM phosphate buffer (pH 7.4), 175 mM NaCl, 1.25 mM MgCl2). The individual DNA components (H1, H2 and I1) and complexes (I2 + protector) were heated to 95°C for 2 min and allowed to cool to room temperature for 1 h. α-Thrombin was diluted to 5 μM working concentration in α-thrombin binding buffer (10 mM pH 7.4 phosphate buffer, 50 mM KCl). Equal volume of each component was mixed to obtain a final reaction concentration of 1 μM, unless otherwise stated. For control step-ups in which certain DNA components or α-thrombin was not required, the equivalent volume of DNA or α-thrombin binding buffer was added instead. The final reaction condition was kept constant as 10 mM phosphate buffer (pH 7.4), 140 mM NaCl, 10 mM KCl and 1.0 mM MgCl2. The reaction temperature was maintained at 37°C using a thermal cycler, unless otherwise stated.
Analysis was carried out on 3% agarose gel which was pre-stained with 1× SYBR gold nucleic acid stain. The gel was run at 90 V for 50 min in 0.5× TBE running buffer at 4°C. The signal intensity of the hybridization chain reaction (HCR) products relative to the total amount of hairpins inputted was quantified using ImageJ (21).
Kinetics study of individual modules
All DNA components and α-thrombin were prepared and reacted under the same conditions stated above. The trigger for each module, that is, c* b* for (i), t* c* for (ii) and α-thrombin for (iii) was added at specific time points of 0 min, 15 min, 30 min, 40 min and 50 min to separate reaction tubes such that the respective tubes underwent reaction times of 60 min, 45 min, 30 min, 20 min and 10 min at the end of the 1 h kinetics study. The reaction was then immediately quenched on ice and loaded onto the 3% agarose gel for analysis.
Data analysis
Relative signal intensity, denoted as rSI, indicates the percentage of hairpin strands (HP1 and HP2) that polymerized to give the HCR products. This was determined by quantifying the intensity of the HCR products and unpolymerized hairpin bands using ImageJ. The first value (representing positive readout signal) was then divided by the sum of the two values (representing the total amount of hairpin strands inputted) to obtain an rSI parameter. Signal-to-noise ratio, denoted as S/N, was calculated by dividing the rSI obtained in presence of α-thrombin (positive signal) to that in absence of α-thrombin (background noise). This parameter was used to select the optimal molecular switch design.
RESULTS AND DISCUSSION
Overall DNA circuit domain design
The main DNA components involved in the circuit presented in Figure 1 are shown in Figure 2. The key (domain ‘t*’) of initiator 1 (I1) was initially locked up in the stem of a hairpin molecular switch and opened in response to the specific aptamer recognition event of α-thrombin binding by domain ‘a1’. The key (domain ‘b*’) of initiator 2 (I2), originally locked up by a partially complementary protector (P) strand, would then be unlocked by the released key of I1. I2 was bound to thrombin via domain ‘a2’. Through the dynamic signal transduction process, the second key was exposed to trigger the HCR process. First described by Pierce's group, HCR involves the cascaded opening of two partially complementary hairpins which are originally trapped kinetically in their stem-loop structure (22). The overall DNA circuit design proposed is capable of capturing proximity interaction and translating the event into a localized signal comprising of DNA oligomers (HCR products) at the binding site. The domain sequences are provided in Supplementary Table S2.
Figure 2.
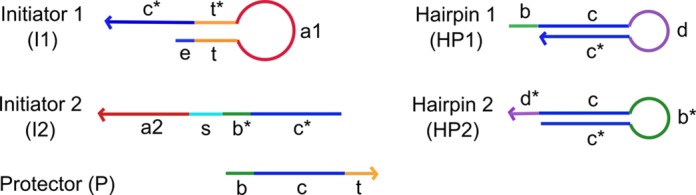
The key DNA components involved in the DNA circuit are shown in terms of their domain specifications. Initiator 1 (I1) and initiator 2 (I2) were bound to α-thrombin via domain ‘a1’ and ‘a2’ Domain ‘t*’ and ‘b*’ are ‘keys’ initiating the signal transduction and HCR process, respectively. Domain ‘c’ and ‘c*’ represents the HCR hairpin stem; while domain ‘b’ and ‘d’ are alternating toeholds mediating the HCR process. A spacer (‘s’) domain was included to signify the possibility of probing over separation distances up to ca. 30 nm by varying its sequence length.
Design of target binding-induced molecular switch
As the two triggering processes, i.e. α-thrombin binding-induced hairpin opening and initiation of HCR, were independent of each other, we could design the initiator sequences individually. The original HCR hairpin sequences reported by Pierce's group were used, leaving the molecular switch sequence for I1 as the only DNA component to be optimized. This highlights the potential for the DNA circuit designed in this work to be a general framework for sensing other biomolecular interactions with known target recognition aptamer sequence without the need to re-design the entire circuit sequences.
Though several hairpin molecular switch designs for α-thrombin binding aptamers were previously reported (23–25), the presence of a toehold sequence could compete to open up the hairpins spuriously. For instances, domain t in P can bind to domain t* in I1 when the end sequence ‘breathes’ (10). Thus, we first optimized the molecular switch design based on two criteria: (i) being able to lock up the ‘key’ in I1 sufficiently against hybridization pressure from the complementary toehold sequence to minimize background leakage while (ii) being able to undergo conformation change and open up in the presence of α-thrombin. As thrombin-binding aptamers are known to form G-quadruplex, a drastic conformation change from the duplex structure held in a hairpin loop was expected to reveal the sequestered toehold domain.
Five molecular switch designs were considered, each varying in terms of the toehold length, clamping sequence length and position of the aptamer sequence. Figure 3a shows the only design that remained locked in absence of α-thrombin yet triggered HCR in presence of α-thrombin is Design iii (lanes 12 and 13). The crucial element of this design lies in the use of a short clamping sequence (2 nt) which could have prevented the transient exposure of the toehold sequence when the end sequence ‘breathed’ as observed for Design i (lanes 6 and 7) and Design ii (lanes 10 and 11). The use of a longer clamping sequence and hence more stable hairpin stem hindered the hairpin opening process as seen from the lack of positive signal in presence of α-thrombin for Design iv (lanes 14 and 15). We also considered placing the aptamer sequence at the end of the hairpin stem in Design v (lanes 16 and 17) to avoid the kinetically slow process of hairpin opening but found it an ineffective strategy. We attributed that to the lack of drastic conformation change to the five nucleotides locked in the stem, resulting in its inability to expose the toehold sequence upon α-thrombin binding. Thus, we selected Design iii for the rest of this study which gave a reasonable signal-to-noise ratio of ca. 3.5 (Figure 3b).
Figure 3.
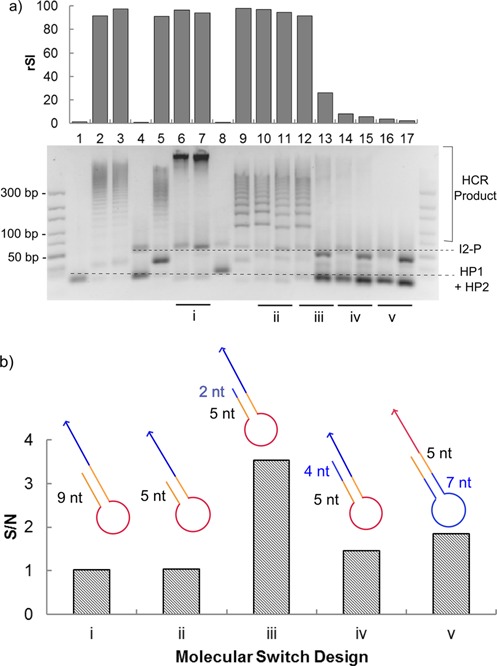
(a) Gel electrophoresis image of the performance of the five molecular switch candidates (denoted as ‘i’ to ‘v’). For each of the two lanes compared, the left lane, i.e. lanes 6, 10, 12, 14 and 16, correspond to the presence of α-thrombin, while the right lane, i.e. lanes 7, 11, 13, 15 and 17, correspond to the absence of α-thrombin. Lane 1 corresponds to hairpins (HP1 and HP2) only, lanes 2 and 3 correspond to the case where initiator 1 was replaced by a strand designed to be fully complementary to the protector strand in presence and absence of α-thrombin, respectively. Lanes 4 and 5 correspond to the negative and positive control for toehold length of 9 nt while lanes 8 and 9 correspond to that for toehold length of 5 nt. A 10–300 bp DNA ladder is shown at both sides of the gel. The relative signal intensity (rSI) of the HCR products formed is indicated above the respective lanes. The identity of each band was indicated with dotted lines. (b) The signal-to-noise ratio (S/N) of the HCR products formed in presence to that in absence of α-thrombin was calculated. The schematic design for each molecular switch is presented above the respective columns and is color-coded as: red—aptamer sequence; yellow—toehold sequence; blue—stem of HCR hairpins or clamping sequence. Design iii gave the best signal-to-noise ratio of ca. 3.5.
Evaluating kinetics of individual circuit modules
The circuit was further re-constructed stepwise according to the three key modules: (i) signal amplification—HCR, (ii) signal transduction and (iii) α-thrombin recognition to evaluate the kinetics of individual modules separately. The basic DNA components involved in each module and the required add-ons when progressing from one module to the next during the re-construction process are shown in Figure 4a. For (i) HCR, c* b* strand sufficed in triggering the cascaded opening of hairpin 1 (HP1) and hairpin 2 (HP2). To progress to (ii) signal transduction, a protector (P) strand was added to c* b* strand to block HCR which was subsequently triggered only in presence of an additional t* c* strand. To ensure that the signal developed only in the presence of (iii) specific target recognition, two aptamer sequences (a1 and a2) were added to the t* c* and c* b* strands to obtain the complete initiator 1 (I1) and initiator 2 (I2) probes. In particular, I1 was locked up in the molecular switch design obtained in ‘Design of target binding-induced molecular switch’ section. The binding of α-thrombin induced the opening of I1 to initiate signal transduction followed by HCR in the final re-constructed circuit.
Figure 4.
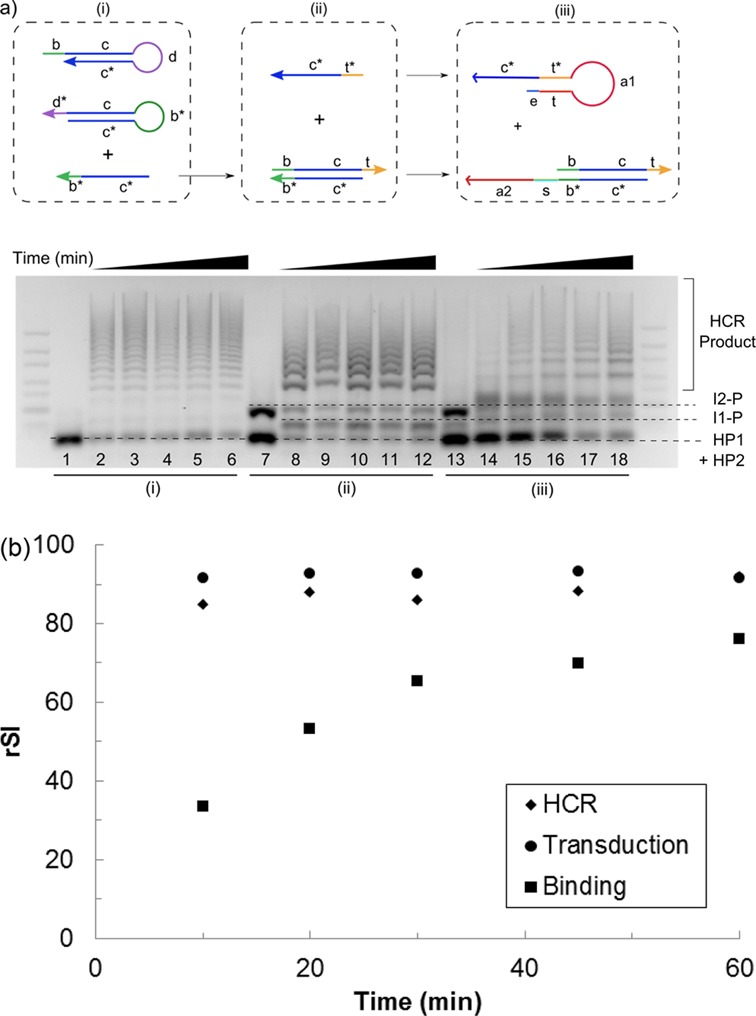
(a) (Top) Kinetics of the individual module was characterized by decomposing each module into the basic DNA components required for the process: (i) a trigger strand (c* b*) to initiate the HCR, (ii) addition of a protector (b c t) and initiator strand (t* c*) to control signal transduction and (iii) the incorporation of the aptamer sequences (a1 and a2) into the two respective initiator strands to achieve α-thrombin binding-induced signal development. (Bottom) Gel electrophoresis image of the kinetics of the three individual modules as labeled below the gel. The reaction was allowed to proceed for various time periods of 10, 20, 30, 45 and 60 min (from left to right of each module) at 37°C. Lanes 1, 7 and 13 correspond to the negative control for each module where the trigger was not added, i.e. c* b* strand for HCR, t* c* strand for signal transduction and α-thrombin for target binding. The identity of each band was indicated with dotted lines. (b) α-thrombin binding, and hence the molecular switch opening step, was found to be still rate limiting although the time required to reach equilibrium was shortened to ca. 30 min at 37°C. rSI, relative signal intensity.
The kinetics of the individual components was first evaluated at room temperature and the molecular switch opening was found to be rate limiting (Supplementary Figure S1). Subsequently, a higher incubation temperature of 37°C was used to speed up the kinetically hindered hairpin opening process (Supplementary Figure S2). Though the reaction rate was still limited by the molecular switch opening step, the time required to reach equilibrium state was greatly reduced from ca. 7 h (at room temperature) to the current 30 min (Figure 4b). It is worth noting that the buffer condition chosen to carry out the reaction was a physiologically relevant condition of 140 mM NaCl, 1 mM MgCl2 and 10 mM KCl in 10 mM phosphate buffer (pH 7.5) for ensuring compatibility of the DNA circuit with other biomolecules or cell systems. In spite of using a considerably lower salt condition than those typically used for pure DNA circuitry studies (26) which could have disadvantaged our reaction kinetics and thermodynamics, the relatively short analysis time of 30 min and ca. 70% of HCR signal developed at equilibrium was encouraging. Nonetheless, we should point out that the time scale involved might not be sufficiently short for monitoring fast kinetic events, such as enzymatic conversion, by our DNA circuit in its current form.
Evaluating overall circuit performance
After characterizing the individual DNA circuit components, the overall circuit performance in terms of detection limit and selectivity was evaluated (Figure 5a). In this proof-of-concept work, gel electrophoresis was used as the main readout tool to visualize the formation of HCR products. The one-pot mixture of our DNA circuit and α-thrombin was loaded directly into agarose gel without further manipulation steps. The lack of distinct HCR products in presence of non-specific protein (1 μM bovine serum albumin (BSA), lane 6) and interference matrix (1% fetal bovine serum (FBS), lane 7, and 10% FBS, lane 8) demonstrated the good selectivity of the DNA circuit. It should be noted that the selectivity is closely linked to the intrinsic performance of the aptamer sequence and may exhibit variable performance for different targets. HCR products could form in the presence of a mixture of α-thrombin with the same set of interferences mentioned above (lanes 9–11), which demonstrated the robustness of the DNA circuit.
Figure 5.
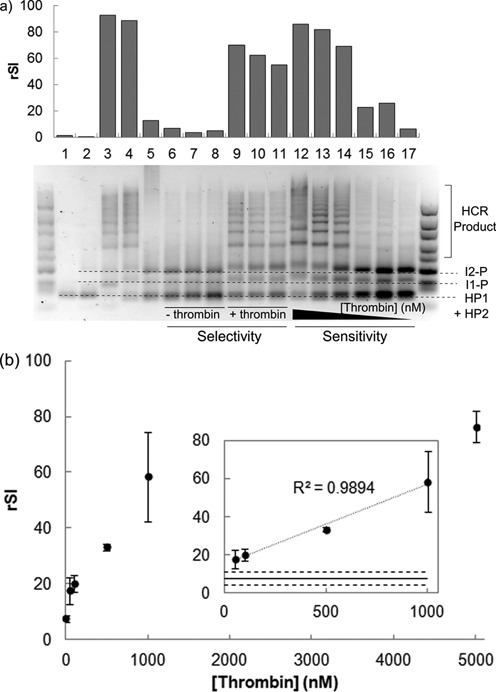
(a) Gel electrophoresis image of the performance of proximity-based sensing circuit evaluated at 37°C. Lanes 6–11 represents the assay selectivity in presence of interference from 1 μM BSA (lanes 6 and 9), 1% FBS (lanes 7 and 10) and 10% FBS (lanes 8 and 11). HCR was not triggered by non-specific protein and interfering matrix (lanes 6–8) and the signal in presence of α-thrombin could develop even with interferences (lanes 9–11). Lanes 12–17 represent the signal developed in presence of 5 μM, 1 μM, 0.5 μM, 0.1 μM, 50 nM and 0 nM of α-thrombin. Lane 1 corresponds to hairpins (HP1 and HP2) only; lane 2 corresponds to hairpins and initiator 2–protector (I2-P) complex; lane 3 corresponds to hairpins, I2-P and t* c* trigger strand; lanes 4 and 5 represent equilibrium signal in presence and absence of 1 μM α-thrombin. A 10–300 bp DNA ladder is shown at both sides of the gel. The relative signal intensity (rSI) of the HCR products formed is indicated above the respective lanes. The identity of each band was indicated with dotted lines. (b) The HCR signal developed exhibited dosage-dependence for a physiologically relevant α-thrombin concentration ranging from 50 nM to 5 μM. Inset: A linear dosage-signal relationship (R2 = 0.99) was obtained for α-thrombin concentration of 50 nM to 1 μM. The solid line represents the mean background noise in absence of α-thrombin while the dashed lines represent ±3 σ of this mean. The upper bound represents the limit of detection of the assay which was found to be 50 nM in this study. All data are shown as mean ± standard deviation (n = 3).
By varying the concentration of α-thrombin added within a physiologically relevant range of 50 nM–5 μM (lanes 12–16) (27), it is evident that the extent of HCR was dosage-dependent. Semi-quantitative analysis was carried out by measuring the relative signal intensity of the HCR products in each gel lane. From Figure 5b, a higher signal intensity was developed with increasing α-thrombin concentration which started to plateau off at 1 μM and above when the initiator strands were exhausted ([I1] = [I2] = 1 μM).
A linear dosage–signal relationship was obtained for α-thrombin concentration of 50 nM–1 μM, suggesting the possibility of using the proximity-based DNA circuit for quantification of biomolecular interaction (Figure 5b). The limit of detection for this assay was found to be 50 nM without further effort to optimize the current performance We believe that a lower limit of detection is achievable by using fluorescence signal as the readout in future works which should also facilitate the in situ visualization of the interaction activities.
Feasibility of enhancing the development of localized signal through dual binding events
Since the ultimate aim of the proposed DNA circuit is for the sensing of biomolecular interaction, it is imperative that localized signal is formed from multiple bio-recognition events. However, we noted the possibility for unbound I2-P complex to undergo signal transduction with bound I1 strand to form freely moving HCR products. From the intention of probing for interaction activities on top of just end-point detection, this possibility poses two challenges to the DNA circuit designed—occurrence of false-positives and development of non-localized signal. However, the co-binding of two initiator strands on a single α-thrombin molecule should increase their local concentrations for a faster rate of signal transduction as compared to freely moving unbound I2. To verify this, we performed a control experiment in which I2 strand was modified to exclude the aptamer sequence so that it could not bind to α-thrombin. We demonstrated that the undesired triggering by unbound I2 can be kinetically subdued by using a lower I2 concentration (Supplementary Figure S3) and shorter analysis time (Figure 6a). Referring to Figure 6a, for analysis time shorter than 30 min, HCR products started forming for bound I2 (first 2 lanes for each time point)in a self-contained format while the signal for unbound I2 (3rd lane for each time point) remained relatively weak. The difference in the rate of signal development for bound and unbound I2 (noApt) was found to be statistically significant using two-factor ANOVA test for α = 0.05 (Figure 6b).
Figure 6.
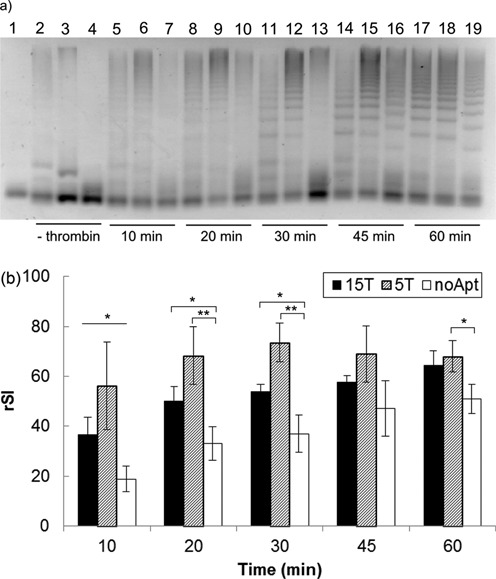
Dual binding events of two initiators on the α-thrombin molecule facilitated the development of localized signal due to the increase in the local DNA concentration. (a) Gel electrophoresis image of the HCR products developed at the indicated time points using three initiator 2 (I2) designs, i.e. (from left to right) 15 nt spacer length (15T), 5 nt spacer length (5T) and absence of aptamer 2 and spacer (‘s’) domain (noApt). Lane 1 corresponds to hairpins (HP1 and HP2) only as reference. Lanes 2–4 correspond to negative controls for the respective I2 designs after 1 h reaction time. Signal developed more rapidly when I2 was bound to α-thrombin, with the effect being most pronounced for shorter analysis time (<30 min). (b) The readout signal increased with time for all three designs. In general, the rate of formation of the localized signal was enhanced kinetically when the two initiators were brought to closer proximity on the binding site through the use of a shorter spacer distance (5T versus 15T). The effect of analysis time and the use of bound/unbound I2 was found to be statistically significant using two-factor ANOVA test (α = 0.05, n = 3). Pair-wise comparisons were made between the unbound (noApt) and bound (15T or 5T) cases at each time points using one-tailed Student's t-test (*P < 0.1, **P < 0.05.). All data are shown as mean ± standard error (n = 3). rSI, relative signal intensity.
Encouraged by this finding, we further differentiated the effect of localization of the bound initiators by using two spacer lengths on I2—15 thymine (15T) and 5 thymine (5T). Figure 6a and b show that the signal developed with the 5T spacer was more pronounced than that with the 15T spacer. It could be that for bound I2, having a longer spacer length resulted in a more flexible structure leading to a slightly longer time for I1 and I2 to come into contact. This suggests that the shortest possible spacer length should be used to enhance the selectivity of localized over non-localized circuit assembly, so long as it is still possible for the I1 and I2 to physically approach each other for the particular separation distance. The maximum separation distance that the circuit is theoretically capable of probing for is ca. 30 nm (or 100 nt) by varying the spacer length.
We envision that our DNA circuit design can be applied for cell-surfacing sensing. The idea of cell typing via DNA computation of cell-surface receptors was conceptualized by Stojanovic's (28) and Tan's (29) groups separately, where a one-target-to-one-signal design was adopted. Together with our proposed localized signal amplification design reported here, the characterization of low abundance cell markers that can serve as disease biomarkers either as a single target, such as CCR5, a co-receptor in HIV-1 infection (30), or by profiling multiple cell-surface markers, such as epithelial cell-adhesion molecule and cytokeratin on circulating tumor cells (31), shall be demonstrated in the near future. We believe that the additional amplification scheme featured in this work can enhance the signal generated to facilitate the direct spatial visualization of the interrogated site under microscopy, thereby expanding the scope of cell-surface sensing.
CONCLUSION
In conclusion, a self-contained DNA circuit capable of sensing proximity-based interaction activity was designed and demonstrated using α-thrombin as a model protein. The circuit performed with good selectivity and reasonable limit of detection (50 nM) which can be further improved by using fluorescence signal for more precise target quantification. More importantly, we demonstrated the concept of a self-contained DNA circuit where the DNA components were engineered to assemble autonomously onto individual targets and enhance the formation of amplified, localized signal. Overall, the designed DNA circuit is versatile and can be used for either end-point detection or for spatial visualization of the interaction site to facilitate upstream biological studies. General design strategies, including target-responsive molecular switching, the use of a spacer domain to modulate the circuitry kinetics and the key reaction conditions controlling various states of the DNA circuit, were proposed and validated. This work provides a basic framework from which other circuit modules can be adapted to sense for other specific biomolecular interaction of interest, e.g. tri-molecular interaction and cell-surface sensing.
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online.
Acknowledgments
Y.S.A. would like to thank the National University of Singapore and Ministry of Education for the President Graduate Fellowship.
FUNDING
Singapore Millennium Foundation and the National Medical Research Council [NMRC/CIRG/1358/2013]. Source of open access funding: Singapore Millennium Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1.Nam J.-M., Stoeva S.I., Mirkin C.A. Bio-bar-code-based DNA detection with PCR-like sensitivity. J. Am. Chem. Soc. 2004;126:5932–5933. doi: 10.1021/ja049384+. [DOI] [PubMed] [Google Scholar]
- 2.Nam J.-M., Thaxton C.S., Mirkin C.A. Nanoparticle-based bio-bar codes for the ultrasensitive detection of proteins. Science. 2003;301:1884–1886. doi: 10.1126/science.1088755. [DOI] [PubMed] [Google Scholar]
- 3.Obernosterer G., Martinez J., Alenius M. Locked nucleic acid-based in situ detection of microRNAs in mouse tissue sections. Nat. Protoc. 2007;2:1508–1514. doi: 10.1038/nprot.2007.153. [DOI] [PubMed] [Google Scholar]
- 4.Piston D.W., Kremers G.-J. Fluorescent protein FRET: the good, the bad and the ugly. Trends Biochem. Sci. 2007;32:407–414. doi: 10.1016/j.tibs.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Chen T.T., Luque A., Lee S., Anderson S.M., Segura T., Iruela-Arispe M.L. Anchorage of VEGF to the extracellular matrix conveys differential signaling responses to endothelial cells. J. Cell Biol. 2010;188:595–609. doi: 10.1083/jcb.200906044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winkler E., Bell R., Zlokovic B. Pericyte-specific expression of PDGF beta receptor in mouse models with normal and deficient PDGF beta receptor signaling. Mol. Neurodegener. 2010;5:1–11. doi: 10.1186/1750-1326-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakano A., Luini A. Passage through the Golgi. Curr. Opin. Cell Biol. 2010;22:471–478. doi: 10.1016/j.ceb.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Soderberg O., Gullberg M., Jarvius M., Ridderstrale K., Leuchowius K.-J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L.-G., et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 9.Zhang D.Y., Seelig G. Dynamic DNA nanotechnology using strand-displacement reactions. Nat. Chem. 2011;3:103–113. doi: 10.1038/nchem.957. [DOI] [PubMed] [Google Scholar]
- 10.Li B., Ellington A.D., Chen X. Rational, modular adaptation of enzyme-free DNA circuits to multiple detection methods. Nucleic Acids Res. 2011;39:e110. doi: 10.1093/nar/gkr504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Picuri J.M., Frezza B.M., Ghadiri M.R. Universal translators for nucleic acid diagnosis. J. Am. Chem. Soc. 2009;131:9368–9377. doi: 10.1021/ja902490x. [DOI] [PubMed] [Google Scholar]
- 12.Zhang D.Y., Turberfield A.J., Yurke B., Winfree E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 2007;318:1121–1125. doi: 10.1126/science.1148532. [DOI] [PubMed] [Google Scholar]
- 13.Jiang Y., Li B., Milligan J.N., Bhadra S., Ellington A.D. Real-time detection of isothermal amplification reactions with thermostable catalytic hairpin assembly. J. Am. Chem. Soc. 2013;135:7430–7433. doi: 10.1021/ja4023978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H., Li X.-F., Le X.C. Binding-induced DNA assembly and its application to yoctomole detection of proteins. Anal. Chem. 2011;84:877–884. doi: 10.1021/ac203207g. [DOI] [PubMed] [Google Scholar]
- 15.Li F., Zhang H., Wang Z., Li X., Li X.-F., Le X.C. Dynamic DNA assemblies mediated by binding-induced DNA strand displacement. J. Am. Chem. Soc. 2013;135:2443–2446. doi: 10.1021/ja311990w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H., Li F., Dever B., Wang C., Li X.-F., Le X.C. DNA-Assemblierung mittels Affinitätsbindung für die ultraempfindliche Proteindetektion. Angew. Chem. 2013;125:10894–10902. [Google Scholar]
- 17.Zhang H., Li F., Dever B., Wang C., Li X.-F., Le X.C. Assembling DNA through affinity binding to achieve ultrasensitive protein detection. Angew. Chem. Int. Ed. 2013;52:10698–10705. doi: 10.1002/anie.201210022. [DOI] [PubMed] [Google Scholar]
- 18.Zhao W., Schafer S., Choi J., Yamanaka Y.J., Lombardi M.L., Bose S., Carlson A.L., Phillips J.A., Teo W., Droujinine I.A., et al. Cell-surface sensors for real-time probing of cellular environments. Nat. Nanotechnol. 2011;6:524–531. doi: 10.1038/nnano.2011.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rinker S., Ke Y., Liu Y., Chhabra R., Yan H. Self-assembled DNA nanostructures for distance-dependent multivalent ligand-protein binding. Nat. Nanotechnol. 2008;3:418–422. doi: 10.1038/nnano.2008.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zadeh J.N., Steenberg C.D., Bois J.S., Wolfe B.R., Pierce M.B., Khan A.R., Dirks R.M., Pierce N.A. NUPACK: analysis and design of nucleic acid systems. J. Comput. Chem. 2011;32:170–173. doi: 10.1002/jcc.21596. [DOI] [PubMed] [Google Scholar]
- 21.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dirks R.M., Pierce N.A. Triggered amplification by hybridization chain reaction. Proc. Natl. Acad. Sci. U.S.A. 2004;101:15275–15278. doi: 10.1073/pnas.0407024101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J.J., Fang X., Tan W. Molecular aptamer beacons for real-time protein recognition. Biochem. Biophys. Res. Commun. 2002;292:31–40. doi: 10.1006/bbrc.2002.6581. [DOI] [PubMed] [Google Scholar]
- 24.Bang G.S., Cho S., Kim B.-G. A novel electrochemical detection method for aptamer biosensors. Biosens. Bioelectron. 2005;21:863–870. doi: 10.1016/j.bios.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 25.Xue L., Zhou X., Xing D. Sensitive and homogeneous protein detection based on target-triggered aptamer hairpin switch and nicking enzyme assisted fluorescence signal amplification. Anal. Chem. 2012;84:3507–3513. doi: 10.1021/ac2026783. [DOI] [PubMed] [Google Scholar]
- 26.Zhang D.Y., Hariadi R.F., Choi H.M.T., Winfree E. Integrating DNA strand-displacement circuitry with DNA tile self-assembly. Nat. Commun. 2013;4:1965. doi: 10.1038/ncomms2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee M., Walt D.R. A fiber-optic microarray biosensor using aptamers as receptors. Anal. Biochem. 2000;282:142–146. doi: 10.1006/abio.2000.4595. [DOI] [PubMed] [Google Scholar]
- 28.Rudchenko M., Taylor S., Pallavi P., Dechkovskaia A., Khan S., Butler V.P., Jr, Rudchenko S., Stojanovic M.N. Autonomous molecular cascades for evaluation of cell surfaces. Nat. Nanotechnol. 2013;8:580–586. doi: 10.1038/nnano.2013.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.You M., Peng L., Shao N., Zhang L., Qiu L., Cui C., Tan W. DNA “nano-claw”: logic-based autonomous cancer targeting and therapy. J. Am. Chem. Soc. 2013;136:1256–1259. doi: 10.1021/ja4114903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simon V., Ho D.D. HIV-1 dynamics in vivo: implications for therapy. Nat. Rev. Micro. 2003;1:181–190. doi: 10.1038/nrmicro772. [DOI] [PubMed] [Google Scholar]
- 31.Deng G., Herrler M., Burgess D., Manna E., Krag D., Burke J. Enrichment with anti-cytokeratin alone or combined with anti-EpCAM antibodies significantly increases the sensitivity for circulating tumor cell detection in metastatic breast cancer patients. Breast Cancer Res. 2008;10:R69. doi: 10.1186/bcr2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.