

Abstract
Phosphatidic acid (PA) is a critical metabolite at the heart of membrane phospholipid biosynthesis. However, PA also serves as a critical lipid second messenger that regulates several proteins implicated in the control of cell cycle progression and cell growth. Three major metabolic pathways generate PA: phospholipase D (PLD), diacylglycerol kinase (DGK), and lysophosphatidic acid acyltransferase (LPAAT). The LPAAT pathway is integral to de novo membrane phospholipid biosynthesis, whereas the PLD and DGK pathways are activated in response to growth factors and stress. The PLD pathway is also responsive to nutrients. A key target for the lipid second messenger function of PA is mTOR, the mammalian/mechanistic target of rapamycin, which integrates both nutrient and growth factor signals to control cell growth and proliferation. Although PLD has been widely implicated in the generation of PA needed for mTOR activation, it is becoming clear that PA generated via the LPAAT and DGK pathways is also involved in the regulation of mTOR. In this minireview, we highlight the coordinated maintenance of intracellular PA levels that regulate mTOR signals stimulated by growth factors and nutrients, including amino acids, lipids, glucose, and Gln. Emerging evidence indicates compensatory increases in one source of PA when another source is compromised, highlighting the importance of being able to adapt to stressful conditions that interfere with PA production. The regulation of PA levels has important implications for cancer cells that depend on PA and mTOR activity for survival.
Keywords: glycolysis, Lipid Metabolism, Mammalian Target of Rapamycin (mTOR), Phosphatidic Acid, Phospholipase D, Lysophosphatidic Acid Acyltransferase, Diacylglycerol Kinase
Introduction
Phosphatidic acid (PA)2 has many diverse roles in cell physiology. Most significantly, PA is at the center of membrane phospholipid biosynthesis (Fig. 1), and as a consequence, the level of PA is carefully controlled to maintain lipid homeostasis (1, 2). In addition, PA has emerged as a critical factor for several key signaling molecules that regulate cell cycle progression and survival, including the protein kinases mTOR (mammalian/mechanistic target of rapamycin) (3) and Raf (4). Of significance, both mTOR and Raf have been implicated in human cancer. Consistent with this emerging role for PA in regulating cell proliferation, elevated expression and/or activity of enzymes that generate PA is commonly observed in human cancer, most notably phospholipase D (PLD) (5, 6), which is elevated especially in K-Ras-driven cancers (7–9). Other enzymes that generate PA (lysophosphatidic acid (LPA) acyltransferase (LPAAT), and diacylglycerol (DG) kinase (DGK) (Fig. 1)) have also been implicated in human cancers (10–14). Importantly, LPAAT and DGK have been shown to stimulate mTOR (14–17), reinforcing the importance of the PA-mTOR axis in the control of cell growth and proliferation. Moreover, there appears to be compensatory production of PA under stressful conditions where one source of PA is compromised (7, 18).
FIGURE 1.
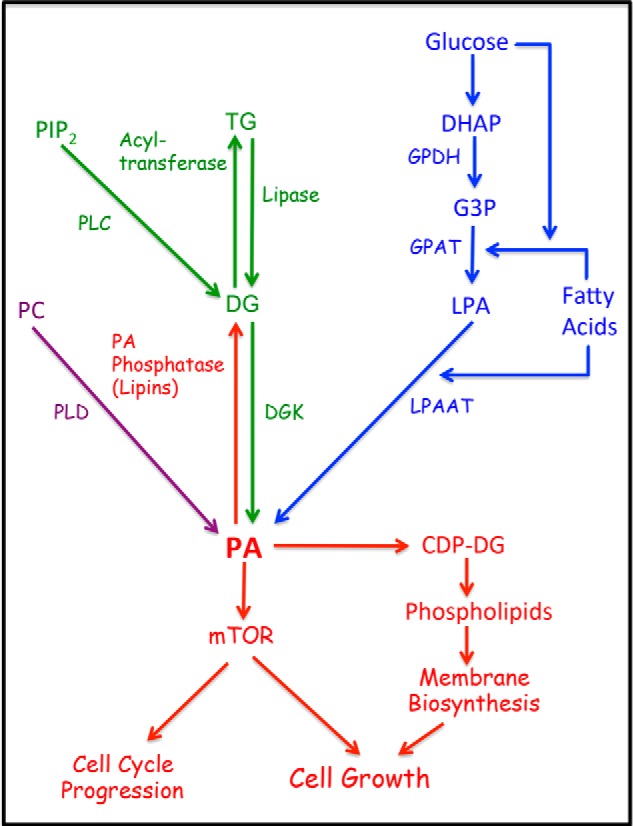
Metabolic pathways for PA production. There are three main pathways leading to the production of PA. For de novo synthesis of membrane phospholipids is the LPAAT pathway where G3P, derived largely from the glycolytic intermediate DHAP, is doubly acylated with a fatty acid, first by G3P acyltransferase (GPAT) to generate LPA, and then by LPAAT to generate PA. The DGK pathway involves the phosphorylation of DG to generate PA. DG can be generated from stored triglycerides (TG) by a lipase, or from phosphatidylinositol 4,5-bisphosphate (PIP2) via growth factor-stimulated phospholipase C. The third mechanism is the hydrolysis of phosphatidylcholine (PC) by PLD. Like PLC, the PLD reaction is commonly stimulated by growth factors. The balance between PA and DG is carefully controlled by both DGK and PA phosphatases that convert PA to DG. Both PA and DG are important intermediates in phospholipid biosynthesis. It is hypothesized that the PA input to mTOR is an indicator of sufficient lipid precursors for cell growth and a signal to promote cell cycle progression. GPDH, G3P dehydrogenase.
The LPAAT pathway, which is an integral part of the de novo pathway for biosynthesis of membrane phospholipids, is likely the most significant source of PA for lipid biosynthesis. However, growth factors (6) and nutrients (19, 20) also stimulate PA production through the action of phospholipases that breakdown membrane phospholipids, potentially leading to high PA concentrations at specific locations and times. This can be accomplished by PLD, or a combination of phospholipase C (PLC), which generates DG, and the subsequent conversion to PA by DGK. The generation of PA from membrane phospholipids by phospholipases produces PA predominantly for second messenger effects on proteins such as mTOR and Raf. mTOR especially is a critical target of PA because of its role as an integrator of both growth factor and nutrient signals (21, 22). Because PA is produced in response to both growth factor signals and synthesized from nutrient sources such as glucose and possibly Gln, PA is ideally positioned as a key regulator of both cell cycle progression and cell growth.
The Role for PA in Cell Cycle Progression
During the mammalian cell cycle, it is in early G1 phase where growth factors exert their influence on whether it is appropriate for a cell to divide or to enter a state of quiescence commonly referred to as G0 (23). After committing to divide in response to growth factor cues, cells must then determine whether there are sufficient nutrients available for the cell to double its mass and divide (24). There must be a supply of essential amino acids, Gln, and glucose available to generate the biological molecules needed to produce two daughter cells. Notably, there are a series of cell cycle checkpoints that sense the presence of essential amino acids and Gln in late G1 that must be passed before cells commit to enter S-phase and replicate the genome (25) (Fig. 2). Suppression of mTOR, like amino acid deprivation, also leads to late G1 arrest (25, 26). Because essential amino acids activate mTOR via Rag GTPases at the lysosomal membrane (27), it was surprising that suppression of mTOR blocked cell cycle progression at a site later in G1 than the checkpoints that monitor the presence of essential amino acids and Gln (25) (Fig. 2). Thus, nutrient input to mTOR for control of G1/S cell cycle progression appears to be more complicated than simply reflecting a need for essential amino acids.
FIGURE 2.
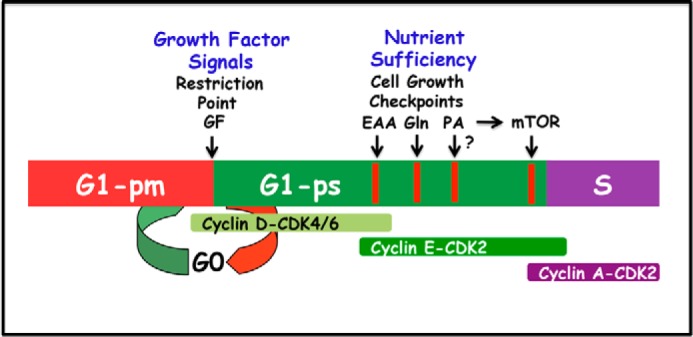
Regulation of G1 cell cycle progression by growth factors and nutrients. G1 can be separated into two phases referred to as G1-pm (post-mitotic) and G1-ps (pre-S) by a growth factor (GF)-dependent restriction point (23). At the restriction point, the cell receives signals signifying that it is appropriate to divide. Later in G1-ps there is a series of metabolic checkpoints that evaluate whether there are sufficient nutrients for the cell to double in mass and divide. There are distinct checkpoints for essential amino acids (EAA), the conditionally essential amino acid Gln, and a later checkpoint mediated by mTOR. The schematic shows the relative order of the checkpoints, but does not reflect an accurate time frame. Because mTOR requires PA for stability of the mTOR complexes (30), this late mTOR checkpoint also requires PA. It is not clear whether there is a separate checkpoint for PA like there is for the essential amino acids (EAA), which are also required for mTOR activity.
We previously proposed that the responsiveness of mTOR to PA evolved as a means for sensing the sufficiency of lipid precursors for membrane phospholipid biosynthesis (28). This was based on the central position of PA in the anabolic synthesis of membrane phospholipids (Fig. 1) and is therefore an ideal indicator of lipid sufficiency. The ability to sense the presence of lipids via interaction with PA was proposed as a complement for the ability of mTOR to sense the presence of essential amino acids and glucose. As indicated in Fig. 2, an mTOR-dependent cell cycle checkpoint maps late in G1 downstream of essential amino acid and Gln checkpoints (25). Because PA interacts directly with mTOR (29) and is required for the stability of both mTORC1 and mTORC2 complexes (30), PA likely works in concert with essential amino acids and possibly Gln to promote cell cycle progression through the late mTOR-dependent checkpoint. Although there is much to be learned about nutrient input into G1 cell cycle progression, it is clear that PA is essential for mTOR activity and mTOR activity is required for progression from G1 into S-phase, indicating that PA, via input to mTOR, is requisite for cell cycle progression.
Sources of PA
Most of the support for a role for PA in the mTOR-dependent cell cycle progression from G1 into S-phase comes from studies linking PLD with cell transformation and cancer (3, 5, 29–31). However, knock-out of both PLD1 and PLD2 yields viable mice (32, 33), whereas mTOR knockouts are embryonic lethal (34, 35). Thus, the PA needed to keep mTOR intact and active must be generated from sources other than the hydrolysis of phosphatidylcholine by PLD. As shown in Fig. 1, there are minimally three sources of PA, perhaps the most significant being the LPAAT pathway where de novo synthesized and dietary fatty acids are acylated onto glycerol 3-phosphate (G3P) derived from dihydroxyacetone phosphate (DHAP), a glycolytic intermediate (Fig. 1). The LPAAT pathway is likely the most significant for sensing lipids needed for cell growth because it is via this pathway where lipids targeted for membrane phospholipid biosynthesis are generated and incorporated into PA. A third pathway for PA production is via DGK, which phosphorylates DG to generate PA (Fig. 1). The source of DG for synthesis of PA is of interest. DG can be generated from stored triglycerides by a triglyceride lipase or from the PLC-mediated hydrolysis of phosphatidylinositol 4,5-bisphosphate. However, it is difficult to imagine generating significant levels of PA via the PLC-DGK pathway because the source of the PLC-generated PA is phosphatidylinositol 4,5-bisphosphate, which is present in very small amounts in the cell and is generated by the action of phosphatidylinositol kinases (36) and is therefore energetically expensive to generate. In contrast, the PLD substrate is phosphatidylcholine, the most abundant membrane phospholipid, and it does not need to be modified to be a substrate, as does phosphatidylinositol. Thus, it is not clear under what conditions the PLC-DGK pathway would be used, but it has been suggested as a compensatory mechanism if PLD is suppressed (18).
Another factor that regulates PA levels are the PA phosphatases, also called lipins, that convert PA to DG (2, 37). The lipins are critical for maintaining lipid homeostasis and may contribute to determining the equilibrium between PA and DG. This equilibrium could have important implications for cell cycle control, with PA and mTOR favoring proliferation and DG promoting cell cycle arrest. DG leads to the activation of protein kinase C isoforms that, with the exception of protein kinase Cϵ, tend to have anti-proliferative effects (38, 39). Thus, the complex interplay of lipid metabolic flux through PA and DG could have profound effects on cell cycle progression and cell growth.
PA as a Broader Indicator of Nutrient Sufficiency
The role of mTOR as a sensor of nutrients is based largely on its dependence on the presence of essential amino acids (21, 40). Much has been learned in the last several years on the mechanistic basis for the sensing of amino acids by mTOR at the lysosomal membrane via Rag GTPases (27, 41). The activation of mTOR in response to amino acids also requires PLD (19, 20, 42). However, very little is known about the dependence of mTOR on glucose, another critical nutrient sensed by mTOR. Although the PA dependence of mTOR that has been proposed represents a means for sensing sufficient lipids for cell growth (17, 28), it is plausible that PA represents a broader indicator of nutrient sufficiency. In dividing cells and cancer cells, there is a metabolic reprograming that shifts from the catabolic generation of reducing power (NADH) that drives mitochondrial ATP generation to anabolic synthetic reactions that generate the biological molecules needed for doubling the cell mass prior to cell division (43). Much of the reprogramming involves diverting glycolytic and TCA cycle intermediates for synthesis of amino acids, nucleotides, and lipids. During glycolysis, glucose is converted to pyruvate in the cytosol. Pyruvate enters the mitochondria and is converted to acetyl-CoA, which condenses with oxaloacetate to form citrate. In dividing cells, citrate exits the mitochondria, and acetyl-CoA and oxaloacetate are regenerated. The acetyl-CoA is then used for fatty acid synthesis, generating palmitoyl-CoA, which can be acylated onto G3P and ultimately become part of PA. The G3P is derived from the glycolytic intermediate DHAP; thus, PA is synthesized from two distinct components derived from glucose and therefore could contribute to the sensing of sufficient glucose. This is shown schematically in Fig. 3.
FIGURE 3.
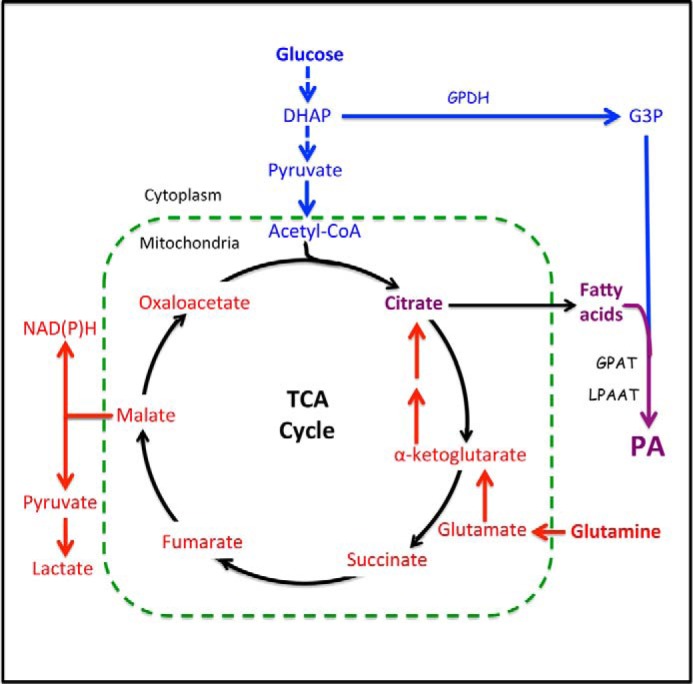
Metabolic pathways from glucose and Gln to PA. Glucose is converted into lipids via two pathways. The first pathway is the conversion of the glycolytic intermediate DHAP to G3P by G3P dehydrogenase (GPDH). G3P is then fatty acylated, first to LPA by G3P acyltransferase (GPAT) and then to PA by LPAAT. The second pathway utilizes the end product of glycolysis, pyruvate. Pyruvate is converted to acetyl-CoA, which condenses with oxaloacetate to form citrate. Citrate leaves the mitochondria and is then converted back to oxaloacetate and acetyl-CoA, which is then used to synthesize the fatty acids that will be used to acylate G3P and generate PA. With the exit of citrate from the TCA cycle, there is a need for anaplerotic replenishment of the carbon provided by citrate. This is provided by the conditionally essential amino acid Gln, which enters the TCA cycle by being deaminated to glutamate and then to α-ketoglutarate by transamination. Via the TCA cycle, most of the Gln is converted to malate and then to pyruvate to generate NADPH for fatty acid synthesis. Gln can also go from malate to oxaloacetate where it can then condense with acetyl-CoA derived from glucose to form citrate and then fatty acids as above. Gln can also be reductively carboxylated to isocitrate and then converted to citrate in a reverse TCA cycle reaction of isocitrate dehydrogenase. In the absence of Gln, glucose cannot be converted to fatty acid synthesis.
The exit of citrate from the TCA cycle and the mitochondria creates a need for anaplerotic replenishment of a TCA cycle intermediate to provide the carbon lost by the exit of citrate. Although there are many possible anaplerotic sources, the most abundant is Gln, which is used as both a carbon and a nitrogen source for dividing cells (44). Gln enters the TCA after being converted first to glutamate and then to α-ketoglutarate (Fig. 3). Gln is designated as a “conditionally” essential amino acid because although it is synthesized under non-proliferative conditions, it becomes essential during proliferation. Of significance, there is a Gln-sensitive G1 cell cycle checkpoint that can be distinguished from an essential amino acid checkpoint in mammalian cells (25). Thus, it may be important for mTOR to sense this critical nutrient input. Because anaplerotic entry of Gln into the TCA cycle is essential for continued exit of citrate for fatty acid synthesis, and as a consequence, PA synthesis via the LPAAT pathway, it is plausible that the presence of both glucose (which generates acetyl-CoA and G3P) and Gln is critical for mTOR function. Most of the anaplerotic Gln is used for NADPH production via the oxidative decarboxylation of malate to pyruvate to generate the NADPH needed for fatty acid synthesis and other anabolic reactions (Fig. 3). However, 25% of the anaplerotic Gln is converted into lipids (45). This observation demonstrates that Gln contributes significantly to the fatty acids incorporated into PA via the LPAAT pathway. The conversion of α-ketoglutarate to citrate can be accomplished by two different mechanisms: first, by traversing the TCA cycle to oxaloacetate, which can condense with acetyl-CoA (derived from glycolysis) to form citrate; and second, by the reductive carboxylation of α-ketoglutarate to isocitrate and then to citrate via a reverse reaction of the TCA cycle (46, 47) (Fig. 3). Thus, the generation of PA from synthesized fatty acids and G3P involves both glycolysis and glutaminolysis, which could represent input to mTOR from both glucose and Gln.
Compensatory Production of PA in Response to Metabolic Stress in Cancer Cells
We previously reported that in response to serum withdrawal there was a substantial increase in PLD activity in cancer cells (7), most notably in cancer cells harboring Ras mutations (9). More recently, we reported that PLD activity is also elevated in response to changing from medium with 10% serum to 10% delipidated serum (48). The effect appears to be a stress response in Ras-driven cancer cells because these cells have a greater need for exogenously supplied lipids (48, 49). Ras-driven cancer cells have a compromised ability to increase levels of stearoyl-CoA desaturase-1 in response to serum withdrawal (48). Thus, newly synthesized fatty acids cannot be desaturated, which is essential for synthesis of phospholipids targeted for membranes. Of interest, Ras-driven cancer cells have increased macropinocytosis (50), which has been shown to be an important source for amino acids derived from proteolytic digestion of scavenged proteins, the most abundant being albumin (51). Albumin is a carrier protein for lipids (52), and thus, the scavenging of albumin also involves the scavenging of lipids. It was recently reported that constitutive mTORC1 activity renders hypoxic cells dependent on exogenous desaturated lipids for survival (53). Although this study did not connect the need for desaturated lipids and the dependence of mTOR on PA, it did provide further evidence for a lipid dependence of mTOR and potentially a dependence on desaturated lipids. Coleman and colleagues (54) recently reported that the mTORC2 complex falls apart in the presence of dipalmitoyl-PA, which consists of two saturated fatty acids. This is in stark contrast to the effect of PA containing palmitate (saturated) and oleate (mono-unsaturated), which stabilized both mTORC1 and mTORC2 complexes in cells where PA production by PLD was suppressed (30). These studies suggest a significant difference between PA with saturated fatty acids and those with some degree of unsaturation on mTOR. Because PLD generates PA from membrane phosphatidylcholine, this PA will most likely consist of a saturated and an unsaturated fatty acid that is typical of membrane glycerophospholipids (55). Thus, the ability of Ras-driven cancer cells to elevate PA levels in the absence of exogenous lipids prevents these cells from undergoing a default apoptotic program and underscores the importance for cells to generate compensatory levels of PA when another source of PA is compromised. It is also of significance that under the stress of serum withdrawal, these cells increase their ability to migrate and invade Matrigel in a PLD-dependent manner (7), indicating a survival program that not only prevents apoptosis, but also promotes migration to a more hospitable environment. This effect in cancer cells suggests a link between the level of PA and metastatic potential in cancer cells.
There are other examples of compensatory changes in PA that go in the opposite direction. Inhibition of PLD activity actually led to increased levels of PA from an undetermined source (18). There is also evidence that endoplasmic reticulum stresses such as low glucose or hypoxia cause the protein kinase PERK (protein kinase R-like endoplasmic reticulum kinase) to phosphorylate DG to generate PA and elevate mTOR activity (56). These results indicate that regulating PA levels, for both membrane phospholipid biosynthesis and second messenger activity that controls cell cycle progression and survival, are carefully controlled. They also point out the potential for targeting PLD and PA metabolism in cancer cells to suppress survival and perhaps migration signals.
An intriguing question with regard to alternative compensatory increases in PA via alternative mechanisms is whether the acyl component of PA is equivalent when coming from different sources. As indicated above, there is an apparent requirement for an unsaturated fatty acid in order to achieve mTOR complex stability (30, 54). Thus, it will be of interest to determine whether there are significant differences in the acyl composition of PA obtained from the different sources. An interesting possibility is the purposeful generation of PA consisting of two saturated fatty acids to suppress mTOR as was shown with dipalmitoyl-PA and mTORC2 (54).
PLD and Intracellular Signals That Target mTOR
Since the seminal finding that PA is critical for the activity of mTOR (29), there has been a substantially increased interest in PLD. However, it is likely that the more primitive pathway for PA generation is the LPAAT pathway, which generates PA targeted for either membrane phospholipid synthesis or lipid storage. The generation of PA for mTOR via PLD likely evolved later in multicellular organisms where nutrient sensing by mTOR became coupled with response to growth factors and insulin. Significantly, PLD activity is elevated in response to platelet-derived growth factor (57), fibroblast growth factor (58), epidermal growth factor (59), insulin-like growth factor 1 (60), and insulin (61). The activation of PLD by insulin is of particular interest because insulin controls the levels of glucose and glucose transporters, and PLD is dependent on mTOR (22), but is not ordinarily associated with mitogenic signals. The dependence of insulin-induced mTOR on PLD suggests that stimulation of PLD is needed because of the need for PA by mTOR, and not just for mitogenic signals. Thus, activation of PLD in mammalian cells can be elevated in response to signals that require mTOR activation, including both growth factors and insulin.
It has been speculated that signals leading to mTOR activation are the most commonly dysregulated in human cancer (47, 62). Because PLD activity is elevated in many human cancers (5, 6), it appears that cancer cells have co-opted the dysregulation of PLD along with dysregulation of other signaling pathways that contribute to mTOR activation, such as the phosphatidylinositol-3-kinase/AKT/Rheb pathway that activates mTORC1 (40). Consistent with the importance of increased PLD activity observed in human cancers, early studies demonstrated that PLD activity is elevated in cells transformed by a variety of transforming oncogenes including v-Src (31), v-Ras (63), v-Fps (64), and v-Raf (65). Thus, there is a strong correlation between cell transformation and elevated PLD activity, a signal that is critical for mTOR activation.
Conclusions and Perspective
In this review we have highlighted the significance of controlling the levels of PA and its impact on mTOR, which requires PA for the stability and activity of both mTOR complexes, mTORC1 and mTORC2 (30). It is proposed that the PA requirement for mTOR evolved as a need to sense the presence of sufficient lipids, and perhaps glucose and Gln, for cell growth and division. However, with evolution to multicellularity, PLD emerged as a critical factor in the ability of mTOR to respond to both nutrients and growth factors/insulin. Many questions remain with regard to the regulation of PA levels and the impact on mTOR. A key issue is the location of PA synthesis. Phospholipid biosynthesis via the LPAAT pathway takes place on subdomains of the endoplasmic reticulum and then is shuttled via vesicles to various cellular destinations (66). mTOR has a strong lysosomal location under conditions where there are sufficient amino acids (27). It is unclear as to whether shuttled PA can impact on lysosomal mTOR. Thus, PLD may be the more likely source of PA on lysosomes, in that PLD, notably PLD1, can shuttle between organelles and has a strong lysosomal distribution (67, 68). It is also of note that forced localization of mTOR to lysosomes activated mTOR in the absence of amino acids if Rheb was present (69). Rheb is one of many GTPases that activate PLD1 (20, 70, 71), indicating that PLD may work in concert with the signaling mechanisms that activate Rheb. The picture that emerges is one where LPAAT-generated PA may be the more critical source for nutrient sensing by mTOR, but that PLD is the more versatile source of PA that can respond locally to growth factor/insulin signals and stress. The PLC/DGK pathway may also provide PA under other less well understood conditions. Given the critical role that mTOR plays in cancer cell survival and proliferation, interfering with PA metabolism could prove to be an effective strategy for targeting what would be a large number of human cancers.
This work was supported, in whole or in part, by National Institutes of Health Grant 1R01-CA046677 (to D. A. F.) from the NCI. Research Centers in Minority Institutions Award RR-03039 from the National Center for Research Resources of the National Institutes of Health, which supports infrastructure and instrumentation in the Biological Sciences Department at Hunter College, is also acknowledged. This is the fourth article in the Thematic Minireview Series “Phospholipase D and Cancer.”
- PA
- phosphatidic acid
- mTOR
- mammalian/mechanistic target of rapamycin
- mTORC
- mTOR complex
- PLD
- phospholipase D
- LPA
- lysophosphatidic acid
- LPAAT
- LPA acyltransferase
- DG
- diacylglycerol
- DGK
- DG kinase
- PLC
- phospholipase C
- G3P
- glycerol 3-phosphate
- DHAP
- dihydroxyacetone phosphate
- TCA
- tricarboxylic acid.
REFERENCES
- 1. Athenstaedt K., Daum G. (1999) Phosphatidic acid, a key intermediate in lipid metabolism. Eur. J. Biochem. 266, 1–16 [DOI] [PubMed] [Google Scholar]
- 2. Pascual F., Carman G. M. (2013) Phosphatidate phosphatase, a key regulator of lipid homeostasis. Biochim. Biophys. Acta 1831, 514–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Foster D. A. (2007) Regulation of mTOR by phosphatidic acid? Cancer Res. 67, 1–4 [DOI] [PubMed] [Google Scholar]
- 4. Kraft C. A., Garrido J. L., Fluharty E., Leiva-Vega L., Romero G. (2008) Role of phosphatidic acid in the coupling of the ERK cascade. J. Biol. Chem. 283, 36636–36645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Foster D. A. (2009) Phosphatidic acid signaling to mTOR: signals for the survival of human cancer cells. Biochim. Biophys. Acta 1791, 949–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Foster D. A., Xu L. (2003) Phospholipase D in cell proliferation and cancer. Mol. Cancer Res. 1, 789–800 [PubMed] [Google Scholar]
- 7. Zheng Y., Rodrik V., Toschi A., Shi M., Hui L., Shen Y., Foster D. A. (2006) Phospholipase D couples survival and migration signals in stress response of human cancer cells. J. Biol. Chem. 281, 15862–15868 [DOI] [PubMed] [Google Scholar]
- 8. Garcia A., Zheng Y., Zhao C., Toschi A., Fan J., Shraibman N., Brown H. A., Bar-Sagi D., Foster D. A., Arbiser J. L. (2008) Honokiol suppresses survival signals mediated by Ras-dependent phospholipase D activity in human cancer cells. Clin. Cancer Res. 14, 4267–4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shi M., Zheng Y., Garcia A., Xu L., Foster D. A. (2007) Phospholipase D provides a survival signal in human cancer cells with activated H-Ras or K-Ras. Cancer Lett. 258, 268–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coon M., Ball A., Pound J., Ap S., Hollenback D., White T., Tulinsky J., Bonham L., Morrison D. K., Finney R., Singer J. W. (2003) Inhibition of lysophosphatidic acid acyltransferase β disrupts proliferative and survival signals in normal cells and induces apoptosis of tumor cells. Mol. Cancer Ther. 2, 1067–1078 [PubMed] [Google Scholar]
- 11. Cheng K. W., Agarwal R., Mills G. B. (2009) Ras-superfamily GTP-ases in ovarian cancer. Cancer Treat. Res. 149, 229–240 [DOI] [PubMed] [Google Scholar]
- 12. Diefenbach C. S., Soslow R. A., Iasonos A., Linkov I., Hedvat C., Bonham L., Singer J., Barakat R. R., Aghajanian C., Dupont J. (2006) Lysophosphatidic acid acyltransferase-β (LPAAT-β) is highly expressed in advanced ovarian cancer and is associated with aggressive histology and poor survival. Cancer 107, 1511–1519 [DOI] [PubMed] [Google Scholar]
- 13. Dominguez C. L., Floyd D. H., Xiao A., Mullins G. R., Kefas B. A., Xin W., Yacur M. N., Abounader R., Lee J. K., Wilson G. M., Harris T. E., Purow B. W. (2013) Diacylglycerol kinase α is a critical signaling node and novel therapeutic target in glioblastoma and other cancers. Cancer Discov. 3, 782–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blaskovich M. A., Yendluri V., Lawrence H. R., Lawrence N. J., Sebti S. M., Springett G. M. (2013) Lysophosphatidic acid acyltransferase β regulates mTOR signaling. PLoS One 8, e78632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Avila-Flores A., Santos T., Rincón E., Mérida I. (2005) Modulation of the mammalian target of rapamycin pathway by diacylglycerol kinase-produced phosphatidic acid. J. Biol. Chem. 280, 10091–10099 [DOI] [PubMed] [Google Scholar]
- 16. Tang W., Yuan J., Chen X., Gu X., Luo K., Li J., Wan B., Wang Y., Yu L. (2006) Identification of a novel human lysophosphatidic acid acyltransferase, LPAAT-θ, which activates mTOR pathway. J. Biochem. Mol. Biol. 39, 626–635 [DOI] [PubMed] [Google Scholar]
- 17. Mérida I., Avila-Flores A., Merino E. (2008) Diacylglycerol kinases: at the hub of cell signalling. Biochem. J. 409, 1–18 [DOI] [PubMed] [Google Scholar]
- 18. Antonescu C. N., Danuser G., Schmid S. L. (2010) Phosphatidic acid plays a regulatory role in clathrin-mediated endocytosis. Mol. Biol. Cell 21, 2944–2952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yoon M. S., Du G., Backer J. M., Frohman M. A., Chen J. (2011) Class III PI-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. J. Cell Biol. 195, 435–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu L., Salloum D., Medlin P. S., Saqcena M., Yellen P., Perrella B., Foster D. A. (2011) Phospholipase D mediates nutrient input to mammalian target of rapamycin complex 1 (mTORC1). J. Biol. Chem. 286, 25477–25486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Loewith R., Hall M. N. (2011) Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics 189, 1177–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zoncu R., Efeyan A., Sabatini D. M. (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zetterberg A., Larsson O., Wiman K. G. (1995) What is the restriction point? Curr. Opin. Cell Biol. 7, 835–842 [DOI] [PubMed] [Google Scholar]
- 24. Foster D. A., Yellen P., Xu L., Saqcena M. (2010) Regulation of G1 cell cycle progression: distinguishing the restriction point from a nutrient-sensing cell growth checkpoint(s). Genes Cancer 1, 1124–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Saqcena M., Menon D., Patel D., Mukhopadhyay S., Chow V., Foster D. A. (2013) Amino acids and mTOR mediate distinct metabolic checkpoints in mammalian G1 cell cycle. PLoS One 8, e74157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fingar D. C., Richardson C. J., Tee A. R., Cheatham L., Tsou C., Blenis J. (2004) mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell. Biol. 24, 200–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sancak Y., Peterson T. R., Shaul Y. D., Lindquist R. A., Thoreen C. C., Bar-Peled L., Sabatini D. M. (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Foster D. A. (2013) Phosphatidic acid and lipid-sensing by mTOR. Trends Endocrinol. Metab. 24, 272–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fang Y., Vilella-Bach M., Bachmann R., Flanigan A., Chen J. (2001) Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 294, 1942–1945 [DOI] [PubMed] [Google Scholar]
- 30. Toschi A., Lee E., Xu L., Garcia A., Gadir N., Foster D. A. (2009) Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol. Cell. Biol. 29, 1411–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Song J. G., Pfeffer L. M., Foster D. A. (1991) v-Src increases diacylglycerol levels via a type D phospholipase-mediated hydrolysis of phosphatidylcholine. Mol. Cell. Biol. 11, 4903–4908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dall'Armi C., Hurtado-Lorenzo A., Tian H., Morel E., Nezu A., Chan R. B., Yu W. H., Robinson K. S., Yeku O., Small S. A., Duff K., Frohman M. A., Wenk M. R., Yamamoto A., Di Paolo G. (2010) The phospholipase D1 pathway modulates macroautophagy. Nat. Commun. 1, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thielmann I., Stegner D., Kraft P., Hagedorn I., Krohne G., Kleinschnitz C., Stoll G., Nieswandt B. (2012) Redundant functions of phospholipases D1 and D2 in platelet α-granule release. J. Thromb. Haemost. 10, 2361–2372 [DOI] [PubMed] [Google Scholar]
- 34. Gangloff Y. G., Mueller M., Dann S. G., Svoboda P., Sticker M., Spetz J. F., Um S. H., Brown E. J., Cereghini S., Thomas G., Kozma S. C. (2004) Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol. Cell. Biol. 24, 9508–9516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guertin D. A., Stevens D. M., Thoreen C. C., Burds A. A., Kalaany N. Y., Moffat J., Brown M., Fitzgerald K. J., Sabatini D. M. (2006) Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCα, but not S6K1. Dev. Cell 11, 859–871 [DOI] [PubMed] [Google Scholar]
- 36. D'Souza K., Epand R. M. (2014) Enrichment of phosphatidylinositols with specific acyl chains. Biochim. Biophys. Acta 1838, 1501–1508 [DOI] [PubMed] [Google Scholar]
- 37. Brindley D. N., Pilquil C., Sariahmetoglu M., Reue K. (2009) Phosphatidate degradation: phosphatidate phosphatases (lipins) and lipid phosphate phosphatases. Biochim. Biophys. Acta 1791, 956–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jackson D. N., Foster D. A. (2004) The enigmatic protein kinase Cδ: complex roles in cell proliferation and survival. FASEB J. 18, 627–636 [DOI] [PubMed] [Google Scholar]
- 39. Mochly-Rosen D., Das K., Grimes K. V. (2012) Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 11, 937–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sengupta S., Peterson T. R., Sabatini D. M. (2010) Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell 40, 310–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Efeyan A., Zoncu R., Sabatini D. M. (2012) Amino acids and mTORC1: from lysosomes to disease. Trends Mol. Med. 18, 524–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wiczer B. M., Thomas G. (2012) Phospholipase D and mTORC1: nutrients are what bring them together. Sci. Signal. 5, pe13. [DOI] [PubMed] [Google Scholar]
- 43. DeBerardinis R. J., Lum J. J., Hatzivassiliou G., Thompson C. B. (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20 [DOI] [PubMed] [Google Scholar]
- 44. DeBerardinis R. J., Cheng T. (2010) Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29, 313–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. DeBerardinis R. J., Mancuso A., Daikhin E., Nissim I., Yudkoff M., Wehrli S., Thompson C. B. (2007) Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. U.S.A. 104, 19345–19350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hensley C. T., Wasti A. T., DeBerardinis R. J. (2013) Glutamine and cancer: cell biology, physiology, and clinical opportunities. J. Clin. Invest. 123, 3678–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ward P. S., Thompson C. B. (2012) Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21, 297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Salloum D., Mukhopadhyay S., Tung K., Polonetskaya A., Foster D. A. (2014) Mutant Ras elevates dependence on serum lipids and creates a synthetic lethality for rapamycin. Mol. Cancer Ther. 13, 733–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kamphorst J. J., Cross J. R., Fan J., de Stanchina E., Mathew R., White E. P., Thompson C. B., Rabinowitz J. D. (2013) Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. U.S.A. 110, 8882–8887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bar-Sagi D., Feramisco J. R. (1986) Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by Ras proteins. Science 233, 1061–1068 [DOI] [PubMed] [Google Scholar]
- 51. Commisso C., Davidson S. M., Soydaner-Azeloglu R. G., Parker S. J., Kamphorst J. J., Hackett S., Grabocka E., Nofal M., Drebin J. A., Thompson C. B., Rabinowitz J. D., Metallo C. M., Vander Heiden M. G., Bar-Sagi D. (2013) Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. van der Vusse G. J. (2009) Albumin as fatty acid transporter. Drug Metab. Pharmacokinet. 24, 300–307 [DOI] [PubMed] [Google Scholar]
- 53. Young R. M., Ackerman D., Quinn Z. L., Mancuso A., Gruber M., Liu L., Giannoukos D. N., Bobrovnikova-Marjon E., Diehl J. A., Keith B., Simon M. C. (2013) Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev. 27, 1115–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang C., Wendel A. A., Keogh M. R., Harris T. E., Chen J., Coleman R. A. (2012) Glycerolipid signals alter mTOR complex 2 (mTORC2) to diminish insulin signaling. Proc. Natl. Acad. Sci. U.S.A. 109, 1667–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shindou H., Shimizu T. (2009) Acyl-CoA:lysophospholipid acyltransferases. J. Biol. Chem. 284, 1–5 [DOI] [PubMed] [Google Scholar]
- 56. Bobrovnikova-Marjon E., Pytel D., Riese M. J., Vaites L. P., Singh N., Koretzky G. A., Witze E. S., Diehl J. A. (2012) PERK utilizes intrinsic lipid kinase activity to generate phosphatidic acid, mediate Akt activation, and promote adipocyte differentiation. Mol. Cell. Biol. 32, 2268–2278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Plevin R., Cook S. J., Palmer S., Wakelam M. J. (1991) Multiple sources of sn-1,2-diacylglycerol in platelet-derived-growth-factor-stimulated Swiss 3T3 fibroblasts. Evidence for activation of phosphoinositidase C and phosphatidylcholine-specific phospholipase D. Biochem. J. 279, 559–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Motoike T., Bieger S., Wiegandt H., Unsicker K. (1993) Induction of phosphatidic acid by fibroblast growth factor in cultured baby hamster kidney fibroblasts. FEBS Lett. 332, 164–168 [DOI] [PubMed] [Google Scholar]
- 59. Song J., Jiang Y. W., Foster D. A. (1994) Epidermal growth factor induces the production of biologically distinguishable diglyceride species from phosphatidylinositol and phosphatidylcholine via the independent activation of type C and type D phospholipases. Cell Growth Differ. 5, 79–85 [PubMed] [Google Scholar]
- 60. Banno Y., Takuwa Y., Yamada M., Takuwa N., Ohguchi K., Hara A., Nozawa Y. (2003) Involvement of phospholipase D in insulin-like growth factor-I-induced activation of extracellular signal-regulated kinase, but not phosphoinositide 3-kinase or Akt, in Chinese hamster ovary cells. Biochem. J. 369, 363–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Standaert M. L., Bandyopadhyay G., Zhou X., Galloway L., Farese R. V. (1996) Insulin stimulates phospholipase D-dependent phosphatidylcholine hydrolysis, Rho translocation, de novo phospholipid synthesis, and diacylglycerol/protein kinase C signaling in L6 myotubes. Endocrinology 137, 3014–3020 [DOI] [PubMed] [Google Scholar]
- 62. Blagosklonny M. V. (2011) Molecular damage in cancer: an argument for mTOR-driven aging. Aging 3, 1130–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jiang H., Lu Z., Luo J. Q., Wolfman A., Foster D. A. (1995) Ras mediates the activation of phospholipase D by v-Src. J. Biol. Chem. 270, 6006–6009 [DOI] [PubMed] [Google Scholar]
- 64. Jiang Y. W., Song J., Zang Q., Foster D. A. (1994) Phosphatidylcholine-specific phospholipase D activity is elevated in v-Fps-transformed cells. Biochem. Biophys. Res. Commun. 203, 1195–2003 [DOI] [PubMed] [Google Scholar]
- 65. Frankel P., Ramos M., Flom J., Bychenok S., Joseph T., Kerkhoff E., Rapp U. R., Feig L. A., Foster D. A. (1999) Ral and Rho-dependent activation of phospholipase D in v-Raf-transformed cells. Biochem. Biophys. Res. Commun. 255, 502–507 [DOI] [PubMed] [Google Scholar]
- 66. Lagace T. A., Ridgway N. D. (2013) The role of phospholipids in the biological activity and structure of the endoplasmic reticulum. Biochim. Biophys. Acta 1833, 2499–2510 [DOI] [PubMed] [Google Scholar]
- 67. Jang Y. H., Min do S. (2012) The hydrophobic amino acids involved in the interdomain association of phospholipase D1 regulate the shuttling of phospholipase D1 from vesicular organelles into the nucleus. Exp. Mol. Med. 44, 571–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Arneson L. S., Kunz J., Anderson R. A., Traub L. M. (1999) Coupled inositide phosphorylation and phospholipase D activation initiates clathrin-coat assembly on lysosomes. J. Biol. Chem. 274, 17794–17805 [DOI] [PubMed] [Google Scholar]
- 69. Sancak Y., Bar-Peled L., Zoncu R., Markhard A. L., Nada S., Sabatini D. M. (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sun Y., Fang Y., Yoon M. S., Zhang C., Roccio M., Zwartkruis F. J., Armstrong M., Brown H. A., Chen J. (2008) Phospholipase D1 is an effector of Rheb in the mTOR pathway. Proc. Natl. Acad. Sci. U.S.A. 105, 8286–8291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Durán R. V., Hall M. N. (2012) Regulation of TOR by small GTPases. EMBO Rep. 13, 121–128 [DOI] [PMC free article] [PubMed] [Google Scholar]