

Background: Bone type I collagen is glycosylated.
Results: The major glycosylation sites are involved in intermolecular cross-linking. The extent and pattern of glycosylation vary depending on the site, type, and maturation of cross-links.
Conclusion: Glycosylation may control collagen cross-linking in bone type I collagen.
Significance: The results provide important insight into the role of glycosylation in collagen stability in bone.
Keywords: Bone, Collagen, Glycosylation, Hydroxylysine (Hyl), Mass Spectrometry (MS), Post-translational Modification (PTM)
Abstract
Fibrillar type I collagen is the major organic component in bone, providing a stable template for mineralization. During collagen biosynthesis, specific hydroxylysine residues become glycosylated in the form of galactosyl- and glucosylgalactosyl-hydroxylysine. Furthermore, key glycosylated hydroxylysine residues, α1/2-87, are involved in covalent intermolecular cross-linking. Although cross-linking is crucial for the stability and mineralization of collagen, the biological function of glycosylation in cross-linking is not well understood. In this study, we quantitatively characterized glycosylation of non-cross-linked and cross-linked peptides by biochemical and nanoscale liquid chromatography-high resolution tandem mass spectrometric analyses. The results showed that glycosylation of non-cross-linked hydroxylysine is different from that involved in cross-linking. Among the cross-linked species involving α1/2-87, divalent cross-links were glycosylated with both mono- and disaccharides, whereas the mature, trivalent cross-links were primarily monoglycosylated. Markedly diminished diglycosylation in trivalent cross-links at this locus was also confirmed in type II collagen. The data, together with our recent report (Sricholpech, M., Perdivara, I., Yokoyama, M., Nagaoka, H., Terajima, M., Tomer, K. B., and Yamauchi, M. (2012) Lysyl hydroxylase 3-mediated glucosylation in type I collagen: molecular loci and biological significance. J. Biol. Chem. 287, 22998–23009), indicate that the extent and pattern of glycosylation may regulate cross-link maturation in fibrillar collagen.
Introduction
Collagens comprise a large family of structurally related extracellular matrix proteins distributed throughout the body (1, 2). Of these, type Ι collagen is the most abundant and is the major organic matrix component of most connective tissues, including bone. In bone, it plays a crucial role in organizing the deposition and growth of mineralization (3–6). One of the functionally important characteristics of collagen is its unique post-translational modifications, including hydroxylation of specific Pro and Lys residues, O-glycosylation of hydroxylysine (Hyl),3 and covalent intermolecular cross-linking (1). The critical importance of these post-translational modifications in bone formation is evident from the consequences of mutations in genes encoding post-translational modification-associated enzymes (7–11). O-Linked glycosylation of Hyl is catalyzed by hydroxylysyl galactosyltransferase (EC 2.4.1.50) and galactosylhydroxylysyl glucosyltransferase (EC 2.4.1.66), resulting in formation of galactosylhydroxylysine (G-Hyl) and glucosylgalactosylhydroxylysine (GG-Hyl), respectively (12). It has been reported that glycosylation might play a role in collagen fibrillogenesis (13–17), cross-linking (2, 18–23), mineralization (24), and collagen-cell interaction (25, 26). We have recently reported the helical cross-linking residue α1-87 (87th residue from the N terminus of the triple helix on an α1 chain) as the major glycosylation site in type I collagen synthesized by murine-derived osteoblastic cells and its potential role in cross-link maturation (24).
Covalent intermolecular cross-linking of collagen is initiated by the conversion of Lys/Hyl residues located in the non-helical C- and N-telopeptides (C- and N-telo) of the molecule to the respective aldehydes Lysald/Hylald, catalyzed by lysyl oxidase. In type I collagen, there are two Lys/Hyl residues in the C-telo part (two of the α1-16C; i.e. 16th residue from the N terminus in the C-telo) and three in the N-telo domains (two α1-9N and one α2-5N; i.e. 9th and 5th residue from the N terminus in the α1 and α2 N-telo, respectively). The aldehyde can then form an iminium bond to a juxtaposed ϵ-amino group of Lys/Hyl in the helical domain of the staggered adjacent molecule. The possible combinations are as follows: α1-16C × α1-87, α1-16C × α2-87, α1-9N × α1-930, α1-9N × α2-933, α2-5N × α1-930, and α2-5N × α2-933. Some of these combinations are preferred for cross-linking (27). In skeletal tissues, these divalent cross-links can then mature into trivalent cross-links, pyridinoline (Pyr), deoxypyridinoline (d-Pyr), and pyrrole (Prl) (1, 28), tying two or three collagen molecules (29).
Despite current knowledge regarding type I collagen glycosylation (14, 30), the precise molecular loci and the extent/type of glycosylation in cross-linked and non-cross-linked residues are still not well characterized. This information, however, is essential to understand the role of glycosylation in collagen biosynthesis and function. Mass spectrometry (MS) has become increasingly used in the structural characterization of collagens from different sources, and several studies have utilized MS to characterize collagen cross-linked peptides (30–34). Although these studies pioneered the MS-based characterization of collagen cross-linked species, their major limitation is the use of low resolution mass analyzers. This renders accurate characterization of species bearing naturally occurring heterogeneity (e.g. incomplete hydroxylation of Lys or Pro) or of species with similar mass/charge ratio (m/z), difficult. In the present study, we used high performance/high resolution nanoscale liquid chromatography-tandem mass spectrometry (nanoLC/MS/MS) to comprehensively characterize the glycosylation at various molecular loci in non-cross-linked and cross-linked peptides in bovine bone type I collagen. A multistep chromatographic approach was employed to obtain highly purified cross-linked tryptic peptides. The analytical challenges associated with these large cross-linked species were overcome with the use of an alternative enzyme. The molecular distribution of glycosylation in non-cross-linked Hyl and immature and mature cross-links was quantitatively determined by nanoLC/MS. The results revealed a differential glycosylation pattern, depending on the involvement in cross-linking, molecular loci, and type and maturational stage of cross-linking.
EXPERIMENTAL PROCEDURES
Collagen Preparation
Fresh femoral bone samples from 2–3-year-old bovine animals were obtained commercially (Aries Scientific, Dallas, TX). After removing the surrounding connective tissues, both ends of the bone, and the bone marrow, the bones were cut into small pieces. All operations were carried out at 4 °C. The bone pieces were pulverized to a fine powder under liquid nitrogen using a Spex Freezer Mill (Spex, Inc., Metuchen, NJ). Pulverized samples were washed several times with cold phosphate-buffered saline (PBS), and cold distilled water, centrifuged at 4000 × g for 30 min, and lyophilized. Bone powder was then demineralized with 0.5 m EDTA, pH 7.5, for 2 weeks with several changes of the EDTA solution by centrifugation at 4000 × g. The EDTA-insoluble residue was thoroughly washed with cold distilled water by repeated centrifugation at 4000 × g and lyophilized.
Reduction with NaB3H4
Demineralized bone (∼2.0 g) was suspended in buffer containing 0.15 m N-trismethyl-2-aminoethanesulfonic acid and 0.05 m Tris-HCl, pH 7.4, and reduced with standardized NaB3H4. The specific activity of the NaB3H4 was determined by the method described previously (35, 36). The reduced samples were washed with cold distilled water several times by repeated centrifugation at 4000 × g and lyophilized. Upon reduction, the dehydrodihydroxylysinonorleucine (dehydro-DHLNL) and dehydrohydroxylysinonorleucine (dehydro-HLNL) and their respective keto amine forms are reduced to stable secondary amines, DHLNL and HLNL, and radiolabeled simultaneously (+2 Da molecular mass increase). Hereafter, the terms DHLNL and HLNL will be used for both the unreduced and reduced forms.
Cross-link Analysis
Reduced collagen was hydrolyzed with 6 n HCl and subjected to cross-link analysis as described previously (37). The reducible cross-links were analyzed as their reduced forms (i.e. DHLNL and HLNL, respectively). The levels of the immature reducible (DHLNL and HLNL) and mature non-reducible cross-links (Pyr and d-Pyr) were quantified and expressed in mol/mol of collagen.
Digestion with Trypsin
Digestion with trypsin of reduced bone collagen was prepared by the procedure described previously (37) with slight modifications. Briefly, the reduced collagen was heated at 65 °C for 15 min, digested with 1% (w/w) trypsin for 16 h at 37 °C, reheated to 65 °C for 10 min, and retreated with 0.5% (w/w) trypsin for 3 h at 37 °C. Over 99% of the starting material was recovered in the supernatant of the trypsin digest.
Molecular Sieve Chromatography
Molecular sieve chromatography of the bone tryptic digest was performed on a HiLoad Superdex 75 preparative scale column (1.6 × 60 cm) equilibrated with 0.05 m ammonium bicarbonate (pH 7.9) at room temperature. Aliquots of the reduced bone tryptic digest (∼200 mg) were injected and separated at a flow rate of 0.75 ml/min. Fractions of 1.5 ml were collected, and their absorbance (230 nm), radioactivity, and fluorescence (excitation 330 nm and emission 390 nm) were measured. Fluorescence measurement was employed to detect Pyr and d-Pyr, and radioactivity was used to detect DHLNL and HLNL. The main fluorescent (F1–F3) and radioactive (R1 and R2) fractions were recovered. An equal aliquot from each fraction was lyophilized and subjected to cross-link analysis as described above. The majority of aliquots were subjected to further purification by reversed phase chromatography or digestion with chymotrypsin (see below). To identify fractions containing the Prl cross-link, aliquots from each fraction were subjected to Ehrlich's chromogen (EC) analysis following reported methods (30, 38). Briefly, the aliquots were lyophilized and resuspended in distilled water, and 35 μl of a 5% solution (w/v) of p-dimethylaminobenzaldehyde in 4 m perchloric acid were added. The reaction was incubated for 5 min at room temperature. The absorbance (572 nm) was monitored with a F-2000 fluorescence spectrophotometer (Hitachi, Tokyo, Japan). The main EC-positive fractions (P1–P3) eluted at similar positions as F1–F3 and were further characterized by mass spectrometry (see below).
Chymotrypsin Digestion
Chymotrypsin digestion of the fractions F1–F3, R1, and R2 collected after molecular sieve chromatography was performed as follows. The lyophilized fractions were dissolved in 100 μl of 25 mm ammonium bicarbonate (pH 7.4). Aliquots of 50 μl from each fraction were incubated with 2 μl of chymotrypsin solution (0.5 μg/μl in 25 mm ammonium bicarbonate) overnight at room temperature. These digests were further analyzed by nanoLC/MS/MS.
Reversed Phase High Performance Liquid Chromatography
The lyophilized fluorescent and radioactive fractions resolved by molecular sieve chromatography were further fractionated by reversed phase on a SOURCE 5RPC ST 4.6/150 column (GE Healthcare) using a Varian HPLC system (Prostar 240/310, Varian, Walnut Creek, CA). The solvents employed were 2% (v/v) acetonitrile in 10 mm ammonium acetate (solvent A) and 70% acetonitrile in deionized water (v/v) (solvent B). The samples were dissolved in solvent A and eluted with a linear gradient from 0 to 15% solvent B for the first 10 min, followed by a linear gradient from 15 to 50% solvent B over the next 50 min at a flow rate of 1.0 ml/min at room temperature. The effluent was monitored for absorbance (230 nm) and fluorescence (excitation 330 nm, emission 390 nm). Fractions of 1 ml were collected, and aliquots were subjected to radioactivity and fluorescence measurements. The radioactive and fluorescent fractions were pooled, lyophilized, and subjected to further analysis.
Ion-Exchange Chromatography
Ion-exchange chromatography of fractions collected after reversed phase separation was performed on a TSK-GEL DEAE-5PW column (8.0 × 75 mm, Tosoh Bioscience LLC, Montgomeryville, PA). The column was equilibrated with 0.01 m NH4HCO3 containing 1% isopropyl alcohol. Elution was carried out with a linear gradient from 0.01 to 0.25 m NH4HCO3 at a flow rate of 1 ml/min, monitoring the absorbance at 230 nm. One-minute fractions were collected, and those corresponding to fluorescent and/or radioactive signals were pooled and lyophilized. Their structural characterization was performed by mass spectrometry.
Nanoscale Liquid Chromatography, Mass Spectrometry, and Data Analysis
Glycosylated, non-cross-linked collagen peptides were characterized by nanoLC/MS/MS from a bovine bone tryptic digest on a nanoACQUITY UPLC-Q-Tof Premier mass spectrometer (Waters, Milford, MA). Their structure- and site-specific quantitative analyses were performed as described previously (24).
Flow Injection Analyses of Purified Cross-linked Tryptic Peptides
Flow injection analyses of purified cross-linked tryptic peptides were performed by positive ion nanoelectrospray (ESI+) on a Waters Micromass Q-Tof Micro mass spectrometer (Waters). The samples were desalted using a C18 ZipTip pipette tip (EMD Millipore, Billerica, MA) and reconstituted in 50% acetonitrile with 0.1% formic acid (v/v). Dilutions were performed as needed to ensure optimal signal intensity. Mass spectrometer parameters were as follows: capillary voltage, 3.8 kV; sampling cone, 30; source temperature, 80 °C; desolvation temperature, 20 °C.
NanoLC/MS/MS Analyses
NanoLC/MS/MS analyses of cross-linked peptides were carried out on a nanoACQUITY UPLC-Q-Tof Global mass spectrometer (Waters) with data-dependent acquisition and charge state selection of the top four ions. Separations of chymotryptic digests of fractions after molecular sieve chromatography were carried out on a C18 BEH column (1.7 μm, 75 μm × 100 mm) at 0.3 μl/min, with a gradient from 1 to 50% solvent B over 30 min. The solvents were as follows: solvent A (0.1% formic acid in deionized water) and solvent B (0.1% formic acid in acetonitrile). Mass spectrometer settings for (+)-nano-ESI were as follows: capillary voltage, 3.4 kV; sampling cone, 30; source temperature, 80 °C. To ensure optimal fragmentation, the collision energies were optimized over the range 20–30 V.
Data Analysis
Data analysis was performed using the MassLynx software, version 4.1 (Waters), including the embedded deconvolution algorithms MaxEnt 1 and 3. The glycoform distribution of C-telo-derived cross-linked peptides was determined from the LC/MS data of chymotryptic digests. Quantitative glycosylation analyses (percentage of free, galactosyl (G), and glucosylgalactosyl (GG)) of cross-linked peptides were performed as described for non-cross-linked peptides by integrating the extracted ion chromatograms (EICs) generated post-data acquisition for each charge state of individual glycoforms (24).
Glycosylation of Type II Collagen-derived Cross-links
Articular cartilage from tibial plateau of 0–1-year bovine knees were obtained using a 5-mm biopsy punch within 24 h of slaughter. The cartilage pieces were rinsed in cold PBS and stored at −20 °C in a protease inhibitor solution until use. The cartilage pieces were then pulverized, washed several times with cold PBS and cold distilled water thoroughly, and lyophilized. The dried samples were reduced with NaB3H4, digested with trypsin, and fractionated by molecular sieve chromatography as described above. The cross-link-containing fractions were identified by cross-link analysis and then subjected to nanoLC/MS/MS as described above.
RESULTS
Glycosylation of Non-cross-linked Peptides
To identify glycosylation sites in type I collagen, a tryptic digest of reduced bone collagen was analyzed by nanoLC/MS/MS on a Waters nanoACQUITY UPLC-Q-Tof Premier mass spectrometer (24, 39). Five glycosylation sites were characterized, as follows: α1-/α2–87, observed in glycopeptides α1-/α2-(76–90); α1-/α2-174 in glycopeptides α1-(145–183) and α2-(145–192), respectively; and α2-219 in glycopeptide α2-(193–237). The distribution of Lys, Hyl, and G- and GG-Hyl at each site was determined as described previously (24), and the results are summarized in Table 1. The highest relative extent of glycosylation was found for residues α1-/α2-87 (i.e. 68% G and 13% GG form for α1-87, and 63.1% G and 18.5% GG form for α2-87, respectively). Site α2–174 was found mostly in the form of G (56%) with a minor amount of GG (2.9%) and with a considerable level of non-hydroxylated/non-glycosylated Lys (29.3%). Site α2-219 was found to contain 20% (Lys), 42.6% (Hyl), 29.9% (G-Hyl), and (7.5% GG-Hyl). Site α1-174 was found minimally modified by glycosylation, with only 1.5% G-Hyl and noise level GG-Hyl, with 49.6% non-glycosylated/non-hydroxylated Lys. Residues α1-/α2-87, the major glycosylation sites (24), are also major helical cross-linking sites, whereas the other three glycosylated residues are not involved in cross-linking.
TABLE 1.
Quantitative site-specific modification analysis by LC/MS/MS of non-cross-linked, glycosylated residues in bovine bone type Ι collagen
| Chain residue no. | Site occupancy |
|||
|---|---|---|---|---|
| Lys | Hyl | G-Hyl | GG-Hyl | |
| % | % | % | % | |
| α1-87 | 12.6 ± 3.1 | 6.3 ± 1.1 | 68.1 ± 4.2 | 13.0 ± 1.9 |
| α1-174 | 49.6 ± 5.0 | 48.9 ± 4.6 | 1.5 ± 0.4 | n/a |
| α2-87 | 15.4 ± 0.9 | 3.0 ± 0.3 | 63.1 ± 1.7 | 18.5 ± 2.6 |
| α2-174 | 29.3 ± 1.2 | 11.8 ± 0.7 | 56.0 ± 1.1 | 2.9 ± 0.3 |
| α2-219 | 20.0 ± 1.1 | 42.6 ± 0.4 | 29.9 ± 0.2 | 7.5 ± 1.0 |
Two additional helical Hyl residues involved in cross-linking are α1-930 and α2-933. The tryptic peptide containing α1-930 spans the residues 928GI-Hyl930-GHR933. This peptide was not observed by LC/MS/MS, most likely due to its short length. Residue α2-933 was observed in peptide α2-(928–963) and assigned based on the MS/MS of m/z 677.281 (5+) (supplemental Fig. S1). The following modified residues are consistent with this fragment ion spectrum: Hyp-930, -942, and -954, Hyl-933, and deamidated Asn-936 (Fig. 1). Residue α2-933 was observed non-glycosylated. Both Asn-936 and the deamidated form were observed in peptide α2-(928–963), in an approximate Asp/Asn ratio of 3:1. This is relevant for the analysis of cross-linked peptides containing α2-(928–963) (see below).
FIGURE 1.

Deconvoluted MS/MS spectrum of the precursor ion of m/z 677.281 (5+), assigned to peptide α2-(928–963). The following modifications are consistent with the fragment ion spectrum: Hyl-933, Hyp-930, Hyp-942, Hyp-954, and deamidated Asn-936 (indicated as N → D). Site α2-Hyl-933 is involved in intermolecular cross-linking. Within the non-cross-linked structures containing residue α2-Hyl-933 (i.e. spectrum shown above), no glycosylation was observed in bovine bone.
Isolation, Molecular Characterization, and Glycosylation of Cross-linked Peptides
Using a NaB3H4-reduced collagen tryptic digest as starting material, a multistep chromatographic approach was employed to obtain highly purified tryptic cross-linked peptides. Three sequential chromatographic steps were performed: 1) molecular sieve; 2) reversed phase; and 3) ion exchange. After each step, the fractions containing immature and mature cross-linked peptide were detected based on radioactivity and fluorescence, respectively (Fig. 2), whereas Prl cross-links were detected by reaction with EC (see “Experimental Procedures”) (40). The chromatographic profiles are shown in Fig. 2, with the major fluorescent and radioactive peaks designated as F1–F3 and R1 and R2, respectively. Cross-link analysis performed after molecular sieve separation was in agreement with the profiles shown in Fig. 2 (i.e. Pyr/d-Pyr were found in fractions F1–F3, whereas DHLNL/HLNL were found in R1 and R2). Quantitative cross-link analysis on bone collagen (Table 2) indicated that DHLNLs are the most abundant (1.58 mol/mol of collagen), followed by HLNL (0.50 mol/mol) and Pyr (0.28 mol/mol), whereas d-Pyr was found in low amounts (0.04). Based on these numbers, the molecular loci identified in F1–F3 and R1 and R2 (i.e. R1 and F1 contain the C-telo derived cross-links; see below), and direct cross-link analyses of these fractions, we estimated that within DHLNLs, the C-telo-derived (α1/2-87-involved) cross-link was ∼4-fold higher than that of the N-telo-derived (∼1.3 versus ∼0.3), whereas the levels of C- and N-telo-derived HLNLs were comparable. Pyr was almost equally located between C- and N-telo, whereas d-Pyr was enriched in the N-telo (∼80% of total d-Pyr). The relative small fluorescence of the N-telo Pyr/d-Pyr (F2 and F3) in Fig. 2 is probably due to the fluorescence quenching (30). From F1 and R1 fractions, after the final chromatographic step, one fluorescent (a) and two radioactive peaks (b and c) were collected (Fig. 2, D–F) and were analyzed by flow injection nano-ESI-MS.
FIGURE 2.

A, molecular sieve elution profile of a tryptic digest of NaB3H4-reduced mature bone collagen. Eight peaks were collected, as follows: F1–F3 (fluorescent), R1 and R2 (radioactive content), and P1–P3 (EC-reactive species, absorbance at 572 nm). B and C, reversed phase purifications of peaks F1 and R1, resulting in peak 1 (fluorescent) and peaks 2 and 3 (radioactive), respectively. D–F, ion exchange purification of peaks 1, 2, and 3, resulting in fluorescent peak a and radioactive peaks b and c, respectively.
TABLE 2.
Quantitative cross-link analysis (in mol/mol of collagen) of immature and mature cross-links in bovine bone type I collagen and estimated values of the C-telo-derived cross-links
| DHLNL | HLNL | Pyr | d-Pyr | |
|---|---|---|---|---|
| mol/mol | mol/mol | mol/mol | mol/mol | |
| Total bovine digest | 1.58 (100%) | 0.50 (100%) | 0.28 (100%) | 0.04 (100%) |
| C-telo-derived | ∼1.3 (∼80%) | ∼0.25 (∼50%) | ∼0.11 (∼40%) | ∼0.007 (∼20%) |
Flow Injection Analysis-Mass Spectrometry of Peaks a, b, and c
The theoretical molecular weights of collagen cross-linked peptides were determined using the equations listed in Table 3. The raw mass spectrum of the cross-linked species contained in peak a is shown in Fig. 3A. The charge state envelope containing the ions of m/z 2532.62 (4+), 2026.27 (5+), 1688.75 (6+), 1447.65 (7+), 1266.81 (8+), and 1126.14 (9+) was deconvoluted to the average mass of 10,126.4 Da (Fig. 3A, inset). This value is consistent with the theoretical average molecular weight of G-Pyr cross-linked tryptic peptides α1-(993–22C) × α1-(993–22C) × α1-(76–90). The peptide identities were confirmed by MS/MS (not shown). Ion clusters of species with a molecular mass of 162 Da lower or higher than G-Pyr, corresponding to Pyr and GG-Pyr, respectively, were observed with very low relative abundance, indicating that G is the predominant glycoform of the C-telo-derived Pyr cross-link.
TABLE 3.
Theoretical molecular weight (Mr) determination of collagen cross-linked peptides in bone
| Mr (theoretical) | Equation |
|---|---|
| Immaturea | |
| Mr(DHLNL) = Mr(peptide 1) + Mr(peptide 2) − 16 | 1 |
| Mr(HLNL) = Mr(DHLNL) − 16 | 2 |
| Mature | |
| Mr(Pyr) = Mr(peptide 1) + Mr(peptide 2) + Mr(peptide 3) − 57 | 3 |
| Mr(d-Pyr) = Mr(Pyr) − 16 | 4 |
| Mr(pyrrole) = Mr(Pyr) − 15 | 5 |
a The Mr of immature cross-linked peptides was determined by accounting for the incorporation of two 3H atoms, as a result of reduction with NaB3H4.
FIGURE 3.
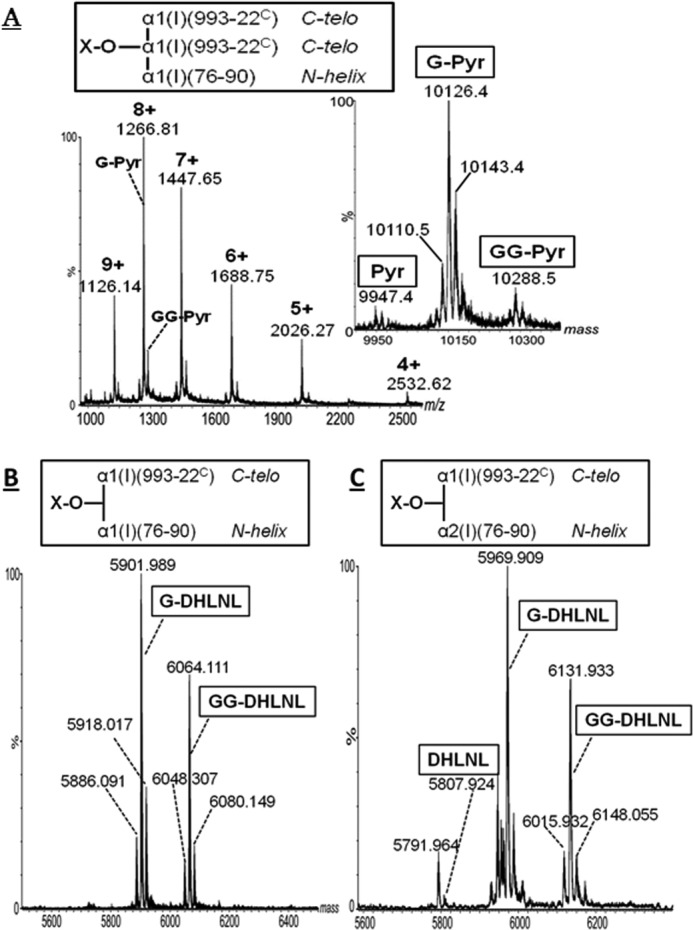
MS spectra of pyridinoline cross-linked tryptic peptides α1-(993–22C) × α1-(993–22C) × α1-(76–90) (A) (inset shows the masses obtained by deconvoluting the m/z range 1000–2600); DHLNL α1-(993–22C) × α1-(76–90) (B); and DHLNL α1-(993–22C) × α2-(76–90) (C). Glycosylation is indicated by G and GG. These spectra were obtained by flow injection analysis of fractions purified by multistage chromatography.
The deconvoluted mass spectra of the DHLNL cross-linked peptides α1-(993–22C) × α1-(76–90) and α1-(993–22C) × α2-(76–90) observed in peaks b and c are shown in Fig. 3, B and C, and Supplemental Fig. S2, respectively. The ratio of the former to the latter was ∼3:1 (Fig. 2C), which is consistent with our previous report (36). In contrast to Pyr, both the G and GG glycoforms of α1-/α2-87-containing DHLNLs represent major species. As shown in Fig. 3, B and C, extended molecular heterogeneity of the cross-linked peptides was observed as ±16 Da species. The +16 Da might be due to oxidized Met-86 in the α1h peptide, whereas the −16 Da could be attributed to either incomplete Pro hydroxylation or HLNL species, respectively. The occurrence of the latter was confirmed in the fractions b and c (i.e. when the fractions were hydrolyzed and directly analyzed for cross-links, HLNL was present as a minor species in both fractions). These low abundance species are difficult to assign because their MS/MS lack informative fragment ions. Nevertheless, the species of molecular mass 5791.96 Da (Fig. 3C) probably represent the HLNL (α1-16C × α2-87). This would be in agreement with a previous study suggesting higher abundance of HLNL (α1-16C × α2-87) compared with HLNL (α1-16C × α1-87) (41).
Quantitative Determination of Glycosylation in the C-telopeptide Containing Immature and Mature Cross-linked Peptides
For LC/MS-based quantitative glycosylation analysis of the C-telo cross-linked peptides, the multistage chromatographic approach was modified as follows. 1) the NaB3H4-reduced bone tryptic digest was separated by molecular sieve chromatography as described above. Fractions were collected based on fluorescence (F1–F3) and radioactivity (R1 and R2). 2) the tryptic peptide fractions F1–F3, R1, and R2 were digested with chymotrypsin to reduce the size of the cross-linked peptides. 3) the resulting double digests were analyzed by nanoLC/MS/MS.
The major chymotryptic product was formed by cleavage in the C-telopeptide between Tyr and Asp, resulting in the α1C sequence DLSFLPQPPQEKAHDGGR, whereas the helical peptides (residues 76–90) remained undigested. All mature cross-link species, including C-telo Pyr/Prl (α1C × α1C × α1h/α2h) were identified in the chymotryptic digests of F1/P1 by nanoLC/MS/MS. Prl (α1C × α1C × α2h) was identified as G-Prl, but, due to the low abundance, its relative glycoforms (free, G-, and GG-) were not quantitatively estimated (Table 4A). The average molecular mass of G-Pyr (α1C × α1C × α1h) (10,126.4 Da; Fig. 3) was reduced to 5614.501 Da, as determined from the charge state envelope of m/z 802.964 (7+), 936.618 (6+), 1123.777 (5+), and 1404.487 (4+). The MS/MS of m/z 1123.74 (5+) (Fig. 4A and supplemental Fig. S3A) confirms the sequences of both C-telo and N-helical peptides, and the molecular ion is consistent with the presence of galactose. Consistent with the above data, G-Pyr (α1C × α1C × α1h) was the most abundant glycoform (81%), whereas GG-Pyr (m/z 963.634 (6+)) and non-glycosylated Pyr (m/z 909.607 (6+)) had markedly lower abundances (13 and 6%, respectively; Table 4A). The G-Prl (α1C × α1C × α1h) species, having a 15 Da lower molecular mass than G-Pyr, was observed as the ion of m/z 934.292 (6+). Furthermore, G- and GG-Pyr (α1C × α1C × α2h) were observed in chymotryptic fraction F1/P1 as the ions of m/z 947.969 (6+) (supplemental Fig. S3B) and 974.951 (6+), respectively. The MS/MS of the ion of m/z 947.969 (6+) is shown in Fig. 4B.
TABLE 4.
Glycoform distribution (free, G, and GG)
A, C-telopeptide immature and mature bone cross-links. B, relative abundance of α1h/α2h containing Pyr and of α1h-Pr1/Pyr. Values represent mean (S.D. in parentheses) from three independent experiments.

a ND, not detected.
b Identified, but due to the low abundance, the relative glycoforms were not estimated (see “Results”).
FIGURE 4.

Deconvoluted MS/MS spectra of m/z 1123.74 (5+), assigned to G-Pyr (α1C × α1C × α1h) (A); m/z 947.969 (5+), assigned to G-Pyr (α1C × α1C × α2h) (B); m/z 912.939 (4+), assigned to G-DHLNL (α1C × α1h) (C); and m/z 929.966 (4+), assigned to G-DHLNL (α1C × α2h) (D). These ions were observed in the LC/MS/MS analysis of chymotrypsin digests of fractions F1 and R1 collected after molecular sieve chromatography. The superscripts C and h indicate the C-telopeptide and the helical peptides, respectively.
The C-telo DHLNLs were well characterized in both tryptic and chymotryptic forms. The MS/MS spectra of the G-DHLNL α1C × α1h/α2h protonated molecules (Fig. 4, C and D, and supplemental Fig. S3, C and D) confirm their structural assignment. In MS/MS, no neutral loss of G was observed, suggesting increased stability of the glycosidic bond of cross-linked peptides. Lower abundance glycosylated ions G- and GG-HLNLs were assigned based on the 16 Da lower mass compared with the corresponding DHLNL glycoforms.
To determine the distribution of the free, G, and GG forms in immature/mature C-telo-containing cross-linked species, glycoform EICs were generated from the LC/MS of F1- and R1-chymotryptic digests and were normalized to the most abundant species within a cross-link type (Fig. 5). Quantitative analyses of technical triplicates are shown in Table 4. Within mature cross-links Pyr/Prl α1C × α1C × α1h/α2h, the G glycoform is the most abundant, whereas GG is minimal. The relative amounts of α1C free Pyr are higher in α2h compared with α1h. In contrast to mature cross-links, both the G and GG glycoforms of the DHLNL cross-links α1C × α1h/α2h are highly abundant, with minute amounts of free DHLNLs. The glycosylation patterns of HLNL α1C × α1h/α2h are comparable with those of DHLNLs. However, quantitative analyses of HLNLs and of α2h-Prl glycoforms were not performed because of poor signal/noise ratio.
FIGURE 5.

A–G, EICs showing the relative glycoform distribution (free, G, and GG) for various C-telopeptide-containing immature and mature cross-linked peptides observed in the LC/MS-MS analyses of chymotryptic digests of fractions F1 and R1 collected after molecular sieve separation. For each cross-linked species, the most abundant charge state was used to generate the EICs. For a particular cross-linked species, the EICs were normalized to that of the most abundant glycoform.
The relative abundance of the C-telo-/helical residue 87-based Pyr (i.e. the α1h/α2h ratio) was estimated from the LC/MS data of chymotryptic F1. The EICs of α1h and α2h G-Pyr (Fig. 6A) suggest that α1h-Pyr is far more abundant (∼85%) than α2h-Pyr (∼15%). This might explain why the α2h Pyr was not observed previously by flow injection analysis of peaks a, b, and c. Moreover, we determined the relative abundance of Pyr/Prl containing α1-87 to be ∼4:1 (Fig. 6B and Table 4B).
FIGURE 6.

EICs of C-telopeptide galactosyl-pyridinoline species containing helical residues α1- and α2-87 (A) and C-telopeptide-based Pyr and pyrrole (B). The EICs were generated for the most abundant charge state (6+) of the most abundant glycoforms (G-Pyr and G-pyrrole, respectively) observed in the chymotryptic digest of fraction F1/P1 collected after molecular sieve chromatography.
Characterization of N-telopeptide Containing Immature and Mature Cross-links
By flow injection analysis, the molecular mass of free (non-glycosylated) DHLNL α1N × α2h, determined as 5943.18 Da, is consistent with the structure of the reduced cross-link α1N-(15N–9) × α2h-(928–963) containing pyro-Gln in α1N peptide and three Hyp residues and deamidated Asn-936 in the α2h peptide. This assignment was confirmed from the MS/MS of the ion of m/z 991.530 (6+) (Fig. 7A and supplemental Fig. S4, A and B). The α1N × α2h was observed completely non-glycosylated, consistent with the profile of the non-cross-linked peptide α2-(928–963) (see above).
FIGURE 7.

Deconvoluted MS and MS/MS spectra of m/z 991.530 (6+), assigned to non-glycosylated DHLNL α1-(1N–9) × α2-(928–963) (A); m/z 807.887 (4+), assigned to non-glycosylated HLNL α1-(1N–9) × α1-(928–933) (B); and deconvoluted MS spectrum of cross-linked species α1-(1N–9) × α2-(1N–9) × α2-(928–963) (top) and deconvoluted MS/MS of m/z 1097.83 (7+) (bottom) (C), confirming the identity of the indicated cross-linked peptides.
After molecular sieve separation, the free DHLNL α1N × α1h, consisting of tryptic peptides α1N-(15N–9) × α1h-(928–933), was identified in fraction R2 as the ions of m/z 811.878 (4+) and 649.703 (5+) (data not shown). The MS/MS spectrum of the precursor ion of m/z 807.866 (4+) (Fig. 7B and supplemental Fig. S4, A and B), assigned to reduced non-glycosylated HLNL (α1N × α1h), confirms the identity of peptides α1N-(15N–9) and α1h-(928–933). The cross-links α1N × α1h were also found completely non-glycosylated. The N-telopeptide cross-link α2N × α1h was not observed.
The mature N-telo cross-linked tryptic peptides were observed in fractions F2/P2 and F3/P3 after molecular sieve chromatography, with the major species assigned to non-glycosylated Pyr/Prl α1N × α2N × α2h in fraction F2/P2 and non-glycosylated Pyr (α1N × α1N × α1h and α1N ×α2N × α1h) and Prl (α1N×α2N×α1h) in fraction F3/P3. The average molecular mass of 7678.70 Da (Fig. 7C) is consistent with the calculated mass of α1N-(1N–9) × α2N-(1N–9) × α2h-(928–963), containing pyro-Gln in the α1N/α2N peptides and deamidated Asn-936 and three Hyp residues in the α2h peptide. Additional species at +16/−15 Da most likely represent the Prl (−15 Da), whereas +16 Da might be due to Met oxidation or additional Pro hydroxylation. Peptide identities were established from the MS/MS of m/z 1097.83 (7+) (Fig. 7C and supplemental Fig. S4C). Furthermore, incomplete hydroxylation of Hyp-954 in α2-(928–963) is suggested by the fragment ion of m/z 878.49, which is 16 Da lower than y10h (m/z 894.48). These features and the fragment ion clusters at m/z 6781.37, 6782.18, and 6797.09 suggest increased molecular heterogeneity of Pyr/Prl α1N × α2N × α2h. Hence, the average mass of 7678.70 most likely arises from overlapping isotopic envelopes of Pyr and Prl (+16 Da). Qualitatively, these data suggest that the Prl/Pyr ratio is higher in the N- than in C-telopeptide (Fig. 6). Most importantly, all immature and mature N-telo cross-links were found non-glycosylated.
Glycosylation in the C-telopeptide-containing Immature and Mature Cross-linked Peptides from Type II Collagen
Following the initial molecular sieve chromatography of reduced tryptic digests, the cross-link-containing fractions were characterized by nanoLC/MS as described above. The C-telo (type II collagen)-derived DHLNL (α1(II)-17C × α1(II)-87h)-containing peptide was identified as a larger peptide, α1(II)-(993–26C) × α1h(II)-(76–90). Apparently, the peptide bond R–E in the C-telo was miscleaved due to the presence of an acidic amino acid (E) adjacent to R (42). This fraction was retrypsinized, and the C-telo-containing Pyr peptide was identified and characterized. The DHLNL involving the residue Hyl-87 was identified in the form of non-glycosylated (m/z 1050.17 (6+)) and GG-DHLNL (1104.17 (6+)) (45 and 54%, respectively), whereas G-DHLNL (m/z 1077.17 (6+)) had markedly lower abundance (∼1%; Fig. 8). Non-glycosylated Pyr, G-Pyr, and GG-Pyr (α1C × α1C × α1h) were observed as the ions of m/z 967.67 (4+), 1008.20 (4+), and 1048.71 (4+), respectively. The non-glycosylated Pyr was the most abundant form (93%), with minute amounts of G- and GG-Pyr (<1% and ∼6%, respectively; Fig. 8). Thus, diglycosylated Pyr involving α1(II)-87 in type II collagen was markedly low, whereas it is one of the major forms in its precursor DHLNL. This is consistent with the above bone type I collagen data and is similar to what was described by Eyre et al. (32). The N-telo-derived DHLNL and Pyr cross-linked peptides involving α1(II)-Hyl-930 were also identified, and none was glycosylated (data not shown), which is also the same as for bone type I collagen.
FIGURE 8.

Relative glycoform distribution (free, G, and GG) of C-telopeptide based Pyr (A) and DHLNL (B) cross-linked peptides in type II collagen observed in the LC/MS-MS analyses. For each cross-linked species, the most abundant charge state (4+ for Pyr and 6+ for DHLNL, respectively) was used to generate the EICs. Within each set of glycosylated cross-links, the EICs were normalized to that of the most abundant glycoform.
DISCUSSION
Collagen glycosylation, consisting of the O-glycosides in the form of G- and GG-Hyl, is a key modification involved in collagen cross-linking, fibrillogenesis, mineralization, and collagen-protein interactions (1, 21, 24, 29, 43, 44). Type I collagen, the main organic component in bone, is one of the minimally glycosylated members in the collagen family (45). Even among type I collagen in various tissues, such as skin type I collagen, the extent of glycosylation in bone type I collagen is low (45, 46). Despite numerous studies indicating the functional importance of this modification in type I collagen biosynthesis, the type and distribution of Hyl glycosides and their involvement in cross-linking have not been characterized in a comprehensive manner. In this study, by employing a wide range of analytical methods, we performed residue-specific quantitative glycosylation analysis in both non-cross-linked and cross-linked peptide species from bovine bone type I collagen.
Five Hyl residues were found glycosylated in bovine bone type I collagen (i.e. α1-/α2-87, α1-/α2-174, and α2-219). Among the non-cross-linked peptides, the major glycosylation occurs at the helical cross-linking site, α1-/α2-87, and is present mainly as G-Hyl with lower relative amounts of GG-Hyl. This is in contrast to collagen secreted from mouse calvaria-derived MC3T3-E1 osteoblastic cells, where α1-87 is mostly found as GG-Hyl, and α2-87 is >90% non-glycosylated Hyl (24). Differences in the α2-87 glycosylation between bovine and mouse could be related to species-specific variations in the α2 collagen sequence (GFKGVK in mouse versus GFKGIR in bovine and other species), suggesting a certain substrate specificity for collagen galactosyltransferases (24).
The C- and N-telo immature and mature cross-links were characterized in the form of tryptic/chymotryptic cross-linked peptides. In agreement with previous studies of bone collagen (24, 36), cross-link analysis suggested that the ∼80% of DHLNL was derived from the C-telo site. This might represent a feature conserved among species, because a similar distribution was found in mouse osteoblasts as well (24). Cross-linked peptides containing DHLNL α1-16C × α1/α2-87 and HLNL α1-16C × α1-87 were found mostly glycosylated in the form of both G and GG glycoforms having similar abundances. In contrast, the N-telo DHLNL α1-9N × α1-930, α1-9N × α2-933, and α2-5N × α2-933 were found non-glycosylated (30).
In contrast to the immature cross-links, the mature Pyr/Prl species (α1–16C × α1-16C × α1/2-87) were found glycosylated primarily as G-Pyr, with minute amounts of the free and GG glycoforms. In this study, we identified the previously undescribed α2-87-containing Pyr and Prl peptide. In comparison with the α1–87-involved Pyr and Prl, this is a minute species representing ∼15% of the former. We previously reported the residue α1-87 to be the preferential cross-linking site over α2-87 (>3:1) for the formation of C-telo-derived divalent cross-links in bone. Thus, the low abundance of the mature trivalent cross-links is not surprising. This is probably due to the specific molecular packing in the fibril (35), which could be important for collagen mineralization in an orderly fashion (36).
It is generally accepted that two residues of immature cross-links, DHLNL/HLNL, mature into one residue of trivalent cross-link, Pyr/d-Pyr, with aging (29, 43). However, our in vitro incubation study and the report by Saito et al. (47) showed that the decreased level of immature cross-links is disproportionally higher than the increase of Pyr (24). Most likely, the spontaneous non-enzymatic maturation is controlled by the microenvironment, such as presence of mineral (30, 48), the glycosylation state of the immature cross-links (35), and the presence of collagen-binding proteoglycans around the cross-linking sites. The data in the current study showing abundance of both G- and GG-DHLNL/HLNL but the predominance of G-Pyr and G-Prl forms suggests that GG divalent cross-links may not favor maturation into trivalent cross-links. This was supported by the data on type II collagen. In this heavily hydroxylated and glycosylated collagen type (49), we also found that ∼50% of the Hyl-87 involving DHLNL is in the form of GG-DHLNL, whereas GG-Pyr at the same locus is minimal. Eyre et al. (32) also reported the lack of glycosylated Pyr at this locus (residue 87) in type II collagen. Possibly, the bulky disaccharide structure sterically hinders or delays the condensation reaction to form mature cross-links. Along with this conjecture, it is interesting to note that, for the N-telo-derived cross-links in which no glycosylated forms were found, immature cross-links were significantly lower, and mature cross-links were higher than the heavily glycosylated C-telo-derived cross-links. Possibly, without glycosylation of the helical Hyl, maturation of the N-telo-derived cross-links is accelerated. Clearly, further studies are warranted to determine the fate of glycosylated immature cross-links by employing, for instance, an in vitro maturation study (24) using type I and other types of collagen.
Recently, trivalent Prl cross-links having an identical chain topology with Pyr were identified (30, 50, 51). These were predominantly observed at the N-telo-to-helix site (30, 50, 51), concentrated at the loci α1-9N × α2-5N × α2-933 (30, 50) and α1-9N × α1-9N × α1-930/α2-933 (51). Here, we characterized the Prl species α1-16C × α1-16C × α1-87/α2-87 and α1-9N × α2-5N × α1-930/α2-933. Prl has a glycosylation pattern similar to that of Pyr (i.e. C-telo Prl contains mainly G, whereas N-telo Prl are non-glycosylated). This indicates there is no preference in the glycosylation pattern between Pyr and Prl cross-links.
In conclusion, this study provides a detailed molecular characterization of glycosylation and its involvement in intermolecular cross-linking in bovine bone type I collagen. Specific molecular loci and the differential glycosylation pattern between the immature and mature cross-links suggest that glycosylation might regulate the cross-link maturation in fibrillar collagen.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants DE020909 and AR060978 (to M. Y.) and the Intramural Research Program of National Institutes of Health, NIEHS, Project ES050171 (to K. B. T.).

This article contains supplemental Figs. S1–S4.
- Hyl
- hydroxylysine
- G
- galactosyl
- GG
- glucosylgalactosyl
- C-telo
- C-telopeptide(s)
- N-telo
- N-telopeptide(s)
- Pyr
- pyridinoline
- d-Pyr
- deoxypyridinoline
- Prl
- pyrrole
- DHLNL
- dihydroxylysinonorleucine
- HLNL
- hydroxylysinonorleucine
- EC
- Ehrlich's chromogen
- EIC
- extracted ion chromatogram
- Hyp
- hydroxyproline
- ESI
- electrospray ionization
- nanoLC
- high performance/high resolution nanoscale liquid chromatography.
REFERENCES
- 1. Yamauchi M., Sricholpech M. (2012) Lysine post-translational modifications of collagen. Essays Biochem. 52, 113–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ruotsalainen H., Sipilä L., Vapola M., Sormunen R., Salo A. M., Uitto L., Mercer D. K., Robins S. P., Risteli M., Aszodi A., Fässler R., Myllylä R. (2006) Glycosylation catalyzed by lysyl hydroxylase 3 is essential for basement membranes. J. Cell Sci. 119, 625–635 [DOI] [PubMed] [Google Scholar]
- 3. Veis A. (1993) Mineral-matrix interactions in bone and dentin. J. Bone Miner. Res. 8, S493–S497 [DOI] [PubMed] [Google Scholar]
- 4. Weiner S., Traub W. (1986) Organization of hydroxyapatite crystals within collagen fibrils. FEBS Lett. 206, 262–266 [DOI] [PubMed] [Google Scholar]
- 5. Landis W. J., Song M. J. (1991) Early mineral deposition in calcifying tendon characterized by high voltage electron microscopy and three-dimensional graphic imaging. J. Struct. Biol. 107, 116–127 [DOI] [PubMed] [Google Scholar]
- 6. Silver F. H., Landis W. J. (2011) Deposition of apatite in mineralizing vertebrate extracellular matrices: a model of possible nucleation sites on type I collagen. Connect. Tissue Res. 52, 242–254 [DOI] [PubMed] [Google Scholar]
- 7. Myllyharju J., Kivirikko K. I. (2004) Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 20, 33–43 [DOI] [PubMed] [Google Scholar]
- 8. Myllyharju J, Kivirikko KI. (2001) Collagens and collagen-related diseases. Ann. Med. 33, 7–21 [DOI] [PubMed] [Google Scholar]
- 9. Bank R. A., Robins S. P., Wijmenga C., Breslau-Siderius L. J., Bardoel A. F. J., van der Sluijs H. A., Pruijs H. E. H., TeKoppele J. M. (1999) Defective collagen crosslinking in bone, but not in ligament or cartilage, in Bruck syndrome: indications for a bone-specific telopeptide lysyl hydroxylase on chromosome 17. Proc. Natl. Acad. Sci. U.S.A. 96, 1054–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Byers P. H., Pyott S. M. (2012) Recessively inherited forms of osteogenesis imperfecta. Annu. Rev. Genet. 46, 475–497 [DOI] [PubMed] [Google Scholar]
- 11. Forlino A., Cabral W. A., Barnes A. M., Marini J. C. (2011) New perspectives on osteogenesis imperfecta. Nat. Rev. Endocrinol. 7, 540–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Heikkinen J., Risteli M., Wang C., Latvala J., Rossi M., Valtavaara M., Myllylä R. (2000) Lysyl hydroxylase 3 is a multifunctional protein possessing collagen glucosyltransferase activity. J. Biol. Chem. 275, 36158–36163 [DOI] [PubMed] [Google Scholar]
- 13. Amudeswari S., Liang J. N., Chakrabarti B. (1987) Polar-apolar characteristics and fibrillogenesis of glycosylated collagen. Coll. Relat. Res. 7, 215–223 [DOI] [PubMed] [Google Scholar]
- 14. Bätge B., Winter C., Notbohm H., Acil Y., Brinckmann J., Müller P. K. (1997) Glycosylation of human bone collagen I in relation to lysylhydroxylation and fibril diameter. J. Biochem. 122, 109–115 [DOI] [PubMed] [Google Scholar]
- 15. Notbohm H., Nokelainen M., Myllyharju J., Fietzek P. P., Müller P. K., Kivirikko K. I. (1999) Recombinant human type II collagens with low and high levels of hydroxylysine and its glycosylated forms show marked differences in fibrillogenesis in vitro. J. Biol. Chem. 274, 8988–8992 [DOI] [PubMed] [Google Scholar]
- 16. Torre-Blanco A., Adachi E., Hojima Y., Wootton J. A. M., Minor R. R., Prockop D. J. (1992) Temperature-induced post-translational over-modification of type I procollagen. J. Biol. Chem. 267, 2650–2655 [PubMed] [Google Scholar]
- 17. Yang C. L., Rui H., Mosler S., Notbohm H., Sawaryn A., Müller P. K. (1993) Collagen I1 from articular cartilage and annulus fibrosus Structural and functional implication of tissue specific posttranslational modifications of collagen molecules. Eur. J. Biochem. 213, 1297–1302 [DOI] [PubMed] [Google Scholar]
- 18. Rautavuoma K., Takaluoma K., Passoja K., Pirskanen A., Kvist A. P., Kivirikko K. I., Myllyharju J. (2002) Characterization of three fragments that constitute the monomers of the human lysyl hydroxylase isoenzymes 1–3. The 30-kDa N-terminal fragment is not required for lysyl hydroxylase activity. J. Biol. Chem. 277, 23084–23091 [DOI] [PubMed] [Google Scholar]
- 19. Wang C., Risteli M., Heikkinen J., Hussa A. K., Uitto L., Myllyla R. (2002) Identification of amino acids important for the catalytic activity of the collagen glucosyltransferase associated with the multifunctional lysyl hydroxylase 3 (LH3). J. Biol. Chem. 277, 18568–18573 [DOI] [PubMed] [Google Scholar]
- 20. Rautavuoma K., Takaluoma K., Sormunen R., Myllyharju J., Kivirikko K. I., Soininen R. (2004) Premature aggregation of type IV collagen and early lethality in lysyl hydroxylase 3 null mice. Proc. Natl. Acad. Sci. U.S.A. 101, 14120–14125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sricholpech M., Perdivara I., Nagaoka H., Yokoyama M., Tomer K. B., Yamauchi M. (2011) Lysyl hydroxylase 3 glucosylates galactosylhydroxylysine residues in type I collagen in osteoblast culture. J. Biol. Chem. 286, 8846–8856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suarez K. N., Romanello M., Bettica P., Moro L. (1996) Collagen type I of rat cortical and trabecular bone differs in the extent of posttranslational modifications. Calcif. Tissue Int. 58, 65–69 [DOI] [PubMed] [Google Scholar]
- 23. Moro L., Romanello M., Favia A., Lamanna M. P., Lozupone E. (2000) Posttranslational modifications of bone collagen type I are related to the function of rat femoral regions. Calcif. Tissue Int. 66, 151–156 [DOI] [PubMed] [Google Scholar]
- 24. Sricholpech M., Perdivara I., Yokoyama M., Nagaoka H., Terajima M., Tomer K. B., Yamauchi M. (2012) Lysyl hydroxylase 3-mediated glucosylation in type I collagen: molecular loci and biological significance. J. Biol. Chem. 287, 22998–23009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vogel W., Gish G. D., Alves F., Pawson T. (1997) The discoidin domain receptor tyrosine kinases are activated by collagen. Mol. Cell 1, 13–23 [DOI] [PubMed] [Google Scholar]
- 26. Myers L. K., Myllyharju J., Nokelainen M., Brand D. D., Cremer M. A., Stuart J. M., Bodo M., Kivirikko K. I., Kang A. H. (2004) Relevance of posttranslational modifications for the arthritogenicity of type II collagen. J. Immunol. 172, 2970–2975 [DOI] [PubMed] [Google Scholar]
- 27. Yamauchi M., Katz E. P. (1993) The post-translational chemistry and molecular packing of mineralizing tendon collagens. Connect. Tissue Res. 29, 81–98 [DOI] [PubMed] [Google Scholar]
- 28. Eyre D. R., Weis M. A. (2013) Bone collagen: new clues to its mineralization mechanism from recessive osteogenesis imperfecta. Calcif. Tissue Int. 93, 338–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eyre D. R. (1984) Cross-linking in collagen and elastin. Annu. Rev. Biochem. 53, 717–748 [DOI] [PubMed] [Google Scholar]
- 30. Hanson D. A., Eyre D. R. (1996) Molecular site specificity of pyridinoline and pyrrole cross-links in type I collagen of human bone. J. Biol. Chem. 271, 26508–26516 [DOI] [PubMed] [Google Scholar]
- 31. Eyre D. R., Weis M. A., Wu J. J. (2010) Maturation of collagen ketoimine cross-links by an alternative mechanism to pyridinoline formation in cartilage. J. Biol. Chem. 285, 16675–16682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eyre D. R., Weis M. A., Wu J. J. (2008) Advances in collagen cross-link analysis. Methods 45, 65–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Henkel W., Dreisewerd K. (2007) Cyanogen bromide peptides of the fibrillar collagens I, III, and V and their mass spectrometric characterization: detection of linear peptides, peptide glycosylation, and cross-linking peptides involved in formation of homo- and heterotypic fibrils. J. Proteome Res. 6, 4269–4289 [DOI] [PubMed] [Google Scholar]
- 34. Eyre D. R., Pietka T., Weis M. A., Wu J. J. (2004) Covalent cross-linking of the NC1 domain of collagen type IX to collagen type II in cartilage. J. Biol. Chem. 279, 2568–2574 [DOI] [PubMed] [Google Scholar]
- 35. Yamauchi M., Katz E. P., Mechanic G. L. (1986) Intermolecular cross-linking and stereospecific molecular packing in type I collagen fibrils of the periodontal ligament. Biochemistry 25, 4907–4913 [DOI] [PubMed] [Google Scholar]
- 36. Yamauchi M., Katz E. P., Otsubo K., Teraoka K., Mechanic G. L. (1989) Cross-linking and stereospecific structure of collagen in mineralized and nonmineralized skeletal tissue. Connect. Tissue Res. 21, 159–167; discussion 168–169 [DOI] [PubMed] [Google Scholar]
- 37. Kuboki Y, Tsuzaki M, Sasaki S, Liu CF, Mechanic GL. (1981) Location of the intermolecular cross-links in bovine dentin collagen, solubilization with trypsin, and isolation of cross-link peptides containing dihydroxylysinonorleucine and pyridinoline. Biochem. Biophys. Res. Commun. 102, 119–126 [DOI] [PubMed] [Google Scholar]
- 38. Scott J. E., Hughes E. W., Shuttleworth A. (1981) A collagen-associated Ehrlich chromogen: a pyrrolic cross-link? Biosci. Rep. 1, 611–618 [Google Scholar]
- 39. Perdivara I., Perera L., Sricholpech M., Terajima M., Pleshko N., Yamauchi M., Tomer K. B. (2013) Unusual fragmentation pathways in collagen glycopeptides. J. Am. Soc. Mass Spectrom. 24, 1072–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Scott J. E., Hughes E. W., Shuttleworth A. (1983) An “affinity” method for preparing polypeptides enriched in the collagen-associated Ehrlich chromogen. J. Biochem. 93, 921–925 [DOI] [PubMed] [Google Scholar]
- 41. Eriksen H. A., Sharp C. A., Robins S. P., Sassi M. L., Risteli L., Risteli J. (2004) Differently cross-linked and uncross-linked carboxy-terminal telopeptides of type I collagen in human mineralised bone. Bone 34, 720–727 [DOI] [PubMed] [Google Scholar]
- 42. Glatter T., Ludwig C., Ahrné E., Aebersold R., Heck A. J., Schmidt A. (2012) Large-scale quantitative assessment of different in-solution protein digestion protocols reveals superior cleavage efficiency of tandem Lys-C/trypsin proteolysis over trypsin digestion. J. Proteome Res. 11, 5145–5156 [DOI] [PubMed] [Google Scholar]
- 43. Eyre D. R., Oguchi H. (1980) The hydroxypyridinium crosslinks of skeletal collagens: their measurement, properties and a proposed pathway of formation. Biochem. Biophys. Res. Commun. 92, 403–410 [DOI] [PubMed] [Google Scholar]
- 44. Yamauchi M., Mechanic G. L. (1988) Cross-linking of collagen. in Collagen, Vol. 1 (Nimni M. E., ed) pp. 157–172, CRC Press, Inc., Boca Raton, FL [Google Scholar]
- 45. Segrest J. P., Cunningham L. W. (1970) Variations in human urinary O-hydroxylysyl glycoside levels and their relationship to collagen metabolism. J. Clin. Invest. 49, 1497–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Royce P. M., Barnes M. J. (1977) Comparative studies collagen glycosylation in chick skin and bone. Biochim. Biophys. Acta 498, 132–142 [DOI] [PubMed] [Google Scholar]
- 47. Saito M., Marumo K., Fujii K., Ishioka N. (1997) Single-column high-performance liquid chromatographic-fluorescence detection of immature, mature, and senescent cross-links of collagen. Anal. Biochem. 253, 26–32 [DOI] [PubMed] [Google Scholar]
- 48. Eyre D. R., Dickson I. R., Van Ness K. (1988) Collagen cross-linking in human bone and articular cartilage: age-related changes in the content of mature hydroxypyridinium residues. Biochem. J. 252, 495–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Song E., Mechref Y. (2013) LC-MS/MS identification of the O-glycosylation and hydroxylation of amino acid residues of collagen α-1(II) chain from bovine cartilage. J. Proteome Res. 12, 3599–3609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kuypers R., Tyler M., Kurth L. B., Jenkins I. D., Horgan D. J. (1992) Identification of the loci of the collagen-associated Ehrlich chromogen in type I collagen confirms its role as a trivalent cross-link. Biochem. J. 283, 129–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brady J. D., Robins S. P. (2001) Structural characterization of pyrrolic cross-links in collagen using a biotinylated Ehrlich's reagent. J. Biol. Chem. 276, 18812–18818 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.