

Background: Rad17 is a key DNA damage response protein that undergoes ubiquitylation-mediated degradation.
Results: USP20 is a deubiquitylase that interacts with and stabilizes Rad17 in a proteasome-dependent manner, and it is required for Chk1 phosphorylation.
Conclusion: USP20 is a novel regulator of the DNA damage response.
Significance: USP20 role sheds more light on the ubiquitylation events associated with DNA damage, and may predict chemotherapy response.
Keywords: Checkpoint Control, Deubiquitylation (Deubiquitination), DNA Damage Response, DNA Repair, Homologous Recombination, USP20, chk1, Cisplatin, rad17
Abstract
Rad17 is a subunit of the Rad9-Hus1-Rad1 clamp loader complex, which is required for Chk1 activation after DNA damage. Rad17 has been shown to be regulated by the ubiquitin-proteasome system. We have identified a deubiquitylase, USP20 that is required for Rad17 protein stability in the steady-state and post DNA damage. We demonstrate that USP20 and Rad17 interact, and that this interaction is enhanced by UV exposure. We show that USP20 regulation of Rad17 is at the protein level in a proteasome-dependent manner. USP20 depletion results in poor activation of Chk1 protein by phosphorylation, consistent with Rad17 role in ATR-mediated phosphorylation of Chk1. Similar to other DNA repair proteins, USP20 is phosphorylated post DNA damage, and its depletion sensitizes cancer cells to damaging agents that form blocks ahead of the replication forks. Similar to Chk1 and Rad17, which enhance recombinational repair of collapsed replication forks, we demonstrate that USP20 depletion impairs DNA double strand break repair by homologous recombination. Together, our data establish a new function of USP20 in genome maintenance and DNA repair.
Introduction
Replication forks encounter DNA lesions that arise from internal processes such as reactive oxygen species (ROS),3 or external factors including UV light and harmful chemicals. Stalled forks activate the DNA damage response (DDR), which enhances the repair process, arrests the cell cycle to allow time for repair, or leads to apoptosis activation in the case of severe damage to avoid genomic instability and cancer formation (1). After fork arrest, the replication helicases continue unwinding DNA beyond the blocking lesion thus generating a long stretch of single-strand DNA (ssDNA) (2). Replication protein A (RPA) binds ssDNA and initiates the damage response signal by recruiting ATR-interacting protein (ATRIP)/ATR (3). Ataxia telangiectasia and Rad3-related (ATR) is a kinase that plays an essential role in DDR activation in response to fork arrest or collapse (4). To activate ATR, a protein complex, consisting of Rad17 and the Replication Factor C (RFC) subunits, loads the PCNA-like Rad9-Hus1-Rad1 (9-1-1) clamp into the DNA ends at the arrested fork. 9-1-1 clamp recruits topoisomerase-binding protein-1 (TopBP1), which activates the nearby ATR to phosphorylate Chk1and other repair proteins (4). Chk1 in turns phosphorylates a collection of substrates to stabilize the fork and arrest the cell cycle. This orchestrated process is under strict regulation by a plethora of protein post-translational modifications including phosphorylation, de-phosphorylation, ubiquitylation, sumoylation, among others (1, 4). Ubiquitylation events are prevalent in the vicinity of DNA damage, and these events play principle roles in the recruitment and stability of the repair proteins (5). Little is known about which deubiquitylases (Dubs) participate in the DDR in response to replication arrest or stress. There are ∼100 human Dubs that have to deal with thousands of ubiquitylated proteins to regulate their stability, subcellular localization, and interactions with other proteins (6). Rad17 is a conserved protein from yeast to human, and Rad17 gene deletion in mice results in embryonic lethality (7). It is constitutively associated with chromatin and is required for normal DNA replication (7, 8). Rad17 protein was reported to be regulated by the ubiquitin-proteasome system (UPS) (9). The anaphase-promoting complex (APC) was identified as the ubiquitin E3 ligase that mediates Rad17 degradation (9). In this report we implicate a novel Dub, ubiquitin-specific peptidase 20 (USP20), in the DDR by interacting with and regulating the protein level and function of Rad17.
EXPERIMENTAL PROCEDURES
Cell Lines and siRNA Treatment
U2OS, HEK293, and A549 cells were obtained from the American Type Culture Collection (ATCC) and maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum and a mixture of antibacterial and antifungal antibiotics. Cells were incubated at 37 °C in 5% CO2. siRNAs specific for different coding regions of USP20 mRNA (si USP20#9: GGA CAA UGA UGC UCA CCU A UU, si USP20#7: CCA UAG GAG AGG UGA CCA A UU) were obtained from Dharmacon and transfected into the cells using Lipofectamine RNAi/Max reagent according to the manufacturer's instructions (Invitrogen). Other siRNA used in this study (BRCA1, Rad17, Plk1, Chk1) were obtained from Dharmacon through their Design Center and tested by Western blot. siRNAs were delivered at 100 nm concentration. Scrambled siRNA controls were used in all experiments. Cells were treated with DNA-damaging agents 72 h after siRNA exposure.
Reagents and Plasmids
The proteasome inhibitor MG132 and the protein synthesis inhibitor cycloheximide were purchased from Sigma-Aldrich. USP20 and Rad17 coding regions were amplified from a placenta cDNA library and cloned into the PcDNA3.1 vector (Invitrogen). USP20 deletions were generated by PCR amplification and cloned into PcDNA3.1. Untagged USP20 clone in PCMV6 vector was obtained from Origene and mutated in the catalytic domain by changing the cysteine 154 within the conserved catalytic triad into alanine (C154A) using the Quickchange Lightning Site-directed Mutagenesis Kit from Agilent technologies. The WT and mutant clones were rendered siRNA resistant by introducing multiple synonymous changes in the nucleotide sequence using the following sense primer (5′-tgccggacgccagagccag ataacgacgcccatctacgcagctcctctcg-3′) also by utilizing the site-directed mutagenesis kit. All clones were verified by Sanger DNA sequencing.
Cell Viability Assays
Cells were treated with control or USP20 siRNA and distributed into the wells of a 96-well plate. After 72 h, cisplatin was added continuously at the specified concentrations. Cells were analyzed 48 h later for viability utilizing the Cell Titer 96® Non-Radioactive Cell Proliferation Assay (MTT) (Promega) according to the manufacturer's instructions. To determine ultraviolet radiation sensitivity, cells were treated with the appropriate siRNA for 72 h, distributed into a 96-well plates, irradiated with 254 nm UV light (UVX radiometer, UVP Inc; San Gabriel, CA) for the indicated doses. The cells were evaluated for viability 48 h later using the same MTT assay.
Immunoprecipitation, Western Blotting, and Antibodies
Cells were collected with IP lysis buffer (50 mm Tris-HCl. pH 8, 150 mm NaCl, EDTA 1 mm, 1 mm Dithiothreitol (DTT), 0.5% Nonidet P-40, with a mixture of protease and phosphatase inhibitors (Sigma)), sonicated, and centrifuged at high speed for 30 min. Samples were pre-cleared and incubated with protein G-conjugated magnetic beads (Dynabeads, Invitrogen) that had been bound to the corresponding antibodies. The beads were washed extensively with the IP lysis buffer and PBS and boiled in Laemmli sample buffer. For Flag-USP20 IP, a stable HEK293 clone expressing Flag-USP20 was established by infecting the cells with retroviral particles expression Flag-HA USP20 in MSCV-N-Flag-HA-IRES-PURO vector (Addgene), followed by selection in a medium containing 2 μg/ml puromycin. The viral particles were generated by a standard protocol in Phoenix packaging cells. Nuclear extracts were obtained by swelling cells with hypotonic buffer (10 mm HEPES-KOH pH 7.9, 10 mm KCl, 0.1 mm EDTA, and 1 mm DTT) for 10 min, followed by the addition of Nonidet P-40 to a final concentration of 0.5%. After vortexing for 2 min, nuclear pellets were collected, lysed with IP lysis buffer, and sonicated. The nuclear lysate was pre-cleared and incubated with Flag-agarose beads (Sigma) for 2 h, then extensively washed and the USP20 complex was eluted with Flag peptides. For the in vivo ubiquitylation experiment, USP20 was knocked down in HEK293 cells by siRNA, and 24 h later V5-Rad17, and HA-Ubiquitin plasmids were introduced by Ca Phosphate method. 48 h later, cells were incubated with 20 μm of the proteasome inhibitor MG132 for 6 h, and collected in denaturing lysis buffer containing 1% SDS and promptly boiled and sonicated. The lysate was diluted for a final SDS concentration of 0.1% and centrifuged at high speed. IP was done on Dynabeads pre-incubated with V5 rabbit antibody from GeneScript.
For Western blot analysis, proteins were separated on SDS-PAGE gels, transferred to PVDF membranes, and blocked with 4% milk or 2% BSA before primary antibodies were added. Antibody-antigen complexes were detected using horseradish peroxidase (HRP)-conjugated secondary antibodies and the enhanced chemiluminescence (ECL) substrate from Santa Cruz Biotechnology. For measuring the rate of protein degradation, the Western blot bands were quantified using Image J software (NIH Image), and all bands were normalized to actin. The following antibodies were used: Rabbit Phospho-Chk1 (Ser-345), Rabbit Phospho-Chk1 (Ser-317), Rabbit GST (91G1), Rabbit P-ATR/ATM substrate, and Rabbit Chk1 from Cell Signaling technology, Mouse Rad17, and Mouse KU-70 from Santa Cruz Biotechnology, Mouse V5 from Invitrogen, Rabbit USP20 from Abcam, Mouse HA from Covance, Rabbit V5 from Genetex, Mouse Anti-Flag and Mouse actin from Sigma-Aldrich.
Glutathione S-Transferase (GST)-Rad17 Expression
Rad17 was cloned into the pGEX vector from GE Healthcare. The GST-Rad17 fusion protein was expressed in Escherichia coli BL21 strain after induction with 1 mm isopropyl β-d-1-thiogalactopyranoside (IPTG) for 5 h at 30 °C. Bacteria were lysed with the B-PER buffer (Pierce) in the presence of a protease inhibitor mixture, and the lysate was added to glutathione-agarose beads (Sigma), followed by extensive washing with the cold lysis buffer and PBS.
In Vitro De-ubiquitylation
For the in vitro de-ubiquitylation of Rad17 by USP20, purified GST-Rad17 protein from bacteria was ubiquitylated in vitro in the presence of Ubiquitin, E1, E2, ATP-generating system, and ubiquitylation buffer (25 mm Tris-HCl pH 7.6, 5 mm MgCl2, 100 mm NaCl, 1 mm DTT, with protease and Dub inhibitors mixture). The ubiquitylated Rad17 was washed and incubated with USP20 protein (purchased from Abnova) in a de-ubiquitylation buffer (100 mm Tris-HCl, pH 8.0, 1 mm EDTA, and protease inhibitor mixture). After 1 h, the reaction was stopped by boiling in SDS-based loading buffer, and the ubiquitylated species was detected by anti-ubiquitin chain FK2 antibody.
The HR Reporter Assay
U2OS-DR-GFP and HELA-DR-GFP cells harboring an integrated reporter substrate to measure HR were described elsewhere (10). These cells were subjected to the indicated siRNA in a 12-well plate format and 48 h later transfected with a plasmid encoding I-Sce I using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). Cells were analyzed 72 h later for GFP expression by FACS analysis at the University of New Mexico Cancer Center Flow Cytometry and High Throughput Shared Resource.
RT-PCR
U2OS cells were treated with siRNA and 72 h later total RNA was extracted using the RNeasy mini kit (Qiagen). cDNA was synthesized from equal amounts of mRNA using the reverse transcriptase SuperScript III (Invitrogen). Triplicates of cDNA samples were assembled in a 96-well plate, and real-time PCR was conducted utilizing Maxima Cybergreen master mix (Thermo Scientific). The amplification reactions were followed on the Step One Plus real-time PCR machine (Applied Biosystem). The results were normalized to mRNA levels of 18 S and β-actin.
Cell Cycle Analysis
Asynchronized U2OS cells were treated with control or USP20 siRNA and fixed 72 h later with 70% ETOH at 4 °C overnight. Fixed cells were treated with RNase H and propidum iodide at 50 μg/ml concentration, and analyzed by Caliber FACS Scan for DNA content.
RESULTS
USP20 Interacts with Rad17
Given the regulation of Rad17 protein levels by the proteasome (9), we reasoned that Rad17 would be stabilized through deubiquitylation. To identify the responsible Dub, we mined a large-scale proteomic study (6) and identified Rad17 peptides in a USP20 pull-down. To investigate a potential association between the two proteins, we performed immunoprecipitation (IP) using an antibody that recognizes the endogenous USP20 and identified Rad17 in the immunoprecipitate by Western blot in two cancer cell lines (Fig. 1, A and B). The association between the two proteins was enhanced following UV exposure. We also established a stable non-cancerous HEK293 cell line overexpressing Flag-USP20, and performed IP on Flag-agarose beads and again identified RAD17 in the immunoprecipitate. However, the association in this case was not promoted by UV exposure, possibly due to the non-physiologic overexpression of USP20 (Fig. 1C). To confirm an interaction between the two proteins, we expressed GST-tagged Rad17 in bacteria, and performed GST pull-down on glutathione beads. Fig. 2A demonstrates specific pull-down of V5-tagged USP20, expressed in a human cell lysate, by GST-Rad17.
FIGURE 1.

USP20 association with Rad17. A, demonstration of the endogenous USP20 and Rad17 interaction in the lung cancer line A549, which is enhanced after UV exposure. B, similar interaction in U2OS osteosarcoma cells. C, Flag-USP20 associates with Rad17 in the non-cancerous HEK293 embryonic kidney cells.
FIGURE 2.

Mapping the interaction between USP20 and Rad17. A, GST-Rad17 immobilized on glutathione beads pulled down V5-USP20 expressed in HEK293 lysate. B, depiction of the USP20 fragments corresponding to the protein domains that were cloned into V5-tagged PcDNA3.1. C, GST-Rad17 pulls down D1 segment, which harbors the UBP-type zinc finger and to a lesser extent D2 and D3 that are parts of the catalytic domain.
Mapping the Interaction between USP20 and Rad17
USP20 is a cysteine hydrolase that also harbors UBP-type zinc finger and two C terminus domains in ubiquitin-specific proteases (DUSP) (Fig. 2B). To identify the area of USP20 that interacts with Rad17 we generated USP20 deletions corresponding to the three recognizable domains within this protein (Fig. 2B), and we investigated which of the domains would interact in vitro with the GST-Rad17. GST pull-down demonstrated an interaction with the N terminus UBP-type zinc finger motif of USP20, and to a lesser extent with the catalytic domain (Fig. 2C). This indicates that USP20 binds Rad17 through more than one domain, but the strongest interaction is mediated by the USP20 N terminus.
USP20 Stabilizes Rad17 in a Proteasome-dependent Manner
Rad17 was reported to be ubiquitylated by the APC E3 ligase (9). Thus we investigated whether USP20 would stabilize Rad17 in the face of this ubiquitylation-associated degradation. As shown in Fig. 3, USP20 depletion results in decreased Rad17 protein levels in two malignant cell lines. This decrease is more prominent after DNA damage in the normal human fibroblasts (Fig. 3C). To rule out off-target effect of the siRNA and to identify the mechanism by which USP20 impacts Rad17 protein levels, we performed rescue experiments utilizing siRNA-resistant wild type and catalytically inactive USP20. USP family of Dubs shares a catalytic amino acid triad that relies on a critical cysteine for its activity (11). To impair the catalytic activity of USP20, we changed the corresponding cysteine (No 154) into alanine. We show that the wild type USP20, but not the catalytically inactive version, rescues the decrease in Rad17 protein level (Fig. 3D). To rule out a transcriptional or translational effect of USP20 depletion on Rad17, we blocked new protein generation by cycloheximide and measured the rate of Rad17 degradation. Rad17 protein levels decline faster in cells depleted of USP20 compared with control cells as shown in Fig. 4, A and B. We have also demonstrated that the decrease in Rad17 protein levels after USP20 depletion is mediated by the UPS since incubating the cells with the proteasome inhibitor MG132 reverses this phenotype (Fig. 4C). When we examined the amount of ubiquitylated Rad17 in vivo, we observed increased Rad17 ubiquitylation after USP20 knock-down (Fig. 4D). To confirm direct de-ubiquitylation of Rad17 by USP20, we performed an in vitro de-ubiquitylation reaction using purified and in vitro ubiquitylated Rad17, and as shown in Fig. 4E, the addition of USP20 protein abolished Rad17 ubiquitin smear. The mRNA level of Rad17 was not lower in USP20 depleted cells arguing against a transcriptional effect (Fig. 4F). Altogether, our data demonstrate that USP20 regulates Rad17 at the protein level by deubiquitylation.
FIGURE 3.
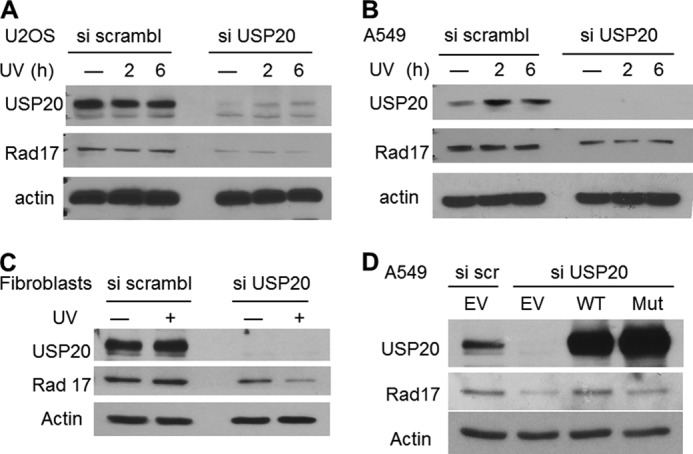
USP20 depletion decreases Rad17 protein levels in a catalytic domain-dependent manner. A, effects of USP20 depletion on Rad17 levels in U2OS osteosarcoma line. B, similar phenotype of USP20 depletion in A549 lung cancer line. C, USP20 depletion results in Rad17 instability in normal human fibroblasts particularly after UV exposure. D, wild type but not catalytically-inactive, and siRNA resistant, USP20 rescues the decrease in Rad17 protein levels. EV, empty vector. Mut, catalytically mutant.
FIGURE 4.

USP20 depletion results in an accelerated Rad17 protein degradation in a proteasome-dependent manner. A, decline of Rad17 protein levels in cells treated with USP20 siRNA versus control cells treated with scrambled siRNA in the presence of cycloheximide. U2OS cells were treated with scrambled or USP20 siRNA for 72 h. followed by cycloheximide (50 mg/ml) for the indicated time points. B, representation of the ratio of Rad17/actin blotted against cycloheximide exposure time. C, decrease in Rad17 protein levels after USP20 depletion is reversed by the proteasome inhibitor MG132. U2OS cells were treated with scramble and USP20 siRNA for 72 h followed by the proteasome inhibitor MG132 (20 μm) for 6 h before or after UV exposure. D, increased rad17 in vivo ubiquitylation after USP20 depletion. V5-Rad17 was overexpressed with HA-ubiquitin and the ubiquitylated Rad17 species was detected by immunoprecipitating V5-Rad17 followed by immunoblotting with HA antibody. E, purified Rad17 protein from bacteria was ubiquitylated in vitro and subsequently incubated with USP20 protein in a de-ubiquitylation reaction as described under “Experimental Procedures.” F, Rad17 mRNA levels measured by RT-PCR in cells treated with USP20 or scrambled siRNA.
USP20 Depletion Results in Defective Chk1 Activation after DNA Damage
A recognizable role of Rad17 in the DDR is to facilitate Chk1 activation by phosphorylation as described above. Thus, we investigated the status of Chk1 phosphorylation in USP20-depleted cells. We observed defective Chk1 phosphorylation on two key serine residues in U2OS osteosarcoma cells and in normal human fibroblasts after UV exposure (Fig. 5, A and D). Other DNA-damaging agents that arrest or block replication forks such as Hydroxyurea (HU) and cisplatin failed to induce adequate Chk1 phosphorylation in USP20 depleted cells as well (Fig. 5B). To rule out off-target effects, we used two distinct siRNAs that complement different regions of the USP20 mRNA, and we reproduced the same phenotype of defective Chk1 activation in the lung cancer line A549 (Fig. 5C). The forced expression of Rad17 rescued the decrease in Chk1 phosphorylation after USP20 depletion, attesting to the role of Rad17 in mediating USP20 effect on this phosphorylation (Fig. 5E).
FIGURE 5.

Chk1 phosphorylation is defective when USP20 is depleted. A, USP20 depletion in U2OS cells resulted in a significant decrease in Chk1 phosphorylation by ATR on serine 317 and serine 345 after UV exposure. B, similar defects in chk1 phosphorylation after the incubation with hydroxyurea (2 mm for 8 h) or cisplatin (20 μm for 10 h). C, two distinct siRNAs that target different sequences in the coding area of USP20 result in similar phenotype of defective chk1 phosphorylation after USP20 depletion in A549 lung cancer cells. D, defective Chk1 activation is also noted in normal fibroblasts after USP20 depletion, and an increase in USP20 protein level is observed after UV exposure in the control cells. E, Rad17 overexpression reverses the decrease in Chk1 phosphorylation after USP20 depletion. HEK293 cells were treated with scrambled or USP20 siRNA, and 48 h later were transfected with an empty vector (EV) or untagged Rad17. Cells were treated with UV light and collected 48 h after Rad17 introduction. F, USP20 is phosphorylated after DNA damage. V5-USP20 was immunoprecipitated and phospho-USP20 was detected by an antibody that was raised against a phospho-peptide that represents an ATR target.
USP20 Is Phosphorylated and Its Protein Level Increases after DNA Damage
Many DNA repair proteins become phosphorylated after DNA damage (12), therefore, we aimed to investigate a potential USP20 phosphorylation in this setting. We immunoprecipitated V5-tagged USP20, and demonstrated its phosphorylation after UV exposure by utilizing a commercial anti phosphoserine antibody that was raised against a phospho-peptide targeted by the ATR/ATM kinases (Fig. 5F). We also observed an increase in USP20 protein level after DNA damage in human fibroblasts consistent with a function of this protein in DNA repair (Fig. 5D).
USP20 Depletion Sensitizes Human Cancer Cells to DNA Damage
A cellular consequence of defective DNA repair is the increased toxicity of DNA-damaging agents. USP20 depletion in two cancer cell lines results in increased sensitivity to UV light and to the widely used DNA damaging drug cisplatin (Fig. 6), adding further evidence of USP20 involvement in the DDR and repair. siRNA resistant and WT USP20 rescued the cisplatin sensitivity after endogenous USP20 depletion, while a catalytically-defective USP20 failed to do so (Fig. 7A). Forced Rad17 expression also rescued this cisplatin sensitivity (Fig. 7B) consistent with what we observed with Rad17 rescue of Chk1 phosphorylation (Fig. 5E). We used the MTT assay to measure cellular viability after DNA damage since USP20-depleted cells failed to form colonies after 14 days of incubation in the clonogenic survival assay. This failure to form colonies is likely due to increased cellular death in USP20-depleted cells because we did not observe a significant change in cell cycle phase distribution after USP20 depletion (Fig. 8, A and B).
FIGURE 6.

Increased sensitivity to DNA damage after USP20 depletion. A, enhanced sensitivity of U2OS cells to cisplatin after using USP20 siRNA. Cells were exposed to the specified doses of cisplatin 72 h after the addition of siRNA, and viability was checked 48 h later by MTT assay as described under “Experimental Procedures.” B, similar phenotype in A549 cells. C, increased UV sensitivity after USP20 depletion in U2OS, and A549 (D) cancer cells.
FIGURE 7.
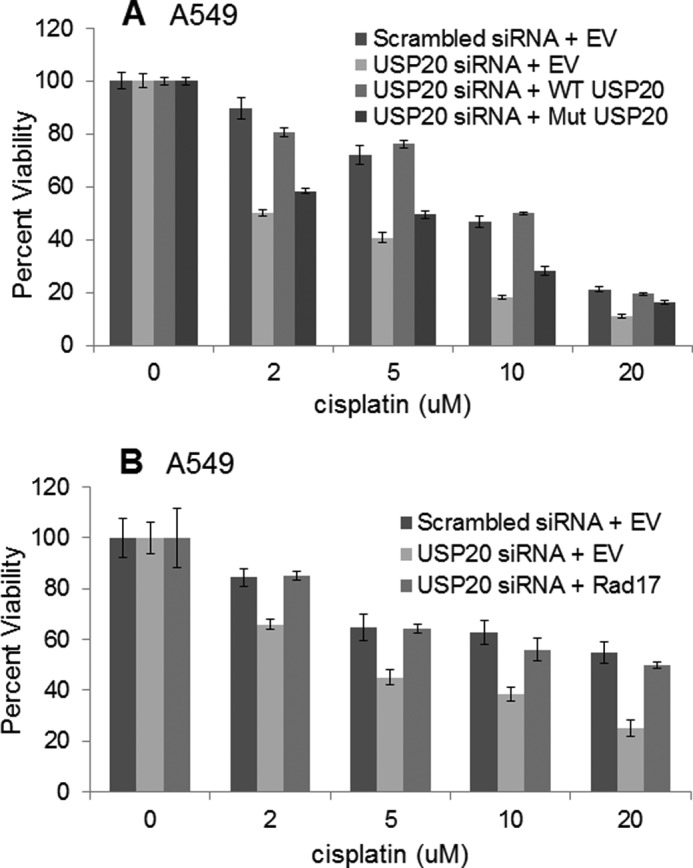
Rescue of the cisplatin sensitivity after USP20 depletion by USP20 or Rad17 overexpression. A, siRNA-resistant and wild-type USP20 (generated by synonymous mutations within untagged USP20 plasmid) rescued the cisplatin sensitivity after endogenous USP20 depletion, while a catalytically-defective USP20 did not. B, Rad17 overexpression rescued the cisplatin sensitivity after USP20 depletion as well. EV, empty vector.
FIGURE 8.

Cell cycle distribution and G2/M checkpoint in cells depleted of USP20. A, cell cycle phase distribution in USP20 depleted cells versus control. B, percentage of cells in each cell cycle phase. C, G2/M checkpoint analysis. Cells were exposed to siRNA and then treated with 6 Gs of IR followed an hour later by 200 ng/ml of nocodazole to trap them in mitosis. Mitotic cells were identified by being positive in histone 3, serine 10 phosphorylation (y axis), against propidium iodide (PI) DNA stain (x axis). D, G2/M checkpoint is assessed by measuring the percentage of cells that entered mitosis after IR compared with those that enter mitosis during the same period and without IR exposure. E, PLK1 down-regulation following USP20 depletion. Ku70 is utilized to show equal protein loading.
USP20 Is Required for the Homologous Recombination (HR) Repair of DNA Double Strand Breaks (DSBs)
One recognizable outcome of Chk1 phosphorylation is the activation of the G2/M cell cycle checkpoint after DNA damage. When we investigated this checkpoint in USP20-depleted cells, by measuring the mitotic index after DNA damage, we did not observe a difference when compared with control cells (Fig. 8, C and D). We attempted to explain this conundrum by searching for other potential USP20 targets. Polo-Like Kinase 1 (PLK1) satisfies criteria for a high confidence interacting protein (HCIP) with USP20 (6). Notably, PLK1 is required for recovery from the G2/M checkpoint post DNA damage (13) and in that manner opposes the function of Chk1 in checkpoint activation. We found decreased PLK1 protein levels in USP20-depleted cells (Fig. 8E) that would explain the lack of G2/M checkpoint defect in USP20-depleted cells. We also identified an interaction between USP20 and PLK1 by IP (data not shown). We then reasoned that USP20 might impact another function mediated by Chk1 phosphorylation besides the G2/M checkpoint activation. One such function is to enhance DNA DSB repair by HR. Arrested replication forks collapse after prolonged exposure to damaging agents (14). The collapsed forks generate one-ended DSBs that are repaired by the HR pathway (15). Each of Rad17 (7, 16), the 9-1-1 clamp (17), Chk1 (18), and PLK1 (19) has been reported to be involved in the HR repair of DSBs. The 9-1-1 clamp interacts with Rad51 (17), and Chk1 phosphorylates this key HR protein (18). HR proceeds when the DSB end is processed by exonucleases to generate ssDNA that binds Rad51 protein and forms a nucleofilament, which invades the homologous sequence to repair the damage (15). We asked if USP20 is involved in HR similar to Rad17, 9-1-1, Chk1, and PLK1. To answer this, we used derivatives of the U2OS and Hela cell lines with an integrated reporter substrate to measure DSB-induced HR. The reporter substrate consists of two copies of green fluorescent protein (GFP) (Fig. 9A), one interrupted by the rare-cutting restriction enzyme I Sce-1 recognition sequence, and the other without a promoter and with defective ends. When I Sce-1 is expressed it causes a DSB that, if repaired by HR using the other GFP sequence, will generate a functional GFP. The number of cells that express GFP, which is quantitated by flow cytometry, reflects the efficiency of HR. As shown in Fig. 9, E, F, and G depletion of the key HR protein BRCA1 by siRNA resulted in a dramatic decrease in the percentage of GFP-positive cells as expected. Depletion of USP20 also decreased the rate of HR to a significant level (Fig. 9, D, F, and G). Decreasing the protein levels of Rad17, Chk1, and PKL1 by siRNA also impaired HR as reported in the literature (Fig. 9, F and G). Our data demonstrate that one of USP20 functions is to enhance HR repair of DSBs likely by stabilizing Rad17 and PLK1 and activating Chk1.
FIGURE 9.

Defective HR in cells depleted of USP20. A, depiction of the HR substrate. Details of the assay are provided in the text. B, U2OS-DR-GFP cells in the absence of the restriction enzyme I Sce-1. No GFP-positive cells are detected. C, GFP-positive cells are observed after treatment with scrambled siRNA and I Sce-1. D, lower percentage of GFP cells when USP20 is depleted. E, dramatic decrease of GFP-positive cells after the depletion of the key HR protein BRCA1. F, representation of percentage of GFP-positive cells after the depletion of USP20. Rad17, PLK1, Chk1, and BRCA1 in U2OS, and (G) Hela cells. The means of three experiments with the standard deviations are shown. H, extent of the protein depletion is shown.
DISCUSSION
DNA insults are frequent and the DNA repair machinery has to deal with many on daily basis. Activating the DNA damage response is an ancient phenomenon that has been conserved from yeast to humans. Rad17 is a key protein that is involved in mediating the signal between the DNA injury and Chk1 activation. Rad17 has also been implicated in carcinogenesis as a tumor suppresser gene that functions in a haplo-insufficient manner (20). We have identified USP20 as a new player in the DDR by stabilizing Rad17. The enhanced interaction between USP20 and Rad17 after DNA damage is probably required to protect RAD17 from the excessive ubiquitylation that occurs in this situation (9). The increased in vivo Rad17 ubiquitylation when USP20 is depleted, the in vitro activity of USP20 on Ub-Rad17, the reversal of Rad17 instability in the presence of a proteasome inhibitor, and the requirement of USP20 catalytic domain for Rad17 stability; all point to a direct enzymatic effect of USP20 on Rad17 protein. Similar to other repair and damage response proteins, USP20 is phosphorylated after DNA injury. Consistent with our finding, USP20 has been identified as one of the hits in large-scale phospho-proteomic analysis post DNA damage (21), and multiple USP20 residues turned out to be phosphorylated in this study. Identifying the importance of these phosphorylated residues in USP20 function will be the focus of future efforts. We have found that USP20 depletion impairs Chk1 phosphorylation and thus activation. Chk1 exerts multiple functions in response to DNA damage including stabilizing the stalled forks, activating the G2/M checkpoint, and enhancing fork repair by HR (2, 4). The lack of G2/M checkpoint defect when we depleted USP20 was surprising, and further investigation suggested that USP20 impact on the checkpoint is likely to be complex since it involves more than one pertinent protein (Chk1 and PLK1). The exact role of USP20 in relation to the chronology of events preceding or following checkpoint activation may not be easily uncovered by the non-physiologic depletion using siRNA. It is interesting that USP20 is required for the function of two kinases (Chk1 and PLK1) that play opposing roles when it comes to G2/M checkpoint activation, while both kinases are required for the completion of of DNA DSB repair by HR through the phosphorylation of Rad51 (18, 19). Helleday and co-workers reported that inhibition of Chk1 leads to increased accumulation of replication-associated DSBs and failure to repair DSBs after replication arrest by HR (18). Plk1 phosphorylates Rad51 at serine 14 in response to DNA damage, and this phosphorylation facilitates Rad51 recruitment to damage sites (19). Similar to what we found with USP20, Rad17-deficient embryonic stem (ES) cells have intact checkpoints after DNA damage, but they display impaired homologous recombination as evidenced by a strongly reduced gene targeting efficiency (7).
The identification of Rad17 as a frequent target of inactivation in an in vivo siRNA-screen of mouse tumor formation strongly implicates this pathway in tumor suppression (20). The locus of RAD17 gene was reported to be deleted in a significant percentage of human cancers (20). Because USP20 stabilizes Rad17, we reason that USP20 itself may represent a novel tumor suppressor protein. By searching public databases including the Sanger Cosmic and the Cancer Genome Atlas, we have found frequent point mutations and deletions of USP20 in a variety of malignancies. Lastly, we have demonstrated that USP20 depletion sensitizes cancer cell lines to DNA damaging agents including the widely used chemotherapy drug cisplatin, which renders USP20 a potential target for cancer therapy.
Acknowledgment
We thank W. Arap for critical review of the manuscript.
This work was supported, in whole or in part, by NIH Grant 5K01HL103182-03 (to M. S.) and start-up funds from the UNM Cancer Center.
- ROS
- reactive oxygen species
- DDR
- DNA damage response
- ssDNA
- single-strand DNA
- RPA
- replication protein A
- ATRIP
- ATR-interacting protein
- ATR
- Ataxia telangiectasia and Rad3-related
- TopBP1
- topoisomerase-binding protein 1
- Dub
- deubiquitylase
- PLK
- Polo-like kinase
- UPS
- ubiquitin-proteasome system
- APC
- anaphase-promoting complex.
REFERENCES
- 1. Ciccia A., Elledge S. J. (2010) The DNA damage response: making it safe to play with knives. Mol. Cell 22, 179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nam E. A., Cortez D. (2011) ATR signalling: more than meeting at the fork. Biochem. J. 436, 527–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zou L., Elledge S. J. (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548 [DOI] [PubMed] [Google Scholar]
- 4. Cimprich K. A., Cortez D. (2008) ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9, 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jackson S. P., Durocher D. (2013) Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 49, 795–807 [DOI] [PubMed] [Google Scholar]
- 6. Sowa M. E., Bennett E. J., Gygi S. P., Harper J. W. (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Budzowska M., Jaspers I., Essers J., de Waard H., van Drunen E., Hanada K., Beverloo B., Hendriks R. W., de Klein A., Kanaar R., Hoeijmakers J. H., Maas A. (2004) Mutation of the mouse Rad17 gene leads to embryonic lethality and reveals a role in DNA damage-dependent recombination. EMBO J. 23, 3548–3558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zou L., Cortez D., Elledge S. J. (2002) Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 16, 198–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang L., Park C. H., Wu J., Kim H., Liu W., Fujita T., Balasubramani M., Schreiber E. M., Wang X. F., Wan Y. (2010) Proteolysis of Rad17 by Cdh1/APC regulates checkpoint termination and recovery from genotoxic stress. EMBO J. 29, 1726–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weinstock D. M., Nakanishi K., Helgadottir H. R., Jasin M. (2006) Assaying double-strand break repair pathway choice in mammalian cells using a targeted endonuclease or the RAG recombinase. Methods Enzymol. 409, 524–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hu M., Li P., Li M., Li W., Yao T., Wu J. W., Gu W., Cohen R. E., Shi Y. (2002) Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell 111, 1041–1054 [DOI] [PubMed] [Google Scholar]
- 12. Matsuoka S., Ballif B. A., Smogorzewska A., McDonald E. R., 3rd, Hurov K. E., Luo J., Bakalarski C. E., Zhao Z., Solimini N., Lerenthal Y., Shiloh Y., Gygi S. P., Elledge S. J. (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166 [DOI] [PubMed] [Google Scholar]
- 13. van Vugt M. A., Brás A., Medema R. H. (2004) Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol Cell 15, 799–811 [DOI] [PubMed] [Google Scholar]
- 14. Hanada K., Budzowska M., Modesti M., Maas A., Wyman C., Essers J., Kanaar R. (2006) The structure-specific endonuclease Mus81-Eme1 promotes conversion of interstrand DNA crosslinks into double-strands breaks. EMBO J. 25, 4921–4932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Petermann E., Helleday T. (2010) Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol. 11, 683–687 [DOI] [PubMed] [Google Scholar]
- 16. Nishino K., Inoue E., Takada S., Abe T., Akita M., Yoshimura A., Tada S., Kobayashi M., Yamamoto K., Seki M., Enomoto T. (2008) A novel role for Rad17 in homologous recombination. Genes Genet. Syst. 83, 427–431 [DOI] [PubMed] [Google Scholar]
- 17. Pandita R. K., Sharma G. G., Laszlo A., Hopkins K. M., Davey S., Chakhparonian M., Gupta A., Wellinger R. J., Zhang J., Powell S. N., Roti J. L., Lieberman H. B., Pandita T. K. (2006) Mammalian Rad9 plays a role in telomere stability, S- and G2-phase-specific cell survival, and homologous recombinational repair. Mol. Cell. Biol. 26, 1850–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sørensen C. S., Hansen L. T., Dziegielewski J., Syljuåsen R. G., Lundin C., Bartek J., Helleday T. (2005) The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 7, 195–201 [DOI] [PubMed] [Google Scholar]
- 19. Yata K., Lloyd J., Maslen S., Bleuyard J. Y., Skehel M., Smerdon S. J., Esashi F. (2012) Plk1 and CK2 act in concert to regulate Rad51 during DNA double strand break repair. Mol Cell 45, 371–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bric A., Miething C., Bialucha C. U., Scuoppo C., Zender L., Krasnitz A., Xuan Z., Zuber J., Wigler M., Hicks J., McCombie R. W., Hemann M. T., Hannon G. J., Powers S., Lowe S. W. (2009) Functional identification of tumor-suppressor genes through an in vivo RNA interference screen in a mouse lymphoma model. Cancer Cell 16, 324–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Beli P., Lukashchuk N., Wagner S. A., Weinert B. T., Olsen J. V., Baskcomb L., Mann M., Jackson S. P., Choudhary C. (2012) Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 46, 212–225 [DOI] [PMC free article] [PubMed] [Google Scholar]