

Background: Phosphatidylinositol 4,5-bisphosphate (PIP2) regulates a variety of ion channels including Kv7 channels.
Results: Several Kv7.1 mutations impair functional interaction with PIP2.
Conclusion: PIP2 interacts with several Kv7.1 amino acids, indicating the existence of at least two binding pockets.
Significance: Impaired molecular interaction between mutant Kv7.1 and PIP2 may underlie heart disease.
Keywords: Electrophysiology, Heart, Molecular Modeling, Phosphatidylinositol, Phosphoinositide, Phospholipid, Potassium Channel, Structural Biology, Xenopus
Abstract
Kv7.1 to Kv7.5 α-subunits belong to the family of voltage-gated potassium channels (Kv). Assembled with the β-subunit KCNE1, Kv7.1 conducts the slowly activating potassium current IKs, which is one of the major currents underlying repolarization of the cardiac action potential. A known regulator of Kv7 channels is the lipid phosphatidylinositol 4,5-bisphosphate (PIP2). PIP2 increases the macroscopic current amplitude by stabilizing the open conformation of 7.1/KCNE1 channels. However, knowledge about the exact nature of the interaction is incomplete. The aim of this study was the identification of the amino acids responsible for the interaction between Kv7.1 and PIP2. We generated 13 charge neutralizing point mutations at the intracellular membrane border and characterized them electrophysiologically in complex with KCNE1 under the influence of diC8-PIP2. Electrophysiological analysis of corresponding long QT syndrome mutants suggested impaired PIP2 regulation as the cause for channel dysfunction. To clarify the underlying structural mechanism of PIP2 binding, molecular dynamics simulations of Kv7.1/KCNE1 complexes containing two PIP2 molecules in each subunit at specific sites were performed. Here, we identified a subset of nine residues participating in the interaction of PIP2 and Kv7.1/KCNE1. These residues may form at least two binding pockets per subunit, leading to the stabilization of channel conformations upon PIP2 binding.
Introduction
The voltage-gated potassium channel Kv7.1 (also called KCNQ1 and KvLQT1) is one of five members of the Kv7 subfamily. It is foremost expressed in the heart and epithelial tissues of many organs (1, 2). Although Kv7.1 can assemble with all five members of the KCNE β-subunit family, its interaction with KCNE1 (also called minK or IsK) is best described (1, 3–7). The noninactivating Kv7.1/KCNE1 channel complex shows slow activation and deactivation kinetics (1, 4, 8, 9). These channels mediate the slowly activating cardiac potassium current IKs (1, 3), which contributes to the repolarization of myocyte membranes during phase 3 of the cardiac action potential (10, 11). Many mutations in Kv7.1- and KCNE1-encoding genes have been reported to result in loss of function and to cause long QT syndrome (LQTS),2 which is associated with cardiac arrhythmia, syncope, and sudden cardiac death (12–18).
All Kv7 channels are inhibited after stimulation of Gq- or G11-protein-coupled receptors (19–22). This is assumed to be primarily due to depletion of the plasma membrane lipid phosphatidylinositol 4,5-bisphosphate (PIP2) upon activation of phospholipase C (23–25). PIP2 is important for the function of many proteins, including trafficking molecules such as small GTPases (26, 27) and cytoskeletal proteins (28, 29), as well as transporters and ion channels (30).
Among the ion channels regulated by PIP2 are transient receptor potential channels, (32), inward-rectifier potassium (Kir) channels (33), and voltage-gated potassium channels such as Kv7.1. The lipid presumably exerts its effect on the latter by stabilizing the open channel conformation, thereby slowing the deactivation kinetics and shifting the activation curve to more negative potentials (34).
To date, the interaction has been localized to the C terminus of Kv7.1, in which four amino acids have been identified to interact with PIP2 (35). However, it is not known whether the interaction site is confined to these amino acids. The aim of the present study, therefore, was the identification of residues within Kv7.1 responsible for the interaction with PIP2. Our experiments were based on the assumption that direct interaction is caused by electrostatic forces between negatively charged PIP2 head groups and positively charged amino acid residues. Hence, we generated neutralizing mutants of 13 positively charged amino acids. These were coexpressed with KCNE1 in Xenopus laevis oocytes to form slowly activating (IKs) channels. Effects of injected diC8-PIP2 on wild type and mutant channels was investigated by means of the two-electrode voltage-clamp. Included in the charge neutralization scan were the residues identified by Thomas et al. (35). Nine of the thirteen amino acids proved positive for a functional interaction with the lipid. Five of these had been identified previously as LQTS mutants. Electrophysiological analysis revealed an impaired PIP2 interaction with two of these. Two regions containing examined amino acids bound PIP2 and other lipids in lipid binding assays. To gain insight into the underlying structural mechanisms, we conducted molecular dynamics simulations of Kv7.1 tetramers in complex with four KCNE1 β-subunits, both with and without PIP2 molecules. The results implied that binding of PIP2 to at least eight specific binding sites causes the stabilization of open and closed channel conformations.
EXPERIMENTAL PROCEDURES
Molecular Biology
The molecular biological procedures were previously described (36). A mutant channel based on human Kv7.1 (GenBankTM accession number NM_000218, isoform 0; the N terminus is truncated compared with isoform 1) in which all cysteines were mutated to alanines (Kv7.1 clone without cysteines, here called Kv7.1 −cys) was described before and kindly provided by G. N. Tseng (Virginia Commonwealth University, Richmond, VA) (37). The clone was subcloned into the oocyte expression vector pSGEM. Further, human Kv7.1 wild type, isoform 1 was subcloned into pXOOM. Human KCNE1 (GenBankTM accession number NM_000219) was subcloned into pSP64. The Kv7.1 mutations H126C, R181C, K183C, R190C, R192C, R195C, K196C, R249C, R259C, K354C, K358C, R360C, and K362C were introduced into the Kv7.1 −cys clone, the Kv7.1 mutations R190L, R192H, R195W, K354R, R360M, R360T, and K362R were introduced into the Kv7.1 WT clone by site-directed mutagenesis (Agilent Technologies, Santa Clara, CA). All constructs were confirmed by automated DNA sequencing. The plasmids were linearized using XbaI (Kv7.1 −cys), NheI (Kv7.1 WT), and EcoRI (KCNE1). In vitro synthesis of cRNA was performed using the T7 (Kv7.1) or SP6 (KCNE1) mMessage mMachine kit (Ambion, Austin, TX) according to the manufacturer's instructions.
Electrophysiology
X. laevis oocytes were harvested in accordance with German laws as previously described (36). Ovarian lobes from tricaine-anesthetized X. laevis were digested with collagenase (Type II, Worthington, 1 mg/ml in calcium-free Barth's solution) for ∼120 min. Stage V and VI oocytes were injected with ∼20 nl of cRNA solution. Oocytes were injected with 2.5 ng of Kv7.1 WT or mutant cRNA and 2.5 ng of KCNE1 WT cRNA. The oocytes were stored for 3–4 days at ∼18 °C in Barth's solution containing 88 mmol/liter NaCl, 1.1 mmol/liter KCl, 2.4 mmol/liter NaHCO3, 0.3 mmol/liter Ca(NO3)2, 0.3 mmol/liter CaCl2, 0.8 mmol/liter MgSO4, 15 mmol/liter HEPES-NaOH, 31 mg/liter penicillin-G, 50 mg/liter gentamicin, 20 mg/liter streptomycin sulfate, and 80 mg/liter theophylline, pH 7.6. Two-electrode voltage-clamp recordings were performed at ∼22 °C using a Turbo Tec-10CX amplifier (NPI, Tamm, Germany) equipped with a Digidata 1322A AD/DA interface and pCLAMP 9.0 software (Axon Instruments Inc./Molecular Devices, Sunnyvale, CA). The data were analyzed with Clampfit 9.0 (Axon Instruments Inc.), Prism 4.00 (GraphPad Software, La Jolla, CA), and QtiPlot (Ion Vasilief, Craiova, Romania). Recording pipettes were filled with 3 m KCl and had resistances of 0.4–1 MΩ. Channel currents were recorded in ND96 recording solution containing 96 mmol/liter NaCl, 4 mmol/liter KCl, 1.8 mmol/liter MgCl2, 1.0 mmol/liter CaCl2, and 5 mmol/liter HEPES, pH 7.6. Some oocytes were injected with 13.8 ng (−cys mutants) or 27.6 ng (LQTS mutants and comparison WT/−cys) of diC8-PIP2 (P-4508; Mobitec, Göttingen, Germany) at least 30 min prior to the recordings. All other reagents were purchased from Sigma-Aldrich.
Lipid Binding Assay
The assay was recently described in detail (31). In brief, PIP Strips from Molecular Probes were used (P23750). The peptides were FITC-labeled and had the following amino acid sequences: Q1-S2/S3, (FITC)-CRSKYVGLWGRLRFARK; Q1-S4/S5, (FITC)-GTWRLLGSVVFIHRQEL; and Q1-S6, (FITC)-GSGFALKVQQKQRQKHF.
PIP Strips membranes were blocked with TBS-T containing 10 mmol/liter Tris-HCl, pH 8.0, 150 mmol/liter NaCl (containing 0.1% (v/v) Tween 20 detergent) + 3% fatty acid-free BSA and gently agitated for 1 h at room temperature protected from light. The peptides (50 μg/ml) were each solved in TBS-T + 3% fatty acid-free BSA and incubated on the membranes for 1 h at room temperature protected from light, using gentle agitation. The membranes were then washed three times for 10 min each with TBS-T + 3% fatty acid-free BSA, protected from light, and gently agitated. Peptide binding was detected by analysis of fluorescence signal.
Molecular Dynamics Simulations
The Kv7.1/KCNE1 structural models from Kang et al. (38) were used. Eight PIP2 molecules were introduced in proximity to the eight residue clusters identified by electrophysiology. Structures with and without PIP2 were inserted into a membrane (composition PEA/PSE/PCH = 1/1/1) and incorporated into a simulation box filled with NaCl-H2O (0.9%). All-atoms-mobile simulations were performed to reach stable conformations using YASARA Structure version 10.1 (YASARA Biosciences GmbH, Vienna, Austria) as previously described (39).
Data Analysis
Mean current-voltage curves were analyzed using the Boltzmann equation multiplied with the driving force, considering increased current amplitudes caused by increased applied voltages,
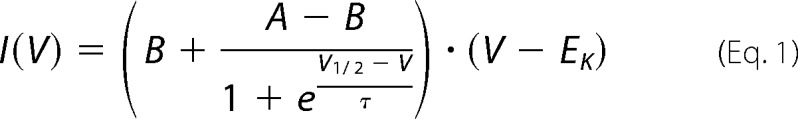 |
where I is the current amplitude, V is the applied voltage, EK is the potassium equilibrium potential, V½ is the half-maximal activation voltage, τ is the time constant, and A and B are the maximal and minimal current amplitudes, respectively.
The variation of structures during the molecular dynamics simulations is presented as the root mean square deviation, which is a measure of the deviation of the atom positions compared with the first structure,
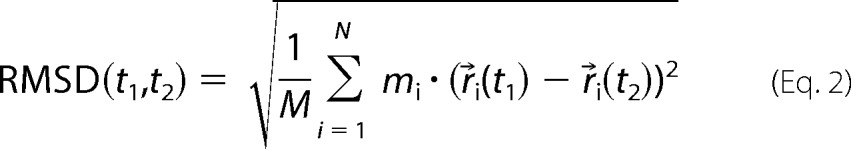 |
where t1 = 0, N is the number of atoms, r→i is the position of atom i at time t, mi is the atom mass, and M is the sum of the atom masses.
RESULTS
Electrophysiological Examination of Kv7.1 Mutants
Wild type (cysteine-free Kv7.1, −cys) or mutated Kv7.1/KCNE1 channels were expressed in X. laevis oocytes. The examined mutations are located in transmembrane segment S1 (H126C), the intracellular linker between S2 and S3 (R181C, K183C, R190C, R192C, R195C, and K196C), the intracellular linker between S4 and S5 (R249C and R259C), and the intracellular extension of helix S6 (K354C, K358C, R360C, and K362C; Fig. 1A). All channels yielded characteristic, noninactivating, slowly activating and deactivating, voltage-gated potassium currents (Fig. 1B). Current-voltage relationships (Fig. 1C) showed WT (−cys) or mutant channel activation upon depolarization to approximately −30 mV. The shapes of the curves and hence kinetic parameters appeared nearly unchanged, but some mutations led to significantly increased current amplitudes compared with WT (−cys) currents (Fig. 1, B, red curves, and D).
FIGURE 1.

Intracellularly located mutations modulate Kv7.1/KCNE1 channel currents. Currents were recorded 3 days after injection. A, topology model of Kv7.1, showing all six transmembrane segments S1–S6 with pore loop and selectivity filter sequence GYG. Shown are all amino acids included in this study. Red, necessary for interaction with PIP2; black, not relevant for interaction with PIP2 (according to our electrophysiological data; see Fig. 2). The cysteine-free clone used in this study is based on isoform 0, whose N terminus is 95 amino acids shorter than isoform 1 and in which the first 11 amino acids are altered (green). B, representative current traces and voltage protocol with steps from −100 to 40 mV to −120 mV. Current of mutant H126C (red) was slightly decreased compared with wild type (black), whereas the current of mutant R181C was increased. C, mean data ± S.E. of current-voltage relationships between wild type (emphasized) and mutants are similar. The arrow in voltage-step protocol marks the time point of analysis. D, currents at 40 mV were normalized to the corresponding wild type. The mean data ± S.E. (all wild type recordings averaged) show significantly increased (R181C, K183C, R190C, R192C, K196C, R249C, R259C, and R360C) or unchanged (H126C, R195C, K354C, K358C, and K362C) current amplitudes. n, number of oocytes. Asterisks indicate significance: ***, p < 0.001; **, p < 0.01; *, p < 0.05; ns, not significant.
Because oocyte batches may vary in their endogenous PIP2 levels and hence binding sites may be occupied to different extents, we included only those batches in which increased WT currents were observed after PIP2 application. In these batches, currents of the mutants were increased, decreased, or unchanged (Fig. 2A). To discover different properties upon diC8-PIP2 application, we analyzed current amplitudes. The amplitudes at 40 mV were normalized to the respective current without the PIP2 analog diC8-PIP2 of each individual batch (Fig. 2B). diC8-PIP2 significantly increased the WT (−cys) current amplitude to ∼160% on average, as well as the amplitudes of four mutants shown in gray (H126C, R195C, K196C, and K358C). Supposedly, the stimulation indicates that diC8-PIP2 was able to stabilize the open channel conformation of these mutants. Four mutants showed reduced stimulation (marked in blue; K183C, R190C, R249C, and K354C) to values between 120 and 150%. In contrast, currents of mutants shown in red (R181C, R192C, R259C, R360C, and K362C) were either not affected or even significantly decreased by up to 25% upon diC8-PIP2 injection, indicating an impaired functional interaction with PIP2. Further analysis of the diC8-PIP2 effects on mutant channel currents compared with wild type emphasized these differences (Fig. 2C). Here, current amplitudes were normalized to the WT (−cys) current with diC8-PIP2 of each individual batch. This analysis indicated no significant differences between current amplitudes of wild type and mutants shown in gray, suggesting that these residues do not play a significant role in the protein-lipid interaction. In contrast, current amplitudes of mutants depicted in blue were stimulated to a lesser extent compared with WT channels, implying an involvement in the interaction. Intriguingly, currents carried by several mutants shown in red were unchanged or reduced by diC8-PIP2 injection, suggesting an important role for these residues in PIP2 coordination.
FIGURE 2.

Kv7. 1/KCNE1 channel currents were changed in oocytes upon injection of the water soluble PIP2 analog diC8-PIP2. Currents were recorded 3–4 days after injection. diC8-PIP2 was injected at least 30 min prior to the recordings. A, representative current traces recorded with the voltage protocol shown in Fig. 1B. Black, without diC8-PIP2; red, with diC8-PIP2. Currents were increased (wild type and H126C), decreased (R181C), or unchanged (R192C) by diC8-PIP2. B, current amplitudes recorded in the presence of diC8-PIP2 were normalized to the corresponding currents without diC8-PIP2. The mean data ± S.E. at 40 mV show changes in currents amplitude by diC8-PIP2. Currents of wild type (all recordings averaged), H126C, K183C, R190C, R195C, K196C, R249C, K354C, and K358C were increased by diC8-PIP2. Currents of R181C, R192C, R259C, R360C, and K362C were unchanged or decreased upon diC8-PIP2 application. C, results with diC8-PIP2 were scaled and normalized to the wild type amplitude of the individual batch of oocytes also with diC8-PIP2. Hence, the value of 100% represents the increase of current amplitude for WT channels by diC8-PIP2, a value of 0 reflects no change in current amplitude by diC8-PIP2 at all, and values below 0 show decreased current amplitudes. Mutants equally increased as wild type (H126C, R195C, K196C, and K358C) are colored in gray. Mutants with reduced diC8-PIP2 stimulation compared with wild type (K183C, R190C, R249C, and K354C) are colored in blue, and those mutants whose function is completely inhibited upon diC8-PIP2 application (R191C, R192C, R259C, R360C, and K362C) are colored in red. n, number of oocytes. Asterisks indicate significance: ***, p < 0.001; **, p < 0.01; *, p < 0.05; ns, not significant.
Next we analyzed current-voltage relationships of the neutralizing mutants after injection of diC8-PIP2. To examine differences in voltage dependence of activation caused by diC8-PIP2, we fit our data according to the Boltzmann equation multiplied by the driving force (representative fits in Fig. 3A). Analysis of the half-maximal activation voltage V½ showed that the values of the various mutants were distributed between −20 and 10 mV compared with −4 mV of the WT (−cys) (Fig. 3B). The voltage dependences of WT (−cys) and mutants R181C, R190C, R249C, R259C, and K358C were unchanged by diC8-PIP2. The voltage dependences of mutants R192C, K354C, and R360C were shifted by diC8-PIP2 to more negative potentials. Half-maximal activation voltages of mutants H126C, K183C, R195C, K196, and K362C were shifted to more positive potentials. However, significances could not be determined. Comparison between half-maximal activation voltage upon diC8-PIP2 injection and current-amplitude changes revealed no correlation (Fig. 3C).
FIGURE 3.
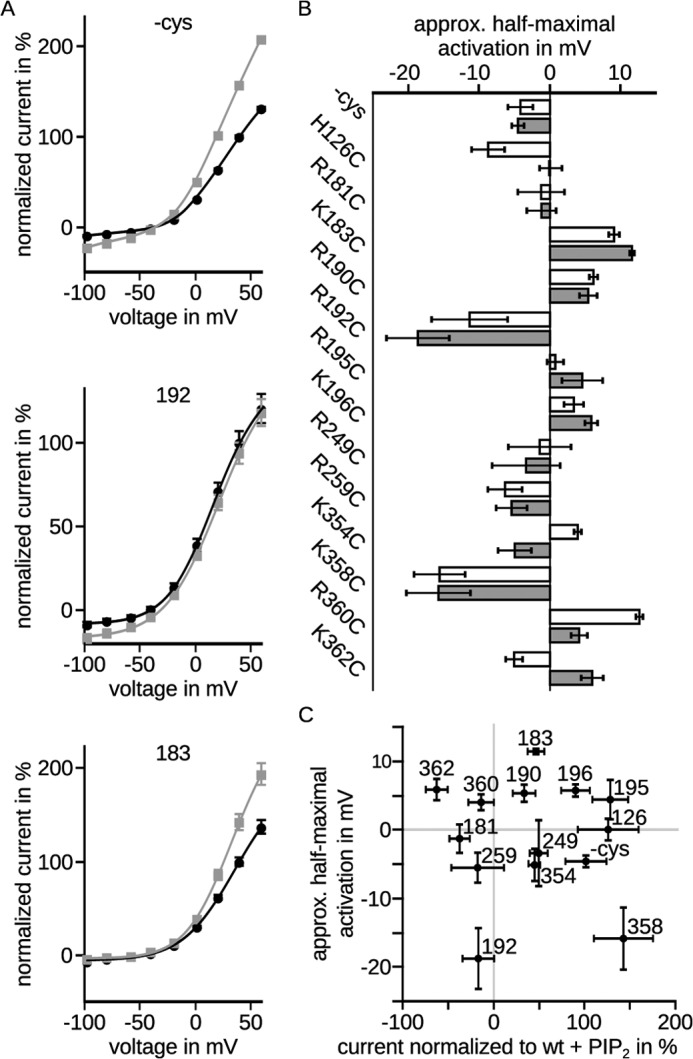
Conductivity-voltage relationships were differently affected by diC8-PIP2. Currents were recorded 3 days after injection. diC8-PIP2 was injected at least 30 min prior to the recordings. A, channel conductivity was calculated from current-voltage relationships obtained by the voltage protocol shown in Fig. 1B. The mean data ± S.E. were fit according to the Boltzmann equation multiplied with the driving force. Black, without diC8-PIP2; gray, with diC8-PIP2. Representative fits showed that saturation was not reached. B, half-maximal activation voltages ± fit error were unchanged (WT, R181C, R190C, R249C, R259C, and K358C) or shifted to positive (H126C, K183C, R195C, K196C, and K362C) or negative potentials (R192C, K354C, and R360C) by diC8-PIP2. White, without diC8-PIP2; gray, with diC8-PIP2. Significance could not be determined. C, half-maximal activation voltage ± fit error is compared with normalized current amplitudes ± S.E. shown in Fig. 2C. Clearly, no correlation between half-maximal activation voltage and normalized current amplitudes could be detected.
As a control, we compared the effect of diC8-PIP2 on Kv7.1 WT and −cys (Fig. 4A). Both current amplitudes were augmented, however, with −cys augmented to a slightly but significantly lesser extent.
FIGURE 4.

A, LQTS mutants impaired PIP2 regulation. Comparison of Kv7.1 −cys and WT. Left panel, representative current amplitude (black, WT; green, −cys). Right panel, current amplitudes recorded in the presence of diC8-PIP2 were normalized to the corresponding currents without diC8-PIP2. The mean data ± S.E. at 40 mV showed similar changes in currents amplitude. B, LQTS mutants were measured with and without diC8-PIP2. The results with diC8-PIP2 were scaled and normalized to the wild type amplitude of the individual batch of oocytes also with diC8-PIP2. Three mutants were lesser increased (blue). n, number of oocytes. Asterisks indicate significance: ***, p < 0.001; **, p < 0.01; *, p < 0.05; ns, not significant. C, the S2-S3 linker and S6 fragment bound PIP2 in vitro. Representative lipid binding assays of the S2-S3 linker (CRSKYVGLWGRLRFARK), S4-S5 linker (GTWRLLGSVVFIHRQEL), and part of S6 (ALKVQQKQRQKHFNR). Binding of the S2-S3 linker and S6 fragment was demonstrated with PtdIns3P, PtdIns4P, PtdIns5P, PtdIns(3,4)P2, PtdIns(3,5)P2, PtdIns(4,5)P2, and PtdInsP3. S6 also bound to PS. No binding was observed for the S4-S5 linker. Positive results are emphasized. LPA, lysophosphatidic acid; LPC, lysophosphatidylcholine; PtdInsP, phosphatidylinositol phosphate; PE, phosphatidylethanolamine; PC, phosphatidylcholine; S1P, sphingosine 1-phosphate; PtdInsP2, phosphatidylinositol bisphosphate; PtdInsP3, phosphatidylinositol trisphosphate; PA, phosphatidic acid; PS, phosphatidyl serine.
Notably, there are known LQTS mutants at amino acid positions 190, 192, 195, 354, 360, and 362. Because we could observe an impaired diC8-PIP2 regulation when neutralizing these residues, we hypothesized that the impaired regulation is the underlying cause for channel dysfunction. Therefore, we also investigated these mutations electrophysiologically under the influence of diC8-PIP2 (Fig. 4B). Indeed, mutations R190L, R192H, and R195W showed less increased current amplitudes. Mutations K354R and K362R showed even enhanced current amplitude increases (not significantly and significantly, respectively). Mutations R360T and R360M did not yield functional currents (data not shown).
Lipid Binding Assay
The S2-S3 linker, the S4-S5 linker, and the C-terminal part of S6, all containing the examined amino acids, were subjected to lipid binding assays (Fig. 4C). Both the S2-S3 linker and S6 were shown to bind to all phosphatidylinositol (PtdIns) phospho derivatives, including PIP2. Additionally, the S6 fragment was positive for phosphatidyl serine binding. The S4-S5 linker did not bind to any of the tested lipids.
Molecular Dynamics Simulation
We based molecular dynamics simulations on Kv7.1/KCNE1 models (4:4 relation because of the ongoing discussion on stoichiometry (40)) in a closed and an open conformation as described by Kang et al. (38) and introduced eight PIP2 molecules into the models. Conventional molecular dynamics simulations on channels in a membranous environment were performed until stable structures were reached. The last nanosecond of each simulation was averaged (Fig. 5A, cyan without PIP2 and yellow with PIP2) and compared with the starting structure (Fig. 5A, green). In the open conformation, differences between the starting structure and the average without the lipid were slightly larger than the differences between the starting structure and the average with PIP2. In the closed conformation, the structural differences without PIP2 were much larger than in presence of PIP2.
FIGURE 5.

Molecular dynamics simulations of Kv7.1/KCNE1 in closed and open conformations showed stabilization of the structures by PIP2. I, assumed inner binding site; II, assumed outer binding site. Four Kv7.1 subunits in complex with four KCNE1 subunits were incorporated into a membrane and NaCl-H2O. A, first structures (green) and mean structures of the last ns of the closed (left panel) and open (right panel) conformation without (cyan) and with (yellow) PIP2, shown from the intracellular side. Yellow, carbon; white, hydrogen; red, oxygen; orange: phosphor. B, RMSD during the simulations show that a stable conformation was reached. C, residue-resolved mean RMSD, averaged over time and subunits. Numbering of residues was in accordance with Kv7.1 variant 1 (GenBankTM accession number NM_000218). Gray rectangles show the approximate transmembrane segments. Large variations were seen in the voltage sensor. Small variations were seen in the S4-S5 linker and helix S6. D, mean RMSD (time, residue, and subunit) for the S4-S5 linker (residues 249–261) and pore helix to helix S6 (residues 305–343). The mean RMSD were increased in the absence of PIP2 in both closed and open conformation. Asterisks indicate significance: ***, p < 0.001. E, stereo view of one PIP2 molecule (sticks) surrounded by several positively charged residues (spheres) in the closed (left panel) and open (right panel) conformation in the averaged structure. Yellow, Kv7.1 backbone and carbon; violet, carbon of PIP2; white, hydrogen; red, oxygen; blue, nitrogen; orange, phosphor.
Root mean square deviation (RMSD) analysis, which indicates the structural variation over time, showed that all structures were in equilibrium for at least the last nanosecond of simulation (Fig. 5B). In equilibrium, the RMSD of all simulations were ∼4 Å, except for the closed conformation with PIP2 with an RMSD of ∼3 Å, agreeing with the direct visual comparison of the structures. The RMSD values of the simulations with and without PIP2 in the open conformation (Fig. 5C, green and blue) were almost identical, whereas there was a large difference of ∼1 Å in the closed conformation (Fig. 5C, black and violet). Residue-resolved RMSD values were averaged over time and all four subunits (Fig. 5C). These values confined these results to the pore region, beginning with the S4-S5 linker, because the voltage sensor itself is much more flexible. Both regions seemed more flexible when all four subunits were averaged, although this was not the case for each individual subunit (data not shown). The results were confirmed by the averaged RMSD of the S4-S5 linker (residues 249–261) and pore helix to the upper part of helix S6 region (residues 305–343; Fig. 4D). RMSD values without PIP2 were enlarged by factors of ∼1.8 and 1.3 (linker) and 1.5 and 1.2 (S6) in the closed and open conformation, respectively.
In addition, both PIP2-free structures were distorted. That is, α-helices were bent or in part degraded, hinting at a destabilization of the channel in the absence of PIP2. Recently, Hansen et al. (41) and Whorton and MacKinnon (42) demonstrated electrostatic interactions as the basis for PIP2 binding in Kir channels, underscoring our assumption. Our results agreed with these data, because some identified residues surrounded the PIP2 head groups (Fig. 5E).
DISCUSSION
PIP2 is a known regulator of diverse ion channels and transporters (30). Recently, two crystal structures of ion channels in complex with PIP2 have been resolved. Key to the interaction of channels with PIP2 are positively charged residues that enable electrostatic interactions with the negatively charged phosphates of PIP2 (41, 42). There are several charged residues at the predicted membrane border of the inner membrane leaflet, which potentially represent PIP2 interaction sites. In this study, we scanned for amino acids in Kv7.1 that functionally interact and potentially bind to PIP2. Wild type current amplitudes were up-regulated by PIP2 to different extents, probably depending on the amount of endogenous PIP2 and whether its concentration was saturating.
First of all, we observed some mutants (R181C, K183C, R190C, R192C, K196C, R249C, R259C, and R360C) with increased current amplitudes compared with WT channels without the influence of diC8-PIP2. Second, we identified residues Lys-183, Arg-190, Arg-249, and Lys-354 to be necessary for PIP2 regulation, because their currents were increased by diC8-PIP2 to a lesser extent than WT currents. Finally, amino acids Arg-181, Arg-192, Arg-259, Arg-360, and Lys-362 seemed even more important because diC8-PIP2 failed to stimulate channel currents or even inhibited function. In the latter case, binding of PIP2 to the mutants may be so unfavorable that the equilibrium shifts toward the closed instead of the open state. Interestingly, these mutants were the same as those that showed increased current amplitudes compared with WT currents with only three exceptions (K196C, K354C, and K362C). This suggests that endogenous PIP2 significantly influences current amplitudes.
To determine whether changed effects on current amplitudes of the neutralizing mutants can be explained by differences in channel kinetics, we analyzed the half-maximal activation voltage of WT and mutant channels. First of all, half-maximal activation voltages of many mutants differ largely from the wild type, which might be influenced by changed interaction with endogenous PIP2. Half-maximal activation voltage of WT was unchanged by application of diC8-PIP2. However, half-maximal activation voltages of several mutants were either shifted to more positive or negative potentials. Nevertheless, these V½ shifts do not correlate with the current-amplitude change upon diC8-PIP2 application. One factor may be weak analysis of the V½ values, because Boltzmann fits were difficult to obtain, since conductivity-voltage relationships did not reach saturation (43), compromising detailed interpretation. The lack of V½-IPIP2 correlation suggests either complete or partial independence and too complex interaction of PIP2 molecules to be analyzed by the techniques used here.
Mutations of several amino acids that we identified as PIP2 interaction sites have been reported to be associated with LQTS (R190L, R190W, and R190Q; R192H and R192P; R195W; R259H, R259L, and R259C; K354R; R360G, R360T, and R360M; and K362R) (51–58). It is plausible that disturbed PIP2 interaction may contribute to an impaired channel function responsible for LQTS. For all of these residues, except for Arg-195, we demonstrated that the current amplitude was not changed by diC8-PIP2 or significantly less increased than WT current amplitude, further supporting the notion that impaired PIP2 binding, and hence regulation is involved in the pathogenesis of LQTS. To test this hypothesis, we investigated some of the mentioned mutations. Indeed, in mutants R190L, R192H, R195W, and R259C, regulation by PIP2 was disturbed, corroborating our assumption. It is noteworthy that also R195W was up-regulated to a lesser extent, although we could not see a difference between R195C and the wild type. Therefore, it is presumable that not the neutralization of the arginine is responsible for the disturbed regulation but rather conformational changes introduced by tryptophan. Analysis of both conserved mutations K354R and K362R, on the other hand, revealed preserved PIP2 regulation, which can be easily explained by the conservation of positive charges. Therefore, a disturbed PIP2 regulation cannot explain channel dysfunction and LQTS development in these cases.
To date, PIP2 binding sites have been identified mainly in the C terminus of ion channels. That is, residues in the C terminus of various inward rectifier potassium (Kir) channels have been shown to participate in PIP2 binding (33, 29, 44–48). Recently, the binding site in Kir channels has been located to the interface between transmembrane domain and C terminus (41, 42). Also, a cluster of basic amino acids in the C terminus of Kv7.2 and Kv7.3 has been demonstrated as a PIP2 binding site (49). Moreover, the C-terminal residues Arg-539 and Arg-555 within Kv7.1 have been shown to participate in the interaction with PIP2 (50). The amino acids identified here are located in the C-terminal portion of helix S6 beneath the membrane (Lys-354, Arg-360, and Lys-362) but also in the intracellular linkers between S2 and S3 (Arg-181, Lys-183, Arg-190, and Arg-192), as well as between S4 and S5 (Arg-249 and Arg-259; Fig. 2C). Unspecific binding to PtdIns was shown for both the S2-S3 linker and the C-terminal fragment of S6. This is in contrast to an earlier study by Thomas et al. (35), who reported that PIP2 binds exclusively in the C terminus, because they did not observe lipid binding of the C-terminally truncated protein in lipid binding assays. A possible explanation for this conflict might be an incorrect folding of the truncated protein. However, Zaydman et al. (59) analyzed the same regions as in this study and also concluded that these regions form PIP2 binding sites. In contrast to the electrophysiological data, the S4-S5 linker did not bind to any of the tested lipids. A possible explanation is that the investigated region is not sufficient for PIP2 binding but requires further protein parts, for example Arg-243, which was already demonstrated to reduce PIP2 affinity (50). However, amino acids Arg-249 and Arg-259 may affect PIP2 regulation allosterically.
Residues outside of the C terminus have been detected before, for instance in the N terminus of Kir channels (42, 46, 48) and at the end of S4 in Kv7.1 (50). Thus, it is possible that all basic residues in a certain distance to the membrane mediate interaction with PIP2 without forming a specific binding site. However, in our model obtained from the molecular dynamics simulations, the identified residues were clustered and might form distinct binding pockets because of their tertiary structure. The PIP2 molecules were in direct contact with most identified residues. These residues might form two binding pockets per subunit together with noncharged residues or the protein backbone. The positions of these proposed binding sites are indicated in Fig. 6. Binding site I in Fig. 6 seemed to be close to the pore domain, which is in line with recent work by Hansen et al. (41), as well as Whorton and MacKinnon (42) showing PIP2 molecules in this region in Kir channel crystal structures. Our results suggest a second binding site in each subunit located further outside at the interface between voltage sensor and plasma membrane.
FIGURE 6.

Proposed positions of PIP2 molecules around voltage-gated potassium channels. The first PIP2 binding site per subunit (light gray, labeled I) is located at the pore domain. We propose the second binding site (dark gray, labeled II) to be located at the voltage sensor.
We found that the absence of PIP2 led to a higher structural flexibility of the pore domain and a distortion of the channel in both the open and closed conformation, suggested by conventional molecular dynamics simulations (Fig. 5). Thus, PIP2 presence might result in a stabilization of the overall channel structure in both conformations. However, in the absence of the lipid, the structural stability of the closed conformation was decreased as suggested by the ∼1 Å larger RMSD values compared with the structure in the presence of PIP2. Consequently, the closed conformation might be structurally more stable in the absence of PIP2. Hence, the thermodynamic equilibrium may be shifted to the closed state, increasing V½ and thereby decreasing the open probability of the channel.
Thomas et al. (35) have previously shown that four amino acids within Kv7.1 helix S6, Lys-354, Lys-358, Arg-360, and Lys-362, directly interact with PIP2. Based on double mutant analysis, they identified residues Lys-358 and Arg-360 as the most important ones. This is largely in agreement with our results, confirming the putative participation of Lys-354, Arg-360, and Lys-362 in PIP2 interactions. Moreover, the importance of Arg-360 is underlined by the observation of completely abolished PIP2 regulation when mutated. However, we could not find any significant participation of the residue Lys-358, because we observed no difference in PIP2 sensitivity compared with WT. A possible explanation for this contradictory finding might be a weak interaction between PIP2 and Lys-358 that can be functionally compensated by other amino acids interacting with PIP2. However, if both residues are lost, compensation might not be possible anymore, increasing the impairment in PIP2 regulation. However, these interpretations require further investigation. Furthermore, Zaydman et al. (59) included the same amino acids as in our study. However, comparison of both studies reveals discrepancies as to which amino acids participate in the interaction and which do not. However, it has to be considered that Zaydman et al. (59) did not analyze the influence of PIP2 directly but rather hypothesized endogenous PIP2 as the cause for loss of function of the mutant channels. Therefore, other influences cannot be excluded.
In summary, we showed that several Kv7.1 amino acids participate in the PIP2 regulation of the cardiac IKs channel Kv7.1/KCNE1. These are located in both the S2-S3 and S4-S5 linker, as well as in the intracellular region of helix S6. Together with molecular dynamics simulations, our results suggested the existence of at least two specific PIP2 binding sites per subunit. Disturbed binding caused by mutations of these amino acids may contribute to the manifestation of LQTS.
Acknowledgment
We thank G. N. Tseng (Virginia Commonwealth University, Richmond, VA) for providing the cysteine-free Kv7.1 clone.
This work was supported by Deutsche Forschungsgemeinschaft Grant 1077/3-1 and by student grants of the International Graduate School of Neuroscience (Ruhr University Bochum) and the Ruhr University Bochum Research School (to K. E.).
- LQTS
- long QT syndrome
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- Kir
- inward rectifier potassium channel
- PtdIns
- phosphatidylinositol
- RMSD
- root mean square deviation.
REFERENCES
- 1. Sanguinetti M. C., Curran M. E., Zou A., Shen J., Spector P. S., Atkinson D. L., Keating M. T. (1996) Coassembly of KvLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature 384, 80–83 [DOI] [PubMed] [Google Scholar]
- 2. Demolombe S., Franco D., de Boer P., Kuperschmidt S., Roden D., Pereon Y., Jarry A., Moorman A. F., Escande D. (2001) Differential expression of KvLQT1 and its regulator IsK in mouse epithelia. Am. J. Physiol. Cell. Physiol. 280, C359–C372 [DOI] [PubMed] [Google Scholar]
- 3. Barhanin J., Lesage F., Guillemare E., Fink M., Lazdunski M., Romey G. (1996) KvLQT1 and lsK (minK) proteins associate to form the IKs cardiac potassium current. Nature 384, 78–80 [DOI] [PubMed] [Google Scholar]
- 4. Schroeder B. C., Waldegger S., Fehr S., Bleich M., Warth R., Greger R., Jentsch T. J. (2000) A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature 403, 196–199 [DOI] [PubMed] [Google Scholar]
- 5. Tinel N., Diochot S., Borsotto M., Lazdunski M., Barhanin J. (2000) KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. EMBO J. 19, 6326–6330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Angelo K., Jespersen T., Grunnet M., Nielsen M. S., Klaerke D. A., Olesen S.-P. (2002) KCNE5 induces time- and voltage-dependent modulation of the KCNQ1 current. Biophys. J. 83, 1997–2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grunnet M., Jespersen T., Rasmussen H. B., Ljungstrøm T., Jorgensen N. K., Olesen S.-P., Klaerke D. A. (2002) KCNE4 is an inhibitory subunit to the KCNQ1 channel. J. Physiol. 542, 119–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pusch M. (1998) Increase of the single-channel conductance of KvLQT1 potassium channels induced by the association with minK. Pflügers. Arch. 437, 172–174 [DOI] [PubMed] [Google Scholar]
- 9. Tristani-Firouzi M., Sanguinetti M. C. (1998) Voltage-dependent inactivation of the human K+ channel KvLQT1 is eliminated by association with minimal K+ channel (minK) subunits. J. Physiol. 510, 37–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tamargo J., Caballero R., Gómez R., Valenzuela C., Delpón E. (2004) Pharmacology of cardiac potassium channels. Cardiovasc. Res. 62, 9–33 [DOI] [PubMed] [Google Scholar]
- 11. Nerbonne J. M., Kass R. S. (2005) Molecular physiology of cardiac repolarization. Physiol. Rev. 85, 1205–1253 [DOI] [PubMed] [Google Scholar]
- 12. Jespersen T., Grunnet M., Olesen S. (2005) The KCNQ1 potassium channel: from gene to physiological function. Physiology (Bethesda) 20, 408–416 [DOI] [PubMed] [Google Scholar]
- 13. Jervell A., Lange-Nielsen F. (1957) Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am. Heart. J. 54, 59–68 [DOI] [PubMed] [Google Scholar]
- 14. Romano C., Gemme G., Pongiglione R. (1963) Rare cardiac arrhythmias of the pediatric age. I. repetitive paroxysmal tachycardia. Minerva Pediatr. 15, 1155–1164 [PubMed] [Google Scholar]
- 15. Fraser G., Froggatt P., Murphy T. (1964) Genetical aspects of the cardio-auditory syndrome of Jervell and Lange-Nielsen (congenital deafness and electrocardiographic abnormalities). Ann. Hum. Genet. 28, 133–157 [DOI] [PubMed] [Google Scholar]
- 16. Ward O. C. (1964) A new familial cardiac syndrome in children. J. Ir. Med. Assoc. 54, 103–106 [PubMed] [Google Scholar]
- 17. Moss A. J., Schwartz P. J., Crampton R. S., Tzivoni D., Locati E. H., MacCluer J., Hall W. J., Weitkamp L., Vincent G. M., Garson A., Jr., et al. (1991) The long QT syndrome. Prospective longitudinal study of 328 families. Circulation 84, 1136–1144 [DOI] [PubMed] [Google Scholar]
- 18. Moss A. J., Schwartz P. J., Crampton R. S., Locati E., Carleen E. (1985) The long QT syndrome: a prospective international study. Circulation 71, 17–21 [DOI] [PubMed] [Google Scholar]
- 19. Pfaffinger P. (1988) Muscarine and t-LHRH suppress M-current by activating an IAP-insensitive G-protein. J. Neurosci. 8, 3343–3353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lopez H. S., Adams P. R. (1989) A G protein mediates the inhibition of the voltage-dependent potassium M current by muscarine, LHRH, substance P and UTP in bullfrog sympathetic neurons. Eur. J. Neurosci. 1, 529–542 [DOI] [PubMed] [Google Scholar]
- 21. Selyanko A. A., Hadley J. K., Wood I. C., Abogadie F. C., Jentsch T. J., Brown D. A. (2000) Inhibition of KCNQ1–4 potassium channels expressed in mammalian cells via M1 muscarinic acetylcholine receptors. J. Physiol. 522, 349–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Delmas P., Brown D. (2005) Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat. Rev. Neurosci. 6, 850–862 [DOI] [PubMed] [Google Scholar]
- 23. Berridge M. (1984) Inositol trisphosphate and diacylglycerol as second messengers. Biochem. J. 220, 345–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Suh B. C., Hille B. (2002) Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 35, 507–520 [DOI] [PubMed] [Google Scholar]
- 25. Zhang H., Craciun L. C., Mirshahi T., Rohács T., Lopes C. M., Jin T., Logothetis D. E. (2003) PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37, 963–975 [DOI] [PubMed] [Google Scholar]
- 26. Huijbregts R. P., Topalof L., Bankaitis V. A. (2000) Lipid metabolism and regulation of membrane trafficking. Traffic 1, 195–202 [DOI] [PubMed] [Google Scholar]
- 27. Martin T. (2001) PI(4,5)P2 regulation of surface membrane traffic. Curr. Opin. Cell Biol. 13, 493–499 [DOI] [PubMed] [Google Scholar]
- 28. Nebl T., Oh S. W., Luna E. J. (2000) Membrane cytoskeleton: PIP2 pulls the strings. Curr. Biol. 10, R351–R354 [DOI] [PubMed] [Google Scholar]
- 29. Sechi A. S., Wehland J. (2000) The actin cytoskeleton and plasma membrane connection: PtdIns(4,5)P2 influences cytoskeletal protein activity at the plasma membrane. J. Cell Sci. 113, 3685–3695 [DOI] [PubMed] [Google Scholar]
- 30. Hilgemann D. W., Feng S., Nasuhoglu C. (2001) The complex and intriguing lives of PIP2 with ion channels and transporters. Sci. STKE 2001, re19. [DOI] [PubMed] [Google Scholar]
- 31. Seebohm G., Wrobel E., Pusch M., Dicks M., Terhag J., Matschke V., Rothenberg I., Ursu O., Hertel F., Pott L., Lang F., Schulze-Bahr E., Hollmann M., Stoll R., Strutz-Seebohm N. (2014) Structural basis of PI(4,5)P2-dependent regulation of GluA1 by phosphatidylinositol-5-phosphate 4-kinase, type II, alpha (PIP5K2A). Pflügers Arch., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chuang H. H., Prescott E. D., Kong H., Shields S., Jordt S. E., Basbaum A. I., Chao M. V., Julius D. (2001) Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 411, 957–962 [DOI] [PubMed] [Google Scholar]
- 33. Huang C. L., Feng S., Hilgemann D. W. (1998) Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature 391, 803–806 [DOI] [PubMed] [Google Scholar]
- 34. Loussouarn G., Park K.-H., Bellocq C., Baró I., Charpentier F., Escande D. (2003) Phosphatidylinositol-4,5-bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage-gated potassium channels: a functional homology between voltage-gated and inward rectifier K+ channels. EMBO J. 22, 5412–5421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thomas A. M., Harmer S. C., Khambra T., Tinker A. (2011) Characterisation of a binding site for anionic phospholipids on KCNQ1. J. Biol. Chem. 286, 2088–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Seebohm G., Strutz-Seebohm N., Baltaev R., Korniychuk G., Knirsch M., Engel J., Lang F. (2005) Regulation of KCNQ4 potassium channel prepulse dependence and current amplitude by SGK1 in Xenopus oocytes. Cell. Physiol. Biochem. 16, 255–262 [DOI] [PubMed] [Google Scholar]
- 37. Xu X., Jiang M., Hsu K.-L., Zhang M., Tseng G.-N. (2008) KCNQ1 and KCNE1 in the IKs channel complex make state-dependent contacts in their extracellular domains. J. Gen. Physiol. 131, 589–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kang C., Tian C., Sönnichsen F. D., Smith J. A., Meiler J., George A. L., Jr., Vanoye C. G., Kim H. J., Sanders C. R. (2008) Structure of KCNE1 and implications for how it modulates the KCNQ1 potassium channel. Biochemistry 47, 7999–8006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Strutz-Seebohm N., Pusch M., Wolf S., Stoll R., Tapken D., Gerwert K., Attali B., Seebohm G. (2011) Structural basis of slow activation gating in the cardiac IKs channel complex. Cell. Physiol. Biochem. 27, 443–452 [DOI] [PubMed] [Google Scholar]
- 40. Wrobel E., Tapken D., Seebohm G. (2012) The KCNE Tango: how KCNE1 interacts with Kv7.1. Front. Pharmacol. 3, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hansen S. B., Tao X., MacKinnon R. (2011) Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 477, 495–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Whorton M. R., MacKinnon R. (2011) Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell. 147, 199–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Seebohm G., Lerche C., Busch A. E., Bachmann A. (2001) Dependence of IKs biophysical properties on the expression system. Pflügers Arch. 442, 891–895 [DOI] [PubMed] [Google Scholar]
- 44. Fan Z., Makielski J. C. (1997) Anionic phospholipids activate ATP-sensitive potassium channels. J. Biol. Chem. 272, 5388–5395 [DOI] [PubMed] [Google Scholar]
- 45. Zhang H., He C., Yan X., Mirshahi T., Logothetis D. E. (1999) Activation of inwardly rectifying K+ channels by distinct PtdIns(4,5)P2 interactions. Nat. Cell Biol. 1, 183–188 [DOI] [PubMed] [Google Scholar]
- 46. Shyng S. L., Cukras C. A., Harwood J., Nichols C. G. (2000) Structural determinants of PIP2 regulation of inward rectifier KATP channels. J. Gen. Physiol. 116, 599–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lopes C. M., Zhang H., Rohacs T., Jin T., Yang J., Logothetis D. E. (2002) Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron. 34, 933–944 [DOI] [PubMed] [Google Scholar]
- 48. Thomas A. M., Brown S. G., Leaney J. L., Tinker A. (2006) Differential phosphoinositide binding to components of the G protein-gated K+ channel. J. Membr. Biol. 211, 43–53 [DOI] [PubMed] [Google Scholar]
- 49. Hernandez C. C., Zaika O., Shapiro M. S. (2008) A carboxy-terminal inter-helix linker as the site of phosphatidylinositol 4,5-bisphosphate action on Kv7 (M-type) K+ channels. J. Gen. Physiol. 132, 361–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Park K. H., Piron J., Dahimene S., Mérot J., Baró I., Escande D., Loussouarn G. (2005) Impaired KCNQ1-KCNE1 and phosphatidylinositol-4,5-bisphosphate interaction underlies the long QT syndrome. Circ. Res. 96, 730–739 [DOI] [PubMed] [Google Scholar]
- 51. Donger C., Denjoy I., Berthet M., Neyroud N., Cruaud C., Bennaceur M., Chivoret G., Schwartz K., Coumel P., Guicheney P. (1997) KVLQT1 C-terminal missense mutation causes a forme fruste long-QT syndrome. Circulation 96, 2778–2781 [DOI] [PubMed] [Google Scholar]
- 52. Chouabe C., Neyroud N., Richard P., Denjoy I., Hainque B., Romey G., Drici M. D., Guicheney P., Barhanin J. (2000) Novel mutations in KvLQT1 that affect IKs activation through interactions with Isk. Cardiovasc. Res. 45, 971–980 [DOI] [PubMed] [Google Scholar]
- 53. Kubota T., Shimizu W., Kamakura S., Horie M. (2000) Hypokalemia-induced long QT syndrome with an underlying novel missense mutation in S4-S5 linker of KCNQ1. J. Cardiovasc. Electrophysiol. 11, 1048–1054 [DOI] [PubMed] [Google Scholar]
- 54. Napolitano C., Priori S. G., Schwartz P. J., Bloise R., Ronchetti E., Nastoli J., Bottelli G., Cerrone M., Leonardi S. (2005) Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA 294, 2975–2980 [DOI] [PubMed] [Google Scholar]
- 55. Millat G., Chevalier P., Restier-Miron L., Da Costa A., Bouvagnet P., Kugener B., Fayol L., Gonzàlez Armengod C., Oddou B., Chanavat V., Froidefond E., Perraudin R., Rousson R., Rodriguez-Lafrasse C. (2006) Spectrum of pathogenic mutations and associated polymorphisms in a cohort of 44 unrelated patients with long QT syndrome. Clin. Genet. 70, 214–227 [DOI] [PubMed] [Google Scholar]
- 56. Kapplinger J. D., Tester D. J., Salisbury B. A., Carr J. L., Harris-Kerr C., Pollevick G. D., Wilde A. A., Ackerman M. J. (2009) Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 6, 1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang T., Chung S.-K., Zhang W., Mullins J. G., McCulley C. H., Crawford J., MacCormick J., Eddy C.-A., Shelling A. N., French J. K., Yang P., Skinner J. R., Roden D. M., Rees M. I. (2009) Biophysical properties of 9 KCNQ1 mutations associated with long-QT syndrome. Circ. Arrhythm. Electrophysiol. 2, 417–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kanovsky J., Novotny T., Kadlecova J., Gaillyova R. (2010) A new homozygous mutation of the KCNQ1 gene associated with both Romano-Ward and incomplete Jervell Lange-Nielsen syndromes in two sisters. Heart Rhythm. 7, 531–533 [DOI] [PubMed] [Google Scholar]
- 59. Zaydman M. A., Silva J. R., Delaloye K., Li Y., Liang H., Larsson H. P., Shi J., Cui J. (2013) Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proc. Natl. Acad. Sci. U.S.A. 110, 13180–13185 [DOI] [PMC free article] [PubMed] [Google Scholar]