

Background: DNA repair on mitotic chromosomes is attenuated. However, the response of specific chromosomal domains has not been studied.
Results: Damage to a single chromosome tip, but not arms, during mitosis results in a localized DNA damage response and anaphase onset delay.
Conclusion: DNA damage induced at chromosome tips and arms in mitosis has different consequences.
Significance: These findings provide the first evidence of a chromosome locus-specific DNA damage response in mitosis.
Keywords: Chromosomes, Cytokinesis, DNA Damage, DNA Damage Response, Mitosis, Anaphase, Laser Microirradiation
Abstract
Chromosome ends contain nucleoprotein structures known as telomeres. Damage to chromosome ends during interphase elicits a DNA damage response (DDR) resulting in cell cycle arrest. However, little is known regarding the signaling from damaged chromosome ends (designated here as “TIPs”) during mitosis. In the present study, we investigated the consequences of DNA damage induced at a single TIP in mitosis. We used laser microirradiation to damage mitotic TIPs or chromosome arms (non-TIPs) in PtK2 kidney epithelial cells. We found that damage to a single TIP, but not a non-TIP, delays anaphase onset. This TIP-specific checkpoint response is accompanied by differential recruitment of DDR proteins. Although phosphorylation of H2AX and the recruitment of several repair factors, such as Ku70-Ku80, occur in a comparable manner at both TIP and non-TIP damage sites, DDR factors such as ataxia telangiectasia mutated (ATM), MDC1, WRN, and FANCD2 are specifically recruited to TIPs but not to non-TIPs. In addition, Nbs1, BRCA1, and ubiquitin accumulate at damaged TIPs more rapidly than at damaged non-TIPs. ATR and 53BP1 are not detected at either TIPs or non-TIPs in mitosis. The observed delay in anaphase onset is dependent on the activity of DDR kinases ATM and Chk1, and the spindle assembly checkpoint kinase Mps1. Cells damaged at a single TIP or non-TIP eventually exit mitosis with unrepaired lesions. Damaged TIPs are segregated into micronuclei at a significantly higher frequency than damaged non-TIPs. Together, these findings reveal a mitosis-specific DDR uniquely associated with chromosome ends.
Introduction
Unrepaired DNA damage can lead to mutation, chromosomal fragmentation, and genomic rearrangements (1–3). DNA damage, in the form of double-stranded breaks (DSBs)4 or single-stranded breaks, can be generated by many processes, such as the collapse of the replication fork, reactive oxygen species, and radiomimetic compounds (4–8). The DNA damage response (DDR) pathways recognize DNA lesions and recruit proteins to these sites to promote repair. The ends of linear chromosomes, which contain telomeres (also referred to as “TIPs” in this work), can also be recognized as damaged DNA. Telomeres are normally protected from recognition as DNA damage or inappropriate repair processes by a nucleoprotein structure composed of repetitive DNA and the shelterin protein complex (9–13). Removal of this protein complex from chromosome ends results in local activation of the DDR and recruitment of repair proteins to the telomeres (6, 12, 14, 15).
Shelterin proteins TIN2, TRF1, RAP1, TRF2, TPP1, and POT1 act to prevent activation of the canonical DDR. Specifically, TRF2 binds to double-stranded DNA ends to prevent its recognition as a DSB and subsequent activation of the ataxia telangiectasia mutated (ATM) kinase. On the other hand, POT1 binds to single-stranded DNA in the telomere, preventing ATR kinase (ATM and Rad3-related) activation (16). DNA lesions and uncapping of the telomere during interphase of the cell cycle both activate ATM and ATR kinases, phosphorylation of downstream kinases Chk1 and Chk2, and the transcription factor p53, which leads ultimately to cell cycle arrest with a persistent DDR (17–19). In vertebrate cells, uncapped telomeres induce G2 arrest through the inactivation and degradation of cyclin B by activating the phosphatase Cdc25C (20) as well as the up-regulation of the Cdk inhibitor p21 (17). In addition to activation of DDR kinases, unprotected or damaged telomeres are also marked by phosphorylation of H2AX (γ-H2AX) and recruit a number of repair proteins, such as MDC1 and 53BP1, which form telomere dysfunction-induced foci (17, 18).
Our understanding of how the cell responds to telomere damage in mitosis is limited. Cells with defective ATM or p53 escape from G2 arrest and enter mitosis with persisting telomere dysfunction-induced foci on mitotic chromosomes (20). However, recent studies indicated that DDR is attenuated in mitosis compared with interphase (21–23), and thus cells may respond differently to telomere damage induced during mitosis (24). A recent study demonstrated that the forced mitotic arrest results in telomere uncapping with γ-H2AX focus formation and ATM activation (25). However, the cellular response to telomere damage in mitosis under normal cell cycling conditions has not been explored in detail.
In this study, we asked how cells respond to damage induced specifically at telomere-containing chromosome ends (TIPs) in comparison with damage induced at chromosome arms (non-TIPs) in mitosis using laser microirradiation. We performed systematic comparison of the DDR and repair factor recruitment at TIP and non-TIP damage sites and the effect on the progression of mitosis. Laser microirradiation has been shown to produce predominantly DSBs, akin to ionizing radiation, eliciting the DDR in mammalian interphase cells, mitotic chromosomes, and anaphase telomeres of PtK2 cells (26–33). Previous studies have shown that laser-induced DNA breaks on mitotic chromosomes do not result in chromosome fragmentation, and DSBs are only introduced at the laser focal spot (27, 33). The spatio-temporal control of laser ablation permits the generation of damaged chromosome tips without genetic perturbation or long term mitotic arrest. Moreover, cells from the rat kangaroo (Potorous tridactylus kidney, PtK2) are ideally suited to study cell division and checkpoint signaling because they have a small number of large metacentric chromosomes (2n = 12) (34), and close sequence identity with humans, mice, and rats (80–90%) (35). Additionally, a single chromosome tip-containing telomere can be targeted by laser microirradiation in these cells (33, 36). This facilitates the investigation of signaling and protein recruitment on a single damaged chromosome tip. Here, we report that laser-induced damage at TIPs recruits a distinct set of DDR factors compared with damage at non-TIPs. Remarkably, the damage to a single TIP results in a delay in the transition from metaphase to anaphase. This delay was found to be dependent on the DNA damage checkpoint kinases ATM and Chk1 and the spindle assembly checkpoint (SAC) kinase Mps1. Despite the damage-induced delay, cells with a single damaged chromosome tip eventually exited mitosis with persistent DNA lesions forming micronuclei in the G1 phase. Thus, our results uncover a mitotic DDR specifically associated with telomere-containing chromosome ends.
MATERIALS AND METHODS
Cell Lines and Cell Culture
Long nosed potoroo (rat kangaroo), P. tridactylus (PtK2 (male) and PtK1 (female)) kidney epithelial cells (American Type Culture Collection (ATCC), CCL 56 and CCL 35) and TRF2-AID-EYFP PtK2 (where AID represents an auxin-inducible degron that has been shown to degrade the AID-tagged target protein upon the addition of a plant hormone; in these experiments, activation of AID was not induced) (37) were grown in Invitrogen Advanced Minimum Essential Medium (Invitrogen) supplemented with l-glutamine, 4% fetal bovine serum (FBS), and antibiotics. PtK1 cells stably expressing a green fluorescent protein (GFP)-tagged Nbs1 previously generated (27) were incubated with Advanced F-12/DMEM supplemented with l-glutamine, 4% FBS, and antibiotics. All cell types were incubated at 37 °C with 5% CO2. Three days before experiments, cells were trypsinized (TrypLETM Express, Invitrogen) and plated on round 35-mm gridded imaging dishes (MatTek, Ashland, MA) at ∼20,000 cells/dish as shown previously (33). Before laser microirradiation, the medium was replaced with Hanks' balanced salt solution (1×) to prevent the absorption of the laser light by the phenol red and to facilitate the monitoring of the cells after damage via live fluorescence imaging.
Generation of Stable PtK Cell Lines
To generate PtK2 cells stably expressing eGFP-53BP1 or TRF2-AID-EYFP, we transiently transfected retroviral plasmids eGFP-53BP1 pLPC (kindly donated by the Denchi laboratory, Scripps Research Institute) and pBABEneo TRF2-AID-EYFP (kindly donated by the Cleveland laboratory, University of California, San Diego) (37) into Phoenix amphotropic packing cell line, using Effectene transfection reagent (Qiagen) according to the manufacturer's instructions. Viral particles were generated using a modified protocol (38, 39). For 53BP1 and TRF2 infections, PtK2 cells were plated in growth medium containing 4 μg ml−1 Polybrene (Sigma) and viruses. Cells were infected at a multiplicity of infection of 3. Forty-eight hours after infection, cells were split and were incubated with medium containing 2 mg ml−1 puromycin for 53BP1 and 2 mg ml−1 neomycin for TRF2. Cells were selected for 5 days. Stable cell lines were further selected using fluorescence-activated cell sorting (FACS) (City of Hope, Duarte, CA).
siRNA Transfection and Sequences
The partial PtK2 ATM sequence was identified by high-throughput sequencing (Illumina) of a commercially generated PtK2 cDNA library (Express Genomics). Duplexes targeting ATM PtK protein were designed and synthesized (Invitrogen). The sequences of the duplexes for ATM siRNA were as follows: 1) sense (5′-GCAGCUUGGUUAAAUACUUTT-3′) and antisense (5′-AAGUAUUUAACCAAGCUGCTT-3′); 2) sense (5′-GCUACUUAUGGAGCGGAUUTT-3′) and antisense (5′-AAUCCGCUCCAUAAGUAGCTT-3′). Scramble siRNA was used as a control. Both siRNAs (1 and 2) were transfected into PtK2 cells using Lipofectamine RNAiMAX (Invitrogen). To obtain 70% transfection efficiency, two consecutive rounds of two-siRNA duplex (1 and 2) transfection were carried out according to the manufacturer's protocol. After 48 h, transfected PtK2 cells were microirradiated and monitored for 15–30 min. Cells were fixed and subjected to immunofluorescent staining using antibodies specific for ATM and γ-H2AX.
Laser Exposure and Dosimetry
A previously described custom RoboLase ablation system was used in these studies (27, 33, 40). Briefly, the system uses a femtosecond pulsed titanium-sapphire near-infrared (NIR) laser (Coherent Inc., Santa Clara, CA) coupled to a motorized inverted Zeiss microscope (Axiovert 200 M) with a 37 °C culture dish stage (Warner Instruments, LLC). LabView software was developed for use with the automated microscope system and laser (41). Single chromosome tips (TIPs) and chromosome arms (non-TIPs) of live unsynchronized metaphase cells were microirradiated using the 200-fs pulse width NIR emission wavelength of 800 nm with a diffraction-limited (0.7-μm diameter) focal spot as shown previously (33). Due to the ultrashort pulse duration, damage was confined to the focal spot (26, 27). Individual laser exposure (irradiation) to single TIP and non-TIP of cells was performed at a dose range of 2.43–2.65 ×10+11 watts/cm2. DNA breaks were assayed by using that DSB marker H2AX, which becomes phosphorylated on serine 139 (γ-H2AX) upon damage. In addition, to check the recruitment of DDR and repair factors at microirradiated DNA, eGFP-53BP1 PtK2 cells were used as controls.5 DNA breaks were created in interphase cells. However, at irradiances above 2.65 ×10+11 watts/cm2, 53BP1 protein was not recruited, which may be due to optical breakdown and microscopic thermoelastic stress waves (28). Thus, the lower irradiance (2.43 ×10+11 watts/cm2) was used for optimal mitotic protein recruitment and kinetics and has recently been shown to produce DSBs (33).
Immunofluorescence and Imaging
Cells grown on gridded dishes were fixed with 3% formaldehyde Tris-buffered saline (TBS) for 10 min at room temperature and placed on ice after microirradiation. Cells were washed three times in PBS and permeabilized with 0.5% Triton X-100 for 10 min at room temperature. Cells were later washed twice with PBS for 5 min at room temperature and incubated with blocking solution (10% calf serum, 1% BSA in PBS) for 1 h at room temperature. Cells were later washed once in PBS for 5 min at room temperature. Next, cells were incubated with primary antibodies in PBS with 3% BSA. After the incubation, cells were washed twice in PBS, 0.05% Tween 20 for 5 min at room temperature and incubated with secondary antibodies (1:1,000; Invitrogen) for 1 h at room temperature. Cells were washed twice with PBS, 0.05% Tween 20 for 5 min at room temperature, and DNA was stained with 4,6-diamidino-2-phenylindole (1:500 in PBS) for 5 min at room temperature. A final wash was performed with PBS for 5 min. Samples were visualized on a Zeiss inverted microscope (Axiovert 200 M) equipped with a Hamamatsu Orca cooled CCD camera. Images were analyzed using ImageJ software (National Institutes of Health, Bethesda, MD). Immunofluorescent staining was repeated at least five times for each antibody in TIP- and non-TIP-damaged cells, and consistent localization results were obtained with all of the cells examined (n > 5). Localization of the DDR signal at damaged TIPs was observed in all of the cells tested.
Antibodies
The following antibodies were used: anti-γ-H2AX (07-164, Millipore) anti-Nbs1 (NB100-143, Novus Biological), anti-Mre11 (NB1000-142, Novus Biologicals), anti-ATM (NB100-104, Novus Biologicals), anti-FANCD2 (NB100-182, Novus Biologicals), anti-MDC1 (NB100-395, Novus Biologicals), anti-XRCC1 (NB100-532, Novus Biologicals), anti-WRN (ab200, Abcam), anti-CtIP (ab70163, Abcam), RNF8 (ab4183, Abcam), RNF168 (ab58063, Abcam), anti-γ-H2AX (9718, Cell Signaling), phospho-Chk1 Ser-345 (2348, Cell Signaling), phospho-Chk2 Thr-68 (2661, Cell Signaling), anti-proliferating cell nuclear antigen (PCNA) (2586, Cell Signaling), anti-GFP (2956, Cell Signaling), anti-Ku70-Ku80 (sc-71471, Santa Cruz Biotechnology, Inc.), phospho-p53 Ser-15 (sc-101762, Santa Cruz Biotechnology), anti-PARP1 (42), anti-ubiquitin (Ub) (spa-205, StressGen), and anti-BRCA1 (GTX50692, GeneTex).
Nbs1 Kinetics
GFP-Nbs1 PtK1 mitotic cells grown in coverglass bottom 35-mm dishes (World Precision Instruments, Inc. Sarasota, FL) were microirradiated at chromosome ends (TIPs) and chromosome arms (non-TIPs), as described above. GFP-Nbs1 recruitment in individual mitotic cells was monitored by taking fluorescent images at 10-s intervals for 120 s with a Hamamatsu Orca cooled CCD camera. Images were analyzed using ImageJ software (National Institutes of Health).
TUNEL Labeling Assay
Microirradiated mitotic PtK2 cells on gridded dishes were fixed with 3.7% formaldehyde in TBS for 10 min at room temperature. Cells were permeabilized twice with PBS, 0.2% Triton X-100 for 10 min at room temperature. Cells were later washed three times in PBS-EDTA for 5 min and washed once with PBS. Cells were incubated with 1:10 enzyme/label solution mix (TUNEL, Roche Applied Science) in a humidified chamber at 37 °C for 1 h. After the reaction, cells were washed three times on a shaker in PBS-EDTA for 5 min to reduce background staining.
Immunofluorescence in Situ Hybridization (FISH) Assay
Microirradiated mitotic PtK2 cells on gridded dishes were fixed and stained with DDR antibodies as described above. Samples were fixed with 3.7% formaldehyde in TBS for 2 min, and telomeres were visualized with a Cy3-congugated (TTAGGG)-PNA probe (DAKO, Carpinteria, CA) as explained in the manufacturer's instructions.
Cell Division Progression
To monitor individual mitotic PtK2 cells aligned at the metaphase plate, individual prophase cells were followed until the alignment of chromosomes at the metaphase plate was complete. DNA breaks were introduced either at a single chromosome end (TIP) or on a chromosome arm (non-TIP) when the chromosomes were aligned at the metaphase plate. Cells were monitored using a Zeiss microscope coupled to a CCD camera as described above. Cells were monitored via time lapse microscopy imaging at 120-s intervals. Time lapse images were processed with ImageJ, and the durations of mitotic progression were quantitatively measured until the onset of chromosome separation in anaphase.
Inhibitor Studies
PtK2 cells grown on gridded dishes were incubated 1 h prior to microirradiation with either one of the following inhibitors: 1) ATMi, ku-60019 or 2) ATMi, ku-55993 at 10 μm. Chk1 inhibitor (Chk1i, AZD7762) was used at 1 μm. Immediately after irradiation, progression through division was microscopically monitored. For experiments with Mps1 inhibitor (Msp1i), cells were incubated with Mps1i at 1 μm (reversine, R3904) immediately after microirradiation, and subsequent progression through division was monitored microscopically (43). To identify the ideal concentration of all of the inhibitors, cells were incubated with concentrations ranging from 100 nm to 50 μm; the readout was the division time of irradiated and non-irradiated samples.
Criteria for Scoring Micronuclei
Damaged mitotic cells were monitored 8–10 h postmicroirradiation using the laser and microscope system described above. After 8–10 h postdamage, cells were fixed with 3% formaldehyde in TBS, stained with DDR antibodies, co-stained with DAPI, and imaged with a Hamamatsu Orca cooled CCD camera. Structures that were morphologically identical but smaller than the nucleus were classified as micronuclei as described previously (44).
Data Analysis
The statistical analysis was performed using a two-tailed, unpaired Student's t test to obtain p values at each time point, comparing the fluorescent signals of DDR proteins at TIP and non-TIP damage sites (normalized by the fluorescent signals before damage in the same region). Statistically significant differences are indicated by asterisks (p < 0.05). Analysis of variance tests were used to determine significant differences (p < 0.05) between GFP-Nbs1 at TIP and non-TIP damage sites and to determine the division time when cells were incubated with and without inhibitors. A χ2 test was used to determine the significance of the presence of micronuclei due to the production of DNA breaks at TIP and non-TIP sites. Tests were performed with GraphPad Prism version 6.00 (GraphPad Software Inc.).
RESULTS
Damage to a Single Chromosome Tip Results in Anaphase Onset Delay
Localized DNA breaks were induced in metaphase cells by laser microirradiation using a focused femtosecond laser source as described previously (27, 28, 33). Recently, we have shown that focal point laser microirradiation can be used to produce sub-μm DNA lesions. This results in the activation of the DDR at single chromosome tips (containing telomeres) and internal chromosomal sites (chromosome arms) of anaphase PtK2 cells (33). To further characterize the response of DDR at chromosome tips, we created a stable PtK2 cell line expressing TRF2-YFP. Telomere damage in the irradiated PtK2 cells was monitored by the loss of TRF2-YFP signal (Fig. 1A). Laser microirradiation was used to specifically ablate a single telomere while leaving the sister telomere intact (Fig. 1, A and B). To verify that the loss of signal represents generation of DSBs and not photobleaching, the appearance of γ-H2AX was examined in relationship to telomeres (detected by a 5′-TTAGGG-3′ FISH probe) (Fig. 1B, TIP). Damage at a chromosome arm (non-TIP) and TIP induced both γ-H2AX and the recruitment of the non-homologous end joining (NHEJ) factor Ku70-Ku80 in a comparable fashion (Fig. 1, B and C). Fluorescent measurement of the γ-H2AX signal at TIP and non-TIP damage sites indicates that comparable amounts of damage were induced at both locations (Fig. 1D).
FIGURE 1.

Laser-induced damage activates the DDR in mitotic chromosomes. A, mitotic PtK2 cell stably expressing YFP-TRF2. Images were taken before microirradiation (pre-NIR) and at 5 s postmicroirradiation (post-NIR). White arrows point to a single telomere before (0 s) and after laser microirradiation (5 s) (also indicated with an asterisk). B, arrows point to the accumulation of γ-H2AX (green) at damaged non-TIP and TIP sites at 5 min after laser microirradiation. Telomeres are detected by the 5′-TTAGGG-3′ FISH probe (red). C, the arrow points to the accumulation of Ku70-Ku80 complex at non-TIP and TIP damage sites. The insets show 2-fold magnification of the images in the dotted boxes. Scale bar, 10 μm. D, quantitative measurements (mean ± S.D. (error bars), p = 0.08) after 120-s accumulation of γ-H2AX at TIP and non-TIP damage sites. Statistics are for five independent values (n = 5). a.u., arbitrary units.
Having established the activation of the DDR at both TIP and non-TIP damage sites on metaphase chromosomes, we investigated their potential effects on mitotic progression. Damage at a non-TIP site failed to cause any significant delay in anaphase onset, as determined by the initiation of chromosome separation, compared with no damage (29.7 ± 16.2 min (S.D, n = 24) and 26.4 ± 8.94 min (S.D, n = 18), respectively) (Fig. 2). In contrast, anaphase onset was significantly delayed by induction of damage at a single TIP (73.2 ± 29.3 min (S.D, n = 17), p < 0.0001) (Fig. 2). There was no apparent chromosome breakage or segregation defect (Fig. 2A). Thus, the results indicate that chromosome TIP-specific DDR triggers a delay in anaphase transition.
FIGURE 2.

TIP damage results in cell cycle delay. A, time lapse microscopy images (in minutes) of mitotic non-irradiated cell (control) and cells irradiated at non-TIP and TIP. The white arrow points to the irradiated chromosome. Scale bar, 10 μm. B, duration of metaphase progression to anaphase was measured in individual cells (non-irradiated or microirradiated at non-TIP or at TIP) using time lapse microscopy imaging with 120-s intervals (see “Materials and Methods” for details) (p < 0.0001 TIP compared with the control). Error bars, S.D.
Damage to a Single TIP Recruits a Unique Set of DDR Proteins
We next examined whether there are any differences in the recruitment of DDR factors between damaged TIP and non-TIP sites that may explain the TIP damage-specific mitotic delay. We detected γ-H2AX, Mre11, Ku70-Ku80, XRCC1, PCNA, phospho-Chk1, and phospho-p53 at both TIP and non-TIP damage sites (Fig. 3 and Table 1). We found, however, that ATM and MDC1 are detected only at damaged TIPs but not non-TIPs (Figs. 4 and 5 and Table 1). Both ATM and MDC1 were detectable at damaged TIPs as early as 15 min after damage induction (Figs. 4A and 5, n = 9 and n = 10), whereas no recruitment was observed at damaged non-TIPs even after 30 min postirradiation (Figs. 4B and 5, n = 9 and n = 10) (data not shown). Fanconi anemia protein FANCD2 and the RecQ (Werner syndrome) DNA helicase (WRN) were also recruited exclusively to damaged TIPs but were undetectable at non-TIP damage sites in all of the cells tested (Fig. 5, n = 10). In contrast, ATR and 53BP1 were not detected at either TIP or non-TIP damage sites (Figs. 4 and 5) (data not shown). Lack of 53BP1 recruitment to uncapped telomeres and DNA damage sites in mitotic cells is consistent with previous studies (21, 45).
FIGURE 3.

Recruitment of DDR and repair proteins to damaged TIP and non-TIP sites. Recruitment of DDR factors along γ-H2AX at damaged TIP (A) and non-TIP (B) sites. Dashed circles indicate DDR foci at TIP and non-TIP damage sites. Cells were fixed at 15 min after irradiation and were stained with antibodies specific for Mre11, Ku70-Ku80, PCNA, XRCC1, phosphorylated Chk1 at serine 345 (pser345), and phosphorylated p53 at serine 15 (pser15) and counterstained with DAPI (blue). Nbs1 and γ-H2AX antibodies were used as controls to detect DNA breaks. Insets show 2-fold magnification of damage signal at TIP and non-TIP sites. Scale bar, 10 μm. The results are summarized in Table 1.
TABLE 1.
DNA damage response factors and repair proteins that form foci at DNA breaks of mitotic cells
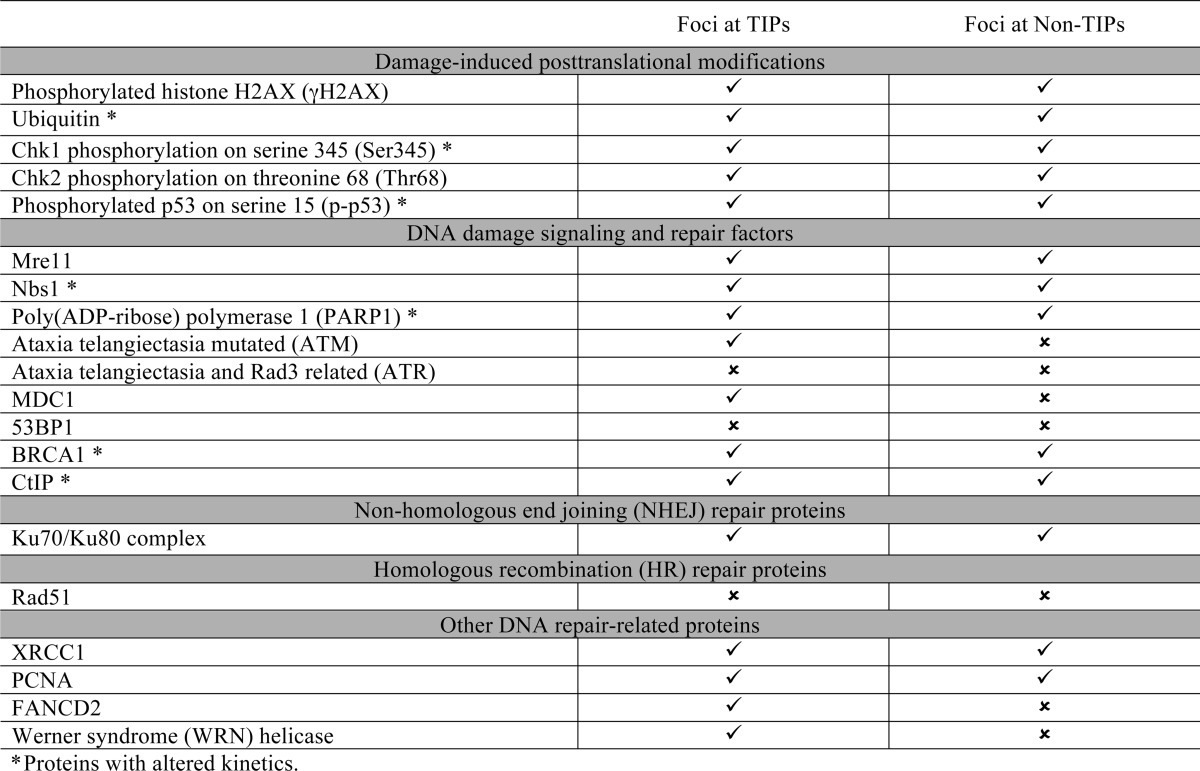
FIGURE 4.

ATM, MDC1, FANCD2, and WRN are preferentially recruited to damaged TIPs. Cells were damaged at a single TIP (A) or non-TIP (B) and fixed at 15 min after damage induction. Damaged samples were stained with antibodies specific for DDR factors and DSB marker γ-H2AX, as indicated. Damage sites are indicated with white arrows in the phase image, and dashed circles indicate corresponding immunofluorescent signals. Asterisks indicate the presence of DDR factors at damaged TIPs. Insets show 2-fold magnification of damage signal at TIP and non-TIP sites. Scale bar, 10 μm. The results are summarized in Table 1.
FIGURE 5.
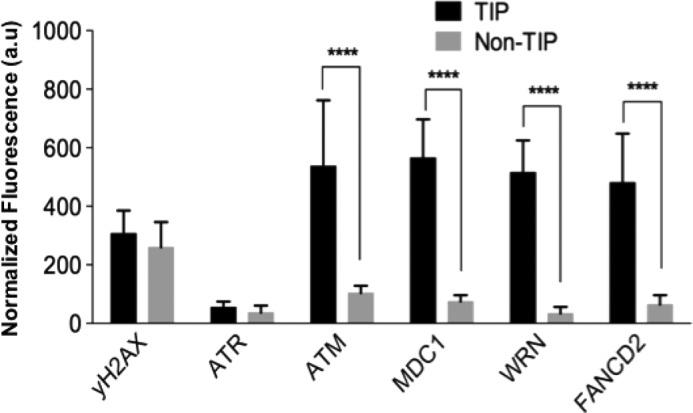
ATM, MDC1, FANCD2, and WRN are preferentially recruited to damage TIPs. Cells were damaged at TIPs and non-TIPs and were fixed at 15 min after damage induction. Cells were subjected to immunofluorescent staining using antibodies specific for γ-H2AX and the indicated DDR factors as in Figs. 3 and 4. Fluorescent signals were quantified by ImageJ software, normalized by the fluorescence signal of the background. n = 10 cells. Two-tailed, unpaired Student's t test was used to obtain p values. a.u., arbitrary units. Error bars, S.D.
We also found that some DDR proteins are recruited to TIP and non-TIP damage sites with different kinetics. PtK2 cells fixed at similar time points after laser damage demonstrated that the recruitment of PARP1 (Fig. 6A, p = 0.005, n = 8) and TUNEL (Fig. 6B, p < 0.003, n = 5) signals persisted for longer periods of time at damaged TIPs compared with non-TIP sites, suggesting the different repair efficiencies. CtIP foci accumulated faster at damaged non-TIP sites compared with damaged TIPs (Fig. 6C, p < 0.05, n = 8). Interestingly, phosphorylation of Chk2, a target of ATM kinase, showed an increased signal at non-TIP sites compared with TIP sites (Fig. 6D, p = 0.006, n = 8). This may be due to the activity of DNA-PK, which was shown to phosphorylate ChK2 in mitosis (46, 47). Similarly, we found that BRCA1 accumulates more efficiently at damaged TIP sites compared with non-TIPs (Fig. 6E, p < 0.05, n = 5). Because BRCA1 recruitment to damage sites is intimately linked to ubiquitylation (48–50), we also examined the Ub signal at TIP and non-TIP damage sites. Previous work has shown that ubiquitylation at IR-induced DNA breaks is absent in mitosis (21). In contrast, we found that the Ub signal is present at laser-induced damage sites in mitosis although with different kinetics at TIP and non-TIP sites (Figs. 3 (bottom) and 6F, p < 0.05). We failed, however, to detect RNF8 and RNF168, the major Ub E3 ligases at damage sites (23, 50), at either TIP or non-TIP damage sites (data not shown). This suggests an alternative mechanism of Ub induction in mitotic PtK2 cells. In addition, GFP-Nbs1 (27) began to accumulate at DNA damage sites 17 ± 4.5 s (S.D.) after microirradiation to a TIP (n = 16), whereas its accumulation was delayed at a non-TIP damage site (27 ± 3.3 s (S.D, p < 0.005, n = 10)) (Fig. 7). The results reveal a faster recruitment and increased accumulation of GFP-Nbs1 at damaged TIPs compared with non-TIPs (Fig. 7B, p < 0.05). Taken together, the results indicate mitosis-specific differential recruitment of repair and checkpoint proteins to TIP and non-TIP damage sites.
FIGURE 6.

DDR factors and repair proteins show altered kinetics at damaged TIPs and non-TIPs. After microirradiation at a single non-TIP or TIP site, cells were fixed at the indicated time points and were subjected to immunofluorescence staining using antibodies specific for PARP1 (A), TUNEL assay (B), CtIP (C), phosphorylated Chk2 (pChk2) (D), BRCA1 (E), and ubiquitin (F). Quantitative measurements (mean ± S.D.) of the fluorescent signals at damaged non-TIP (red) and TIP (blue) sites are plotted. Two-tailed, unpaired Student's t test was used to obtain p values for the differences of fluorescent signals at TIP and non-TIP damage sites at each time point. Asterisks indicate the time points with significant p values. A, PARP1 persists longer at damaged TIP compared with non-TIP sites. Statistics are for eight independent experiments (n = 8; *, p = 0.005). B, TUNEL persists at DNA breaks of mitotic TIP compared with non-TIP sites (n = 5; *, p < 0.002). C, CtIP accumulates more rapidly at non-TIP breaks than at TIP sites (n = 8, p = 0.03; *, p = 0.01). D, pChk2 accumulates faster at non-TIP breaks than at TIPs (n = 8; *, p = 0.03). E, BRCA1 accumulates more at damage TIPs (n = 5; *, p < 0.05). F, ubiquitin accumulates more at damage TIPs (n = 7; *, p < 0.05). a.u., arbitrary units. Error bars, S.D.
FIGURE 7.

Nbs1 accumulates faster at damaged chromosome tip. A, live cell imaging of GFP-Nbs1 accumulation (asterisk shows when Nbs1 starts to accumulate, and white arrow points to the accumulation of Nbs1) at non-TIP and TIP sites of PtK2 cells after microirradiation. Pre-NIR, image taken before microirradiation at time 0 s; Post-NIR, image taken immediately after microirradiation. The white arrows indicate where damage was induced on a single chromosome (Phase image). Fluorescent Nbs1 recruitment at non-TIP and TIP sites was captured at 10-s intervals for 120 s after irradiation. The fluorescent signals were normalized by subtracting the GFP signals in the corresponding regions before damage induction (the pre-laser image (Pre-NIR)). The numbers at the bottom right of each image indicate elapsed time after damage induction (s). B, quantitative measurements (mean ± S.D. (error bars)) at 10-s intervals of GFP-Nbs1 accumulation at TIP (n = 16) and non-TIP (n = 10) sites (p = 0.0048, analysis of variance). a.u., arbitrary units.
Damage-induced Anaphase Delay Requires ATM, Chk1 Activity, and the SAC
During interphase, ATM and Chk1-mediated checkpoint activation can delay cell cycle progression (51). Given the specific localization of ATM at damaged TIPs, we investigated whether the anaphase delay is due to ATM activation. Treatment with ATM and Chk1 inhibitors resulted in the loss of TIP damage-induced anaphase delay (24.9 ± 10.1 min (S.D., p < 0.0001) and 20.5 ± 7.9 min (S.D., p < 0.001), respectively) when compared with untreated cells with TIP damage (73.2 ± 29.3 min (S.D., p < 0.0001) (Fig. 8A). ATM or Chk1 inhibition did not cause any significant change in the timing of anaphase onset in cells with non-TIP damage (Fig. 8B, p > 0.05) and in the absence of damage (data not shown). Similarly, ATM depletion by siRNAs suppressed TIP damage-induced anaphase delay (23.9 ± 5.72 min (S.D., p < 0.0001; Fig. 8A). siRNA transfection had no effect on chromosome segregation (data not shown). ATM depletion was verified by immunofluorescence staining of siRNA-transfected cells (Fig. 8C).
FIGURE 8.

Damaged TIPs activate an ATM-dependent cell cycle arrest. A, the effect of the inhibition of ATM, Chk1, and Mps1 on the anaphase onset delay in the cells with TIP damage. Cells were either untreated or treated for 1 h with the indicated inhibitors or transfected with siRNAs for 48 h before the TIP damage induction in metaphase. Damaged cells were monitored, and the duration of metaphase progression to anaphase was measured as in Fig. 2. Cells were treated with 1 μm Msp1i (reversine) immediately after microirradiation. An analysis of variance test revealed a significant difference among TIP damage-treated cells with inhibitors and siRNAs (p < 0.0001). B, the same analysis as in A for cells with damage at a non-TIP site (p > 0.05). C, immunofluorescence analysis of cells transfected with control scramble siRNA and with two ATM (ATM siRNA 1 and 2) siRNA duplexes. Interphase cells were irradiated 48 h postransfection and fixed at 10 min postmicroirradiation. Cells were stained with ATM (red) and γ-H2AX (green) and counterstained with DAPI (blue). Scale bar, 10 μm. Error bars, S.D.
It is well established that anaphase onset is mediated by the SAC (52). Administration of a small molecule inhibitor of the SAC kinase Mps1, which results in abrogation of the SAC (53), also suppressed the anaphase delay caused by laser-induced TIP damage (23.9 ± 6.3 min (S.D., p < 0.0001) (Fig. 8A). We found no significant acceleration of anaphase onset by the inhibitor treatment in cells with non-TIP damage (Fig. 8B, p > 0.05). The inhibitor did not affect the anaphase onset of undamaged cells (data not shown). Taken together, these results indicate that the anaphase delay caused by TIP damage is dependent on the activity of ATM, Chk1, and Mps1.
Damage at Chromosome Tips Results in the Formation of Micronuclei
Cells with the damaged TIPs eventually undergo anaphase, albeit with delayed kinetics. By monitoring PtK2 cells with a damaged TIP and non-TIP until G1, we observed a significant increase in the presence of micronuclei in cells that were damaged at a single chromosome TIP (54.8%, n = 31) compared with cells that were damaged at a non-TIP (19.2%, n = 26, p < 0.002) or non-irradiated control cells (0%, n = 26, p < 0.002) (Fig. 9A). Damaged TIPs were consistently found within micronuclei, as confirmed by the presence of the telomere repeat sequence using FISH (Fig. 9B, inset b′). Damaged TIPs continued to be marked by γ-H2AX, CtIP (Fig. 9C, TIP panel, inset c′), and PCNA (Fig. 9D, TIP panel, inset d′). Unlike during mitosis, however, 53BP1 associates with damaged TIPs in the micronuclei in G1 (Fig. 10, TIP panel, inset a′). The results indicate that damaged TIPs marked by DDR factors are preferentially encapsulated into micronuclei in the subsequent G1 phase.
FIGURE 9.

Damaged TIPs segregate into micronuclei at G1. A, mitotic cells (damaged at a TIP or non-TIP or no damage) were monitored until G1, fixed, and stained with DAPI to score micronuclei. χ2 test revealed a significant difference between DNA breaks produced at TIP compared with non-TIP and non-irradiated control cells (p = 0.002). Cells with damaged TIPs showed a higher percentage of micronuclei (54.8%). B, microirradiated mitotic cells were monitored until G1, fixed, and stained with Cy3–5′-TTAGGG-3′ probe along with DAPI. Damaged TIP containing telomere repeats is found inside micronuclei (inset b′, white square, *). C, irradiated mitotic cells were monitored until G1, fixed, and stained with antibodies against γ-H2AX and CtIP counterstained with DAPI. CtIP persists into G1 at both non-TIP and TIP DNA breaks (indicated by white squares). Damaged TIP forms a micronucleus colocalizing with CtIP and γ-H2AX (indicated by white squares and asterisks) (2-fold magnification in inset c′). n = 3 independent experiments. D, microirradiated mitotic cells were monitored until G1, fixed, and stained with anti-PCNA and counterstained with DAPI. PCNA is present inside the micronuclei of TIP breaks (inset d′, *). n = 3 independent experiments. Scale bar, 10 μm.
FIGURE 10.
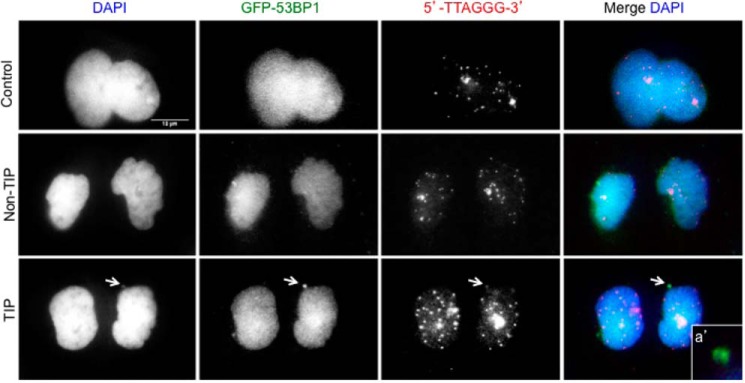
Damaged chromosome tips accumulate 53BP1 in micronuclei. A, PtK2 cells stably expressing GFP-53BP1 were irradiated at TIP and non-TIP during mitosis and monitored until G1. Non-irradiated cells served as controls. Cells were fixed and stained with anti-GFP, Cy3–5′-TTAGGG-3′ probe along with DAPI. Persistent accumulation of 53BP1 colocalizing with Cy3–5′-TTAGGG-3′ is detected inside micronuclei in response to TIP damage (white arrows and inset a′). Insets represent 2-fold magnification. Scale bar, 10 μm.
DISCUSSION
In this study, we show that induction of damage to a single mitotic telomere-containing chromosome tip leads to an ATM/Chk1-dependent anaphase delay that may act through the SAC. In addition, we found that a single damaged chromosome tip specifically recruits ATM, MDC1, FANCD1, and WRN proteins, which are not detected at damaged chromosome arms (Table 1). The mitotic cells with damaged TIPs eventually exit mitosis with an increased occurrence of micronuclei that encapsulate the DNA lesions. Altogether, these results reveal a unique signaling mechanism associated with damaged chromosome ends in mitosis.
ATM and SAC-dependent Mitotic Delay in Response to TIP Damage
In this study, damage to a single telomere-containing chromosome TIP in metaphase cells, as opposed to non-TIP damage sites, delays progression into anaphase and the subsequent separation of chromosomes into daughter cells. Telomere deprotection-induced damage in interphase initiates an ATM/Chk1-dependent damage checkpoint pathway, culminating in cell cycle arrest or delay in G2/M (20) or p53-dependent G1 arrest (24). In mitosis, we also found that ATM and phosphorylated Chk1 at damage sites in the telomere-containing chromosome TIP and subsequent mitotic delay can be suppressed by ATM inhibition, indicating that the similar DDR signaling remains active during mitosis. The delay in anaphase onset caused by TIP damage can be suppressed by inhibition of Mps1, suggesting the involvement of the SAC in this process. ATM activation is known to be involved in the phosphorylation of many SAC components, including SAC kinases, such as Bub1, and core SAC signaling proteins, such as Mad1 and p31comet (54). A recent study has shown that ATM and MDC1 modulate the assembly of SAC components at the kinetochores (55), further supporting the role of ATM signaling in SAC regulation. Recent evidence indicates that the uncapping of telomeres in mitosis results in activation of ATM in human cells (25) and an ATM and SAC-mediated delay in anaphase onset in Drosophila embryos (51, 56). Telomere uncapping-induced damage also results in inhibition of mitotic exit in Saccharomyces cerevisiae (57). Thus, although laser microirradiation may also damage the neighboring subtelomeric regions, the observed ATM/SAC-dependent TIP-specific damage checkpoint signaling is consistent with the disruption of telomere integrity, as also evidenced by the loss of TRF2.
Lack of Checkpoint Activation by Non-TIP Damage in Mitosis
We failed to observe any significant mitotic delay after non-TIP damage in PtK cells. This is consistent with the previous study in which random high irradiance laser-mediated DNA damage evoked no mitotic delay in PtK cells (58). In contrast, in mammalian cells, Plk1 deactivation and subsequent Cdc25C degradation have been reported in response to DNA damage in mitosis (59). Mitotic reversal into a G2-like state, with high cyclin A levels, has also been reported in response to DNA damage (60). In both cases, however, damage was induced genome-wide during nocodazole-induced prometaphase block with SAC activation. Previous work in Drosophila also demonstrated that mitotic damage indirectly activates the SAC (61, 62). A similar mitotic delay was seen in budding yeast, where mitotic DNA damage resulted in Pds1/securin stabilization, preventing Esp1/separase activation and anaphase onset (63). This was recently shown to rely on SAC proteins (64, 65). Additionally, high irradiance laser-mediated ablation to prophase cells reported by Mikhailov et al. (58) demonstrated a SAC-mediated delay in cell cycle progression in human cells. Taken together, these studies indicate the presence of mitotic damage-induced DDR mechanisms. In these studies, damage was induced randomly; thus, whether the DDR signaling was specifically triggered by telomere damage is unclear. It is important to note that PtK cells, like many rodent cells, do not have a strong spindle checkpoint (66). Thus, it is possible that SAC-dependent DDR against non-TIP damage may be attenuated in PtK cells. Further investigation to distinguish TIP and non-TIP damage responses in human cells is important. Nevertheless, our results highlight a distinct mechanism of mitotic surveillance of the telomeres.
Damage at Chromosome Tips Recruits a Distinct Set of Proteins
Our results indicate the selective recruitment of MDC1 at telomere-containing TIP damaged sites but not non-TIP sites of metaphase cells. Although MDC1 focus formation was observed previously following IR damage in mitosis (21), it was unclear whether MDC1 foci are restricted at telomeres. The failure of MDC1 to accumulate at non-TIP damage sites in the current study is consistent with previous studies that suggest that DDR protein recruitment is attenuated in mitosis (67). In addition, our results also show that the accumulation of WRN helicase, FANCD2, and ATM is found specifically at the mitotic TIP and not non-TIP damage sites. WRN is a RecQ helicase involved in DNA repair and is specifically implicated in telomere DDR (68, 69). WRN recruitment to telomeres is also associated with an alternative lengthening pathway of telomeres (70) and requires the activity of FANCD2 (71). It is possible that damaged TIPs are more fragile than non-TIP sites, leading to a more robust DDR at chromosome ends, resulting in the apparent selective recruitment of these factors to damaged TIPs. However, γ-H2AX signals and TUNEL staining suggest that the presence of DNA strand breaks and the DDR are comparable at TIP and non-TIP sites. Thus, our results suggest that these factors make unique contributions to telomere DDR even in mitosis.
It was reported previously that the Ub signal and 53BP1 accumulation are inhibited during mitosis following IR damage in human cells (21). A recent study demonstrated that mitotic kinases phosphorylate RNF8 Ub ligase and 53BP1 to specifically inhibit their damage site recruitment and the NHEJ pathway in mitosis, preventing the telomere fusion in human U2OS cells following IR damage (23). Although we also failed to observe RNF8 and 53BP1 at both TIP and non-TIP damage sites, significant Ub signals were detected at laser-induced damage sites in mitotic PtK cells. This raises the possibility that RNF8-independent Ub signaling is activated by laser-induced damage in mitosis. Furthermore, despite the lack of MDC1, both Ub and BRCA1 were also detectable at non-TIP damage sites, suggesting that Ub signaling and BRCA1 recruitment can occur in an MDC1-independent manner in mitotic PtK cells. This apparent discrepancy may be explained by the species difference (marsupial versus human cells) and/or the different method of damage induction. High-density DNA breaks induced by laser microirradiation at the focal spot of the chromosome may result in the rapid detection of DDR factors not observed by other systems (33, 72). Interestingly, a recent study indicated that the Ub response and BRCA1 recruitment can be mediated by the BBAP Ub ligase independent of ATM and MDC1 at laser-induced damage sites in human cells (73). Thus, a Ub ligase other than RNF8 may also be responsible for Ub and BRCA1 signals at non-TIP damage sites in mitotic PtK cells. The fact that the Ub and BRCA1 signals are stronger at TIPs suggests that the presence of MDC1 further enhances Ub and BRCA1 signals specifically at TIPs. Taken together, the Ub and BRCA1 accumulation at damage sites at TIPs and non-TIPs is complex and may involve multiple mechanisms.
Damage to Chromosome Tips Results in Micronucleus Formation
Despite the recruitment of some of the DDR and repair factors and cell cycle delay, most of the damaged cells eventually enter the G1 phase still carrying unrepaired DNA lesions. Although we observed Ku70-Ku80 recruitment to both TIP and non-TIP damage sites, damage persists in the G1 phase, suggesting that the repair activity is impaired, most likely due to the absence of 53BP1 that facilitates NHEJ. This is consistent with the inhibition of the NHEJ pathway in mitotic U2OS cells (23). There is no discernable recruitment of Rad51 at mitotic damage sites, indicating that the HR pathway is also not active. Interestingly, the single damaged TIP is encapsulated to form a micronucleus in the subsequent G1 at a higher frequency than the damaged non-TIP site. Persistent damage in the micronucleus in G1 is marked by 53BP1, which was absent during mitosis. Micronuclei are characteristic of chromosome instability (44). They are often present in cells with defective DDR or in cells with disrupted cell cycle checkpoint machinery (74). Although micronuclei are often formed as a result of spindle abnormality and chromosome segregation defects (23, 74, 75), telomere erosion has also been reported to result in the formation of micronuclei (23). Interestingly, reactivation of the DDR response at bulk chromatin in mitotic cells by the expression of unphosphorylated 53BP1 and RNF8 mutants also resulted in whole chromosome micronuclei (23). Currently, it is unclear how the specific chromosome or the subregion of the chromosome containing the damaged telomere (or more generally DNA lesions) is selectively encapsulated into a micronucleus. It is also not known whether damaged DNA in the micronuclei are eventually repaired. Inefficient repair of DNA lesions contained in micronuclei can contribute to genome instability (74, 75). It is possible, therefore, that formation of damaged telomere-containing micronuclei may lead to genome instability. Alternatively, segregating the damaged telomere from the rest of the nucleus may help to protect the cell. Further study is necessary to follow the fate of these cells.
Conclusion
Although DDR was thought to be attenuated in mitosis, we found that damage at telomere-containing chromosome tips evokes significant DDR signaling and checkpoint activation during mitosis, followed by encapsulation of damaged telomeres into micronuclei in G1 phase in PtK cells. This may be an important telomere surveillance mechanism to protect cells from transmitting dysfunctional telomeres.
Acknowledgments
We are grateful to Dr. Michelle Duquette-Huber (University of California, San Diego) for anti-ATR antibody; Dr. Eros Lazzerini Denchi (Scripps Research Institute) for the eGFP-53BP1 construct and for several helpful discussions; and Dr. Andrew Holland, Dr. Daniele Fachinetti, and Dr. Don Cleveland (University of California, San Diego) for the TRF2-AID-EYFP construct.
This work was supported, in whole or in part, by National Institutes of Health, NIGMS, Grant R01GM077238 (to J. V. S.). This work was also supported by Air Force Office of Scientific Research Grant FA9550-04-1-0101), the Beckman Laser Institute Inc. Foundation (to M. W. B.), the Beckman Laser Institute Foundation Faculty Fellowship (to J. V. S.), and the National Academy of Sciences Ford Foundation Fellowship Program (to B. A. S.).
B. Alcarez Silva, J. R. Stambaugh, K. Yokomori, J. V. Shah, and M. W. Berns, unpublished data.
- DSB
- double-stranded break
- TIP
- chromosome end/telomere
- DDR
- DNA damage response
- ATM
- ataxia telangiectasia mutated
- SAC
- spindle assembly checkpoint
- NIR
- near-infrared
- Ub
- ubiquitin
- NHEJ
- non-homologous end joining
- PCNA
- proliferating cell nuclear antigen
- EYFP
- enhanced yellow fluorescent protein
- eGFP
- enhanced green fluorescent protein.
REFERENCES
- 1. Berns M. W. (1974) Directed chromosome loss by laser microirradiation. Science 186, 700–705 [DOI] [PubMed] [Google Scholar]
- 2. Phillips J. W., Morgan W. F. (1994) Illegitimate recombination induced by DNA double-strand breaks in a mammalian chromosome. Mol. Cell. Biol. 14, 5794–5803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Richardson C., Jasin M. (2000) Frequent chromosomal translocations induced by DNA double-strand breaks. Nature 405, 697–700 [DOI] [PubMed] [Google Scholar]
- 4. Pfeiffer P., Goedecke W., Obe G. (2000) Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Mutagenesis 15, 289–302 [DOI] [PubMed] [Google Scholar]
- 5. Noon A. T., Goodarzi A. A. (2011) 53BP1-mediated DNA double strand break repair: insert bad pun here. DNA Repair 10, 1071–1076 [DOI] [PubMed] [Google Scholar]
- 6. Takai H., Smogorzewska A., de Lange T. (2003) DNA damage foci at dysfunctional telomeres. Curr. Biol. 13, 1549–1556 [DOI] [PubMed] [Google Scholar]
- 7. Xiao W., Samson L. (1993) In vivo evidence for endogenous DNA alkylation damage as a source of spontaneous mutation in eukaryotic cells. Proc. Natl. Acad. Sci. U.S.A. 90, 2117–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shigenaga M. K., Gimeno C. J., Ames B. N. (1989) Urinary 8-hydroxy-2′-deoxyguanosine as a biological marker of in vivo oxidative DNA damage. Proc. Natl. Acad. Sci. U.S.A. 86, 9697–9701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blackburn E. H. (2001) Switching and signaling at the telomere. Cell 106, 661–673 [DOI] [PubMed] [Google Scholar]
- 10. de Lange T. (2002) Protection of mammalian telomeres. Oncogene 21, 532–540 [DOI] [PubMed] [Google Scholar]
- 11. Greider C. W. (1999) Telomeres do D-loop-T-loop. Cell 97, 419–422 [DOI] [PubMed] [Google Scholar]
- 12. Palm W., de Lange T. (2008) How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42, 301–334 [DOI] [PubMed] [Google Scholar]
- 13. Wright W. E., Shay J. W. (2005) Telomere-binding factors and general DNA repair. Nat. Genet. 37, 116–118 [DOI] [PubMed] [Google Scholar]
- 14. Hockemeyer D., Sfeir A. J., Shay J. W., Wright W. E., de Lange T. (2005) POT1 protects telomeres from a transient DNA damage response and determines how human chromosomes end. EMBO J. 24, 2667–2678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bailey S. M., Meyne J., Chen D. J., Kurimasa A., Li G. C., Lehnert B. E., Goodwin E. H. (1999) DNA double-strand break repair proteins are required to cap the ends of mammalian chromosomes. Proc. Natl. Acad. Sci. U.S.A. 96, 14899–14904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang Q., Zheng Y. L., Harris C. C. (2005) POT1 and TRF2 cooperate to maintain telomeric integrity. Mol. Cell. Biol. 25, 1070–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. d'Adda di Fagagna F., Reaper P. M., Clay-Farrace L., Fiegler H., Carr P., Von Zglinicki T., Saretzki G., Carter N. P., Jackson S. P. (2003) A DNA damage checkpoint response in telomere-initiated senescence. Nature 426, 194–198 [DOI] [PubMed] [Google Scholar]
- 18. Verdun R. E., Crabbe L., Haggblom C., Karlseder J. (2005) Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Mol. Cell 20, 551–561 [DOI] [PubMed] [Google Scholar]
- 19. Fumagalli M., Rossiello F., Clerici M., Barozzi S., Cittaro D., Kaplunov J. M., Bucci G., Dobreva M., Matti V., Beausejour C. M., Herbig U., Longhese M. P., d'Adda di Fagagna F. (2012) Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 14, 355–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thanasoula M., Escandell J. M., Suwaki N., Tarsounas M. (2012) ATM/ATR checkpoint activation downregulates CDC25C to prevent mitotic entry with uncapped telomeres. EMBO J. 31, 3398–3410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Giunta S., Belotserkovskaya R., Jackson S. P. (2010) DNA damage signaling in response to double-strand breaks during mitosis. J. Cell Biol. 190, 197–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peterson S. E., Li Y., Chait B. T., Gottesman M. E., Baer R., Gautier J. (2011) Cdk1 uncouples CtIP-dependent resection and Rad51 filament formation during M-phase double-strand break repair. J. Cell Biol. 194, 705–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Orthwein A., Fradet-Turcotte A., Noordermeer S. M., Canny M. D., Brun C. M., Strecker J., Escribano-Diaz C., Durocher D. (2014) Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 344, 189–193 [DOI] [PubMed] [Google Scholar]
- 24. Cesare A. J., Hayashi M. T., Crabbe L., Karlseder J. (2013) The telomere deprotection response is functionally distinct from the genomic DNA damage response. Mol. Cell 51, 141–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hayashi M. T., Cesare A. J., Fitzpatrick J. A., Lazzerini-Denchi E., Karlseder J. (2012) A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat. Struct. Mol. Biol. 19, 387–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ferrando-May E., Tomas M., Blumhardt P., Stöckl M., Fuchs M., Leitenstorfer A. (2013) Highlighting the DNA damage response with ultrashort laser pulses in the near infrared and kinetic modeling. Front. Genet. 4, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gomez-Godinez V., Wu T., Sherman A. J., Lee C. S., Liaw L. H., Zhongsheng Y., Yokomori K., Berns M. W. (2010) Analysis of DNA double-strand break response and chromatin structure in mitosis using laser microirradiation. Nucleic Acids Res. 38, e202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kong X., Mohanty S. K., Stephens J., Heale J. T., Gomez-Godinez V., Shi L. Z., Kim J. S., Yokomori K., Berns M. W. (2009) Comparative analysis of different laser systems to study cellular responses to DNA damage in mammalian cells. Nucleic Acids Res. 37, e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Botchway S. W., Reynolds P., Parker A. W., O'Neill P. (2010) Use of near infrared femtosecond lasers as sub-micron radiation microbeam for cell DNA damage and repair studies. Mutat. Res. 704, 38–44 [DOI] [PubMed] [Google Scholar]
- 30. Träutlein D., Deibler M., Leitenstorfer A., Ferrando-May E. (2010) Specific local induction of DNA strand breaks by infrared multi-photon absorption. Nucleic Acids Res. 38, e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mari P. O., Florea B. I., Persengiev S. P., Verkaik N. S., Brüggenwirth H. T., Modesti M., Giglia-Mari G., Bezstarosti K., Demmers J. A., Luider T. M., Houtsmuller A. B., van Gent D. C. (2006) Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc. Natl. Acad. Sci. U.S.A. 103, 18597–18602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hartlerode A. J., Guan Y., Rajendran A., Ura K., Schotta G., Xie A., Shah J. V., Scully R. (2012) Impact of histone H4 lysine 20 methylation on 53BP1 responses to chromosomal double strand breaks. PLoS One 7, e49211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Silva B. A., Stambaugh J. R., Berns M. W. (2013) Targeting telomere-containing chromosome ends with a near-infrared femtosecond laser to study the activation of the DNA damage response and DNA damage repair pathways. J. Biomed. Opt. 18, 095003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Altmann S. C. A., Ellery M. E. W. (1925) The chromosomes of four species of marsupials. Q. J. Microsc. Sci. 69, 463–469 [Google Scholar]
- 35. Stout J. R., Rizk R. S., Kline S. L., Walczak C. E. (2006) Deciphering protein function during mitosis in PtK cells using RNAi. BMC Cell Biol. 7, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baker N. M., Zeitlin S. G., Shi L. Z., Shah J., Berns M. W. (2010) Chromosome tips damaged in anaphase inhibit cytokinesis. PLoS One 5, e12398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Holland A. J., Fachinetti D., Han J. S., Cleveland D. W. (2012) Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 109, E3350–E3357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dimitrova N., Chen Y. C., Spector D. L., de Lange T. (2008) 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 456, 524–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shah J. V., Botvinick E., Bonday Z., Furnari F., Berns M., Cleveland D. W. (2004) Dynamics of centromere and kinetochore proteins: implications for checkpoint signaling and silencing. Curr. Biol. 14, 942–952 [DOI] [PubMed] [Google Scholar]
- 40. Duquette M. L., Zhu Q., Taylor E. R., Tsay A. J., Shi L. Z., Berns M. W., McGowan C. H. (2012) CtIP is required to initiate replication-dependent interstrand crosslink repair. PLoS Genet. 8, e1003050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Botvinick E. L., Berns M. W. (2005) Internet-based robotic laser scissors and tweezers microscopy. Microsc. Res. Tech. 68, 65–74 [DOI] [PubMed] [Google Scholar]
- 42. Heale J. T., Ball A. R., Jr., Schmiesing J. A., Kim J. S., Kong X., Zhou S., Hudson D. F., Earnshaw W. C., Yokomori K. (2006) Condensin I interacts with the PARP-1-XRCC1 complex and functions in DNA single-strand break repair. Mol. Cell 21, 837–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Santaguida S., Tighe A., D'Alise A. M., Taylor S. S., Musacchio A. (2010) Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J. Cell Biol. 190, 73–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Terradas M., Martín M., Tusell L., Genescá A. (2009) DNA lesions sequestered in micronuclei induce a local defective-damage response. DNA Repair 8, 1225–1234 [DOI] [PubMed] [Google Scholar]
- 45. Thanasoula M., Escandell J. M., Martinez P., Badie S., Muñoz P., Blasco M. A., Tarsounas M. (2010) p53 prevents entry into mitosis with uncapped telomeres. Curr. Biol. 20, 521–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shang Z. F., Huang B., Xu Q. Z., Zhang S. M., Fan R., Liu X. D., Wang Y., Zhou P. K. (2010) Inactivation of DNA-dependent protein kinase leads to spindle disruption and mitotic catastrophe with attenuated checkpoint protein 2 phosphorylation in response to DNA damage. Cancer Res. 70, 3657–3666 [DOI] [PubMed] [Google Scholar]
- 47. Shang Z., Yu L., Lin Y. F., Matsunaga S., Shen C. Y., Chen B. P. (2014) DNA-PKcs activates the Chk2-Brca1 pathway during mitosis to ensure chromosomal stability. Oncogenesis 3, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang B., Elledge S. J. (2007) Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc. Natl. Acad. Sci. U.S.A. 104, 20759–20763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mailand N., Bekker-Jensen S., Faustrup H., Melander F., Bartek J., Lukas C., Lukas J. (2007) RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 131, 887–900 [DOI] [PubMed] [Google Scholar]
- 50. Doil C., Mailand N., Bekker-Jensen S., Menard P., Larsen D. H., Pepperkok R., Ellenberg J., Panier S., Durocher D., Bartek J., Lukas J., Lukas C. (2009) RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 136, 435–446 [DOI] [PubMed] [Google Scholar]
- 51. Zachos G., Black E. J., Walker M., Scott M. T., Vagnarelli P., Earnshaw W. C., Gillespie D. A. (2007) Chk1 is required for spindle checkpoint function. Dev. Cell 12, 247–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kops G. J., Shah J. V. (2012) Connecting up and clearing out: how kinetochore attachment silences the spindle assembly checkpoint. Chromosoma 121, 509–525 [DOI] [PubMed] [Google Scholar]
- 53. Kwiatkowski N., Jelluma N., Filippakopoulos P., Soundararajan M., Manak M. S., Kwon M., Choi H. G., Sim T., Deveraux Q. L., Rottmann S., Pellman D., Shah J. V., Kops G. J., Knapp S., Gray N. S. (2010) Small-molecule kinase inhibitors provide insight into Mps1 cell cycle function. Nat. Chem. Biol. 6, 359–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Matsuoka S., Ballif B. A., Smogorzewska A., McDonald E. R., 3rd, Hurov K. E., Luo J., Bakalarski C. E., Zhao Z., Solimini N., Lerenthal Y., Shiloh Y., Gygi S. P., Elledge S. J. (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166 [DOI] [PubMed] [Google Scholar]
- 55. Eliezer Y., Argaman L., Kornowski M., Roniger M., Goldberg M. (2014) Interplay between the DNA damage proteins MDC1 and ATM in the regulation of the spindle assembly checkpoint. J. Biol. Chem. 289, 8182–8193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Musarò M., Ciapponi L., Fasulo B., Gatti M., Cenci G. (2008) Unprotected Drosophila melanogaster telomeres activate the spindle assembly checkpoint. Nat. Genet. 40, 362–366 [DOI] [PubMed] [Google Scholar]
- 57. Valerio-Santiago M., de Los Santos-Velázquez A. I., Monje-Casas F. (2013) Inhibition of the mitotic exit network in response to damaged telomeres. PLoS Genet. 9, e1003859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mikhailov A., Cole R. W., Rieder C. L. (2002) DNA damage during mitosis in human cells delays the metaphase/anaphase transition via the spindle-assembly checkpoint. Curr. Biol. 12, 1797–1806 [DOI] [PubMed] [Google Scholar]
- 59. Smits V. A., Klompmaker R., Arnaud L., Rijksen G., Nigg E. A., Medema R. H. (2000) Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat. Cell Biol. 2, 672–676 [DOI] [PubMed] [Google Scholar]
- 60. Chow J. P., Siu W. Y., Fung T. K., Chan W. M., Lau A., Arooz T., Ng C. P., Yamashita K., Poon R. Y. (2003) DNA damage during the spindle-assembly checkpoint degrades CDC25A, inhibits cyclin-CDC2 complexes, and reverses cells to interphase. Mol. Biol. Cell 14, 3989–4002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sibon O. C., Kelkar A., Lemstra W., Theurkauf W. E. (2000) DNA-replication/DNA-damage-dependent centrosome inactivation in Drosophila embryos. Nat. Cell Biol. 2, 90–95 [DOI] [PubMed] [Google Scholar]
- 62. Su T. T., Jaklevic B. (2001) DNA damage leads to a cyclin A-dependent delay in metaphase-anaphase transition in the Drosophila gastrula. Curr. Biol. 11, 8–17 [DOI] [PubMed] [Google Scholar]
- 63. Tinker-Kulberg R. L., Morgan D. O. (1999) Pds1 and Esp1 control both anaphase and mitotic exit in normal cells and after DNA damage. Genes Dev. 13, 1936–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dotiwala F., Harrison J. C., Jain S., Sugawara N., Haber J. E. (2010) Mad2 prolongs DNA damage checkpoint arrest caused by a double-strand break via a centromere-dependent mechanism. Curr. Biol. 20, 328–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kim E. M., Burke D. J. (2008) DNA damage activates the SAC in an ATM/ATR-dependent manner, independently of the kinetochore. PLoS Genet. 4, e1000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Albrecht D. R., Underhill G. H., Resnikoff J., Mendelson A., Bhatia S. N., Shah J. V. (2010) Microfluidics-integrated time-lapse imaging for analysis of cellular dynamics. Integr. Biol. (Camb.) 2, 278–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yu B., Dalton W. B., Yang V. W. (2012) CDK1 regulates mediator of DNA damage checkpoint 1 (MDC1) during mitotic DNA damage. Cancer Res. 72, 5448–5453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Imamura O., Fujita K., Itoh C., Takeda S., Furuichi Y., Matsumoto T. (2002) Werner and Bloom helicases are involved in DNA repair in a complementary fashion. Oncogene 21, 954–963 [DOI] [PubMed] [Google Scholar]
- 69. Eller M. S., Liao X., Liu S., Hanna K., Bäckvall H., Opresko P. L., Bohr V. A., Gilchrest B. A. (2006) A role for WRN in telomere-based DNA damage responses. Proc. Natl. Acad. Sci. U.S.A. 103, 15073–15078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mendez-Bermudez A., Hidalgo-Bravo A., Cotton V. E., Gravani A., Jeyapalan J. N., Royle N. J. (2012) The roles of WRN and BLM RecQ helicases in the alternative lengthening of telomeres. Nucleic Acids Res. 40, 10809–10820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fan Q., Zhang F., Barrett B., Ren K., Andreassen P. R. (2009) A role for monoubiquitinated FANCD2 at telomeres in ALT cells. Nucleic Acids Res. 37, 1740–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Reynolds P., Botchway S. W., Parker A. W., O'Neill P. (2013) Spatiotemporal dynamics of DNA repair proteins following laser microbeam induced DNA damage: when is a DSB not a DSB? Mutat. Res. 756, 14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yan Q., Xu R., Zhu L., Cheng X., Wang Z., Manis J., Shipp M. A. (2013) BAL1 and its partner E3 ligase, BBAP, link poly(ADP-ribose) activation, ubiquitylation, and double-strand DNA repair independent of ATM, MDC1, and RNF8. Mol. Cell. Biol. 33, 845–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Crasta K., Ganem N. J., Dagher R., Lantermann A. B., Ivanova E. V., Pan Y., Nezi L., Protopopov A., Chowdhury D., Pellman D. (2012) DNA breaks and chromosome pulverization from errors in mitosis. Nature 482, 53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Pampalona J., Soler D., Genescà A., Tusell L. (2010) Whole chromosome loss is promoted by telomere dysfunction in primary cells. Genes Chromosomes Cancer 49, 368–378 [DOI] [PubMed] [Google Scholar]