

Background: Enzymes and transporters may contain regulatory nucleotide-binding cystathionine β-synthase domains.
Results: Both substrate and adenine nucleotides bind cooperatively to four bacterial cystathionine β-synthase domain-containing pyrophosphatases.
Conclusion: Cystathionine β-synthase domains internally inhibit catalysis and impart cooperativity; both effects are modulated by bound nucleotides.
Significance: The observed regulatory complexity may be characteristic of cystathionine β-synthase domain-containing proteins involved in hereditary diseases.
Keywords: Allosteric Regulation, Bacterial Signal Transduction, Cooperativity, Enzyme Inhibitor, Ligand-binding Protein, CBS Domain, Pyrophosphatase
Abstract
Regulated family II pyrophosphatases (CBS-PPases) contain a nucleotide-binding insert comprising a pair of cystathionine β-synthase (CBS) domains, termed a Bateman module. By binding with high affinity to the CBS domains, AMP and ADP usually inhibit the enzyme, whereas ATP activates it. Here, we demonstrate that AMP, ADP, and ATP bind in a positively cooperative manner to CBS-PPases from four bacteria: Desulfitobacterium hafniense, Clostridium novyi, Clostridium perfringens, and Eggerthella lenta. Enzyme interaction with substrate as characterized by the Michaelis constant (Km) also exhibited positive catalytic cooperativity that decreased in magnitude upon nucleotide binding. The degree of both types of cooperativity increased with increasing concentration of the cofactor Mg2+ except for the C. novyi PPase where Mg2+ produced the opposite effect on kinetic cooperativity. Further exceptions from these general rules were ADP binding to C. novyi PPase and AMP binding to E. lenta PPase, neither of which had any effect on activity. A genetically engineered deletion variant of D. hafniense PPase lacking the regulatory insert was fully active but differed from the wild-type enzyme in that it was insensitive to nucleotides and bound substrate non-cooperatively and with a smaller Km value. These results indicate that the regulatory insert acts as an internal inhibitor and confers dual positive cooperativity to CBS domain-containing PPases, making them highly sensitive regulators of the PPi level in response to the changes in cell energy status that control adenine nucleotide distribution. These regulatory features may be common among other CBS domain-containing proteins.
Introduction
Pyrophosphate (PPi) is formed as a by-product of nearly 200 biosynthetic reactions in which nucleoside triphosphates are converted to nucleoside monophosphates and PPi (1). Many of these PPi-generating reactions are inhibited by PPi, highlighting the importance of mechanisms that strictly control the free cellular concentration of PPi. This control is generally achieved by the hydrolysis of PPi to phosphate via the action of specific enzymes, soluble inorganic pyrophosphatases (PPases4; EC 3.6.1.1), belonging to three non-homologous families (2–4).
Of these PPases, family II soluble PPases, found in prokaryotes, are the most active (kcat ≈ 104 s−1) and structurally diverse. These homodimeric enzymes are Mn2+ or Co2+ metalloproteins but additionally require Mg2+ for catalysis (5), which occurs by direct attack of a hydroxide ion coordinated with three metal ions (6). Each subunit is formed by two domains connected by a flexible linker with the active site located between the domains (7, 8), making the enzyme ideally suited for allosteric regulation. Indeed, a quarter of the nearly 500 putative family II PPase sequences generally contain a ∼250-residue insert comprising a pair of cystathionine β-synthase (CBS) domains (Bateman module (9)), which have regulatory functions in several other proteins (10, 11–14), and a DRTGG domain of unknown function (see Fig. 1). CBS domains are quite common in nature and are found, for instance, in 75 human proteins and eight proteins of Escherichia coli. Mutations in CBS domains of human enzymes and membrane channels are associated with hereditary diseases, including homocystinuria, retinitis pigmentosa, Bartter syndrome, osteopetrosis, and others (13). CBS domains are self-sufficient units (see Fig. 1B) that retain structure after being removed from functional cores. Moreover, some regulatory proteins consist of only a Bateman module (15, 16). The majority of CBS domain-containing proteins form homodimers with the CBS domains participating in subunit interface interactions. Consequently, the two CBS domain pairs form a disklike structure (10). Interestingly, PPase appears to be the only Bateman module-containing protein in which a DRTGG domain intercalates the CBS domains (see Fig. 1). However, in rare cases, for example Moorella thermoacetica PPase (mtPPase), the enzyme contains only CBS domains in the regulatory part of the molecule. Typically, CBS domains bind adenine nucleotides, such as S-adenosylmethionine, AMP, ADP, and ATP, as regulatory ligands (10, 11–14). Each CBS domain contains a potential binding cavity for nucleotides; however, with rare exceptions (16, 17), each pair of CBS domains generally binds only one adenylate ligand. The structural and kinetic basis for regulation through CBS domains is still unknown for any protein, and CBS domain-containing PPases represent a good model enzyme for exploring this general phenomenon.
FIGURE 1.
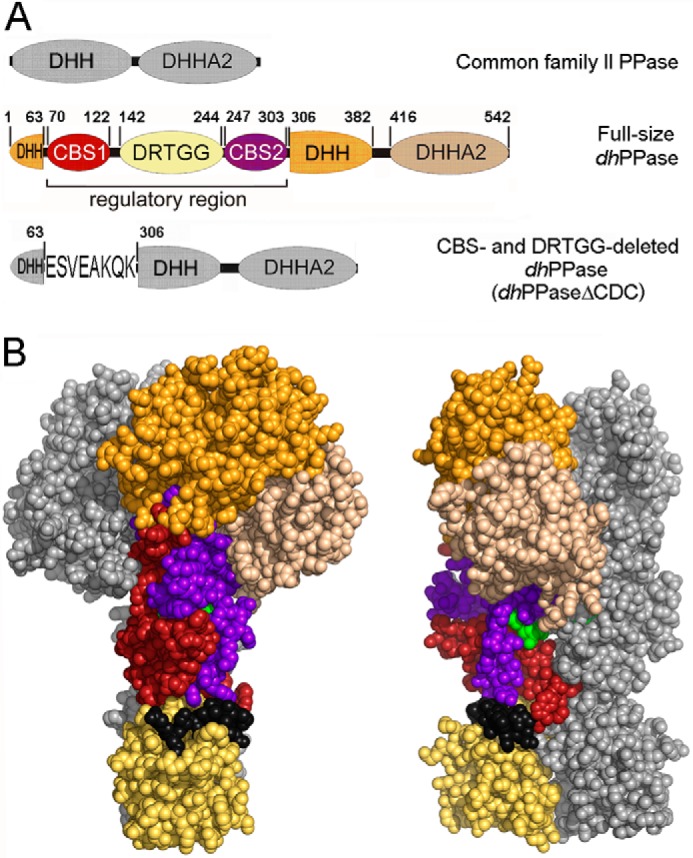
CBS-PPase structure. A, domain arrangement in the primary structures of wild-type dhPPase and its deletion variant. The five domains that form one subunit are shown in different colors and are labeled; short linker regions are depicted in black. DHH and DHHA2 are catalytic domains. Numbers indicate residues that start/end domains or their parts or precede/follow the deleted sequences. Residue numbering is based on the sequence of the full-length protein. For dhPPaseΔCDC, the linker sequence is shown using single letter notation. B, two views of the modeled three-dimensional structure of the cpCBS-PPase homodimers (19) in a sphere representation. Domain colors of one subunit are the same as in A; the other subunit is colored gray. The AMP molecules bound between CBS domains are shown in green.
We reported previously that mtPPase (18) and the CBS domain-containing PPase of Clostridium perfringens (cpPPase) (19) are highly sensitive to micromolar concentrations of adenine nucleotides. Remarkably, AMP and ADP acted as inhibitors, whereas ATP and AP4A were activators. We also determined the three-dimensional structures of an isolated regulatory insert of cpPPase comprising two CBS domains and one DRTGG domain in complexes with AMP and AP4A (19). Rates of AMP-induced conformational transitions were much faster in cpPPase (20) than in mtPPase (21), suggesting a role for the DRTGG domain in regulatory site performance.
Here, we show that the regulatory part of the molecule, comprising two CBS domains with or without an intercalating DRTGG domain, binds adenine nucleotides cooperatively and confers catalytic cooperativity to cpPPase and three newly isolated family II PPases of Desulfitobacterium hafniense (dhPPase), Clostridium novyi (cnPPase), and Eggerthella lenta (elPPase). We found that both types of cooperativity depend on Mg2+ concentration and the identity of the bound nucleotide, further expanding our understanding of the mechanisms involved in regulating cellular PPi level. These findings were corroborated by studies of a genetically engineered dhPPase deletion variant corresponding to the isolated catalytic part of the enzyme. CBS domain-containing PPases are thus the first known oligomeric PPases whose subunits exhibit cooperative behavior.
EXPERIMENTAL PROCEDURES
Cloning
Genomic DNA extracted from D. hafniense, C. novyi, and E. lenta (DSM numbers 10664, 14992, and 2243, respectively) was obtained from DSMZ (Deutsche Sammlung von Microorganismen und Zellkulturen GmbH). The sequences of full-length dhPPase (residues 1–544; YP_002458003), cnPPase (residues 1–548; YP_878001), and elPPase (residues 1–455; YP_003181314) were amplified by PCR and ligated into NdeI and XhoI restriction sites of the pET28bTEV plasmid (Novagen) for dhPPase and pET46 Ek/LIC plasmid (Novagen) for cnPPase and elPPase. A synthetic gene encoding the catalytic part of dhPPase (dhPPaseΔCDC) containing codons for dhPPase fragments 1–63 and 306–544 connected by the linker ESVEAKQK was obtained from GeneArt. The linker sequence was identical to that found in the family II PPase from Desulfosporosinus acidiphilus SJ4 that most closely resembles the catalytic part of dhPPase but has no regulatory element. The gene was subcloned into the pET28bTEV plasmid using NdeI and XhoI restriction sites. The plasmid constructs thus obtained expressed dhPPase, dhPPaseΔCDC, cnPPase, elPPase, or dhCDC with a cleavable His6 tag at their N termini. All constructs were confirmed by sequencing.
Protein Expression and Purification
Full-length dhPPase and dhPPaseΔCDC were produced in the E. coli strain BL21(DE3)-RIL (Stratagene) by cultivating at 37 °C in Terrific Broth medium supplemented with 30 μg/ml each of kanamycin and chloramphenicol. Gene expression was induced by incubating with 1 mm isopropyl β-thiogalactopyranoside for 3 h. Collected cells were suspended in lysis buffer (50 mm Tris-HCl, 500 mm NaCl, 20 mm imidazole-HCl, pH 7.8) and disrupted with an MSE 100-watt ultrasonic disintegrator. The cell lysate was applied to a 2-ml TALON Superflow gravity column (GE Healthcare), and dhPPase was eluted using a buffer containing 50 mm Tris-HCl, 500 mm NaCl, and 0.2 m imidazole-HCl (pH 7.8). Imidazole was removed from eluate fractions containing dhPPase by passing through a HiPrep 26/10 column (GE Healthcare) pre-equilibrated with buffer consisting of 50 mm Tris-HCl (pH 7.8) and 500 mm NaCl. The His6 tag was removed by digesting overnight with tobacco etch virus (TEV) protease at 20 °C as described by the manufacturer after which the mixture was passed through a TALON Superflow gravity column to remove His6-containing peptides from the sample. The flow-through was further purified by size exclusion chromatography on a Superdex 200 26/60 (GE Healthcare) column pre-equilibrated with 50 mm MOPS-KOH (pH 7.2), 150 mm KCl, 0.1 mm CoCl2, and 20 μm diethylenetriaminepentaacetate. cnPPase and elPPase were expressed and purified similarly except that the His6 tag was not removed, and the buffer in size exclusion chromatography was 25 mm MOPS-KOH (pH 7.2), 50 mm KCl, and 0.1 mm CoCl2. cpPPase was expressed and purified as described previously (19). Protein solutions were stored at −40 to −80 °C.
The purity of the isolated proteins was assessed by SDS-PAGE using Phast system 8–25% gradient gels (GE Healthcare). Protein concentrations were determined with a Nanodrop spectrophotometer (Thermo Scientific) using A2800.1% values of 0.478 for dhPPase, 0.419 for dhPPaseΔCDC, 0.548 for cnPPase, 0.426 for cpPPase, and 0.493 for elPPase calculated from the corresponding amino acid compositions with ProtParam. Molar concentrations were calculated on the basis of the subunit molecular masses of 60.4, 34.5, 63.6, 60.8, and 52.5 kDa, respectively.
Kinetic Assays
The activity assay medium contained 140 μm PPi (yielding 50 μm MgPPi complex), 5 mm MgCl2, and 0.1 m Tes-KOH (pH 7.2) except where specified otherwise. In measurements done at higher Mg2+ concentrations, buffer concentration was decreased appropriately to maintain constant ionic strength. The reaction was initiated by adding enzyme, and Pi accumulation due to PPi hydrolysis was continuously recorded for 2–3 min at 25 °C using an automatic Pi analyzer (22). The analyzer utilizes the principle of color change of methyl green in the presence of phosphomolybdate and consists of a three-channel peristaltic pump and a flow-through photometer. The pump delivers a constant flow of sample, acid molybdate, and methyl green solutions, which are successively combined and directed to the flow photometer. The sensitivity of the analyzer was varied to yield full-scale recorder values of 4 or 20 μm Pi using an appropriate choice of pump tubing diameters; corresponding sample consumption rates were 5 and 1 ml/min. The dead time of the analyzer (i.e. the time from feeding the sample into the analyzer to mixing with the acid molybdate solution, which quenches the enzymatic reaction) was 10–20 s in the standard mode. The precision of initial velocity estimates for reactions yielding nonlinear time courses of Pi production was increased by decreasing the dead time to 1 s using an appropriate modification of the inlet system (23). The initial velocity values obtained in replicate measurements agreed within 5–10% and were strictly proportional to enzyme concentration.
Calculations and Data Analysis
The values of the apparent dissociation constants for the various metal ion complexes used to calculate the free metal ion concentrations at pH 7.2 were as follows: MgPPi, 112 μm (24); Mg2PPi, 2.84 mm (24); MgAMP, 10.5 mm (25); MgADP, 0.42 mm (25); and MgATP, 0.034 mm (25). The values for adenine nucleotides were derived from published values of the corresponding pH-independent constants. Nonlinear least square fits were performed using the program Scientist (Micromath).
Initial velocities of PPi hydrolysis were typically estimated graphically from the slopes of the tangents to the initial portion of hydrolysis time courses recorded with the Pi analyzer. The time courses generated by truncated PPase were markedly nonlinear because of gradual inactivation of the enzyme upon dilution into the reaction medium. In this case, the initial velocities (v) were obtained by fitting the time courses to Equation 1,
where kd is the rate constant for enzyme inactivation, t is time, and a is the background signal of the instrument.
The dependence of hydrolysis rate on nucleotide concentration ([N]) at fixed substrate concentration was fit to Equation 2,
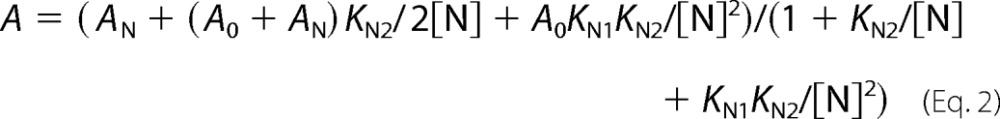 |
where A0 and AN are activities of free and nucleotide-saturated enzyme, respectively, and KN1 and KN2 are the apparent dissociation constants for the enzyme-nucleotide complexes. Equation 2 was derived for Scheme 1, which describes successive binding of nucleotide to two regulatory sites per enzyme molecule. Here, E2 refers to a mixture of free and substrate-bound dimeric enzyme. The activity of E2N is intermediate between the activities of E2 and E2N2 (i.e. activities of two subunits respond independently to nucleotide binding). Equation 2 is equally valid if N is an inhibitor or activator.
SCHEME 1.
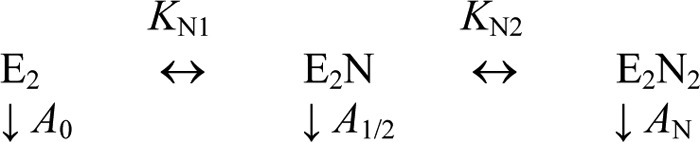
Modulation of PPase activity by nucleotide binding at a fixed substrate concentration.
For non-cooperative binding, Equation 3 with a single KN value was used.
Notably, if Equation 2 instead of Equation 3 is applied to a non-cooperative binding reaction, the fitted values of KN1 and KN2 should differ 4-fold: KN1 = KN/2 and KN2 = 2KN (26). Consequently, the ratio 4KN1/KN2 can be used to recognize cooperativity: it is greater than 1 for positive cooperativity and less than 1 for negative cooperativity. In terms of generally accepted nomenclature, KN is a “microscopic” binding constant, and KN1 and KN2 are “macroscopic” binding constants (26). The merit of using 4KN1/KN2 instead of h to characterize cooperativity is that the former parameter is directly related to the free energy change associated with cooperativity, that is the effect of a ligand binding at one site on the binding affinity of the other site (27).
Alternatively, nucleotide binding data were fit to the Hill equation (Equation 4) to determine the Hill coefficient h.
Cooperative kinetics of substrate hydrolysis was analyzed in terms of Scheme 2, which assumes different Michaelis constants (Km1 and Km2) and equal kcat values for two active sites in the dimer (E2); [E]0 is total enzyme concentration. The values of Km1, Km2, and kcat were obtained by fitting Equation 5 to the dependence of hydrolysis rate (v) on substrate (MgPPi) concentration at fixed Mg2+ concentrations.
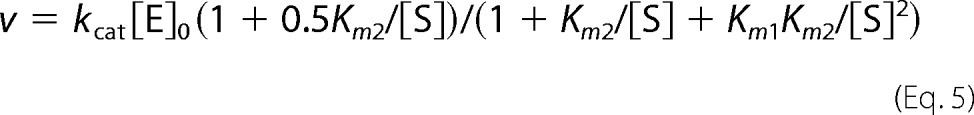 |
As was the case for Equation 2, the ratio 4Km1/Km2 can be used to recognize catalytic cooperativity: its value is greater than 1 for positive cooperativity and less than 1 for negative cooperativity (26). The applicability of the microscopic/macroscopic constant concept to Michaelis constants results from the formal similarity of the Michaelis-Menten equation to the equilibrium binding equation (28) despite the fact that they are not equilibrium substrate binding constants in a general case.
SCHEME 2.
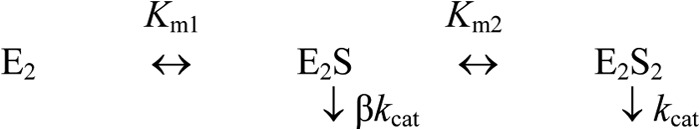
Substrate binding and hydrolysis at a fixed Mg2+ concentration (S = MgPPi).
The dependence of KN on Mg2+ (M) concentration was fit to Equation 6 derived for Scheme 3 where (KN)0 and (KN)M are the limiting values of KN at zero and infinite Mg2+ concentrations, and KM is the metal binding constant. Equations analogous to Equation 6 were used to analyze the Mg2+ concentration dependence of Km.
SCHEME 3.
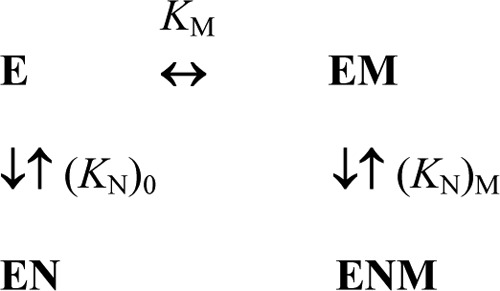
Modulation of nucleotide binding to CBS-PPase by Mg2+.
RESULTS
Production of PPases
The genes for CBS-PPases and the deletion variant of dhPPase (dhPPaseΔCDC; Fig. 1A) were expressed in E. coli, and the proteins were purified with a yield of ∼10 mg of pure protein/liter of medium. Constructs were designed to encode an additional N-terminal MGSSHHHHHHENLYFQGH sequence containing a His6 tag to facilitate isolation by immobilized metal ion affinity chromatography. TEV protease cleaves this sequence between the Gln and Gly residues, leaving only a dipeptide extension on the protein. His6 tag removal did not affect PPase activity but diminished the aggregation of full-size dhPPase during purification and concentration procedures. Tag removal was not performed with dhPPaseΔCDC and other PPases as it did not improve their solubility. Inactive aggregates were separated from soluble active proteins during size exclusion chromatography. The final products were at least 95% pure as estimated by SDS-PAGE.
The final purification step (Superdex 200 chromatography) was conducted in the presence of 0.1 mm Co2+ to stabilize the enzymes; Co2+ was also routinely added to enzyme stock solutions. The stabilizing effect of Co2+ is consistent with data showing that “canonical” family II PPases contain one site that tightly binds transition metal ions (Co2+ or Mn2+) in addition to several more loosely binding sites with broader specificity that are apparently filled with Mg2+ in vivo (5, 29, 30). All of these sites have a role in catalysis, but the tightly bound transition metal ion also has a structural role (7, 8) as it does in many other metalloenzymes. The activities of CBS-PPases preincubated with 0.1 mm Co2+ were somewhat higher than those incubated with Mn2+. Therefore, Co2+ was routinely used as the transition metal cofactor in these studies.
Cooperativity of Substrate Hydrolysis by CBS-PPases
The dependence of CBS-PPase activity on substrate concentration in the presence of 5 mm free Mg2+ exhibited systematic deviations from simple Michaelis-Menten kinetics as illustrated in Fig. 2 for dhPPase. The Hill coefficient h, derived from this dependence using Equation 4 by setting A0 to zero, was 1.37 ± 0.02 (Fig. 2), indicating positive catalytic cooperativity. Therefore, the dependence was analyzed using Equation 5 derived for Scheme 2, which assumes different Michaelis constants (Km1 and Km2) for two active sites. This analysis markedly increased the quality of the fit compared with the Michaelis-Menten equation (Fig. 2) as indicated by a ∼12-fold lower sum of the squares of residuals. The values of Km1 and Km2 were found to be 26 ± 1 and 10 ± 1 μm, respectively; the ratio 4Km1/Km2 (equal to 10) was thus much greater than 1, confirming positive catalytic cooperativity. Importantly, product formation curves were linear, and reaction rates were strictly proportional to enzyme concentration in all cases, ruling out the possibility that the deviation from simple kinetics seen in Fig. 2 resulted from a rapid substrate-dependent change in enzyme oligomeric structure upon dilution into the assay medium.
FIGURE 2.
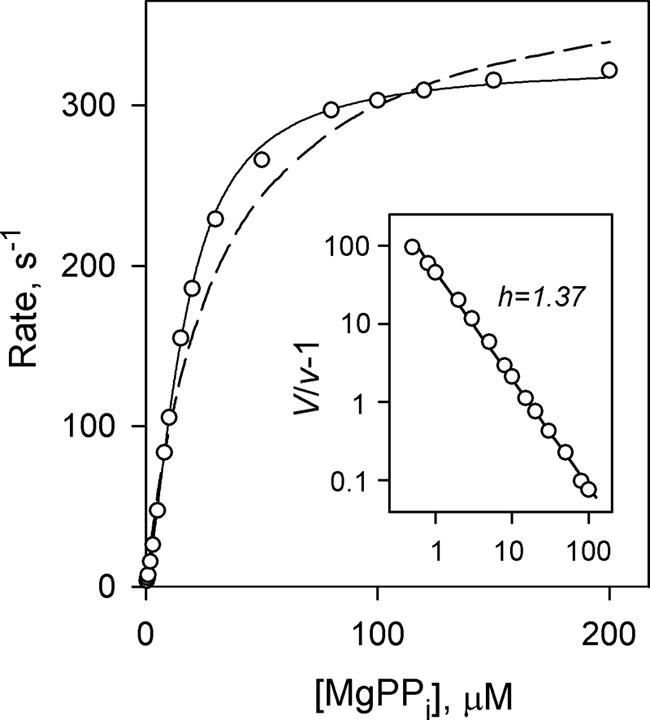
Rate versus substrate concentration profile for dhPPase measured in the presence of 5 mm Mg2+. Enzyme concentration in the assay was 0.1–4 nm. The lines represent the best fits for Equation 5 for cooperative kinetics (solid line) and a simple Michaelis-Menten equation (dashed line). The inset shows a Hill plot of the data where V = kcat[E]0; both axes are scaled logarithmically.
The Hill coefficient h as well as the ratio 4Km1/Km2 for dhPPase were similarly greater than 1 across the entire 0.05–20 mm Mg2+ concentration range (Fig. 3) but decreased at low Mg2+ concentrations, indicating a decrease in the degree of cooperativity. This transition resulted from changes in Km1 but not Km2, which did not vary significantly (Fig. 3). The dependence of Km1 on Mg2+ concentration obeyed Equation 6 with the parameters (KN)0 and (KN)M replaced by the respective parameters (Km1)0 and (Km1)M, the Michaelis constants at zero, and infinite Mg2+ concentration. The fitted (Km1)0, (Km1)M, and KM values were 4.7 μm, 60 μm, and 0.6 mm, respectively (Table 1). The measured value of kcat for dhPPase showed little dependence on Mg2+ concentration (0.05–20 mm) and was in the range of 300–350 s−1.
FIGURE 3.

Dependence of kinetic cooperativity on Mg2+ concentration in four CBS-PPases. The panels show (from top to bottom) the catalytic constant kcat, the Michaelis constants Km1 and Km2, and the Hill coefficient h. The enzyme identities are indicated on the top of the panels. Values of kcat were fit to the equation kcat = kcat,0 + (kcat,M − kcat0)/{1 + (KM/[M])n} (with n equal to 1 and 2 for cnPPase and cpPPase, respectively) where kcat,0 and kcat,M are the limiting values of kcat at zero and infinite Mg2+ concentrations, respectively, and KM is the metal binding constant. The lines for Km1 and Km2 show the best fits for Equation 6. The horizontal dotted lines (h = 1) mark the boundary between positive and negative cooperativity. Km values are measured in terms of the MgPPi complex. Error bars represent S.E.
TABLE 1.
Kinetic parameters for PPi hydrolysis derived from the dependence of kcat, Km1, and Km2 on Mg2+ concentration
| Enzyme | Parameter value |
||||||||
|---|---|---|---|---|---|---|---|---|---|
|
kcat dependence |
Km1 dependence |
Km2 dependence |
|||||||
| kcat,0 | kcat,M | KM | (Km1)0 | (Km1)M | KM | (Km2)0 | (Km2)M | KM | |
| s−1 | s−1 | mm | μm | μm | mm | μm | μm | mm | |
| dhPPase | NAa | 320 ± 20 | NA | 4.7 ± 0.5 | 60 ± 20 | 0.6 ± 0.2 | NA | 11 ± 1 | NA |
| cnPPase | 370 ± 90 | 820 ± 120 | ∼2 | NA | 22 ± 2 | NA | 24 ± 1 | 230 ± 80 | 2.1 ± 0.5 |
| cpPPase | 27 ± 5 | 1310 ± 60 | 1.05 ± 0.05b | <10 | 81 ± 3 | NDc | 80 ± 50 | <1 | ND |
| elPPase | NA | 70 ± 14 | NA | 47 ± 2.5 | 240 ± 10 | 0.14 ± 0.02 | 39 ± 3 | 13 ± 2 | 2.3 ± 1.3 |
a NA, not attendant.
b Estimated assuming binding of two metal ions with the same binding constant (see Fig. 3 legend for details).
c ND, not determined.
A similar kinetic analysis was conducted with three other CBS-PPases. The results are summarized in Fig. 3 and Table 1. With cnPPase, Km1 was virtually independent of Mg2+ concentration, whereas Km2 increased at high [Mg2+]. As a consequence, Mg2+ exerted an opposite effect on cooperativity compared with dhPPase, and the Hill coefficient dropped to slightly below unity at high [Mg2+]. With cpPPase and elPPase, both Km1 and Km2 changed with [Mg2+] concentration but in opposite directions. Consequently, the degree of cooperativity increased with increasing [Mg2+]. The largest effect was observed with cpPPase, which exhibited h values in the range of 1.0–1.8 depending on Mg2+ concentration.
The measured value of kcat was nearly constant over the entire [Mg2+] range for elPPase but changed 2-fold for cnPPase and 50-fold for cpPPase (Fig. 3). For the latter enzyme, kcat depended on the second power of Mg2+ concentration (see Fig. 3 legend), indicating that two metal ions are required for the transition from kcat,0 to kcat,M.
The above analyses were based on the assumption that the kcat value for E2S is half that for E2S2 in Scheme 1. Assuming zero activity for the E2S species increased the sum of the squared residuals by severalfold, indicating significantly worse fits. In contrast, assuming equal activities of E2S and E2S2 decreased the quality of the fits to a lesser extent, making this case a likely alternative. However, if the ratio of the kcat values for E2S and E2S2 was treated as an adjustable parameter, its best fit value was generally closer to 0.5 than 1.0. Importantly, the values of Km1, Km2, and their ratio showed essentially the same trends with the kcat ratio equal to 0.5 and 1, indicating positive cooperativity, which decreased at low [Mg2+] in both cases. We thus conclude that Scheme 1 is the simplest scheme consistent with the data because any value of the kcat ratio greater than 0.5 implies additional negative catalytic cooperativity because of diminished activity of one of the active sites in the dimeric enzyme.
Kinetic Behavior of dhPPaseΔCDC
Remarkably, dhPPaseΔCDC, which is devoid of the regulatory unit, demonstrated Michaelian kinetics in the presence of 5 mm Mg2+ and over the entire 0.1–20 mm Mg2+ concentration range as indicated by a 4Km1/Km2 value for Scheme 2 and a Hill coefficient close to 1 (Fig. 4 and Table 1). The single microscopic Km value describing dhPPaseΔCDC kinetics showed little variation in the metal concentration range used, and its values (3.8–5.0 μm) were markedly less than √Km1Km2 (the geometric mean of the two Km values) values for dhPPase (Fig. 4).
FIGURE 4.
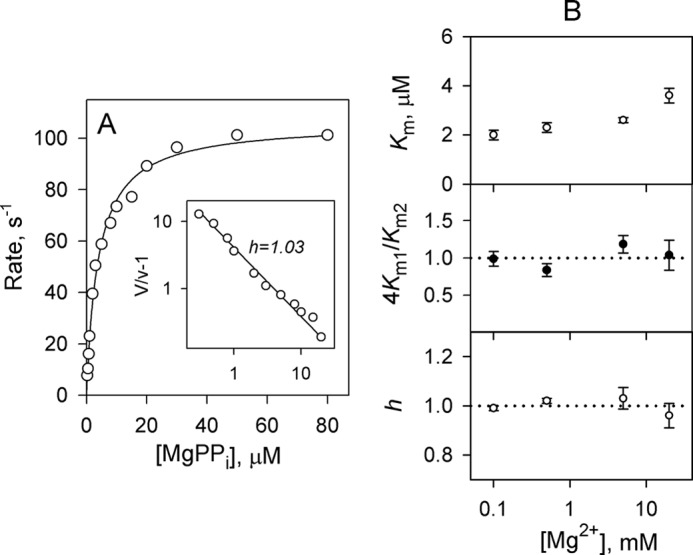
Lack of kinetic cooperativity in dhPPaseΔCDC. Left, rate versus substrate concentration profile measured in the presence of 5 mm Mg2+. Enzyme concentration in the assay was 0.1–4 nm. The line represents the best fit for a Michaelis-Menten equation. The inset shows a Hill plot of the data; both axes are scaled logarithmically. Right, Mg2+ concentration dependence of kinetic parameters. The panels show (from top to bottom) the Michaelis constant Km1, the ratio 4Km1/Km2, and the Hill coefficient h. The horizontal dotted lines (4Km1/Km2 = 1 and h = 1) mark the boundary between positive and negative cooperativity. Error bars represent S.E.
The value of kcat for dhPPaseΔCDC was 100–120 s−1 over the 0.1–20 mm Mg2+ concentration range. The somewhat lower activity of dhPPaseΔCDC compared with dhPPase may indicate that deletion of the regulatory part of the protein caused some distortions in the active site.
Cooperativity of Nucleotide Binding to dhPPase
Fig. 5 shows the concentration dependence of nucleotide effects on activity measured at fixed substrate (MgPPi) and Mg2+ concentrations. Micromolar concentrations of AMP and ADP caused inhibition in most cases, whereas ATP acted as an activator. Qualitatively similar effects of these nucleotides were reported previously for the CBS domain-containing mtPPase (18). The major new finding here is that, in most cases where the size of the effect allowed quantitative analysis, the curves were poorly described in terms of a simple 1:1 binding model (Equation 3) but obeyed Equation 2 for cooperative binding to two sites with concomitant activation or inhibition. The quality of the fit was relatively insensitive to the value of A1/2 in Scheme 1, but better fits were generally observed with A1/2 = (A0 + AN)/2 compared with the limiting cases of A1/2 = 0 or AN. Importantly, most curves generated KN1 values greater than KN2 regardless of the A1/2 value used and Hill coefficients greater than 1 (Table 2). Thus, nucleotide binding also exhibited positive cooperativity. In only two pairs (cpPPase/ADP and elPPase/ATP) did the analysis indicate insignificant cooperativity.
FIGURE 5.
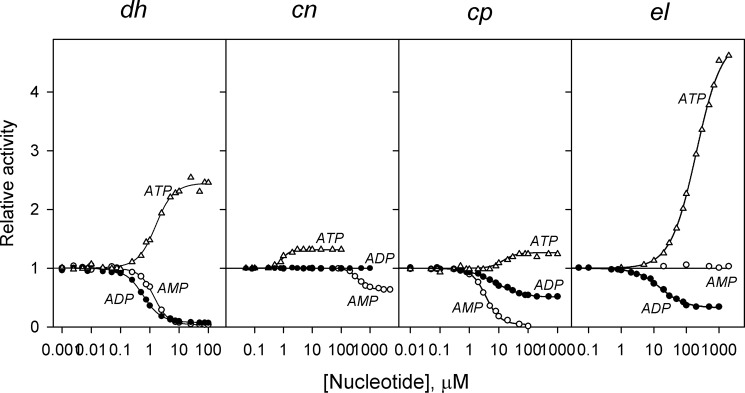
Concentration dependence of adenine nucleotide effects on the activities of four CBS-PPases measured at fixed concentrations of substrate (50 μm MgPPi) and Mg2+ (5 mm). The lines show the best fits to Equation 2 with A1/2 = (A0 + AN)/2. Activities without nucleotides (220, 350, 800, and 35 s−1 for dhPPase, cnPPase, cpPPase, and elPPase, respectively) were taken as unity.
TABLE 2.
Parameters describing nucleotide effects on four CBS-PPases as derived from Figs. 5 and 6 and similar data measured at different substrate concentrations
| Nucleotide | [MgPPi] | vN/v0a | KN1 | KN2 | 4KN1/KN2 | √KN1KN2 | h |
|---|---|---|---|---|---|---|---|
| μm | μm | μm | μm | ||||
| dhPPase | |||||||
| AMP | 5 | 0.13 ± 0.02 | 3 ± 2 | 0.3 ± 0.2 | 40 ± 30 | 0.95 ± 0.05 | 1.75 ± 0.14 |
| AMP | 50 | 0.037 ± 0.001 | 4.8 ± 0.7 | 0.41 ± 0.04 | 48 ± 10 | 1.39 ± 0.04 | 1.68 ± 0.04 |
| AMP | 400 | 0.035 ± 0.006 | 6.7 ± 1.5 | 0.5 ± 0.1 | 53 ± 13 | 1.83 ± 0.05 | 1.74 ± 0.05 |
| ADP | 5 | 0.23 ± 0.01 | 0.38 ± 0.05 | 0.14 ± 0.02 | 11 ± 3 | 0.24 ± 0.01 | 1.51 ± 0.05 |
| ADP | 50 | 0.09 ± 0.01 | 0.99 ± 0.09 | 0.90 ± 0.08 | 4.4 ± 0.8 | 0.95 ± 0.02 | 1.34 ± 0.03 |
| ADP | 400 | 0.05 ± 0.01 | 1.4 ± 0.1 | 2.1 ± 0.2 | 2.6 ± 0.4 | 1.72 ± 0.04 | 1.23 ± 0.04 |
| ATP | 5 | 3.2 ± 0.1 | 2.0 ± 0.3 | 0.63 ± 0.08 | 13 ± 3 | 1.14 ± 0.02 | 1.55 ± 0.04 |
| ATP | 50 | 2.5 ± 0.2 | 2.3 ± 0.8 | 1.5 ± 0.1 | 6 ± 2 | 1.9 ± 0.2 | 1.45 ± 0.13 |
| ATP | 400 | 2.1 ± 0.1 | 4 ± 1 | 1.7 ± 0.5 | 9 ± 5 | 2.5 ± 0.1 | 1.48 ± 0.10 |
| cnPPase | |||||||
| AMP | 50 | 0.63 ± 0.01 | >2000 | <80 | >100 | 390 ± 20 | 2.3 ± 0.2 |
| ADP | 50 | 1.00 | NAb | NA | NA | NA | NA |
| ATP | 50 | 1.32 ± 0.02 | NDc | ND | ND | 0.92 ± 0.07 | ND |
| ADPd | 50 | 0.98 ± 0.02 | ND | ND | ND | 14 ± 1 | ND |
| cpPPase | |||||||
| AMP | 50 | 0.035 ± 0.015 | 12 ± 5 | 1.1 ± 0.5 | 40 ± 20 | 3.7 ± 0.2 | 1.7 ± 0.1 |
| ADP | 50 | 0.50 ± 0.02 | 3.2 ± 0.4 | 18 ± 3 | 0.7 ± 0.2 | 7.7 ± 0.6 | 0.92 ± 0.06 |
| ATP | 50 | 1.26 ± 0.02 | >70 | <3 | >100 | 14 ± 2 | 2.2 ± 0.5 |
| elPPase | |||||||
| AMP | 50 | 1.00 | NA | NA | NA | NA | NA |
| ADP | 50 | 0.32 ± 0.01 | 11.1 ± 1.6 | 24 ± 4 | 1.85 ± 0.6 | 16 ± 1 | 1.14 ± 0.07 |
| ATP | 50 | 5.2 ± 0.3 | 123 ± 14 | 380 ± 70 | 1.3 ± 0.4 | 220 ± 40 | 1.07 ± 0.07 |
| AMPe | 50 | 1.00f | 1100 ± 200 | 1700 ± 300 | 2.3 ± 0.7 | 1350 ± 50 | 1.22 ± 0.06 |
| ADPg | 50 | 0.17 ± 0.01 | 3.9 ± 0.5 | 24 ± 3 | 0.65 ± 0.15 | 10 ± 1 | 0.92 ± 0.05 |
| AMPh | 50 | 0.24 ± 0.01 | >250 | <10 | >100 | 52 ± 3 | 2.2 ± 0.2 |
a Values of vN and v0 are activities extrapolated to infinite concentration of the variable nucleotide and measured in the absence of any nucleotide, respectively.
b NA, not attendant.
c ND, not determined.
d Measured in the presence of 10 μm ATP.
e Measured in the presence of 500 μm ATP.
f Manually fixed parameter value.
g Measured in the presence of 100 μm AMP.
h Measured in the presence of 20 μm ADP.
Interestingly, ADP did not affect cnPPase activity, and AMP was equally ineffective with elPPase (Fig. 5). Activity was nucleotide-insensitive in these enzyme-nucleotide pairs over the entire 0.05–20 mm Mg2+ concentration range.5 To test whether the nucleotides bound to enzyme in these cases, we performed competition experiments (Fig. 6A). In both cases, a “functionally silent” nucleotide modulated the activating effect of ATP, indicating that the former is able to bind. Moreover, although ineffective by itself, AMP synergistically enhanced the effect of ADP on elPPase, shifting the inhibition profile to lower ADP concentrations and decreasing the activity value extrapolated to infinite ADP concentration (Fig. 6B). This profile shift is explained by a 3-fold lower KN1 value for ADP in the presence of 100 μm AMP (Table 2). Accordingly, AMP acted in a concentration-dependent manner to potentiate the inhibitory action of ADP measured at a constant ADP concentration (Fig. 6C). Clearly, binding of AMP to one site greatly enhanced ADP binding to the other site(s) to form a mixed enzyme-AMP-ADP complex with low activity in these conditions. The values in the last line of Table 2 suggest that AMP binding to elPPase is highly cooperative: the first nucleotide molecule binds quite weakly, but the mean binding constant (√KN1KN2) is comparable with that of the other nucleotides. It is unlikely that the extraordinarily high values of 4KN1/KN2 and h for AMP, which are close to their limiting values for two-site binding (27), are somehow associated with the ADP present in the system because the latter nucleotide exhibited quite low binding cooperativity with elPPase (Table 2).
FIGURE 6.
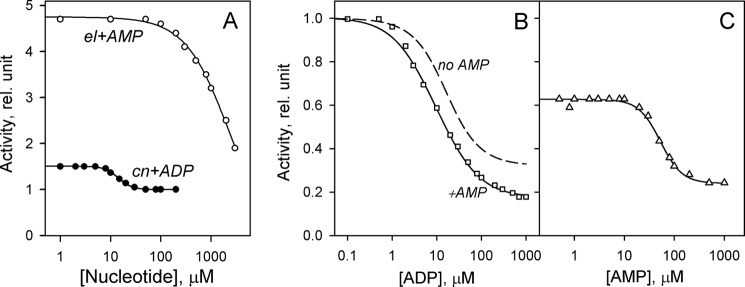
Functionally silent nucleotides as modulators of the effects of “functionally active” nucleotides in CBS-PPases. A, ADP reversal of ATP activation of cnPPase and AMP reversal of ATP activation of elPPase. The fixed ATP concentration was 500 and 10 μm in experiments with elPPase and cnPPase, respectively. B, ADP inhibition of elPPase in the presence of 100 μm AMP. The dependence without AMP (dashed line) was taken from Fig. 5. C, AMP inhibition of elPPase in the presence of 20 μm ADP. The reaction medium contained 50 μm MgPPi and 5 mm Mg2+ in all cases. Activities without any nucleotide were taken as 100%. The lines show the best fits to Equation 2. rel., relative.
The above analysis of nucleotide effects was conducted at a half-saturating substrate concentration. Because the substrate itself also binds cooperatively, the apparent nucleotide binding cooperativity may result from nucleotide effects on substrate binding. Therefore, measurements were extended to low and high substrate concentrations at which the enzyme (dhPPase) predominantly exists in the substrate-free form or as enzyme-substrate complex, respectively. As Table 2 makes clear, the cooperativity of AMP and ATP binding as characterized by 4KN1/KN2 and h did not depend significantly on substrate concentration and showed only a moderate change in the case of ADP binding. Therefore, the observed cooperativity of nucleotide binding is not induced by bound substrate or by modulation of substrate binding. In contrast, the relative final activity at infinite nucleotide concentration (vN/v0) increased with increasing substrate concentration in all cases, whereas the mean binding affinity (√KN1KN2) significantly decreased.
The Mg2+ dependence of AMP, ADP, and ATP binding to dhPPase and of AMP binding to cpPPase was determined (Fig. 7). These enzymes and nucleotides were chosen because they demonstrate profound effects on activity, allowing accurate analysis of the binding curves. Although quite large, the effect of ATP on elPPase was not included in this analysis because ATP chelates Mg2+ appreciably (25) at the high ATP concentrations at which this enzyme operates (Fig. 5), introducing uncertainty in free Mg2+ concentration at the lower values in this range. Varying Mg2+ concentration in the assay changed KN1 and KN2 values for AMP binding to dhPPase and cpPPase in opposite directions (Fig. 7). As a result, the ratio 4KN1/KN2, the Hill coefficient h, and hence the degree of cooperativity increased with increasing Mg2+ concentration. With ATP, KN1, but not KN2, was Mg2+-dependent, resulting in a similar although smaller effect. With ADP, both binding constants changed in parallel without affecting 4KN1/KN2 or h. Notably, dhPPase activity at saturating ATP concentration (470 ± 10 s−1) was independent of [Mg2+].
FIGURE 7.

Mg2+ concentration dependence of nucleotide binding cooperativity in dhPPase and cpPPase. The panels show (from top to bottom) KN1 (○) and KN2 (●) values, the ratio 4KN1/KN2, and the Hill coefficients. The enzyme and nucleotide identities are indicated on the top of the panels. The KN1 and KN2 lines show the best fits for Equation 6. The horizontal dotted lines (4KN1/KN2 = 1 and h = 1) mark the boundary between positive and negative cooperativity. Error bars represent S.E.
The dependences of KN1 and KN2 on Mg2+ concentration ([M]) obeyed Equation 6 derived for a simple model that assumes that metal binding to a site characterized by the dissociation constant KM shifts the dissociation constant for the enzyme-nucleotide complex from (KN)0 to (KN)M. The fitted values of these parameters are summarized in Table 3. Interestingly, KM values were essentially independent of the nucleotide and enzyme used and correlated well with the KM value derived from substrate hydrolysis kinetics for dhPPase (and other PPases) (Table 1). Moreover, in all cases, the KM values derived from KN2 dependence were greater than those derived from the KN1 dependence; in both cases, their ratio was not much different from 4, the value expected for non-cooperative metal binding to two identical sites.
TABLE 3.
Kinetic parameters for nucleotide inhibition derived from the dependencies of KN1 and KN2 on Mg2+ concentration (Fig. 7)
Substrate (MgPPi) concentration was fixed at 50 μm.
| Nucleotide | Parameter value |
|||||
|---|---|---|---|---|---|---|
|
KN1 |
KN2 |
|||||
| KN1,0 | KN1,M | KM | KN2,0 | KN2,M | KM | |
| μm | μm | mm | μm | μm | mm | |
| dhPPase | ||||||
| AMP | 1.7 ± 0.1 | 190 ± 100 | 0.6 ± 0.1 | 7 ± 3 | 0.9 ± 0.3 | 1.4 ± 0.9 |
| ADP | 0.73 ± 0.03 | 1.73 ± 0.06 | 0.5 ± 0.1 | 1.02 ± 0.04 | 3.2 ± 0.2 | 1.3 ± 0.2 |
| ATP | 0.83 ± 0.07 | 2.9 ± 0.2 | 0.3 ± 0.1 | NAa | NA | NA |
| cpPPase | ||||||
| AMP | 0.6 ± 0.1 | 26 ± 11 | 0.3 ± 0.1 | 5.6 ± 0.2 | 2.4 ± 0.2 | 1.6 ± 0.5 |
a NA, not attendant.
Effects of Nucleotides on Kinetic Cooperativity
Table 4 summarizes nucleotide effects on hydrolysis kinetics measured at a Mg2+ concentration of 5 mm. The general trend was that nucleotides decreased kinetic cooperativity as characterized by h values. With cnPPase, which exhibited no cooperativity at this Mg2+ concentration (Fig. 3), ATP conferred slightly negative cooperativity (h = 0.86 ± 0.02). Another general trend was that the activating nucleotide (ATP) increased kcat values up to 2-fold and decreased the mean Km value (√Km1Km2) up to 3-fold for all CBS-PPases except cpPPase in which the latter parameter increased in the presence of ATP. This means that ATP inhibits rather than activates cpPPase at low substrate concentrations.
TABLE 4.
Nucleotide effects on the kinetic parameters for PPi hydrolysis in the presence of 5 mm Mg2+ estimated using Equation 5
| Nucleotide | kcat | Km1 | Km2 | √Km1Km2 | h |
|---|---|---|---|---|---|
| s−1 | μm | μm | μm | ||
| dhPPase | |||||
| None | 350 ± 20 | 26 ± 1 | 10 ± 1 | 20 ± 1 | 1.29 ± 0.02 |
| ATP (10 μm) | 450 ± 10 | 10 ± 1 | 11 ± 1 | 10 ± 1 | 1.24 ± 0.02 |
| cnPPase | |||||
| None | 540 ± 40 | 23 ± 2 | 80 ± 30 | 44 ± 3 | 1.02 ± 0.08 |
| AMP (2 mm) | 400 ± 10 | 25 ± 1 | 65 ± 4 | 42 ± 1 | 1.07 ± 0.01 |
| ADP (1 mm) | 540 ± 30 | 20 ± 1 | 80 ± 20 | 44 ± 5 | 0.95 ± 0.04 |
| ATP (100 μm) | 670 ± 10 | 5.1 ± 0.1 | 45 ± 3 | 15 ± 1 | 0.86 ± 0.02 |
| cpPPase | |||||
| None | 1080 ± 30 | 80 ± 30 | 4 ± 2 | 18 ± 1 | 1.8 ± 0.1 |
| ADP (500 μm) | 1080 ± 40 | 122 ± 19 | 28 ± 7 | 60 ± 7 | 1.50 ± 0.09 |
| ATP (500 μm) | 1700 ± 40 | 63 ± 5 | 32 ± 5 | 47 ± 4 | 1.35 ± 0.06 |
| elPPase | |||||
| None | 89 ± 4 | 220 ± 70 | 18 ± 8 | 63 ± 3 | 1.7 ± 0.1 |
| AMP (1 mm) | 88 ± 4 | 170 ± 20 | 39 ± 9 | 87 ± 10 | 1.47 ± 0.08 |
| ADP (500 μm) | 21 ± 1 | 7.0 ± 0.3 | 32 ± 5 | 15 ± 1 | 0.98 ± 0.03 |
| ATP (1 mm) | 180 ± 10 | 38 ± 4 | 32 ± 7 | 36 ± 4 | 1.27 ± 0.08 |
Of the three inhibition cases documented in Table 4, one (cnPPase/AMP) resulted solely from a decrease in kcat (noncompetitive inhibition), and one (cpPPase/ADP) resulted from an increase in √Km1Km2 (competitive inhibition). In the last case (elPPase/ADP), the inhibitor decreased both kcat and √Km1Km2 to the same degree (uncompetitive inhibition), indicating an absence of inhibition at very low substrate concentrations. Functionally silent nucleotides (ADP with cnPPase and AMP with elPPase; see Fig. 5) did not significantly change kcat or √Km1Km2 (Table 4).
Similar analyses could not be performed in the cases of the most efficient inhibitors (AMP and ADP with dhPPase and AMP with cpPPase). These pairs exhibited quite low (although finite) activities of the enzyme-nucleotide complexes, resulting in a significant relative contribution of nucleotide-free enzyme to activity even at high nucleotide concentrations.
CBS-PPase Activity as a Function of ADP:ATP Ratio
The ADP:ATP ratio is a critical determinant of cellular energy status that regulates many metabolic activities. Fig. 8 shows how the value of this parameter affected activities of the four CBS-PPases. The sum of ADP and ATP concentrations was fixed at 0.5 mm. Although this value is an order of magnitude less than that in a typical cell, this did not markedly affect the profiles because ADP and ATP concentrations exceeded the respective √KN1KN2 values (Table 2) in most cases. Substrate concentration was arbitrarily fixed at 50 μm. Because ATP is an activator and ADP is an inhibitor or an inert species, enzyme activity decreased with increasing ADP:ATP ratios in all cases. However, the sensitivity of CBS-PPases to this parameter varied considerably with dhPPase being the most sensitive and cnPPase being the least sensitive.
FIGURE 8.
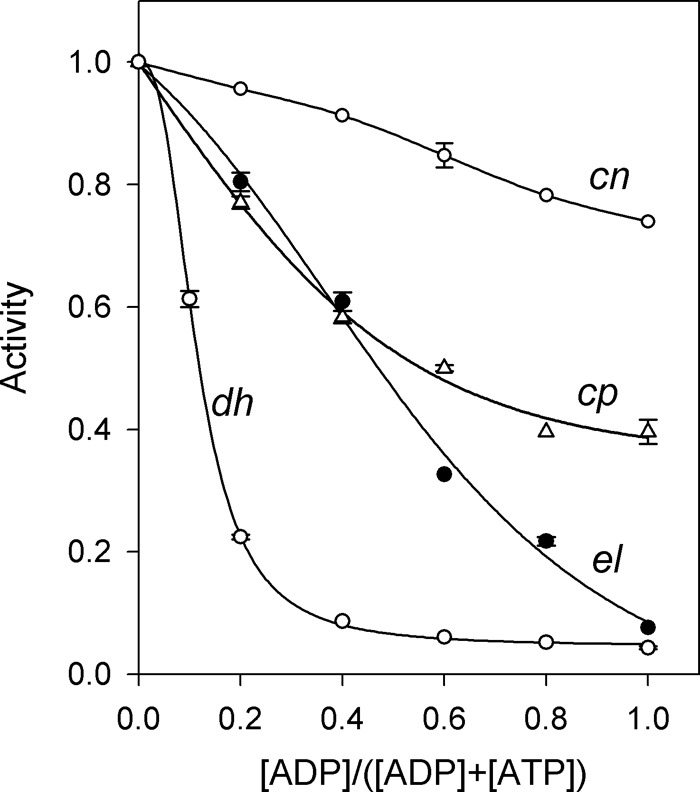
Activities of CBS-PPases as a function of [ADP]:[ATP] ratio. The reaction medium contained 50 μm MgPPi, 5 mm Mg2+, and different concentrations of ADP and ATP such that their sum was 0.5 mm. Activity in the presence of 0.5 mm ATP was taken as unity in all cases. Error bars represent S.D.
DISCUSSION
The organisms that harbor the CBS-PPases used in this study are Gram-positive anaerobic bacteria belonging to the phylum Firmicutes (D. hafniense, C. novyi, C. perfringens) or Actinobacteria (E. lenta). Three of them (C. novyi, C. perfringens, and E. lenta) are pathogenic and are found in the human intestinal tract. D. hafniense is found in soil and is capable of reductive dechlorination of chlorophenols (31). In three of these bacteria (excluding C. perfringens), PPi metabolism depends on soluble and membrane-bound PPases (32), which convert metabolically inert and inhibitory PPi into usable Pi. The soluble PPase converts PPi energy into heat. In contrast, membrane-bound PPases use PPi to establish an H+ gradient (D. hafniense) or Na+ gradient (C. novyi and E. lenta) and thereby mobilize PPi energy to maintain cation gradients and help cells survive under stress conditions (33). This dual utilization of PPi explains the need for regulation of soluble PPases, which has been achieved evolutionarily by the acquisition of a pair of regulatory, energy-sensing CBS domains. Binding of ATP, which prevails under high energy conditions, activates soluble PPases and decreases PPi levels, thereby facilitating biosynthetic reactions that are inhibited by PPi. Under low energy conditions, bound ATP is replaced by ADP and AMP, resulting in inhibition of PPase and an increase in the PPi level, which in turn slows down biosynthesis and provides substrate for membrane PPases that support the membrane H+ or Na+ gradient.
The results reported above reveal added complexity in PPase regulation: the CBS domains introduce dual positive cooperativity to the regulatory mechanism. The first level of cooperativity relates to nucleotide binding and serves to amplify the initial signal (change in adenine nucleotide distribution). Simple calculations for AMP inhibition of dhPPase (Fig. 5) with an h value of 1.68 (Table 2) showed that a change in PPase activity from 10 to 90% of its maximal value occurs when AMP concentration changes 16-fold, whereas the same effect would require an 81-fold change in AMP level if it bound non-cooperatively. Cooperativity of nucleotide binding is not caused by bound substrate as its degree, characterized by the values of 4KN1/KN2 and h in Table 2, does not depend (or only slightly depends) on the degree of enzyme saturation with substrate. Notably, nucleotide binding cooperativity is not uncommon among other CBS domain-containing proteins. Positive cooperativity was reported for S-adenosylmethionine binding to cystathionine β-synthase (34), AMP binding to IMP dehydrogenase (13), and AMP and ATP binding to AMP-activated protein kinase (13). More recently, AMP-activated protein kinase was reported to successively bind nucleotides with decreasing affinity (17, 35), which suggests negative binding cooperativity.
The second level of positive cooperativity in CBS-PPases relates to enzyme interaction with substrate as characterized by the Michaelis constant (Km). This type of behavior is called “catalytic cooperativity” to distinguish it from the more common “binding cooperativity.” One should keep in mind that catalytic cooperativity may not be interpreted in terms of binding insofar as Km is not a thermodynamic binding constant in a general case and is a combination of six individual rate constants in the case of PPase (36). Therefore, a change in Km value may result from a change in any of the rate constants that describe the four catalytic steps in PPase: substrate binding, substrate hydrolysis, and sequential release of two phosphates.
Catalytic cooperativity, which further amplifies the initial signal, is observed in the absence of nucleotides, is modulated by nucleotides (Table 4), and is therefore induced by the regulatory insert itself rather than by the bound nucleotide. Consistent with this notion, removal of the insert abolished the cooperativity. The capacity of CBS domains to impart cooperativity on enzyme or transporter function has not been previously recognized. However, Labesse et al. (37) have recently reported that IMP dehydrogenase binds its substrate, IMP, with positive cooperativity; this cooperativity was abolished by mutation of a single residue in the CBS domain or binding of MgATP to the enzyme. Of note, CBS-PPases are the only cooperative enzymes among different types of oligomeric PPase enzymes.
Mg2+ binding had a strong modulatory effect on both types of cooperativity, but its mode of action varied with enzyme and nucleotide. Increasing Mg2+ concentration generally conferred or increased positive kinetic and nucleotide binding cooperativity (Figs. 3 and 7). However, Mg2+ changed the kinetic cooperativity in cnPPase in the opposite direction (Fig. 3) and did not affect ADP binding cooperativity in dhPPase (Fig. 7). Similar values of the Mg2+ binding constant, KM, derived from measurements of hydrolysis kinetics (Table 1) and nucleotide effects (Table 3) suggest that a single site controls both cooperativity types. Notably, the structure of the AMP-bound regulatory portion of cpPPase crystallized in the presence of Mg2+ contains no bound divalent metal ion (19). In contrast, canonical family II PPases, which represent the catalytic part of CBS-PPases, bind three or four Mg2+ ions in the absence of substrate (5) and up to five metal ions in its presence (6). All metal ions are located in the active site, but one of them is not absolutely required for activity (6). This last activity-modulating site is thus the most likely candidate for the site that modulates cooperativity in CBS-PPases.
All KN values are reported here in terms of total nucleotide concentrations without regard to complexation with Mg2+. Based on the Mg2+ binding constants (see “Experimental Procedures”), 99.8% of ATP, 98% of ADP, and 70% of AMP exist as magnesium complexes at the highest Mg2+ concentration used (20 mm). Thus, it is not unlikely that nucleotide-bound Mg2+ modulates enzyme-nucleotide interactions and therefore contributes to the dependences shown in Fig. 7. However, several lines of evidence rule out the possibility that Mg2+ binding to nucleotides is the sole cause of the Mg2+ effects on KN values. As already mentioned, two molecules of AMP bind to cpPPase as free nucleotide (19). If recalculated in terms of free AMP, KN1 and KN2 values for dhPPase and cpPPase (Fig. 7) would decrease in parallel (up to 3-fold at the highest Mg2+ concentration), resulting in a minor effect on the AMP binding pattern. The crystal structures of the cpPPase fragment (19) suggest that other nucleotides form major contacts with the enzyme through their AMP moieties. Therefore, free and Mg2+-complexed ADP or ATP are likely to have equal or similar affinities for enzyme. This interpretation is consistent with similar mean KN values (√KN1KN2) for AMP, ADP, and ATP observed for dhPPase and cpPPase, which bind all three nucleotides. However, the possibility that Mg2+ binding to ADP and ATP contributes to their binding patterns in Fig. 7 cannot be excluded.
The structural basis for the effects of the regulatory element on the active site and for the modulation of these effects by bound nucleotides still remains to be determined for PPase as well as for other CBS domain-containing enzymes and transporters. Our results pose further questions in this context. Because the two AMP molecules bound per dimeric regulatory fragment of cpCBS-PPase are in proximity to each other (19), one would expect negative rather than positive cooperativity in their binding. Indeed, AMP analog binding was shown to exert negative cooperativity in mtPPase (21). The reversal of nucleotide binding cooperativity in PPase observed in the present study may reflect plasticity of CBS domains and is consistent with the known variability of their amino acid sequences (10). The available crystallographic data show significant opening of the CBS domain interface in the activator (AP4A) complex of cpPPase compared with that in the inhibitor (AMP) complex (19). Lucas et al. (38) observed an open-to-closed conformational change in the CBS domain pair of Methanocaldococcus jannaschii protein MJ0100 upon successive binding of adenosine derivatives. Quite recently, Jeong et al. (39) reported a dramatic AMP-induced structural change in Arabidopsis thaliana protein CBSX2.
The concept of “autoinhibition” was introduced vis-à-vis CBS domains by Janosík et al. (40) to explain cystathionine β-synthase activation by S-adenosyl-l-methionine as well as its limited proteolysis and thermal denaturation. According to this concept, the CBS domain insert acts as an “internal inhibitor,” sensitizing proteins with catalytic or transport functions to structural changes caused by nucleotide binding and permitting either activation or further inhibition. CBS domain-containing PPases provide strong support for this theory. Thus, deletion of the regulatory domains in dhPPase favorably changed its Km1 value (Table 3). A similar deletion in cystathionine β-synthase increased kcat but not Km (41, 42). The concept of autoinhibition is also consistent with the diminished activity (1–3 orders of magnitude) of CBS domain-containing PPases compared with their close homologs lacking CBS domains (5) as well as their ability to be activated by ATP. Many mutations in the CBS domains also activate mtPPase, and some even reverse the effects of nucleotide from inhibition to activation (43), resembling the effects of mutations in cystathionine β-synthase (44) and AMP-dependent protein kinase (45, 46).
Unlike other known CBS domain-containing proteins, dhPPase, cnPPase, and cpPPase contain a DRTGG domain insert within the CBS domain pair. The results of the present work do not shed light on the function of this extra domain in these (and most other) CBS-PPases as we did not observe any fundamental differences in the behavior of these PPases compared with that of elPPase, which has no DRTGG domain in its structure.
In summary, CBS-PPase presents a unique case, demonstrating in one protein the diverse regulatory capabilities of CBS domains as sensors of cell energy status. The regulation of this enzyme is based on a complex interplay among the effects of nucleotides, substrate, and Mg2+ on binding cooperativity. Further work is clearly needed to fully elucidate this interplay and determine its importance for other CBS domain proteins.
This work was supported by Academy of Finland Grant 139031 and Russian Foundation for Basic Research Grant 12-04-01002.
V. A. Anashkin, unpublished observations.
- PPase
- pyrophosphatase
- AP4A
- 5′,5-P1,P4-diadenosine tetraphosphate
- CBS
- cystathionine β-synthase
- cnPPase
- C. novyi pyrophosphatase
- cpPPase
- C. perfringens pyrophosphatase
- dhPPase
- D. hafniense pyrophosphatase
- dhPPaseΔCDC
- dhPPase devoid of regulatory part
- elPPase
- E. lenta pyrophosphatase
- mtPPase
- Moorella thermoacetica pyrophosphatase
- TEV
- tobacco etch virus
- Tes
- 2-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}ethanesulfonic acid.
REFERENCES
- 1. Heinonen J. K. (2001) Biological Role of Inorganic Pyrophosphate, pp. 1–13, Kluwer Academic Publishers, London [Google Scholar]
- 2. Baykov A. A., Cooperman B. S., Goldman A., Lahti R. (1999) Cytoplasmic inorganic pyrophosphatases. Prog. Mol. Subcell. Biol. 23, 127–150 [DOI] [PubMed] [Google Scholar]
- 3. Kajander T., Kellosalo J., Goldman A. (2013) Inorganic pyrophosphatases: one substrate, three mechanisms. FEBS Lett. 587, 1863–1869 [DOI] [PubMed] [Google Scholar]
- 4. Huang H., Patskovsky Y., Toro R., Farelli J. D., Pandya C., Almo S. C., Allen K. N., Dunaway-Mariano D. (2011) Divergence of structure and function in the haloacid dehalogenase enzyme superfamily: Bacteroides thetaiotaomicron BT2127 is an inorganic pyrophosphatase. Biochemistry 50, 8937–8949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Parfenyev A. N., Salminen A., Halonen P., Hachimori A., Baykov A. A., Lahti R. (2001) Quaternary structure and metal-ion requirement of family II pyrophosphatases from Bacillus subtilis, Streptococcus gordonii, and Streptococcus mutans. J. Biol. Chem. 276, 24511–24518 [DOI] [PubMed] [Google Scholar]
- 6. Fabrichniy I. P., Lehtiö L., Tammenkoski M., Zyryanov A. B., Oksanen E., Baykov A. A., Lahti R., Goldman A. (2007) A trimetal site and ground-state substrate distortion mark the active site of family II inorganic pyrophosphatase. J. Biol. Chem. 282, 1422–1431 [DOI] [PubMed] [Google Scholar]
- 7. Merckel M. C., Fabrichniy I. P., Salminen A., Kalkkinen N., Baykov A. A., Lahti R., Goldman A. (2001) Crystal structure of Streptococcus mutans pyrophosphatase: a new fold for an old mechanism. Structure 9, 289–297 [DOI] [PubMed] [Google Scholar]
- 8. Ahn S., Milner A. J., Fütterer K., Konopka M., Ilias M., Young T. W., White S. A. (2001) The “open” and “closed” structures of the type-C inorganic pyrophosphatases from Bacillus subtilis and Streptococcus gordonii. J. Mol. Biol. 313, 797–811 [DOI] [PubMed] [Google Scholar]
- 9. Bateman A. (1997) The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci. 22, 12–13 [DOI] [PubMed] [Google Scholar]
- 10. Baykov A. A., Tuominen H. K., Lahti R. (2011) The CBS domain: a protein module with an emerging prominent role in regulation. ACS Chem. Biol. 6, 1156–1163 [DOI] [PubMed] [Google Scholar]
- 11. Ereño-Orbea J., Oyenarte I., Martínez-Cruz L. A. (2013) CBS domains: ligand binding sites and conformational variability. Arch. Biochem. Biophys. 540, 70–81 [DOI] [PubMed] [Google Scholar]
- 12. Ignoul S., Eggermont J. (2005) CBS domains: structure, function, and pathology in human proteins. Am. J. Physiol. Cell Physiol. 289, C1369–C1378 [DOI] [PubMed] [Google Scholar]
- 13. Scott J. W., Hawley S. A., Green K. A., Anis M., Stewart G., Scullion G. A., Norman D. G., Hardie D. G. (2004) CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Investig. 113, 274–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kemp B. E. (2004) Bateman domains and adenosine derivatives form a binding contract. J. Clin. Investig. 113, 182–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ok S. H., Yoo K. S., Shin J. S. (2012) CBSXs are sensor relay proteins sensing adenosine-containing ligands in Arabidopsis. Plant Signal. Behav. 7, 664–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gómez-García I., Oyenarte I., Martínez-Cruz L. A. (2010) The crystal structure of protein MJ1225 from Methanocaldococcus jannaschii shows strong conservation of key structural features seen in the eukaryal γ-AMPK. J. Mol. Biol. 399, 53–70 [DOI] [PubMed] [Google Scholar]
- 17. Xiao B., Sanders M. J., Underwood E., Heath R., Mayer F. V., Carmena D., Jing C., Walker P. A., Eccleston J. F., Haire L. F., Saiu P., Howell S. A., Aasland R., Martin S. R., Carling D., Gamblin S. J. (2011) Structure of mammalian AMPK and its regulation by ADP. Nature 472, 230–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jämsen J., Tuominen H., Salminen A., Belogurov G. A., Magretova N. N., Baykov A. A., Lahti R. (2007) A CBS domain-containing pyrophosphatase of Moorella thermoacetica is regulated by adenine nucleotides. Biochem. J. 408, 327–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tuominen H., Salminen A., Oksanen E., Jämsen J., Heikkilä O., Lehtiö L., Magretova N. N., Goldman A., Baykov A. A., Lahti R. (2010) Crystal structures of the CBS and DRTGG domains of the regulatory region of Clostridium perfringens pyrophosphatase complexed with the inhibitor, AMP, and activator, diadenosine tetraphosphate. J. Mol. Biol. 398, 400–413 [DOI] [PubMed] [Google Scholar]
- 20. Jämsen J., Baykov A. A., Lahti R. (2012) Fast kinetics of nucleotide binding to Clostridium perfringens family II pyrophosphatase containing CBS and DRTGG domains. Biochemistry 77, 165–170 [DOI] [PubMed] [Google Scholar]
- 21. Jämsen J., Baykov A. A., Lahti R. (2010) Nucleotide- and substrate-induced conformational transitions in the CBS domain-containing pyrophosphatase of Moorella thermoacetica. Biochemistry 49, 1005–1013 [DOI] [PubMed] [Google Scholar]
- 22. Baykov A. A., Avaeva S. M. (1981) A simple and sensitive apparatus for continuous monitoring of orthophosphate in the presence of acid-labile compounds. Anal. Biochem. 116, 1–4 [DOI] [PubMed] [Google Scholar]
- 23. Baykov A. A., Alexandrov A. P., Smirnova I. N. (1992) A two-step mechanism of fluoride inhibition of rat liver inorganic pyrophosphatase. Arch. Biochem. Biophys. 294, 238–243 [DOI] [PubMed] [Google Scholar]
- 24. Baykov A. A., Bakuleva N. P., Rea P. A. (1993) Steady-state kinetics of substrate hydrolysis by vacuolar H+-pyrophosphatase. A simple three-state model. Eur. J. Biochem. 217, 755–762 [DOI] [PubMed] [Google Scholar]
- 25. Smith R. M., Martell A. E., Motekaitis R. J. (1995) NIST Critical Stability Constants of Metal Complexes Database, Version 2.0, National Institute of Standards and Technology, Gaithersburg, MD [Google Scholar]
- 26. Bisswanger H. (2008) Enzyme Kinetics. Principles and Methods, 2nd Ed., Wiley-VCH Verlag, Weinheim, Germany [Google Scholar]
- 27. Forsén S., Linse S. (1995) Cooperativity: over the Hill. TIBS 20, 495–497 [DOI] [PubMed] [Google Scholar]
- 28. Ricard J. (1989) in Allosteric Enzymes (Hervé G., ed) pp. 1–25, CRC Press, Boca Raton, FL [Google Scholar]
- 29. Kuhn N. J., Wadeson A., Ward S., Young T. W. (2000) Methanococcus jannaschii ORF mj0608 codes for a class C inorganic pyrophosphatase protected by Co2+ or Mn2+ ions against fluoride inhibition. Arch. Biochem. Biophys. 379, 292–298 [DOI] [PubMed] [Google Scholar]
- 30. Zyryanov A. B., Vener A. V., Salminen A., Goldman A., Lahti R., Baykov A. A. (2004) Rates of elementary catalytic steps for different metal forms of the family II pyrophosphatase from Streptococcus gordonii. Biochemistry 43, 1065–1074 [DOI] [PubMed] [Google Scholar]
- 31. Madsen T., Licht D. (1992) Isolation and characterization of an anaerobic chlorophenol-transforming bacterium. Appl. Environ. Microbiol. 58, 2874–2878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim S. H., Harzman C., Davis J. K., Hutcheson R., Broderick J. B., Marsh T. L., Tiedje J. M. (2012) Genome sequence of Desulfitobacterium hafniense DCB-2, a Gram-positive anaerobe capable of dehalogenation and metal reduction. BMC Microbiol. 12, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baykov A. A., Malinen A. M., Luoto H. H., Lahti R. (2013) Pyrophosphate-fueled Na+ and H+ transport in prokaryotes. Microbiol. Mol. Biol. Rev. 77, 267–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taoka S., Widjaja L., Banerjee R. (1999) Assignment of enzymatic functions to specific regions of the PLP-dependent heme protein cystathionine β-synthase. Biochemistry 38, 13155–13161 [DOI] [PubMed] [Google Scholar]
- 35. Xiao B., Heath R., Saiu P., Leiper F. C., Leone P., Jing C., Walker P. A., Haire L., Eccleston J. F., Davis C. T., Martin S. R., Carling D., Gamblin S. J. (2007) Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 449, 496–500 [DOI] [PubMed] [Google Scholar]
- 36. Springs B., Welsh K. M., Cooperman B. S. (1981) Thermodynamics, kinetics, and mechanism in yeast inorganic pyrophosphatase catalysis of inorganic pyrophosphate: inorganic phosphate equilibration. Biochemistry 20, 6384–6391 [DOI] [PubMed] [Google Scholar]
- 37. Labesse G., Alexandre T., Vaupré L., Salard-Arnaud I., Him J. L., Raynal B., Bron P., Munier-Lehmann H. (2013) MgATP regulates allostery and fiber formation in IMPDHs. Structure 21, 975–985 [DOI] [PubMed] [Google Scholar]
- 38. Lucas M., Encinar J. A., Arribas E. A., Oyenarte I., García I. G., Kortazar D., Fernández J. A., Mato J. M., Martínez-Chantar M. L., Martínez-Cruz L. A. (2010) Binding of S-methyl-5′-thioadenosine and S-adenosyl-L-methionine to protein MJ0100 triggers an open-to-closed conformational change in its CBS motif pair. J. Mol. Biol. 396, 800–820 [DOI] [PubMed] [Google Scholar]
- 39. Jeong B.-C., Park S. H., Yoo K. S., Shin J. S., Song H. K. (2013) Change in single cystathionine β-synthase domain-containing protein from a bent to flat conformation upon adenosine monophosphate binding. J. Struct. Biol. 183, 40–46 [DOI] [PubMed] [Google Scholar]
- 40. Janosík M., Kery V., Gaustadnes M., Maclean K. N., Kraus J. P. (2001) Regulation of human cystathionine β-synthase by S-adenosyl-L-methionine: evidence for two catalytically active conformations involving an autoinhibitory domain in the C-terminal region. Biochemistry 40, 10625–10633 [DOI] [PubMed] [Google Scholar]
- 41. Banerjee R., Zou C. G. (2005) Redox regulation and reaction mechanism of human cystathionine β-synthase: a PLP-dependent hemesensor protein. Arch. Biochem. Biophys. 433, 144–156 [DOI] [PubMed] [Google Scholar]
- 42. Oliveriusová J., Kery V., Maclean K. N., Kraus J. P. (2002) Deletion mutagenesis of human cystathionine β-synthase. Impact on activity, oligomeric status, and S-adenosylmethionine regulation. J. Biol. Chem. 277, 48386–48394 [DOI] [PubMed] [Google Scholar]
- 43. Jämsen J., Tuominen H., Baykov A. A., Lahti R. (2011) Mutational analysis of residues in the regulatory CBS domains of Moorella thermoacetica pyrophosphatase corresponding to disease-related residues of human proteins. Biochem. J. 433, 497–504 [DOI] [PubMed] [Google Scholar]
- 44. Kery V., Poneleit L., Kraus J. (1998) Trypsin cleavage of human cystathionine β-synthase into an evolutionarily conserved active core: structural and functional consequences. Arch. Biochem. Biophys. 355, 222–232 [DOI] [PubMed] [Google Scholar]
- 45. Adams J., Chen Z. P., Van Denderen B. J., Morton C. J., Parker M. W., Witters L. A., Stapleton D., Kemp B. E. (2004) Intrasteric control of AMPK via the γ1 subunit AMP allosteric regulatory site. Protein Sci. 13, 155–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barnes B. R., Marklund S., Steiler T. L., Walter M., Hjälm G., Amarger V., Mahlapuu M., Leng Y., Johansson C., Galuska D., Lindgren K., Abrink M., Stapleton D., Zierath J. R., Andersson L. (2004) The 5′-AMP-activated protein kinase γ3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. J. Biol. Chem. 279, 38441–38447 [DOI] [PubMed] [Google Scholar]