

Background: Aminolevulinate synthase (ALAS) catalyzes decarboxylative condensation of glycine with succinyl-CoA yielding 5-aminolevulinate.
Results: Unstable ALAS-catalyzed reaction intermediates and conformational changes were characterized using physiological and non-physiological substrates and promiscuous T148A variant.
Conclusion: Rate of ALA release is controlled by a hysteretic kinetic mechanism initiated by ALAS conformational changes.
Significance: Unraveling the ALAS catalytic pathway enhances possible development of therapies for heme synthesis-associated disorders.
Keywords: Enzyme Kinetics, Enzyme Mechanism, Heme, Porphyrin, Pyridoxal Phosphate, 5-Aminolevulinate Synthase, Hysteresis, Oxoamine Synthase, Porphyria, Sideroblastic Anemia
Abstract
5-Aminolevulinate (ALA), an essential metabolite in all heme-synthesizing organisms, results from the pyridoxal 5′-phosphate (PLP)-dependent enzymatic condensation of glycine with succinyl-CoA in non-plant eukaryotes and α-proteobacteria. The predicted chemical mechanism of this ALA synthase (ALAS)-catalyzed reaction includes a short-lived glycine quinonoid intermediate and an unstable 2-amino-3-ketoadipate intermediate. Using liquid chromatography coupled with tandem mass spectrometry to analyze the products from the reaction of murine erythroid ALAS (mALAS2) with O-methylglycine and succinyl-CoA, we directly identified the chemical nature of the inherently unstable 2-amino-3-ketoadipate intermediate, which predicates the glycine quinonoid species as its precursor. With stopped-flow absorption spectroscopy, we detected and confirmed the formation of the quinonoid intermediate upon reacting glycine with ALAS. Significantly, in the absence of the succinyl-CoA substrate, the external aldimine predominates over the glycine quinonoid intermediate. When instead of glycine, l-serine was reacted with ALAS, a lag phase was observed in the progress curve for the l-serine external aldimine formation, indicating a hysteretic behavior in ALAS. Hysteresis was not detected in the T148A-catalyzed l-serine external aldimine formation. These results with T148A, a mALAS2 variant, which, in contrast to wild-type mALAS2, is active with l-serine, suggest that active site Thr-148 modulates ALAS strict amino acid substrate specificity. The rate of ALA release is also controlled by a hysteretic kinetic mechanism (observed as a lag in the ALA external aldimine formation progress curve), consistent with conformational changes governing the dissociation of ALA from ALAS.
Introduction
5-Aminolevulinate synthase (ALAS)2 catalyzes the condensation between glycine and succinyl-CoA to generate CoA, CO2, and ALA (1, 2). This reaction is the first and main regulatory step of heme biosynthesis in mammals. There are two isoforms of the mammalian ALAS gene: a nonspecific and ubiquitously expressed isoform (ALAS1) and an erythroid-specific isoform (ALAS2) (1, 2). Loss-of-function mutations in the human ALAS2 gene result in X-linked sideroblastic anemia (3), whereas gain-of-function mutations lead to X-linked dominant protoporphyria (4).
ALAS belongs to the α-oxoamine synthase family of PLP-dependent enzymes, whose members catalyze a condensation between a small amino acid and an acyl-CoA thioester (1, 5–9). ALAS is a homodimer with its active site located at the dimeric interface (10). At least two different conformations define ALAS: a catalytically incompetent, open conformation and a catalytically competent, closed conformation (11, 12). Interconversion between these two conformations appears closely linked to catalysis (12, 13). Namely, the rate-limiting step of the overall ALAS-catalyzed reaction is proposed to represent a conformational rearrangement from the closed to the open conformation, which is concomitant with the release of ALA from the active site (12, 13). However, how the protein conformational changes are reflected in the last steps of the ALAS kinetic mechanism has not yet been examined.
The proposed sequence of reaction events catalyzed by ALAS is outlined in Scheme 1. The initial catalytic event involves displacement of the Schiff base linkage between a conserved, active site lysine (i.e. Lys-313 in murine ALAS2, Ref. 14) and the PLP cofactor (I) by the incoming glycine substrate to form an external aldimine (II). Removal of one of the two glycine α-carbon hydrogen atoms results in a quinonoid intermediate (III). A Claisen condensation of this intermediate (III) with the succinyl-CoA substrate leads to CoA release and generation of the β-ketoacid aldimine complex (V), which then decarboxylates to yield a second quinonoid intermediate, the quinonoid of the ALA product (VII). Findings from early radiolabeling studies, using Rhodobacter sphaeroides ALAS and stereospecifically tritiated pro-R and pro-S enantiomers of glycine, allowed investigators to reach two major conclusions: 1) ALAS catalyzes the abstraction of the pro-R proton of glycine, giving a stabilized carbanion, the quinonoid intermediate (III), and 2) the postulated ALAS-bound 2-amino-3-ketoadipate intermediate is enzymatically decarboxylated (15, 16). Subsequently, two conserved, active site amino acids, a lysine, which is linked through a Schiff base to PLP, and a histidine, located directly above the plane of the PLP ring, (i.e. Lys-313 and His-207 in murine ALAS2), were identified as the catalytic amino acids involved in the proton abstraction and decarboxylation steps, respectively (13, 17) (Scheme 1).
SCHEME 1.

Proposed chemical mechanism for the reaction catalyzed by ALAS. In the resting state, PLP binds covalently to an active site lysine as an internal aldimine (I) (PLP-K313 internal aldimine in mALAS2). The entry of glycine in the active site proceeds with the formation of an external aldimine (II). Removal of the pro-R proton of glycine results in formation of a first quinonoid intermediate (III), which facilitates the condensation with succinyl-CoA and subsequent release of the CoA moiety (IV). When the resulting 2-amino-3-ketoadipate intermediate (V) is decarboxylated (H207-assisted decarboxylation in the mALAS2 reaction), an enol intermediate is formed (VI), which is in rapid equilibrium with a second quinonoid intermediate (VII). The final reaction intermediate is the ALA external aldimine (VIII).
Despite the efforts made in the last decade toward identifying the specific reaction intermediates (i.e. Scheme 1, III and V) and defining a general reaction mechanism for the α-oxoamine synthase family (1, 5–7, 18, 19), direct evidence of the chemical and spectroscopic nature of these intermediates has been unattainable. Kerbarh et al. (18) used the methyl ester of l-alanine as a pseudo-substrate to establish that the proposed quinonoid intermediate in the 8-amino-7-oxononanoate synthase (AONS)-catalyzed reaction resulted from proton abstraction, and not decarboxylation, of the AONS-alanine external aldimine. Similarly, formation of the initial quinonoid intermediate could be spectroscopically observed upon binding of either glycine or glycine and succinyl-CoA to R. sphaeroides ALAS (20). The monitoring of this quinonoid intermediate was clearly facilitated in the Rhodobacter capsulatus ALAS-catalyzed reaction by substituting glycine with O-methylglycine (21), as the resulting methyl ester of the β-ketoacid-aldimine could not undergo enzymatic decarboxylation, and thus accumulated. However, even with the significant acceleration of the rate of quinonoid intermediate formation upon succinyl-CoA binding to ALAS (12, 22), neither the spectroscopic detection of the first quinonoid intermediate (Scheme 1, III) nor the determination of the chemical nature of the predicted 2-amino-3-ketoadipate intermediate (V) in the mALAS2-catalyzed reaction have yet been achieved, mainly due to the transient and unstable nature of the III and V intermediates (2, 13).
In contrast to the strict specificity for glycine as the only amino acid substrate, ALAS can utilize other acyl-CoA derivatives in addition to succinyl-CoA (23, 24). An important determinant in the specificity of ALAS toward the amino acid substrate is the positioning of a perfectly conserved active site threonine (21, 24). In fact, mutation of this threonine in ALAS from R. sphaeroides (24) or R. capsulatus (21) altered the amino acid substrate specificity of the enzyme. However, the kinetic, mechanistic, and conformational dynamic bases for this alteration remain unknown.
Here we report the direct identification of the 2-amino-3-ketoadipate intermediate, (Scheme 1, V) using glycine and O-methylglycine as substrate and pseudo-substrate, respectively, and a combination of stopped-flow kinetics and liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). This finding, together with the spectroscopic detection of the first quinonoid intermediate (Scheme 1, III) and consequent calculation of the rate constant associated with its formation, supports the occurrence of these two intermediates in the pathway of the ALAS-catalyzed reaction and confirms that the quinonoid intermediate (III) stems from deprotonation rather than decarboxylation of the ALAS-glycine external aldmine (Scheme 1, II). Further, and in contrast to the physiological glycine substrate, mALAS2 displayed hysteresis with l-serine, as indicated by the lag phase in the progress curve for the mALAS2-serine external aldimine production. We propose that this hysteretic behavior emanates from structural rearrangements induced by the different orientation of the bound l-serine in the mALAS2 active site.
EXPERIMENTAL PROCEDURES
Reagents
The following reagents were obtained from Fisher Scientific: glycine, O-methylglycine (also known as glycine methyl ester, glycine O-methyl ester, methyl glycinate, and methyl 2-aminoacetate), potassium phosphate monobasic, potassium phosphate dibasic, HEPES, glycerol, HPLC grade methanol, HPLC grade acetonitrile, and sodium hydroxide. 5-Aminolevulinate hydrochloride was from Acros Organics, and l-serine, pyridoxal 5′-phosphate, deuterated glycine, and trimethylchlorosilane were from Sigma-Aldrich.
Construction and Isolation of the T148A Variant
The T148A variant was obtained by screening a library of mALAS2 variants for ALAS activity. Briefly, the codons for Thr-148 and Asn-150 of mALAS2 were simultaneously randomized to cover all 400 possible combinations of the 20 naturally occurring amino acids.3 To screen the shuffling library for functional variants, we reversed the phenotype of Escherichia coli hemA− (HU227) cells (25), which can survive only if they harbor functional ALAS or if ALA (or hemin) is added to the medium (26). DNA sequencing templates were prepared from colonies recovered on the selective medium and the T184A-encoding mutation was then identified by DNA sequencing.
Overproduction and Purification of Wild-type mALAS2 and T148A Variant
Protein overproduction and purification were performed as described previously (27). Following lysis of the cells, all purification steps were performed in 1 day, and the concentrated, purified protein was stored under liquid nitrogen. Protein purity was assessed by SDS-PAGE. Protein concentrations were determined spectroscopically by monitoring the absorbance at 280 nm and using a molar extinction coefficient of 42860 m−1 cm−1 based on the mALAS2 amino acid sequence.
Synthesis of Deuterated O-Methylglycine
Deuterated O-methylglycine was prepared from deuteroglycine and methanol at room temperature in the presence of trimethylsilane as previously described (28).
mALAS2-catalyzed Reaction with Either Glycine or O-Methylglycine
mALAS2 was reacted with glycine, O-methylglycine or deuterated O-methylglycine and succinyl-CoA in 300 μl of 20 mm HEPES, pH 7.5, containing 10% (w/v) glycerol, at 30 °C for 120 min. The final concentrations in the reaction assays were 20 μm mALAS2, 100 μm succinyl-CoA, and 100 mm glycine, O-methylglycine, or deuterated O-methylglycine. The control assay reactions were run under the above experimental conditions (i.e. same buffer, temperature and time) but solely with 1) mALAS2, 2) mALAS2, and glycine, 3) mALAS2 and O-methylglycine, 4) mALAS2 and deuterated O-methylglycine, and 5) mALAS2 and succinyl-CoA. All of the above experimental and control reactions were run in duplicates, such that the reactions in one of the two sets were stopped by precipitation with 50% (vol) trichloroacetic acid. Briefly, trichloroacetic acid was added to each of these assay reactions (1:1, v:v), the precipitated protein was removed by centrifugation, and the supernatant was stored in liquid nitrogen until the reaction intermediates present in the supernatant were analyzed by LC-MS/MS at the Sanford/Burnham Medical Research Institute, Lake Nona, Orlando, FL.
Sample Preparation for Mass Spectrometry
20 μl of each sample, previously thawed to room temperature, was combined with 250 μl of HPLC grade acetonitrile in a 650-μl micro centrifuge tube and vortexed for 5 min on a multi-tube vortex. The samples were then sonicated for 1 min and centrifuged for 10 min. The supernatant was removed to a clean 96-well plate and evaporated to dryness under high purity N2 gas at 60 °C. Prior to analysis by LC-MS/MS, the samples were reconstituted in 200 μl of HPLC grade water, vortexed and centrifuged.
Synthesis of the Methyl Ester of 2-Amino-3-ketoadipate
5-Amino-6-methoxy-4,6-dioxo-hexanoic acid (i.e. the methyl ester of the putative 2-amino-3-ketoadipate reaction intermediate) was synthesized from benzyl alcohol as described in the supplemental Experimental Procedures.
Data Acquisition
Data acquisition, peak integration, and sample quantitation were performed using a Dell Precision 390 Desktop Computer with an Intel Core 2 Duo Processor, Microsoft Windows XP Professional and Analyst Software Version 1.4.2.
Separation of the Reaction Intermediates by Ultra-high Pressure Liquid Chromatography (UPLC)
UPLC was performed using a Waters Acquity UPLC system (Waters Corp., Milford, MA). Chromatographic separation of the reaction intermediates was achieved using an Acquity BEH C18 1.7 μ 2.1 × 50 mm UPLC column (Waters Corp.). A 0.1% formic acid in water and 0.1% formic acid in acetonitrile solutions were prepared as mobile phases A and B, respectively. The flow rate was set at 0.4 ml/min, and the injection volume was set at 1 μl. A linear gradient separation was used, with 10% of mobile phase B from 0 to 0.50 min, then 10% to 95% of mobile phase B over 0.50 min, holding for 0.80 min, after which returning to 100% A in 2.00 min for initial conditioning.
Detection and Characterization of the Trapped Intermediate by Liquid Chromatography Coupled with Tandem Mass Spectrometry (LC-MS/MS)
LC-MS/MS analysis (i.e. mass detection) was performed using an Applied Biosystems API4000 QTrap LC/MS/MS instrument (AB Sciex, Foster City, CA) equipped with a Turbo Ionspray (TIS) source. The mass spectrometer was operated in a positive ion mode monitoring 4 transitions for ALAS reaction intermediates using optimized analyte-dependent parameters (data not shown). The structure for the proposed “trapped” intermediate, 5-amino-6-methoxy-4,6-dioxo-hexanoic acid, is given in Fig. 1.
FIGURE 1.
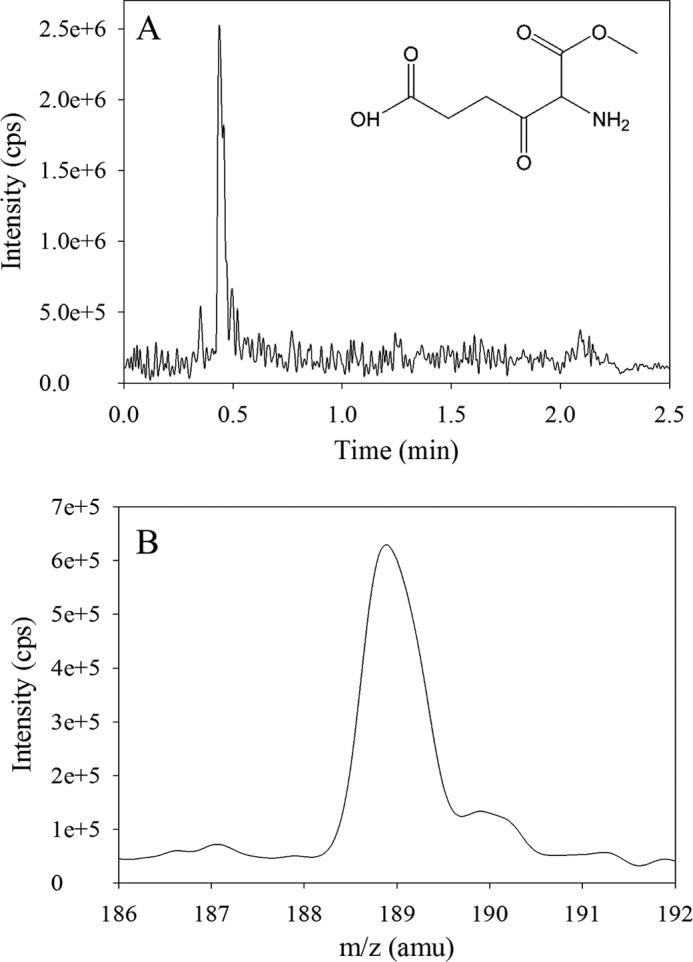
Analysis of the trapped intermediate in the ALAS-catalyzed reaction of O-methylglycine with succinyl-CoA by LC-MS/MS. A, UPLC profile of the reaction sample indicating that the retention time of the major species, which has a mass ion spectrum peak at 188.9 m/z, is 0.44 min. B, MS/MS scan spectrum (abundance versus m/z) of the 0.44 min-fraction derived from the reaction sample indicating that the predominant species, with a peak at 188.9 m/z, is consistent with the molecular mass of 189.17 for the predicted intermediate, 5-amino-6-methoxy-4,6-dioxohexanoic acid (inset in A).
Stopped-flow Absorption Spectroscopy Experiments
A Kintek stopped-flow apparatus (model SF-2001) was used to monitor transient changes in absorbance (at 420 or 510 nm) following rapid mixing of enzyme (mALAS2 or T148A variant) and ligands (glycine, l-serine or ALA). All reactions were performed at 18 °C in 50 mm phosphate (37 mm K2HPO4/13 mm KH2PO4), pH 7.5, containing 10% glycerol (w/v). For the reaction between wild-type mALAS2 and glycine, one of the syringes contained 60 μm wild-type mALAS2 in the above 50 mm phosphate buffer, pH 7.5, whereas the other syringe contained 200 mm glycine in the same buffer. The final concentrations of the reactants upon mixing are reported in the figure legends. The same buffer (composition and concentration) and enzyme and amino acid substrate concentrations were used in the reaction between wild-type mALAS2 (or T148A variant) and l-serine. For the reaction between wild-type mALAS2 and ALA, one of the syringes contained 60 μm wild-type mALAS2 in the above 50 mm phosphate buffer, pH 7.5, while the other syringe contained 20 mm ALA in the same buffer. Prior to reacting mALAS2 with ALA, the 1 m ALA-HCl stock was neutralized with an equal volume of 1 m NaOH. The final concentrations of wild-type mALAS2 and ALA in the observation chamber were 30 μm and 10 mm, respectively. Each time course for the different monitored reactions represents the average of three or more data sets. The time course data were fitted to either a single- or two-exponential equation (Equation 1) using the Kintek stopped-flow software,
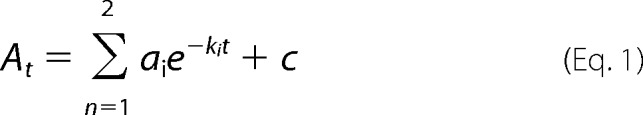 |
where At is the absorbance at time t, a is the amplitude of each phase, k is the observed rate constant for each phase, and c is the final absorbance.
RESULTS
Identification of the Claisen Condensation Intermediate by LC-MS/MS
The detection and characterization of the mALAS2-bound intermediate upon the reaction of mALAS2 with O-methylglycine (or deuterated O-methylglycine) and succinyl-CoA was carried out using LC-MS/MS. Samples were analyzed by full-scan Q1 positive and negative modes and multiple reaction monitoring (MRM) positive mode for the predicted ALAS intermediate (Fig. 1). A signal at 189–190 m/z was observed during the full scan mode in positive ionization, as expected for the methyl ester of the β-ketoacid aldimine complex (i.e. the 5-amino-6-methoxy-4,6-dioxohexanoic acid intermediate; Scheme 1, structure V). To confirm this assignment, 5-amino-6-methoxy-4,6-dioxo-hexanoic acid was synthesized, the analyte-dependent parameters were optimized, and the reaction samples were reanalyzed for the intermediate in the MRM mode (data not shown). The peak corresponding to the trapped methyl ester of the β-ketoacid intermediate was predominantly present in the samples in which mALAS2 was reacted with O-methyl glycine (or deuterated O-methyl glycine) and succinyl-CoA (data not shown). A value of 189.2 m/z was determined for the intermediate in the mALAS2-catalyzed reaction of O-methyl glycine with succinyl-CoA in the full Q1 scan positive ion mode (data not shown).
Reaction between Glycine and Wild-type mALAS2
The reaction between wild-type mALAS2 and glycine was examined by monitoring the changes in absorbance, at either 420 nm or 510 nm, as a function of time, using stopped-flow spectroscopy. The absorbance increase at 420 nm was previously assigned to the formation of the glycine external aldimine, while that observed at 510 nm was ascribed to quinonoid intermediate formation (12). Because of the high Km for glycine (i.e. 12 mm, Ref. 29) and the difficulty in reaching mALAS2 concentrations greater than 600 μm, the reaction was performed using multi-turnover conditions, with the substrate concentration in excess over that of the enzyme. The time course monitored at 420 nm for the reaction of 30 μm wild-type mALAS2 with 100 mm glycine, at pH 7.5 and 18 °C, was best described by a monophasic process with an observed rate constant of 0.080 ± 0.001 s−1 (Fig. 2A). Similarly, the time course data for the same reaction, when monitored at 510 nm, were best fit to a single-exponential equation, yielding virtually the same value for the observed rate constant (0.070 ± 0.001 s−1) (Fig. 2B). However, if one assumes that the extinction coefficients of the two quinonoid intermediates are similar, it becomes apparent that the extent of the formed glycine external aldimine and glycine quinonoid intermediates varied considerably. Comparison between the extent of glycine quinonoid intermediate produced in the absence of succinyl-CoA (Fig. 2B), as assessed by the maximum absorbance amplitude at 510 nm, and that in the presence of 100 μm succinyl-CoA (data not shown), indicated that the amount of quinonoid intermediate formed in the absence of succinyl-CoA was ∼4% of that produced in the presence of the succinyl-CoA substrate (i.e. Equation 2).
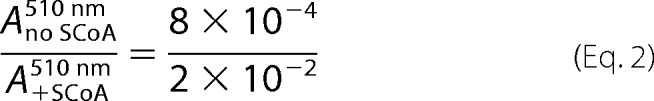 |
These results suggest that even though there is some interconversion between the two intermediates, the external aldimine is the predominant intermediate when the enzyme reacts with glycine in the absence of succinyl-CoA.
FIGURE 2.
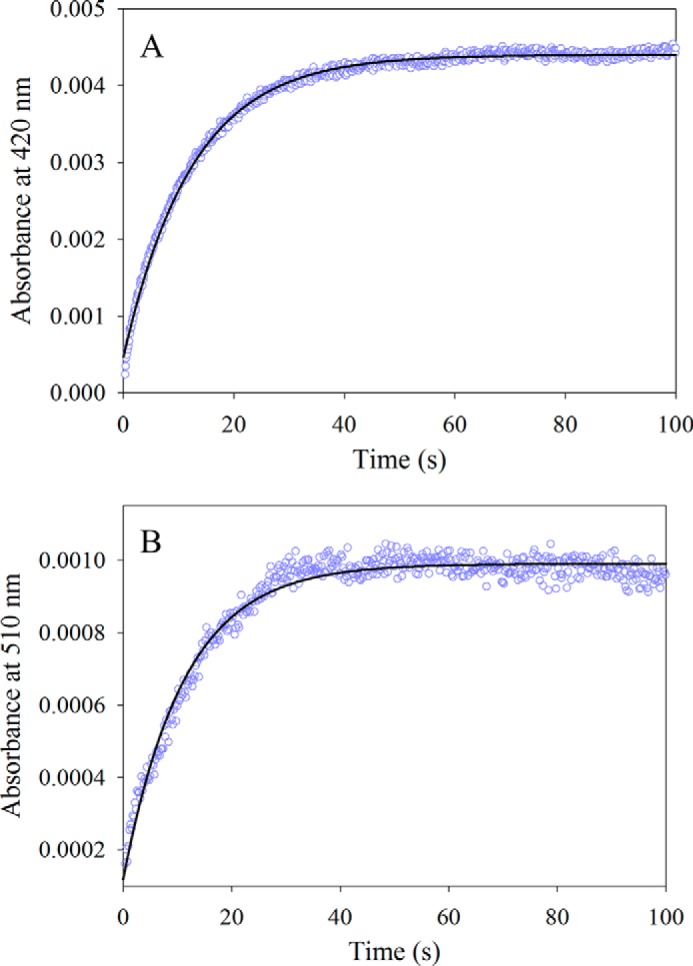
Reaction between wild-type mALAS2 (30 μm) and glycine (100 mm). A, progress curve for the formation of the glycine external aldimine monitored at 420 nm. The data (open purple circles) were fit to a single-exponential equation with an observed rate constant (k1) of 0.080 ± 0.001 s−1 and amplitude (A1) of 4.00 × 10−3 ± 0.01 × 10−3. B, progress curve for the formation of the glycine quinonoid intermediate monitored at 510 nm. The data (open purple circles) were fit to a single-exponential equation with an observed rate constant (k1) of 0.070 ± 0.001 s−1 and amplitude (A1) of 0.80 × 10−3 ± 0.01 × 10−3.
Reaction of l-serine with Either Wild-type mALAS2 or T148A
To address the question of ALAS amino acid substrate specificity, the pre-steady state reaction between 30 μm wild-type mALAS2 and 100 mm l-serine, at pH 7.5, 18 °C, was monitored by following the change in absorbance at 420 nm with time (Fig. 3A). Rather than the monophasic, single-exponential kinetic process associated with the reaction between glycine and the PLP-Lys313 internal aldimine of mALAS2 (Fig. 2A and Scheme I), a distinctive lag phase was observed in the first 1.0 s of the kinetic trace for the external aldimine formation when l-serine was the substrate (Fig. 3A). Further, the reaction with l-serine follows a biphasic kinetic process (Fig. 3A).
FIGURE 3.
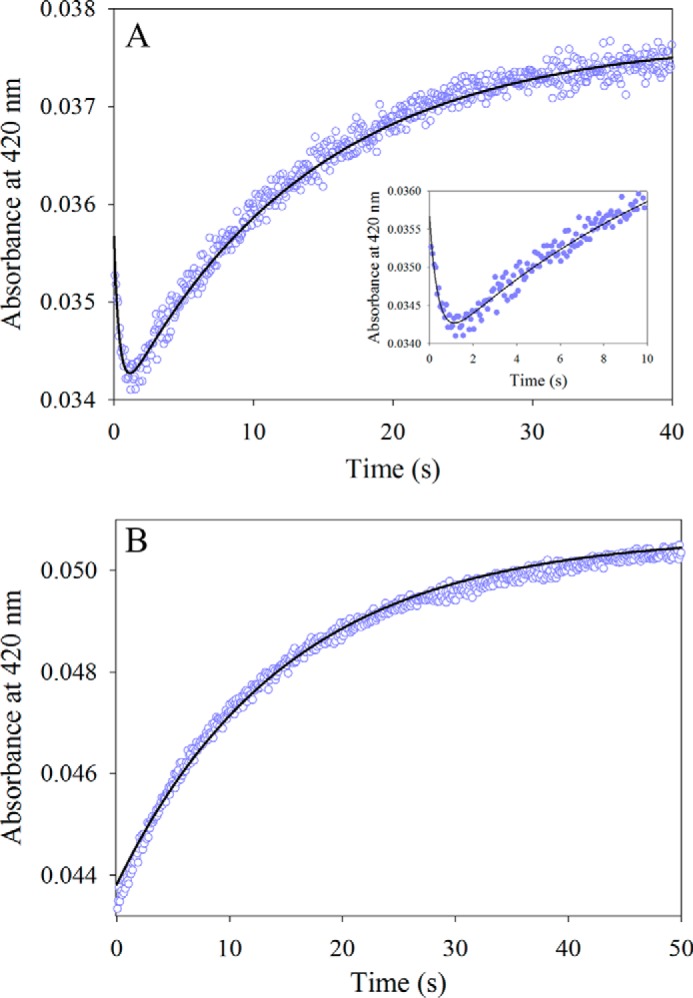
Reaction of either wild-type mALAS2 or T148A variant (30 μm) with l-serine (100 mm). A, progress curve for the formation of the l-serine-mALAS2 external aldimine. The data points were fit to a two-exponential equation yielding the observed rate constants (k1 and k2) and amplitudes (A1 and A2): k1 = 2.5 ± 0.2 s−1and A1 = 1.70 × 10−3 ± 0.01 × 10−3 for the fast phase, and k2 = 0.070 ± 0.001 s−1 and A2 = 3.80 × 10−3 ± 0.01 × 10−3 for the slow phase. The inset shows the first 10 s of the reaction. B, progress curve for the formation of the l-serine-T148A external aldimine. The data were fit to a single-exponential equation yielding the observed rate constant (k1) of 0.070 ± 0.001 s−1 and the amplitude (A1) of 6.80 × 10−3 ± 0.01 × 10−3.
The pre-steady state reaction between 30 μm T148A mALAS2 and 100 mm l-serine (at pH 7.5 and 18 °C) was also investigated by monitoring the progress of external aldimine production, as assessed by the increase in absorbance at 420 nm. The T148A variant, in contrast to wild-type mALAS2, is active when l-serine and succinyl-CoA are the substrates.3 Interestingly, the lag phase observed in the progress curve for the wild-type mALAS2-catalyzed reaction was not observed with the T148A variant under identical experimental conditions (Fig. 3B). This implies that the lag phase observed for the reaction between l-serine and wild-type mALAS2 most likely resulted from unfavorable steric clashes with active site amino acids; thus when the active site architecture is modified, as in the T148A variant, the binding of l-serine can take place, presumably by circumventing any unfavorable steric clashes.
Reaction between ALA and Wild-type mALAS2
The time dependence of the pre-steady state reaction between 30 μm wild-type mALAS2 and 10 mm ALA, at pH 7.5 and 18 °C, was examined by monitoring the change in absorbance at either 420 nm (Fig. 4A) or 510 nm (Fig. 4B). While a distinctive kinetic lag phase of 0.5 s was observed in the progress curve for the formation of the ALA-ALAS external aldimine (Fig. 4A), no kinetic lag phase was detectable during the ALA-quinonoid intermediate formation (Fig. 4B). Formation of the external aldimine and quinonoid intermediates were both best described as biphasic processes. The observed rates for the fast and slow phases associated with the time course at 420 nm were 4.70 s−1 and 0.37 s−1, respectively (Fig. 4A), whereas those pertinent to the time course at 510 nm were 0.61 s−1 and 0.08 s−1 (Fig. 4B).
FIGURE 4.
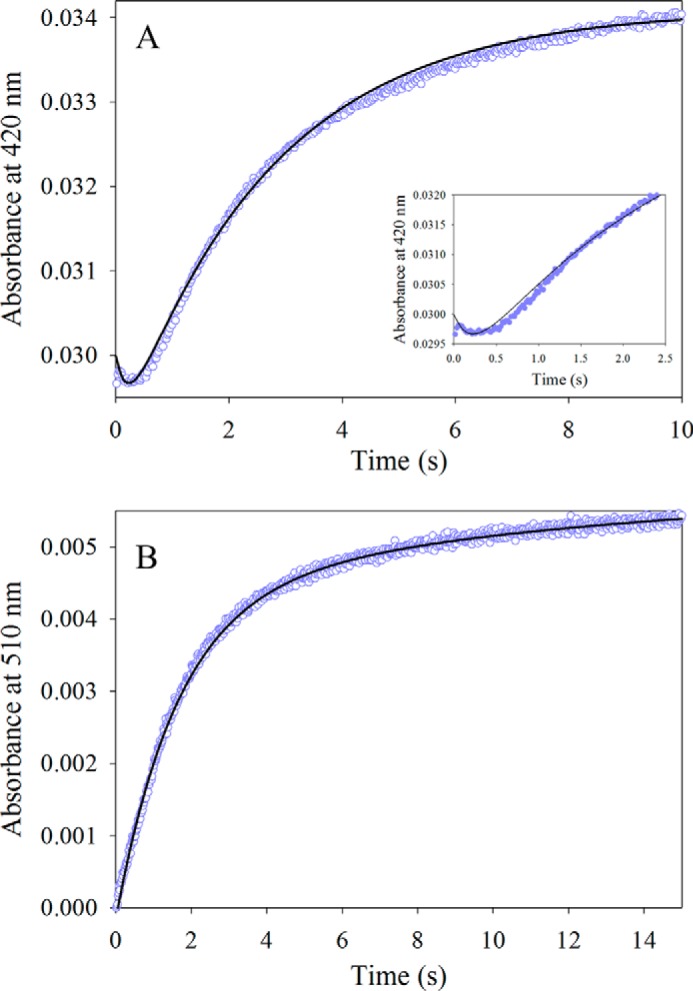
Reaction between wild-type mALAS2 (30 μm) and ALA (10 mm). A, progress curve for the formation of the ALA-mALAS2 external aldimine as monitored at 420 nm. The data points were fit to a two-exponential equation yielding the observed rate constants (k1 and k2) and amplitudes (A1 and A2): k1 = 4.7 ± 0.01 s−1 and A1 = 1.1 × 10−3 ± 0.07 × 10−3 for the fast phase, and k2 = 0.37 ± 0.003 s−1 and A2 = 5.2 × 10−3 ± 0.02 × 10−3 for the slow phase. The inset shows the first 2.5 s of the reaction. B, progress curve for the formation of the ALA quinonoid intermediate as monitored at 510 nm. The observed rate constants (k1 and k2) and amplitudes (A) were: k1 = 0.61 ± 0.01 s−1 and A1 = 4.50 × 10−3 ± 0.07 × 10−3 for the fast phase, and k2 = 0.08 ± 0.01 s−1 and A2 = 1.50 × 10−3 ± 0.04 × 10−3 for the slow phase.
DISCUSSION
In this study, we provide direct evidence for the presence of the first quinonoid and β-ketoacid reaction intermediates in the catalytic pathway of ALAS and examine the amino acid substrate specificity and conformational transitions of ALAS by focusing on the hysteretic behavior of the enzyme. Soon after ALAS was purified (30, 31) and PLP proven to be a cofactor of the enzyme (30, 31), the major question about the ALAS mechanism centered on establishing whether the external aldimine between glycine and ALAS (i.e. II in Scheme 1) would be decarboxylated or deprotonated (15, 30, 32) to give a stabilized carbanion. Findings from radiolabeling studies, using R. sphaeroides and stereospecifically tritiated glycine, clearly pointed to the deprotonation route yielding a quinonoid intermediate (i.e. III in Scheme 1) (15, 32). Further, the deprotonation was stereospecific, with the exclusive removal of the pro-R proton (15) and, ultimately, the pro-S proton of glycine retaining the S-configuration at C-5 of ALA (16). This latter observation indicated not only that the inferred 2-amino-3-ketoadipate intermediate (i.e. V in Scheme 1) was on the catalytic pathway, but also that its decarboxylation occurred on the enzyme surface (16, 33). However, corroborating the presence of the Claisen intermediate, resulting from the condensation of the glycine quinonoid intermediate with succinyl-CoA has proven to be challenging. This is partly because the 2-amino-3-ketoadipate intermediate is very reactive, undergoes a fast decarboxylation, and is difficult to trap (13). Moreover, even if one could successfully trap the 2-amino-3-ketoadipate intermediate on the enzyme, its spontaneous decarboxylation at physiological pH would still be rapid (34).
Because of the chemical liability of the 2-amino-3-ketoadipate intermediate, which precluded its direct detection, we resorted to using O-methylglycine as a pseudo-substrate to study the ALAS-catalyzed reaction. With this pseudo-substrate, the Claisen condensation can still occur, yielding the methyl ester of the 3-ketoacid-aldimine complex, but the reaction cannot proceed beyond this step, as the methyl ester of the 2-amino-3-ketoacid (or methoxy-4,6-dioxo-hexanoic acid) intermediate cannot undergo decarboxylation. Indeed, when we used LC-MS/MS to analyze the mALAS2-catalyzed product resulting from the reaction of mALAS2 with O-methylglycine and succinyl-CoA, a pronounced peak (188.9 m/z) was observed for the molecular ion of the putative, more stable, condensation intermediate (Fig. 1), in good agreement with its predicted molecular weight of 189.17. To verify that the 188.9 m/z peak was due to the mALAS2-catalyzed formation of the Claisen intermediate, 5-amino-6-methoxy-4,6-dioxo-hexanoic acid was synthesized and analyzed using LC-MS/MS. In addition, a shift of +1 m/z was apparent when deuterated methylglycine was the pseudo-substrate, further validating the chemical nature of the condensation intermediate (188.9 m/z) (data not shown). This latter finding probably can be extended to the reaction mechanism of other α-oxoamine synthases, particularly since it supports an earlier report inferring the presence of the AONS-bound methyl ester of the condensation intermediate (18).
Until this study, detection of the first quinonoid intermediate in the mALAS2 catalytic pathway (Scheme 1, III) had not been demonstrated. Clearly, the identification of the Claisen intermediate argues in favor of the first quinonoid (or glycine quinonoid) species (Scheme 1, III), as this intermediate could only have been formed from deprotonation, and not decarboxylation, of the O-methylglycine pseudo-substrate. In prior transient kinetic studies, we had deduced that the formation and decay of the glycine quinonoid intermediate occurred rapidly, i.e. within the dead time of the instrument (2 ms) (12), and we demonstrated that the addition of succinyl-CoA accelerated the rate of the reaction of mALAS2 with glycine by 250,000-fold (22). In the present study we reacted mALAS2 with glycine at 18 °C, instead of 30 °C (12), and monitored the reaction by following the change in absorbance at 420 nm (Fig. 2A) and 510 nm (Fig. 2B). The glycine quinonoid intermediate could be detected at 510 nm, further supporting the conclusion of the mass spectrometry results that wild-type mALAS2 catalyzes the abstraction of a proton from glycine. Even though the formation of the glycine external aldimine and glycine quinonoid intermediates proceeded at similar rates (Fig. 2, A and B), in the absence of succinyl-CoA, the external aldimine outweighs the quinonoid intermediate. In fact, the amplitude value representing the formation of the initial quinonoid intermediate was estimated to be ∼4% of the amplitude value for the same intermediate when produced in the presence of the succinyl-CoA substrate. The significantly reduced amplitude value for the formation of the quinonoid intermediate indicates that this intermediate, in the absence of succinyl-CoA, is catalyzed by a small percentage of the total enzyme population, which can, in part, explain the difficulties associated with the spectroscopic detection of the glycine quinonoid intermediate.
Analysis of the crystal structure of glycine-bound R. capsulatus ALAS offers a plausible explanation for the predominance of the external aldimine over the quinonoid intermediate in the reaction of glycine with mALAS2, by taking into account Dunathan's hypothesis (35). Dunathan (35) proposed that PLP-dependent enzymes favor a specific reaction by orienting the nascent σ orbital of the covalently bound substrate parallel to the π orbitals of the cofactor ring system. Thus, an important question regarding the structure of the external aldimine in the mALAS2 active site relates to the positional stability of the glycine pro-R proton bond with respect to the conjugated π system, in the absence of succinyl-CoA.
Superimposition of the crystallographic structures of different glycine-bound R. capsulatus ALAS monomers indicates that the positioning of the glycine α-carbon varies in relation to the plane of the PLP ring (Fig. 5). These fluctuations affect the length of time during which the pro-R proton of glycine is favorably oriented such that the PLP ring can act as an effective electron sink and the active site lysine-driven abstraction of the pro-R proton can proceed. Therefore, we hypothesize that the ineffective enzymatic removal of the pro-R proton of glycine, observed in the absence of succinyl-CoA, results from the fluctuations affecting the stable positioning of the pro-R bond in a perpendicular orientation relative to the plane of the PLP ring. Moreover, the inability of ALAS to stabilize the positioning of the glycine α-carbon in the absence of succinyl-CoA might be physiologically relevant in preventing unwanted side reactions. For example, slow abortive transamination was reported for AONS, the α-oxoamine synthase that catalyzes the condensation of alanine and pimeloyl-CoA (19, 36). In the absence of the pimeloyl-CoA substrate, AONS reacted with l-alanine to irreversibly convert the PLP cofactor to pyridoxamine 5′-phosphate (36). Therefore, in the absence of succinyl-CoA, ALAS, by not stabilizing the positioning of the glycine α-carbon and the orientation of the bond to be broken, might prevent unwanted side reactions that otherwise would render the enzyme inactive.
FIGURE 5.
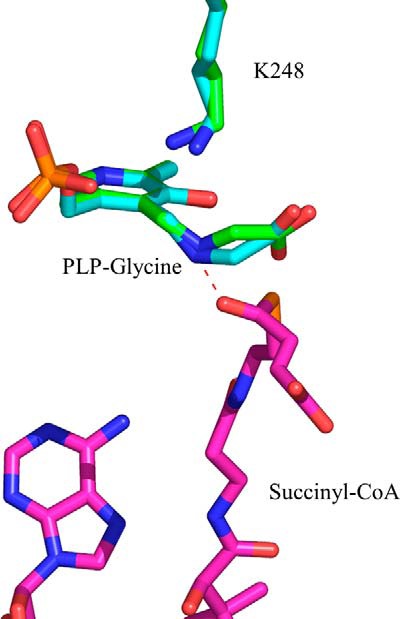
Positioning of the glycine external aldimine in the active site of ALAS. Differences in the positioning of the α-carbon of glycine were detected upon superimposition of the crystal structures for monomers A and E from PDB file 2BWP. The ternary complex formed by the glycine external aldimine and succinyl-CoA was modeled in the active site of ALAS. The succinyl-CoA carbonyl group can strongly interact with the Schiff base nitrogen. The model was built by superimposing the structures with PDB coordinates 2BWP and 2BWO using PyMOL. Amino acid Lys-248 in R. capsulatus ALAS corresponds to Lys-313 in mALAS2.
It is tempting to speculate that the ability of succinyl-CoA to significantly accelerate the rate of the ALAS-catalyzed deprotonation of the glycine external aldimine (12, 22) resides in moving the pro-R proton-α-carbon bond into an optimal position for deprotonation. We modeled the binding of succinyl-CoA to the glycine external aldimine of R. capsulatus ALAS (Fig. 5). A strong interaction between the carbonyl group of the succinyl moiety and the Schiff base nitrogen, which are 1.9 Å apart, is likely. Thus, we propose that succinyl-CoA binding to ALAS can promote the removal of the glycine pro-R proton by 1) interacting with the Schiff base nitrogen, minimizing fluctuations in the positioning of the C-α glycine bond to the pro-R proton and keeping it perpendicular to the plane of the PLP ring, and 2) stabilizing the closed conformation, as previously postulated (12). Moreover, the crystal structure of glycine-bound R. capsulatus ALAS reveals that the positioning of the catalytic lysine varies slightly among the different monomers (Fig. 5). On the other hand, the position occupied by this lysine does not vary in the succinyl-CoA bound monomers of R. capsulatus ALAS (not shown). We therefore also suggest that minimizing fluctuations in the positioning of this lysine is critical for facile and effective deprotonation of the glycine substrate (17).
Unlike the reaction with glycine, the kinetic trace for the reaction between l-serine and wild-type mALAS2 is characterized by a lag phase (Fig. 3A). Typically, a lag phase is observed in the reaction progress curves of enzymes with hysteretic behavior (37). These enzymes undergo conformational changes at a rate that is slower than that of the corresponding chemical reaction (37). We propose that structural rearrangements induced by the entry of l-serine into the ALAS active site precede the formation of the serine-ALAS external aldimine. While the specific structural rearrangements induced by l-serine binding remain to be defined, it is entirely conceivable that the entry of l-serine into the tightly packed ALAS active site results in steric clashes between l-serine and active site amino acids, and the generated conformational strain hinders the ability of the enzyme to properly stabilize the closed conformation. To provide a compatible interpretation for the orientation of l-serine within the active site of ALAS, we modeled the l-serine external aldimine of Sphingomonas paucimobilis SPT (38) in the active site of R. capsulatus ALAS (Fig. 6). There are notable differences in the positioning of the α-carboxyl group of the amino acid substrate: the α-carboxyl group of glycine forms a salt bridge with R439, while the α-carboxyl group of l-serine is predicted to interact with H207 (mALAS2 numbering; R. capsulatus numbering is indicated in Fig. 6). Hence, during initial binding, l-serine can sterically clash with active site amino acids. The resulting conformational strain might be transient, and, once the external aldimine is formed, the enzyme assumes a closed conformation similar to that adopted in the presence of its physiological substrates. However, we cannot exclude the possibility that the binding of l-serine stabilizes a catalytically incompetent conformation, distinct from the previously reported open and closed conformations (11).
FIGURE 6.
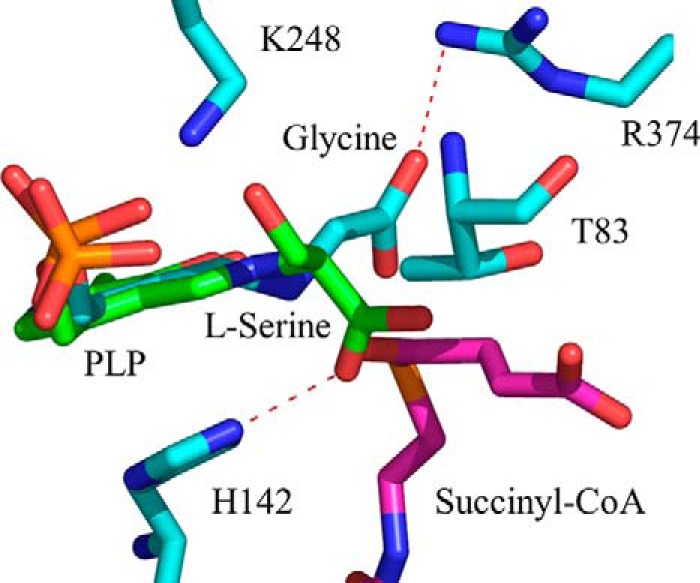
Model depicting the positioning of the l-serine external aldimine in the active site of ALAS. The model was created by superimposing the l-serine external aldimine of S. paucimobilis SPT (PDB file 2W8J) with the glycine external aldimine of R. capsulatus ALAS (PDB file 2BWP). l-Serine is shown in green and glycine in cyan. Succinyl-CoA was subsequently modeled into the active site by superimposing the generated structure with the structure for the R. capsulatus·succinyl-CoA complex (PDB file 2BWO), using PyMOL. R. capsulatus ALAS amino acids Thr-83, His-142, Lys-248, and Arg-374 correspond to mALAS2 Thr-148, His-207, Lys-313, and Arg-439, respectively.
One amino acid within the active site of mALAS2 that appears to sterically clash with l-serine is Thr-148. This amino acid modulates the amino acid substrate specificity of mALAS2, such that its substitution with alanine endows the T148A variant with the capacity to utilize l-serine as substrate. According to our structural model, once the external aldimine is formed, the l-serine α-carboxylate is about 2 Å apart from the Thr-148 side chain methyl group (Fig. 6). This orientation differs from that of the α-carboxylate of the glycine external aldimine, which is oriented away from Thr-148. Consequently, l-serine, by not orienting properly in the active site of mALAS2, clashes with Thr-148 during the formation of the external aldimine. This hypothesis is supported by the absence of a lag phase in the progress curve for the T148A-catalyzed serine external aldimine formation. The considerably smaller side chain of alanine than of threonine provides a more open active site pocket in the T148A variant than in wild-type mALAS2. Thus, in contrast to wild-type mALAS2, not only can l-serine be properly oriented and succinyl-CoA accommodated, but also the condensation reaction can take place in the active site of the T148A variant.
While the formation of the Ser-mALAS2 external aldimine was easily followed (Fig. 3A), the activity of mALAS2 with serine and succinyl-CoA could not be determined under steady state conditions,3 as monitored by CoA production (39). These findings indicate that the mALAS2-catalyzed condensation between l-serine and succinyl-CoA does not proceed beyond external aldimine formation and suggest that succinyl-CoA fails to bind productively to the active site. This inactivity probably stems from improper positioning of l-serine (Fig. 6). For the reaction between the amino acid substrate and succinyl-CoA to occur, the former needs to interact with Arg-439, as demonstrated with the glycine substrate (40). The significance of this interaction in relation to the subsequent correct positioning and productive binding of succinyl-CoA was substantiated when Arg-439 was replaced with lysine. This conservative mutation resulted in a 20-fold decrease in the specificity constant of the enzyme for succinyl-CoA, while the R439L mutation was deleterious (40). According to our structural model, the serine carboxylate does not interact with Arg-439 (Fig. 6), implying that the hindered positioning of this substrate leads to non-productive binding of succinyl-CoA because of steric clashes with the succinyl moiety.
The rate-limiting step in the ALAS-catalyzed reaction has been assigned to a conformational change due to the reversion of the enzyme to the open conformation during the release of ALA (13, 22). Characterization of the reaction between wild-type mALAS2 and ALA indicates that most probably the enzyme starts to return to the open conformation once the ALA-ALAS external aldimine is formed. We arrived to this conclusion based on the presence of a lag in the progress curve for the ALA external aldimine (Scheme 1, VIII) formation and absence of a lag in the progress curve for the ALA quinonoid intermediate (Scheme 1, VII) formation (Fig. 4). We propose that the observed hysteretic behavior arises from structural rearrangements associated with the formation of the ALA external aldimine. It is plausible that either the external aldimine generation or dissociation of ALA from PLP induces a conformational strain within the active site that can destabilize the closed conformation, and thus promote the reversion to the open conformation and facilitate the release of ALA from the enzyme.
Upon close examination of the crystal structure of R. capsulatus ALAS, we identified an alternative access channel to the active site, which possibly provides the path for entry of glycine or exit of ALA (Fig. 7). This channel, distinct from that occupied by succinyl-CoA, can only be seen in the open conformation structures. It covers amino acids from the N-terminal domain, organized in an α-helix and a loop (i.e. R. capsulatus ALAS amino acids 1–24) and amino acids from the C-terminal domain making an α-helix and a linker (i.e. R. capsulatus ALAS amino acids 337–352). The active site glycine-rich loop (i.e. R. capsulatus ALAS amino acids 77–89) from the opposite monomer also contributes to the architecture of this channel. Val-341 and Gly-18, located ∼10Å apart and in the open conformation, outline the entry of the channel (Fig. 8). Actually, the active site loop and the C-terminal helix make up a structural region in R. capsulatus ALAS that undergoes significant conformational changes during the conversion from open to closed conformation (Fig. 8).
FIGURE 7.
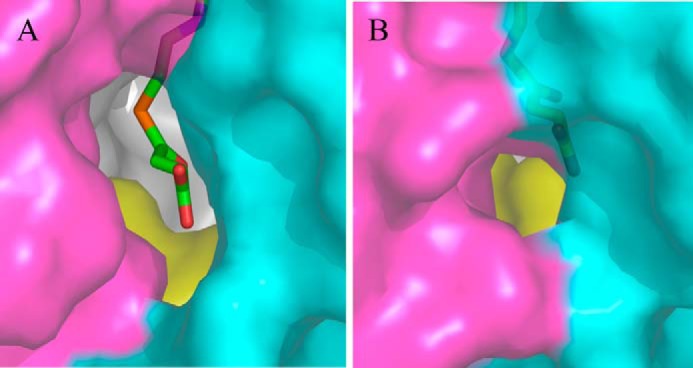
Alternative channel for the entry of substrates into the active site of R. capsulatus ALAS. A, alternative channel in the open and (B) closed conformations (PDB file 2BWN). This channel and the succinyl-CoA-specific channel are distinct. The N-terminal domain is shown in cyan, the catalytic domain in gray, and the C-terminal domain in magenta. The opposite monomer is shown in yellow. Succinyl-CoA is shown in stick representation.
FIGURE 8.
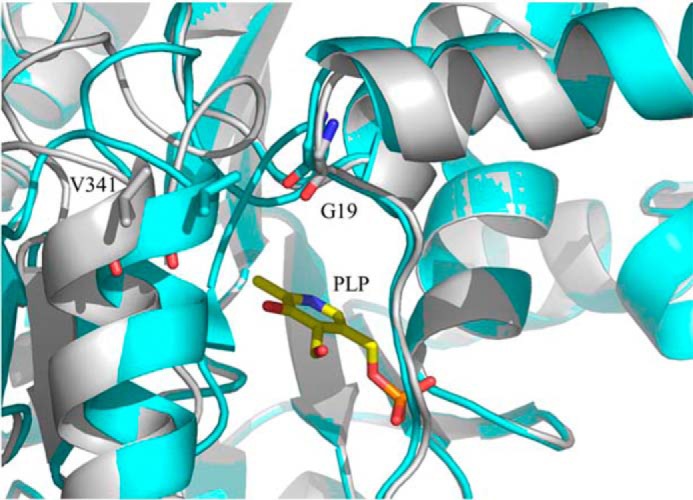
Differences in the positioning of C-terminal α-helix Val-341 in the open and closed conformations. The open and closed conformations are shown in gray and cyan, respectively. Monomers A and B (PDB file 2BWN) were superimposed using PyMOL.
X-linked sideroblastic anemia mutations localized to the human ALAS2 N-terminal α-helix and loop also lead us to suggest a physiological relevance to this structural region of the channel. These mutations [M154I, D159N, D159Y, T161A, and F165L (3, 11)] may perturb the structural integrity of the channel and obstruct the entry of glycine to or ALA exit from the active site. If a protein transporter, as recently suggested for the human SLC25A38 gene product (41), is involved in the import of glycine to or the export of ALA from mitochondria, then it will be important to examine whether the SLC25A38 transporter interacts with ALAS and whether the channel amino acids participate in the protein-protein interaction.
In this work we have shown that ALAS does not favor the catalytic removal of the pro-R proton of glycine in the absence of succinyl-CoA and, due to steric clashes, the binding of the non-physiological substrate l-serine leads to non-productive binding of succinyl-CoA. The biological significance of our findings can be explained in an evolutionary context. It is clear that evolutionary selection did not favor the ALAS sequence that would produce the enzyme with highest catalytic efficiency, since through protein engineering, we have shown that mutations in the active site loop can increase the kcat by 30-fold and the catalytic efficiency for succinyl-CoA by about 100-fold (42). The fact that ALAS did not evolve toward high catalytic turnover is not without physiological relevance if we consider that the product of its reaction, ALA, as well as most subsequent pyrrolic intermediates generated during heme biosynthesis are highly toxic, and therefore it is not desirable to have them in concentrations greatly exceeding that of mitochondrial iron available for heme formation, since it can result in porphyric anomalies. Thus, rather than for catalytic power, ALAS evolved for specificity and efficiency by minimizing undesirable side reactions that can either render the enzyme inactive, or can generate products other than ALA. This is evident from the fact that in the absence of succinyl-CoA, ALAS does not favor deprotonation of glycine, which can lead to inactivation of the enzyme due to abortive transamination; also, the active site of ALAS has been optimized to prevent productive binding of non-physiological amino acid substrates such as serine, since in their presence, succinyl-CoA fails to bind productively, ensuring that products other than ALA are not generated. Hence, through minimizing undesirable side reactions, even with low catalytic turnover, ALAS can meet the high demands for heme during hemoglobinization.
Supplementary Material
Acknowledgments
We thank Dr. Thomas Lendrihas (University of South Florida) for the ALAS reaction samples for the LC-MS/MS analyses, Dr. E. Hampton Sessions (Sanford/Burhnam Institute at Lake Nona, FL) for the synthesis of 5-amino-6-methoxy-4,6-dioxohexanoic acid, and Dr. Michael Vicchiarelli (Sanford/Burhnam Institute at Lake Nona, FL) for the LC-MS/MS analyses.
This work was supported, in whole or in part, by the National Institutes of Health Grant GM080270 and the American Heart Association Greater SE Affiliate Grant 10GRNT4300073, (to G. C. F.).

This article contains supplemental Experimental Procedures and Figs. S1 and S2.
B. M. Stojanovski and G. C. Ferreira, unpublished results.
- ALAS
- 5-aminolevulinate synthase
- ALA
- 5-aminolevulinate
- mALAS2
- murine erythroid ALAS
- LC-MS/MS
- liquid chromatography coupled with tandem mass spectrometry
- MS
- mass spectrometry
- PLP
- pyridoxal 5′-phosphate.
REFERENCES
- 1. Fratz E. J., Stojanovski B. M., Ferreira G. C. (2013) Toward Heme: 5-Aminolevulinate Synthase and Initiation of Porphyrin Synthesis in The Handbook of Porphyrin Science (Ferreira G. C., Kadish K. M., Smith K. M., Guilard R., ed), pp. 1–78, World Scientific Publishing Co., New Jersey [Google Scholar]
- 2. Hunter G. A., Ferreira G. C. (2011) Molecular enzymology of 5-aminolevulinate synthase, the gatekeeper of heme biosynthesis. Biochim. Biophys. Acta 1814, 1467–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bottomley S. S. (2004) Sideroblastic Anemias. in Wintrobe's Clinical Hematology (Greer J., Foerster J., Lukens J. N., Rodgers G. M., Paraskevas R., Glader R. B., ed), pp. 1011–1033, Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 4. Whatley S. D., Ducamp S., Gouya L., Grandchamp B., Beaumont C., Badminton M. N., Elder G. H., Holme S. A., Anstey A. V., Parker M., Corrigall A. V., Meissner P. N., Hift R. J., Marsden J. T., Ma Y., Mieli-Vergani G., Deybach J. C., Puy H. (2008) C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am. J. Hum. Genet. 83, 408–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mann S., Ploux O. (2011) Pyridoxal-5′-phosphate-dependent enzymes involved in biotin biosynthesis: structure, reaction mechanism and inhibition. Biochim. Biophys. Acta 1814, 1459–1466 [DOI] [PubMed] [Google Scholar]
- 6. Ikushiro H., Hayashi H. (2011) Mechanistic enzymology of serine palmitoyltransferase. Biochim. Biophys. Acta 1814, 1474–1480 [DOI] [PubMed] [Google Scholar]
- 7. Schmidt A., Sivaraman J., Li Y., Larocque R., Barbosa J. A., Smith C., Matte A., Schrag J. D., Cygler M. (2001) Three-dimensional structure of 2-amino-3-ketobutyrate CoA ligase from Escherichia coli complexed with a PLP-substrate intermediate: inferred reaction mechanism. Biochemistry 40, 5151–5160 [DOI] [PubMed] [Google Scholar]
- 8. Spirig T., Tiaden A., Kiefer P., Buchrieser C., Vorholt J. A., Hilbi H. (2008) The Legionella autoinducer synthase LqsA produces an α-hydroxyketone signaling molecule. J. Biol. Chem. 283, 18113–18123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jahan N., Potter J. A., Sheikh M. A., Botting C. H., Shirran S. L., Westwood N. J., Taylor G. L. (2009) Insights into the biosynthesis of the Vibrio cholerae major autoinducer CAI-1 from the crystal structure of the PLP-dependent enzyme CqsA. J. Mol. Biol. 392, 763–773 [DOI] [PubMed] [Google Scholar]
- 10. Tan D., Ferreira G. C. (1996) Active site of 5-aminolevulinate synthase resides at the subunit interface. Evidence from in vivo heterodimer formation. Biochemistry 35, 8934–8941 [DOI] [PubMed] [Google Scholar]
- 11. Astner I., Schulze J. O., van den Heuvel J., Jahn D., Schubert W. D., Heinz D. W. (2005) Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to XLSA in humans. EMBO J. 24, 3166–3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hunter G. A., Ferreira G. C. (1999) Pre-steady-state reaction of 5-aminolevulinate synthase. Evidence for a rate-determining product release. J. Biol. Chem. 274, 12222–12228 [DOI] [PubMed] [Google Scholar]
- 13. Hunter G. A., Zhang J., Ferreira G. C. (2007) Transient kinetic studies support refinements to the chemical and kinetic mechanisms of aminolevulinate synthase. J. Biol. Chem. 282, 23025–23035 [DOI] [PubMed] [Google Scholar]
- 14. Ferreira G. C., Neame P. J., Dailey H. A. (1993) Heme biosynthesis in mammalian systems: evidence of a Schiff base linkage between the pyridoxal 5′-phosphate cofactor and a lysine residue in 5-aminolevulinate synthase. Protein Sci. 2, 1959–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zaman Z., Jordan P. M., Akhtar M. (1973) Mechanism and stereochemistry of the 5-aminolaevulinate synthetase reaction. Biochem. J. 135, 257–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Abboud M. M., Jordan P. M., Akhtar M. (1974) Biosynthesis of 5-aminolevulinic acid: involvement of a retention-inversion mechanism. J. Chem. Soc. Chem. Comm. 643–644 [Google Scholar]
- 17. Hunter G. A., Ferreira G. C. (1999) Lysine-313 of 5-aminolevulinate synthase acts as a general base during formation of the quinonoid reaction intermediates. Biochemistry 38, 3711–3718 [DOI] [PubMed] [Google Scholar]
- 18. Kerbarh O., Campopiano D. J., Baxter R. L. (2006) Mechanism of α-oxoamine synthases: identification of the intermediate Claisen product in the 8-amino-7-oxononanoate synthase reaction. Chem. Commun. 60–62 [DOI] [PubMed] [Google Scholar]
- 19. Webster S. P., Alexeev D., Campopiano D. J., Watt R. M., Alexeeva M., Sawyer L., Baxter R. L. (2000) Mechanism of 8-amino-7-oxononanoate synthase: spectroscopic, kinetic, and crystallographic studies. Biochemistry 39, 516–528 [DOI] [PubMed] [Google Scholar]
- 20. Nandi D. L. (1978) Studies on δ-aminolevulinic acid synthase of Rhodopseudomonas spheroides. Reversibility of the reaction, kinetic, spectral, and other studies related to the mechanism of action. J. Biol. Chem. 253, 8872–8877 [PubMed] [Google Scholar]
- 21. Kaufholz A. L., Hunter G. A., Ferreira G. C., Lendrihas T., Hering V., Layer G., Jahn M., Jahn D. (2013) Aminolaevulinic acid synthase of Rhodobacter capsulatus: high-resolution kinetic investigation of the structural basis for substrate binding and catalysis. Biochem. J. 451, 205–216 [DOI] [PubMed] [Google Scholar]
- 22. Zhang J., Ferreira G. C. (2002) Transient state kinetic investigation of 5-aminolevulinate synthase reaction mechanism. J. Biol. Chem. 277, 44660–44669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lendrihas T., Zhang J., Hunter G. A., Ferreira G. C. (2009) Arg-85 and Thr-430 in murine 5-aminolevulinate synthase coordinate acyl-CoA-binding and contribute to substrate specificity. Protein Sci. 18, 1847–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shoolingin-Jordan P. M., Al-Daihan S., Alexeev D., Baxter R. L., Bottomley S. S., Kahari I. D., Roy I., Sarwar M., Sawyer L., Wang S. F. (2003) 5-Aminolevulinic acid synthase: mechanism, mutations and medicine. Biochim. Biophys. Acta 1647, 361–366 [DOI] [PubMed] [Google Scholar]
- 25. Umanoff H., Russell C. S., Cosloy S. D. (1988) Availability of porphobilinogen controls appearance of porphobilinogen deaminase activity in Escherichia coli K-12. J. Bacteriol. 170, 4969–4971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gong J., Ferreira G. C. (1995) Aminolevulinate synthase: functionally important residues at a glycine loop, a putative pyridoxal phosphate cofactor-binding site. Biochemistry 34, 1678–1685 [DOI] [PubMed] [Google Scholar]
- 27. Ferreira G. C., Dailey H. A. (1993) Expression of mammalian 5-aminolevulinate synthase in Escherichia coli. Overproduction, purification, and characterization. J. Biol. Chem. 268, 584–590 [PubMed] [Google Scholar]
- 28. Li J., Sha Y. (2008) A convenient synthesis of amino acid methyl esters. Molecules 13, 1111–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gong J., Kay C. J., Barber M. J., Ferreira G. C. (1996) Mutations at a glycine loop in aminolevulinate synthase affect pyridoxal phosphate cofactor binding and catalysis. Biochemistry 35, 14109–14117 [DOI] [PubMed] [Google Scholar]
- 30. Kikuchi G., Kumar A., Talmage P., Shemin D. (1958) The enzymatic synthesis of δ-aminolevulinic acid. J. Biol. Chem. 233, 1214–1219 [PubMed] [Google Scholar]
- 31. Gibson K. D., Laver W. G., Neuberger A. (1958) Initial stages in the biosynthesis of porphyrins. 2. The formation of δ-aminolaevulic acid from glycine and succinyl-coenzyme A by particles from chicken erythrocytes. Biochem. J. 70, 71–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Akhtar M., Jordan P. M. (1968) The mechanism of action of δ-aminolaevulinate synthetase and the synthesis of stereospecifically tritiated glycine. Chem. Comm. 1691–1692 [Google Scholar]
- 33. Laghai A., Jordan P. M. (1977) An exchange reaction catalysed by δ-aminolaevulinate synthase from Rhodopseudomonas spheroides. Biochem. Soc. Trans. 5, 299–300 [DOI] [PubMed] [Google Scholar]
- 34. Marcus J. P., Dekker E. E. (1993) pH-dependent decarboxylation of 2-amino-3-ketobutyrate, the unstable intermediate in the threonine dehydrogenase-initiated pathway for threonine utilization. Biochem. Biophys. Res. Commun. 190, 1066–1072 [DOI] [PubMed] [Google Scholar]
- 35. Dunathan H. C. (1966) Conformation and reaction specificity in pyridoxal phosphate enzymes. Proc. Natl. Acad. Sci. U.S.A. 55, 712–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ploux O., Marquet A. (1996) Mechanistic studies on the 8-amino-7-oxopelargonate synthase, a pyridoxal-5′-phosphate-dependent enzyme involved in biotin biosynthesis. Eur. J. Biochem. 236, 301–308 [DOI] [PubMed] [Google Scholar]
- 37. Frieden C. (1979) Slow transitions and hysteretic behavior in enzymes. Annu. Rev. Biochem. 48, 471–489 [DOI] [PubMed] [Google Scholar]
- 38. Raman M. C., Johnson K. A., Yard B. A., Lowther J., Carter L. G., Naismith J. H., Campopiano D. J. (2009) The external aldimine form of serine palmitoyltransferase: structural, kinetic, and spectroscopic analysis of the wild-type enzyme and HSAN1 mutant mimics. J. Biol. Chem. 284, 17328–17339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hunter G. A., Ferreira G. C. (1995) A continuous spectrophotometric assay for 5-aminolevulinate synthase that utilizes substrate cycling. Anal. Biochem. 226, 221–224 [DOI] [PubMed] [Google Scholar]
- 40. Tan D., Harrison T., Hunter G. A., Ferreira G. C. (1998) Role of arginine 439 in substrate binding of 5-aminolevulinate synthase. Biochemistry 37, 1478–1484 [DOI] [PubMed] [Google Scholar]
- 41. Guernsey D. L., Jiang H., Campagna D. R., Evans S. C., Ferguson M., Kellogg M. D., Lachance M., Matsuoka M., Nightingale M., Rideout A., Saint-Amant L., Schmidt P. J., Orr A., Bottomley S. S., Fleming M. D., Ludman M., Dyack S., Fernandez C. V., Samuels M. E. (2009) Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat. Genet. 41, 651–653 [DOI] [PubMed] [Google Scholar]
- 42. Lendrihas T., Hunter G. A., Ferreira G. C. (2010) Targeting the active site gate to yield hyperactive variants of 5-aminolevulinate synthase. J. Biol. Chem. 285, 13704–13711 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.