

Background: Trophoblast invasion is regulated by trophoblast-decidual cell interaction.
Results: HtrA1 and HtrA3 expression is up-regulated in decidualization stimuli-treated endometrial cells, which suppresses HtrA4-mediated trophoblast invasion by degradation of HtrA4.
Conclusion: Physical and functional interaction between trophoblastic HtrA4 and decidual HtrA1 and HtrA3 regulates trophoblast invasion.
Significance: This study reveals a novel trophoblast-decidual cell interaction in control of trophoblast invasion.
Keywords: Cell Invasion, Gene Expression, Placenta, Pregnancy, Protease, Trophoblast
Abstract
Human trophoblast invasion of decidualized endometrium is essential for placentation and is tightly regulated and involves trophoblast-decidual cell interaction. High temperature requirement A4 (HtrA4) is a secreted serine protease highly expressed in the invasive extravillous trophoblasts that invade decidua. In contrast, both HtrA1 and HtrA3 have been shown to inhibit trophoblast invasion. Here we provide evidence that decidua-secreted HtrA1 and HtrA3 antagonize HtrA4-mediated trophoblast invasion. We demonstrated that HtrA1 and HtrA3 interact with and degrade HtrA4 and thereby inhibit trophoblast-like JAR cell invasion. Specifically, HtrA1 and HtrA3 expression is up-regulated under decidualization conditions in endometrial stromal and epithelial cells, T-HESCs and Ishikawa cells, respectively. Conditioned media from these two cell lines after decidualization treatment suppress HtrA4-expressing JAR cell invasion in an HtrA1- or HtrA3-dependent manner. Co-culture of the HtrA4-expressing JAR cells with decidualization stimuli-treated T-HESC or Ishikawa monolayer also impairs JAR cell invasion, which can be reversed by HtrA1 or HtrA3 knockdown, supporting that HtrA1 and HtrA3 are crucial for trophoblast-decidual cell interaction in the control of trophoblast invasion. Our study reveals a novel regulatory mechanism of trophoblast invasion through physical and functional interaction between HtrA family members.
Introduction
Human placenta is composed of villous tissues of which floating villi are immersed in maternal blood for gas exchange and nutrient uptake whereas anchoring villi attach and fasten the placenta to the uterus. Human cytotrophoblasts in anchoring villi proliferate into cell columns from where some cells, termed interstitial extravillous trophoblasts (EVTs),2 migrate and invade deeper layers of the decidua. Indeed, EVTs may further invade the uterine myometrium and replace endothelial cells of spiral arteries to ensure sufficient blood flow into the placenta (1). Defective trophoblast invasion results in poor maternal-fetal circulation and placental hypoxia, which are associated with preeclampsia. This condition presents as gestational hypertension, proteinuria, and less than optimal fetal growth. In contrast, excessive invasion of trophoblasts into the endometrium is associated with placenta accreta and invasive choriocarcinoma (1). Therefore, the invasion of EVTs into endometrial stroma is tightly regulated to ensure a successful pregnancy.
The interaction between trophoblasts and decidual cells plays a crucial role in control of EVT invasion. Matrix metalloproteinases produced by trophoblasts and tissue inhibitors of metalloproteinases produced by decidual cells have been reported to regulate trophoblast invasion (2, 3). In addition, adhesion molecules and integrins such as E-cadherin and α6β4 are down-regulated in invasive trophoblasts (4, 5). However, the aforementioned factors and phenomena also participate in tumor cell invasion (6). Unlike uncontrolled tumor cell invasion, EVT invasion is limited both in time and space such that it occurs during early pregnancy and is confined to the proximal third of the uterine myometrium. These observations suggest that additional factors in the placenta and/or uterus may play important roles in the regulation of EVT invasion during placentation.
Placental transcription factor GCM1 (glial cells missing 1) is expressed in the cell columns of anchoring villi and EVTs at the maternal-fetal interface. We recently demonstrated that GCM1 transactivates HtrA4 expression to facilitate JAR and BeWo trophoblast-like cell invasion (7). HtrA proteins are a highly conserved family of serine proteases containing one or two trypsin protease domains and at least a C-terminal PDZ domain (8). Additionally, the human HtrA1, HtrA3, and HtrA4 polypeptides contain a domain with homology to Mac25 and insulin-like growth factor-binding protein and a Kazal protease inhibitor domain, which binds to and inhibits the trypsin protease domain. Members of HtrA family are involved in different biological functions. The Escherichia coli HtrA (DegP) functions as a chaperone in periplasm and as a protease at high temperature (8, 9). Human HtrA2 with an additional transmembrane domain is a mitochondrial serine protease that promotes apoptosis by interaction with and proteolysis of inhibitor of apoptosis (IAP) proteins, which are involved in caspase activation (10). Both HtrA1 and HtrA3 bind to various transformation growth factor-β (TGFβ) family members and inhibit TGFβ signaling in a protease activity-dependent manner (11–13). Interestingly, HtrA1 and HtrA3 are expressed in placenta and decidua and have been reported to inhibit trophoblast invasion (14–16). As invading EVTs encounter an environment of abundant placental and decidual cells, this raises the question of whether HtrA4 activity is modulated by HtrA1 and HtrA3.
In the present study we demonstrate that HtrA4 interacts with HtrA1 and HtrA3 and is subjected to proteolytic cleavage by HtrA1 and HtrA3. Consequently, HtrA4-mediated JAR cell invasion was suppressed in the presence of HtrA1 and HtrA3. We also demonstrated that HtrA1 and HtrA3 expression is stimulated under hormonal conditions mimicking decidualization in endometrial stromal cells (telomerase-immortalized human endometrial stromal cells (T-HESCs)) and endometrial epithelial cells (Ishikawa cells), respectively. The invasion activity of doxycycline-induced HtrA4-expressing JAR cells is decreased in the presence of the conditioned media collected from decidualization stimuli-treated T-HESCs and Ishikawa cells, supporting that HtrA4 activity is suppressed by HtrA1 and HtrA3. To mimic trophoblast-decidual cell interaction, co-culture of doxycycline-induced HtrA4-expressing JAR cells with decidualization stimuli-treated T-HESCs or Ishikawa monolayer resulted in decreased JAR cell invasion, which was reversed by HtrA1 or HtrA3 knockdown. Our results support physical and functional interaction between HtrA family members potentially fine-tuning the regulation of trophoblast invasion by the GCM1-HtrA4 axis.
EXPERIMENTAL PROCEDURES
Plasmid Constructs
The cDNA fragments of HtrA1, HtrA3, and HtrA4 with a C-terminal FLAG tag were cloned into pcDNA vector (Invitrogen) for transient expression. In addition, the cDNA fragment of HtrA4 with a C-terminal hemagglutinin (HA) tag was cloned into pcDNA. For protease-dead mutant (MT) construction, site-directed mutagenesis was performed to change Ser-328, Ser-305, and Ser-326 in HtrA1, HtrA3, and HtrA4 into alanine, respectively. For RNA interference, the lentiviral pLKO.1-Puro short-hairpin RNA (shRNA) expression constructs harboring 5′-CGGTGAAGTGATTGGAATTAA-3′ for HtrA1, 5′-CATCAAGATCCATCCCAAGAA-3′ for HtrA3, and 5′-AAGCTACATACCCAGCCCTCC-3′ for HtrA4 were obtained from the National RNAi Core Facility of Taiwan. The cDNA fragments encoding the Kazal domains of HtrA1, HtrA3, and HtrA4 were cloned into pGEX-6P-1 vector (GE Healthcare) for preparation of recombinant GST-Kazal fusion proteins.
Cell Culture, Transfection, and Lentivirus Transduction
JAR and 293T cells and T-HESCs (CRL-4003) were obtained from the American Type Culture Collection (Manassas, VA). Ishikawa cells were obtained from the Culture Collections of Public Health England (Salisbury, UK). For transient expression, cells were transfected with the indicated expression plasmids using the Lipofectamine 2000 reagent (Invitrogen). For stable expression of exogenous HtrA4, JAR cells were infected with recombinant lentivirus strains harboring empty pCDH vector (SBI, Mountain View, CA) or a pCDH construct encoding HtrA4-FLAG. To establish HtrA-knockdown cells, Ishikawa cells or T-HESCs were infected with recombinant lentivirus strains harboring pLKO.1-Puro expression plasmids containing a scrambled (5′-CCTAAGGTTAAGTCGCCCTCG-3′) or the above-mentioned pLKO.1-Puro constructs for HtrA1, HtrA3, and HtrA4. The infected cells were subjected to antibiotic selection using 10 μg/ml puromycin, and the puromycin-resistant clones were pooled for studies. For Tet-On inducible stable gene expression, JAR cells were infected with a recombinant lentivirus strain harboring a pAS4.1w.Ppuro-aOn (National RNAi Core Facility of Taiwan) construct encoding HtrA4-FLAG. After antibiotic selection, clones were selected for their response to doxycycline (Dox) in terms of HtrA4-FLAG expression.
For decidualization treatment of Ishikawa cells and T-HESCs, cells were precultured in phenol red-free medium and then cultured for 5 days in the presence of 10 nm 17-β-estradiol (Sigma), 1 μm 6α-methyl-17α-hydroxyl-progesterone acetate (Sigma), and 0.5 mm cAMP analog (dibutyryl cAMP, Sigma). The decidualization markers, IGFBP-1 (insulin-like growth factor-binding protein-1), prolactin, and IL-11, were analyzed by immunoblotting for IGFBP-1 (EMD Millipore, Billerica, MA) and ELISA for prolactin and IL-11 (R&D Systems, Minneapolis, MN).
Immunohistochemistry
Normal term placental tissues were mounted in optimal cutting temperature embedding compound (Sakura Finetek, Torrance, CA), frozen at −20 to −80 °C, and sectioned using a cryostat. Sections were fixed in pre-cooled acetone, rinsed with PBS at a neutral pH, and subjected to immunostaining by incubation with HtrA1, HtrA3, HtrA4, and cytokeratin 7 (CK7, EMD Millipore) antibodies at 4 °C for 16 h. After washing, the sections were incubated with secondary antibodies conjugated with Alexa Fluor 568 (for HtrA3), Cy2 (for HtrA4 and CK7), and Rhodamine Red-X (RRX for HtrA1) and DAPI and examined under a fluorescence microscope equipped with a cooled charge-coupled device camera. Antibodies against HtrA1, HtrA3, and HtrA4 were generated from guinea pigs, rabbits, and mice, respectively. The immunogens were recombinant His-tagged human HtrA1, HtrA3, and HtrA4 proteins prepared from bacteria using the pET21 expression vector (EMD Millipore).
Co-immunoprecipitation and in Vitro Proteolysis Assay
To study the interaction between HtrA4 and HtrA1 or HtrA3, 293T cells were transfected with expression constructs for HtrA1MT-FLAG, HtrA3MT-FLAG, and HtrA4MT-HA. At 48 h post-transfection, the culture supernatants were collected for co-immunoprecipitation with FLAG (Sigma) and HA (Roche Applied Science) antibodies. In a separate experiment, 293T cells were transfected with expression constructs for HtrA1-FLAG, HtrA1MT-FLAG, HtrA3-FLAG, HtrA3MT-FLAG, and HtrA4-HA. The culture supernatants were collected for immunoblotting with FLAG and HA antibodies to compare the effect of wild-type or mutant HtrA1-FLAG and HtrA3-FLAG on the HtrA4-HA protein level.
To study the proteolysis of HtrA4 mediated by HtrA1 and HtrA3, recombinant HtrA4MT-FLAG, HtrA4-FLAG, HtrA1-FLAG, HtrA1MT-FLAG, HtrA3-FLAG, and HtrA3MT-FLAG were first immunopurified with FLAG antibody-conjugated agarose beads (Sigma) from 293T cells transfected with their corresponding expression constructs. Recombinant HtrA4MT-FLAG was then incubated with HtrA1-FLAG, HtrA1MT-FLAG, HtrA3-FLAG, or HtrA3MT-FLAG at 37 °C for 1, 3, or 6 h followed by immunoblotting with HtrA4 antibody. Likewise, recombinant HtrA1MT-FLAG or HtrA3MT-FLAG was incubated with HtrA4-FLAG under similar conditions followed by immunoblotting with HtrA1 or HtrA3 antibody. To test the effect of Kazal domain in HtrA1, HtrA3, and HtrA4 on HtrA4 autocatalytic cleavage, recombinant HtrA4-FLAG was incubated with bacterially expressed GST, GST-Kazal1, GST-Kazal3, or GST-Kazal4 at 37 °C for 6 h followed by immunoblotting with FLAG or GST antibody.
Cell Invasion Analysis
JAR cells stably expressing the empty pCDH vector or HtrA4-FLAG were plated in Matrigel-coated transwells (BD Biosciences) and incubated with the conditioned medium collected from 293T cells expressing HtrA1-FLAG, HtrA1MT-FLAG, HtrA3-FLAG, or HtrA3MT-FLAG for 24 h. Invasive cells in the lower surface of filters were fixed with paraformaldehyde, visualized by crystal violet staining, and counted. Four microscopic fields per sample were randomly selected for quantification in each of three independent experiments. Images were prepared for presentation using Adobe Photoshop v7.0.
The tetracycline inducible (Tet-On) HtrA4-FLAG-expressing JAR (HtrA4-FLAG-aOn) cells were pretreated with 2 μg/ml Dox for 24 h before being plated in transwell chambers. The conditioned media from mock- or decidualization stimuli-treated scramble control and HtrA1-knockdown T-HESCs were added to the chambers and incubated for 24 h. A similar experiment was performed using the conditioned media of scramble control and HtrA3-knockdown Ishikawa cells. The invasion of Dox-induced HtrA4-FLAG-aOn cells was visualized and analyzed as described above.
To study trophoblast-decidual cell interaction, mock- or decidualization stimuli-treated scramble control or HtrA1-knockdown T-HESCs were first plated in transwell chambers and incubated for 8 h to form a confluent cell monolayer. The Dox-induced HtrA4-FLAG-aOn cells were preincubated with 10 μg/ml DilC12(3) fluorescent dye (BD Biosciences) for 2 h, and then the cells were trypsinized and plated onto the aforementioned T-HESC monolayer and co-cultured for an additional 24 h. A similar experiment was performed in the scramble control or HtrA3-knockdown Ishikawa monolayer. The invasion activity of the Dox-induced HtrA4-FLAG-aOn cells was analyzed by fluorescence microscopy using a Leica SP5 X laser scanning confocal microscope (Wetzlar) and quantified as described above.
Statistical Analysis
Statistical analysis of the data was performed using Student's t test. Statistical significance was classified as: *, p < 0.05; **, p < 0.01; ***, p < 0.001. A p value of <0.05 was considered statistically significant. A p value of >0.05 was considered no statistical significance and denoted as “ns.”
RESULTS
Expression of HtrA Family Members in Placenta and Decidua
We raised antibodies against HtrA1, HtrA3, and HtrA4 and confirmed their specificities by immunoblotting of 293T cells transiently expressing FLAG-tagged HtrA1, HtrA3, and HtrA4, respectively (Fig. 1A). We then performed immunohistochemistry to examine the expression of HtrA proteins in term human placenta and decidua. For identification of EVTs, CK7 staining was performed in parallel. Although HtrA1 expression was relatively higher in decidua (Fig. 1Ba), HtrA3 expression was relatively higher in placental syncytiotrophoblast layer (Fig. 1Bd). Specifically, HtrA1 expression was detected in fibrinoid, few EVTs, and decidual cells (Fig. 1Bc). Although HtrA3 expression is lower in decidua, its expression was also detected in decidual cells and many EVTs (Fig. 1Bf). HtrA4 expression was detected in placental villi and decidua (Fig. 1Bg). The staining pattern of HtrA4-positive cells is similar to that of CK7-positive cells in consecutive sections (Fig. 1B, h and j). Therefore, it is highly possible that HtrA4 is primarily expressed in EVTs in the decidua, which is also consistent with our previous study that HtrA4 is expressed in EVTs (7). As controls, none of normal guinea pig IgG, rabbit IgG, and mouse IgG detected significant signals (data not shown).
FIGURE 1.

Expression of HtrA proteins in human placenta and decidua. A, characterization of HtrA antibodies. The whole cell lysates of 293T cells transiently expressing HtrA1-FLAG, HtrA3-FLAG, and HtrA4-FLAG were subjected to immunoblotting (IB) with FLAG antibody (Ab), guinea pig (GP) anti-HtrA1 Ab, rabbit anti-HtrA3 Ab, and mouse anti-HtrA4 Ab, respectively. B, immunohistochemistry of HtrA proteins. Term human placental sections were co-stained with Abs to HtrA1 and CK7 (a–c) or HtrA3 and CK7 (d–f). Because both HtrA4 and CK7 Abs are derived from mice, consecutive sections were stained with HtrA4 (g and h) and CK7 (i and j) Abs, respectively. Sections were then stained with secondary Abs labeled with Alexa Fluor 568 (for HtrA3), RRX (for HtrA1), and Cy2 (for HtrA4 and CK7). Nuclei were stained by DAPI. The green fluorescent signals of HtrA4 were converted into a red color for better contrast. In a separate experiment, a consecutive section was stained with hematoxylin and eosin (H&E). D, decidua; V, villi; bar, 100 μm.
Interaction between HtrA Family Members
Because HtrA1, HtrA3, and HtrA4 are secretory proteins and expressed in placenta and decidua, we tested by co-immunoprecipitation analysis the possibility that HtrA4 may interact with HtrA1 or HtrA3 extracellularly. The protease activity of HtrA proteins may compromise the analysis; therefore, we examined the interaction between the protease-dead mutants of HtrA1, HtrA3, and HtrA4, i.e. HtrA1MT-FLAG, HtrA3MT-FLAG, and HtrA4MT-HA. The culture supernatants of 293T cells expressing different combinations of HtrA1MT-FLAG, HtrA3MT-FLAG, and HtrA4MT-HA were collected for reciprocal co-immunoprecipitation analyses using FLAG and HA Abs. As shown in Fig. 2A, specific interaction between HtrA4MT-HA and HtrA1MT-FLAG or HtrA3MT-FLAG was detected. These results suggested that HtrA4 may interact with HtrA1 and HtrA3 extracellularly.
FIGURE 2.
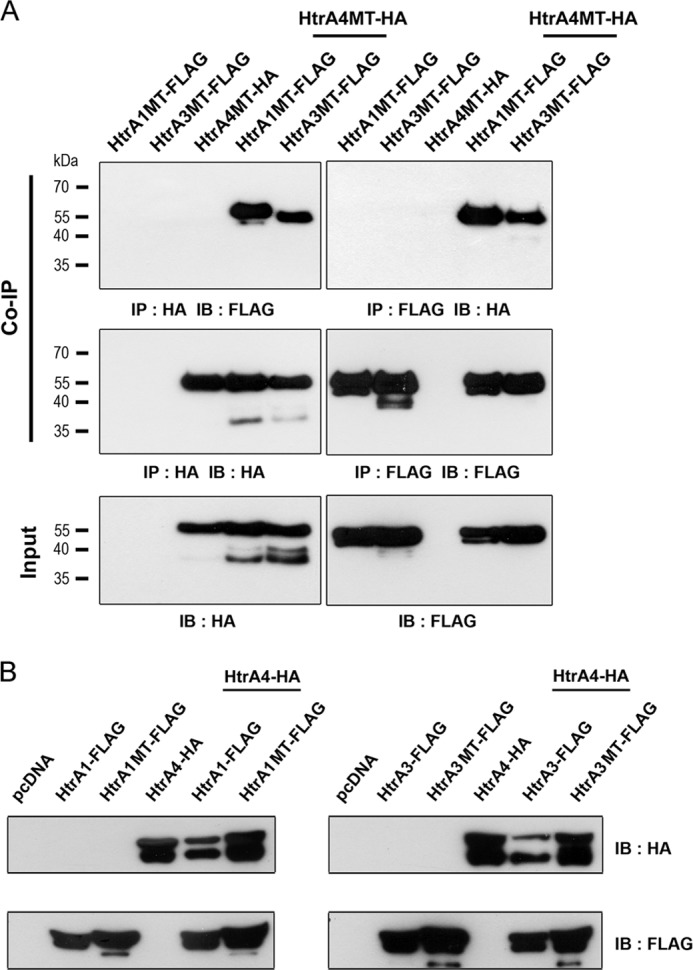
Physical and functional interaction between HtrA4, HtrA1, and HtrA3. A, HtrA4 interacts with HtrA1 and HtrA3. 293T cells were transfected with the indicated combinations of expression plasmids for mutant HtrA-FLAG proteins for 48 h. The culture supernatants were collected for co-immunoprecipitation (Co-IP) with FLAG and HA Abs. As an input control, a portion of the supernatants was also analyzed by immunoblotting with FLAG and HA Abs. B, HtrA1 and HtrA3 decrease HtrA4 protein level. 293T cells were transfected with the indicated combinations of expression plasmids for wild-type and mutant HtrA-FLAG proteins for 48 h. The culture supernatants were collected for immunoblotting (IB) with FLAG and HA Abs.
We then investigated the outcomes of HtrA4-HtrA1 protein-protein interaction by measuring the effects of HtrA1 on the HtrA4 protein level in the culture medium of 293T cells expressing HtrA4-HA and HtrA1-FLAG. Given that wild-type HtrA proteins undergo autocatalytic cleavage, the total level of secretory HtrA4-HA protein decreased in the presence of HtrA1-FLAG (left panel, Fig. 2B). This detrimental effect of HtrA1 on the secretory HtrA4 protein level was dependent on its protease activity as protease-dead mutant HtrA1MT-FLAG did not affect the total level of secretory HtrA4-HA protein (left panel, Fig. 2B). Similarly, co-expression of HtrA3-FLAG, but not HtrA3MT-FLAG, with HtrA4-HA also decreased the total level of secretory HtrA4-HA protein in the culture media (right panel, Fig. 2B). These results suggest that HtrA1 and HtrA3 may interact with HtrA4 and mediate proteolysis of HtrA4.
Proteolytic Cleavage of HtrA4 by HtrA1 and HtrA3
To further characterize the proteolysis of HtrA4 by HtrA1 and HtrA3, we performed in vitro proteolysis assays by incubation of recombinant HtrA4MT-FLAG with recombinant HtrA1-FLAG or HtrA3-FLAG followed by immunoblotting with HtrA4 Ab. Proteolysis of HtrA4MT-FLAG by either HtrA1-FLAG or HtrA3-FLAG was detected at 1 h and was almost completed at 6 h after incubation (Fig. 3A). In contrast, neither HtrA1MT-FLAG nor HtrA3MT-FLAG significantly mediated proteolysis of HtrA4MT-FLAG after incubation for 6 h (Fig. 3A). Therefore, HtrA4 is susceptible to proteolysis by HtrA1 or HtrA3. Similar experiments were performed to test whether HtrA4 proteolyzes HtrA1 and HtrA3. As shown in the lower panels of Fig. 3B, HtrA4-FLAG underwent autocatalytic cleavage into lower molecular weight forms. The protease activity of HtrA4-FLAG might be responsible for the process of HtrA1MT-FLAG into lower molecular weight forms, which were barely detected in HtrA3MT-FLAG (upper panels, Fig. 3B). Importantly, the levels of input HtrA1MT-FLAG and HtrA3MT-FLAG proteins did not significantly change (upper panels, Fig. 3B), suggesting that HtrA1 and HtrA3 are less susceptible to proteolysis by HtrA4. Because the Kazal domain is able to inhibit serine protease, we speculated that the Kazal domain in HtrA1 or HtrA3 may counteract the protease activity of HtrA4 to protect itself from complete degradation by HtrA4. To test this possibility, recombinant HtrA4-FLAG was individually incubated with recombinant GST, GST-Kazal1, GST-Kazal3, and GST-Kazal4. The latter three are GST fusion proteins harboring the HtrA1, HtrA3, and HtrA4 Kazal domains, respectively. Indeed, HtrA4-FLAG autocatalytic cleavage was significantly suppressed in the presence of GST-Kazal1 and GST-Kazal3 but not GST or GST-Kazal4 (Fig. 3C). Therefore, the Kazal domain in HtrA1 and HtrA3 may counteract the protease activity of HtrA4.
FIGURE 3.

Regulation of HtrA4 proteolysis by HtrA1 and HtrA3. A, proteolysis of HtrA4 by HtrA1 and HtrA3. Recombinant HtrA4MT-FLAG was incubated with recombinant wild-type or mutant HtrA1-FLAG and HtrA-3-FLAG for the indicated periods of time followed by immunoblotting (IB) with HtrA4 Ab. B, HtrA1 and HtrA3 are less susceptible to proteolysis by HtrA4. Recombinant HtrA1MT-FLAG or HtrA3MT-FLAG was incubated with recombinant HtrA4-FLAG for the indicated periods of time followed by immunoblotting with HtrA1, HtrA3, and HtrA4 Abs. C, HtrA1 and HtrA3 Kazal domains suppress HtrA4 autocatalytic cleavage. Recombinant HtrA4-FLAG was incubated with GST, GST-Kazal1, GST-Kazal3, or GST-Kazal4 for 6 h and then subjected to immunoblotting with FLAG and GST Abs. The arrows and arrowheads in A–C indicate the positions of unprocessed and proteolytically processed forms of HtrA protein, respectively.
Suppression of HtrA4-mediated Trophoblast Invasion by HtrA1 and HtrA3
Our previous study showed that the endogenous level of HtrA4 is very low in trophoblast-like JAR cells, and HtrA4 overexpression facilitates JAR cell invasion (7). In this study we wished to test whether HtrA1 and HtrA3 affect HtrA4-mediated trophoblast invasion. We generated JAR cells stably expressing HtrA4-FLAG and incubated the cells in Matrigel-coated transwells with conditioned media from 293T cells transiently expressing wild-type or mutant HtrA1-FLAG and HtrA3-FLAG proteins (Fig. 4A). Compared with the mock cells, HtrA4-FLAG-expressing JAR cells exhibited significant invasion activity (Fig. 4B). However, the observed HtrA4-mediated cell invasion was significantly inhibited in the presence of conditioned medium containing HtrA1-FLAG or HtrA3-FLAG but not HtrA1MT-FLAG or HtrA3MT-FLAG (Fig. 4, B and C).
FIGURE 4.

Regulation of HtrA4-mediated cell invasion by HtrA1 and HtrA3. A, stable expression of HtrA4-FLAG in JAR cells. Culture supernatants and whole cell lysates (WCL) were harvested from JAR cells stably expressing the empty pCDH vector or HtrA4-FLAG and subjected to immunoblotting with the indicated Abs. On the other hand, the culture supernatants of 293T cells transiently expressing wild-type or mutant HtrA1-FLAG and HtrA3-FLAG were collected for immunoblotting (IB) with FLAG Ab. B and C, the mock and HtrA4-FLAG-expressing JAR cells were plated in Matrigel-coated chambers and incubated with the conditioned medium (CM) collected from 293T cells expressing HtrA1-FLAG or HtrA3-FLAG mentioned in A. After 24 h, invasive JAR cells in the lower surface of the filters were fixed, stained, and counted. Representative images at 100× magnification from one of three independent experiments are shown. The mean and S.D. from three independent experiments are presented. *, p < 0.05; **, p < 0.01; ns, not significant.
Subsequently, we generated stable Tet-On HtrA4-FLAG-expressing JAR cells (HtrA4-FLAG-aON cells) to further characterize trophoblast invasion mediated by HtrA4. As shown in Fig. 5A, Dox treatment not only induced HtrA4-FLAG expression in HtrA4-FLAG-aON cells but also enhanced cell invasion in Matrigel-coated chambers. Because HtrA1 and HtrA3 are expressed in uterine endometrium, we studied whether HtrA1 and HtrA3 derived from endometrial cells affects HtrA4-mediated trophoblast invasion. The epithelial-like Ishikawa endometrial cells were shown to express HtrA3, which was up-regulated by decidualization stimuli of combined estrogen, progesterone, and cAMP analog (Fig. 5B). Expression of HtrA1 was not detected in Ishikawa cells (data not shown). In addition, stable Ishikawa cells expressing scramble control and HtrA3 shRNA were generated. The effects of decidualization stimuli on Ishikawa cells were measured by morphological changes as well as IGFBP-1, prolactin, and IL-11 expression (Fig. 5B).
FIGURE 5.

Regulation of HtrA4-mediated cell invasion by decidual HtrA1 and HtrA3. A, establishment of tetracycline-inducible (Tet-On) HtrA4-FLAG-expressing JAR cells (HtrA4-FLAG-aON cells). The HtrA4-FLAG-aON cells were treated with or without Dox and then subjected to immunoblotting (IB) with HtrA4, FLAG, and β-actin Abs. In a separate experiment, the mock- and Dox-induced HtrA4-FLAG-aON cells were plated into Matrigel-coated chambers for cell invasion analysis. B, expression of HtrA3 is induced in Ishikawa endometrial cells under decidualization conditions. The scramble control or HtrA3-knockdown Ishikawa cells were treated with 17-β-estradiol (E2), 6α-methyl-17α-hydroxyl-progesterone acetate (MPA) and cAMP analog for 5 days. The effects of decidualization treatment were assessed by morphology and immunoblotting with IGFBP-1 Ab and ELISA analysis of prolactin and IL-11. In addition, the whole cell lysate and culture supernatant of the mock- and decidualization stimuli-treated scramble (Scram.) control and HtrA3-knockdown Ishikawa cells were analyzed by immunoblotting with HtrA3 and HtrA4 Abs. Note that the decidualization treatment also increases HtrA3 expression in Ishikawa cells. C, HtrA3 suppresses HtrA4-mediated trophoblast invasion. The mock- and Dox-induced HtrA4-FLAG-aON cells were plated into Matrigel-coated chambers and incubated with the conditioned medium collected from the mock- and decidualization stimuli-treated scramble control and HtrA3-knockdown Ishikawa cells for 24 h for cell invasion analysis. ***, p < 0.001; ns, not significant. D and E, HtrA1 suppresses HtrA4-mediated trophoblast invasion. The scramble control or HtrA1-knockdown T-HESCs were treated with 17-β-estradiol, 6α-methyl-17α-hydroxyl-progesterone acetate, and cAMP analog for 5 days. Note that HtrA1 gene expression is up-regulated in T-HESCs under decidualization conditions. The mock- and Dox-induced HtrA4-FLAG-aON cells were plated into Matrigel-coated chambers and incubated with the conditioned medium collected from the mock- and decidualization stimuli-treated scramble control and HtrA1-knockdown T-HESCs for 24 h for cell invasion analysis. The mean and S.D. from three independent experiments are presented in B–E. *, p < 0.05; ***, p < 0.001.
We harvested conditioned media from mock- and decidualization stimuli-treated scramble control or HtrA3-knockdown Ishikawa cells and tested their effects on the invasion activity of Dox-induced HtrA4-FLAG-aOn cells. Indeed, the conditioned medium from decidualization stimuli-treated scramble control Ishikawa cells, but not from mock- or decidualization stimuli-treated HtrA3-knockdown Ishikawa cells, significantly inhibited the invasion activity of Dox-induced HtrA4-FLAG-aOn cells (Fig. 5C). We extended our study in endometrial stromal cells, T-HESCs, and found they express HtrA1 (Fig. 5D) but not HtrA3 (data not shown). Moreover, HtrA1 expression was elevated in T-HESCs under decidualization conditions in terms of elevated IGFBP-1, prolactin, and IL-11 expression (Fig. 5D). We tested the conditioned media from mock- or decidualization stimuli-treated scramble control or HtrA1-knockdown T-HESCs for regulation of Dox-induced HtrA4-FLAG-aOn cell invasion. The conditioned medium from decidualization stimuli-treated scramble control T-HESCs significantly inhibited Dox-induced HtrA4-FLAG-aOn cell invasion (Fig. 5E). By contrast, the conditioned media from mock- or decidualization stimuli-treated HtrA1-knockdown T-HESCs seemed to only marginally increase the invasion activity of Dox-induced HtrA4-FLAG-aOn cells (Fig. 5E).
Regulation of Trophoblast Invasion by Trophoblast-Decidual Cell Interaction
Because trophoblasts encounter decidual cells while invading the endometrium, we further studied the role of trophoblast-decidual cell interaction in control of trophoblast invasion by measuring the effects of Ishikawa and T-HESC monolayers on HtrA4-mediated JAR cell invasion. To visualize cell invasion, HtrA4-FLAG-aOn cells were prelabeled with the lipophilic DilC12(3) fluorescent dye and then treated with or without Dox. The DilC12(3)-labeled mock- and Dox-induced HtrA4-FLAG-aOn cells were plated onto mock- and decidualization stimuli-treated scramble control or HtrA3 knockdown Ishikawa monolayers in Matrigel-coated transwell chambers. As shown in Fig. 6, A and B, the Dox-induced HtrA4-FLAG-aOn cells invaded through the mock scramble control Ishikawa monolayer significantly more than the mock-induced HtrA4-FLAG-aOn cells. However, the invasion activity of Dox-induced HtrA4-FLAG-aOn cells was diminished by the decidualization stimuli-treated scramble control Ishikawa monolayer (Fig. 6, A and B). Similar experiments were performed in mock- or decidualization stimuli-treated scramble control or HtrA1-knockdown T-HESC monolayers, and the suppressive effects of decidualization treatment and HtrA1 on the invasion activity of Dox-induced HtrA4-FLAG-aOn cells were observed (Fig. 6C). Interestingly, HtrA1 knockdown further promoted HtrA4-FLAG-aOn cell invasion through the T-HESC monolayer (Fig. 6C). Taken together, these results suggest that HtrA4-HtrA1 and HtrA4-HtrA3 protein-protein interactions may modulate trophoblast invasion into uterine endometrium.
FIGURE 6.

Regulation of trophoblast invasion by trophoblast-decidual cell interaction. A and B, regulation of JAR cell invasion by Ishikawa cells. The mock- and Dox-induced HtrA4-FLAG-aON cells were pretreated with 10 μg/ml DilC12(3) fluorescent dye and then plated onto mock- and decidualization stimuli-treated scramble control or HtrA3-knockdown Ishikawa monolayers for 24 h. Invasive cells in the lower surface of the filters were fixed and visualized by fluorescence microscopy analysis. Four microscopic fields per sample were randomly selected for quantification in each of three independent experiments. Representative images at 100× magnification from one of three independent experiments are shown. Insets are images at 200× magnification. ***, p < 0.001; ns, not significant. C, regulation of JAR cell invasion by T-HESCs. A similar experiment to A was performed in mock- and decidualization stimuli-treated scramble control or HtrA1-knockdown T-HESC monolayers. The mean and S.D. from three independent experiments are presented in B and C. *, p < 0.05; ***, p < 0.001.
DISCUSSION
The physical interaction between trophoblast and uterine decidual cells begins at embryo implantation. Henceforth, placental trophoblasts proliferate and differentiate into invasive EVTs that migrate and invade the decidualized endometrium. Among the many regulatory factors, environmental cues are critical in control of EVT invasion as the process is limited to the proximal third of the myometrium. Given that both GCM1 and HtrA4 are primarily expressed in EVTs, an important function of the GCM1-HtrA4 axis has been shown to facilitate trophoblast-like BeWo and JAR cell invasion (7). Although regulation of HtrA4 expression by GCM1 at the transcriptional level provides an internal measure to control trophoblast invasion, the present study demonstrates that both HtrA1 and HtrA3 function as novel external factors to further modulate HtrA4-mediated trophoblast invasion. Several lines of evidence support this conclusion. First, histological analysis revealed that HtrA1 and HtrA3 are expressed in the placenta and decidua, providing feasible physiological interaction between HtrA4, HtrA1, and HtrA3 during trophoblast invasion. Second, HtrA4 interacts with HtrA1 and HtrA3 extracellularly resulting in a decreased level of secreted HtrA4 protein. Based on in vitro proteolysis assays (Fig. 3), this observation is very likely due to the proteolysis of HtrA4 by HtrA1 and HtrA3 and the lower susceptibility of HtrA1 and HtrA3 to HtrA4-mediated proteolysis. Third, HtrA4-mediated JAR cell invasion is suppressed by HtrA1 and HtrA3. This notion is supported by the observation that exogenous wild-type, but not protease-dead HtrA1 and HtrA3 in conditioned media are able to impede JAR cell invasion mediated by HtrA4.
We further addressed the role of endometrial cells in regulation of trophoblast cell invasion. Ishikawa cells derived from endometrial adenocarcinoma and T-HESCs established from primary HESCs by telomerase immortalization are well accepted for in vitro studies of human endometrium (17–20). Specifically, both cell lines share similar biochemical properties with primary endometrial cells when induced by ovarian steroids and cAMP. The observed elevation of HtrA1 and HtrA3 expression in decidualization stimuli-treated T-HESCs and Ishikawa cells in the present study further suggests that HtrA1 and HtrA3 are of physiological significance in the decidua. We reasoned that the secreted HtrA1 and HtrA3 may regulate the invasion of EVT into the decidua by counteracting the HtrA4 activity of EVT. Indeed, the invasion activity of Dox-induced HtrA4-FLAG-aON cells (tetracycline-inducible HtrA4-FLAG-expressing JAR cells) is suppressed by the culture supernatants of decidualization stimuli-treated Ishikawa cells and T-HESCs but not affected or even modestly elevated when HtrA3 or HtrA1 were knocked down in decidualization stimuli-treated cells. Similar observations were made in the co-culture of Dox-induced HtrA4-FLAG-aON cells with decidualization stimuli-treated T-HESC and Ishikawa monolayers, which mimics trophoblast-decidual cell interaction. Therefore, we believe that decidua-secreted HtrA1 and HtrA3 may modulate the HtrA4 activity of placental trophoblasts to fine-tune their invasion activities. How HtrA1 and HtrA3 expression is up-regulated in endometrial cells during decidualization warrants further investigation.
Chien et al. (21) have demonstrated that an intracellular form of HtrA1 associates with microtubules and suppresses cell motility. Although HtrA1 contains a signal peptide for secretion, it is not clear how the intracellular form of HtrA1 is trafficked to cytosol. Nevertheless, we think that the aforementioned mechanism is unlikely to play a key role in suppression of HtrA4-mediated cell invasion. We attributed the suppression of HtrA4-mediated JAR cell invasion by decidualization stimuli-treated T-HESCs and Ishikawa cells to the possible proteolysis of HtrA4 by HtrA1 or HtrA3. Specifically, our findings were based on the physical and functional interaction between the secretory forms of HtrA4, HtrA1, and HtrA3 from placental and endometrial cells. Moreover, HtrA4 was degraded by HtrA1 or HtrA3, whereas HtrA1 and HtrA3 were relatively resistant to HtrA4-mediated proteolysis in in vitro proteolysis assays. We further attributed this resistance to the inhibitory function of the Kazal domain in HtrA1 and HtrA3. Basic local alignment search tool (BLAST) sequence comparison with the Kazal domain in Mac25 (a follistatin-like protein with activin-binding activity) revealed a higher score for HtrA1 and HtrA3 than HtrA4 (data not shown). Correspondingly, the GST fusion protein containing the HtrA1 or HtrA3 Kazal domain is able to block the autocatalytic cleavage of HtrA4. Therefore, it is feasible to speculate that the Kazal domains of HtrA1 and HtrA3 may protect themselves from proteolysis by HtrA4.
Clinically, elevated HtrA1 and HtrA3 expression has been detected in the serum or placental tissues of patients with preeclampsia (15, 22, 23). Our previous study has demonstrated that HtrA4 expression is decreased in EVTs of preeclamptic placenta (7). As placental hypoxia is associated with preeclampsia, we have also demonstrated that HtrA4 expression is suppressed in hypoxic BeWo cells due to enhanced GCM1 degradation. To the contrary, hypoxia has been shown to enhance HtrA3 expression in first-trimester villous explants and BeWo cells (22). Inagaki et al. (24) have recently reported elevated expression levels of HtrA4, but not HtrA3, in preeclampsia. The discrepancy in the above-mentioned HtrA3 and HtrA4 reports might be due to different assay systems or small patient populations used in the studies. A large scale investigation using highly sensitive and specific assays may resolve the discrepancy. Given that the cause-and-effect relationship between abnormal HtrA protein expression and the pathogenesis of preeclampsia remains elusive, it is feasible to speculate that functional imbalance of HtrA proteins might lead to dysregulated trophoblast invasion with a concomitant development of preeclampsia.
A variety of cellular processes and factors have been implicated in the control of trophoblast-decidual cell interactions, which involve the penetration of trophoblasts through the endometrial epithelial cell layer and subsequent invasion into the endometrial stromal cell layer. For example, epithelial-mesenchymal transition is induced in the Ishikawa monolayer treated with ovarian steroids to promote JAR spheroid implantation (25). In response to growth factors and cytokines, CD82 in decidualized T-HESCs and carcinoembryonic antigen-related cell adhesion molecule-1 in AC-1M88 cells (hybridoma cells of primary EVTs and JEG-3 choriocarcinoma cells) are involved in the regulation of T-HESC and AC-1M88 cell migration and invasion (26, 27). Our study further revealed a new role of decidual HtrA1 and HtrA3 in antagonizing the function of trophoblastic HtrA4, supporting a novel regulatory mechanism for decidua to modulate trophoblast invasion mediated by the GCM1-HtrA4 axis. Of note, the invasion activity of Dox-induced HtrA4-FLAG-aON cells was elevated by the conditioned medium of HtrA1-knockdown T-HESCs or in the HtrA1-knockdown T-HESC monolayer (Figs. 5E and 6C), suggesting that relieving the HtrA1 negative effect may enhance the positive effects of yet-to-be-identified trophoblast and/or endometrial cell factors on cell invasion. It would be intriguing to test whether the aforementioned cellular processes and factors are involved in HtrA4-mediated trophoblast invasion. Finally, in addition to endometrial epithelial and stromal cells, we would like to stress that endometrium contains immune cells and vascular endothelial cells as well as myometrial cells. Therefore, the interactions between EVTs and the latter cell types may also contribute to the fine-tuning of trophoblast invasion.
Acknowledgment
We thank Dr. Chin-Chun Hung for technical assistance in acquiring confocal microscope images.
This work was supported by grants (to H. C.) from the Ministry of Science and Technology (103-2311-B-001-024) and Academia Sinica, Taiwan.
- EVT
- extravillous trophoblasts
- T-HESC
- telomerase-immortalized human endometrial stromal cells
- MT
- mutant
- Dox
- doxycycline
- IGFBP-1
- IGF-binding protein-1
- CK7
- cytokeratin 7
- Ab
- antibody.
REFERENCES
- 1. Benirschke K., Kaufmann P. (2001) Nonvillous parts and trophoblast invasion. In Pathology of the Human Placenta, 4th Ed., pp. 171–272, Springer-Verlag, New York [Google Scholar]
- 2. Irwin J. C., Suen L. F., Faessen G. H., Popovici R. M., Giudice L. C. (2001) Insulin-like growth factor (IGF)-II inhibition of endometrial stromal cell tissue inhibitor of metalloproteinase-3 and IGF-binding protein-1 suggests paracrine interactions at the decidua:trophoblast interface during human implantation. J. Clin. Endocrinol. Metab. 86, 2060–2064 [DOI] [PubMed] [Google Scholar]
- 3. Shimonovitz S., Hurwitz A., Dushnik M., Anteby E., Geva-Eldar T., Yagel S. (1994) Developmental regulation of the expression of 72 and 92 kDa type IV collagenases in human trophoblasts: a possible mechanism for control of trophoblast invasion. Am. J. Obstet. Gynecol. 171, 832–838 [DOI] [PubMed] [Google Scholar]
- 4. Zhou Y., Damsky C. H., Fisher S. J. (1997) Preeclampsia is associated with failure of human cytotrophoblasts to mimic a vascular adhesion phenotype. One cause of defective endovascular invasion in this syndrome? J. Clin. Invest. 99, 2152–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Damsky C. H., Librach C., Lim K. H., Fitzgerald M. L., McMaster M. T., Janatpour M., Zhou Y., Logan S. K., Fisher S. J. (1994) Integrin switching regulates normal trophoblast invasion. Development 120, 3657–3666 [DOI] [PubMed] [Google Scholar]
- 6. Ferretti C., Bruni L., Dangles-Marie V., Pecking A. P., Bellet D. (2007) Molecular circuits shared by placental and cancer cells, and their implications in the proliferative, invasive and migratory capacities of trophoblasts. Hum. Reprod. Update 13, 121–141 [DOI] [PubMed] [Google Scholar]
- 7. Wang L. J., Cheong M. L., Lee Y. S., Lee M. T., Chen H. (2012) High-temperature requirement protein A4 (HtrA4) suppresses the fusogenic activity of syncytin-1 and promotes trophoblast invasion. Mol. Cell. Biol. 32, 3707–3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clausen T., Southan C., Ehrmann M. (2002) The HtrA family of proteases: implications for protein composition and cell fate. Mol. Cell 10, 443–455 [DOI] [PubMed] [Google Scholar]
- 9. Clausen T., Kaiser M., Huber R., Ehrmann M. (2011) HTRA proteases: regulated proteolysis in protein quality control. Nat. Rev. Mol. Cell Biol. 12, 152–162 [DOI] [PubMed] [Google Scholar]
- 10. Vande Walle L., Lamkanfi M., Vandenabeele P. (2008) The mitochondrial serine protease HtrA2/Omi: an overview. Cell Death Differ. 15, 453–460 [DOI] [PubMed] [Google Scholar]
- 11. Tocharus J., Tsuchiya A., Kajikawa M., Ueta Y., Oka C., Kawaichi M. (2004) Developmentally regulated expression of mouse HtrA3 and its role as an inhibitor of TGF-β signaling. Dev. Growth Differ. 46, 257–274 [DOI] [PubMed] [Google Scholar]
- 12. Oka C., Tsujimoto R., Kajikawa M., Koshiba-Takeuchi K., Ina J., Yano M., Tsuchiya A., Ueta Y., Soma A., Kanda H., Matsumoto M., Kawaichi M. (2004) HtrA1 serine protease inhibits signaling mediated by Tgfβ family proteins. Development 131, 1041–1053 [DOI] [PubMed] [Google Scholar]
- 13. Launay S., Maubert E., Lebeurrier N., Tennstaedt A., Campioni M., Docagne F., Gabriel C., Dauphinot L., Potier M. C., Ehrmann M., Baldi A., Vivien D. (2008) HtrA1-dependent proteolysis of TGF-β controls both neuronal maturation and developmental survival. Cell Death Differ. 15, 1408–1416 [DOI] [PubMed] [Google Scholar]
- 14. Nie G., Hale K., Li Y., Manuelpillai U., Wallace E. M., Salamonsen L. A. (2006) Distinct expression and localization of serine protease HtrA1 in human endometrium and first-trimester placenta. Dev. Dyn. 235, 3448–3455 [DOI] [PubMed] [Google Scholar]
- 15. Ajayi F., Kongoasa N., Gaffey T., Asmann Y. W., Watson W. J., Baldi A., Lala P., Shridhar V., Brost B., Chien J. (2008) Elevated expression of serine protease HtrA1 in preeclampsia and its role in trophoblast cell migration and invasion. Am. J. Obstet. Gynecol. 199, 557.e1–557.e10 [DOI] [PubMed] [Google Scholar]
- 16. Singh H., Endo Y., Nie G. (2011) Decidual HtrA3 negatively regulates trophoblast invasion during human placentation. Hum. Reprod. 26, 748–757 [DOI] [PubMed] [Google Scholar]
- 17. Krikun G., Mor G., Alvero A., Guller S., Schatz F., Sapi E., Rahman M., Caze R., Qumsiyeh M., Lockwood C. J. (2004) A novel immortalized human endometrial stromal cell line with normal progestational response. Endocrinology 145, 2291–2296 [DOI] [PubMed] [Google Scholar]
- 18. Nishida M. (2002) The Ishikawa cells from birth to the present. Hum. Cell 15, 104–117 [DOI] [PubMed] [Google Scholar]
- 19. Wang H., Pilla F., Anderson S., Martínez-Escribano S., Herrer I., Moreno-Moya J. M., Musti S., Bocca S., Oehninger S., Horcajadas J. A. (2012) A novel model of human implantation: 3D endometrium-like culture system to study attachment of human trophoblast (Jar) cell spheroids. Mol. Hum. Reprod. 18, 33–43 [DOI] [PubMed] [Google Scholar]
- 20. Wang H., Bocca S., Anderson S., Yu L., Rhavi B. S., Horcajadas J., Oehninger S. (2013) Sex steroids regulate epithelial-stromal cell cross talk and trophoblast attachment invasion in a three-dimensional human endometrial culture system. Tissue Eng. Part C Methods 19, 676–687 [DOI] [PubMed] [Google Scholar]
- 21. Chien J., Ota T., Aletti G., Shridhar R., Boccellino M., Quagliuolo L., Baldi A., Shridhar V. (2009) Serine protease HtrA1 associates with microtubules and inhibits cell migration. Mol. Cell. Biol. 29, 4177–4187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li Y., Puryer M., Lin E., Hale K., Salamonsen L. A., Manuelpillai U., Tong S., Chan W., Wallace E. M., Nie G. (2011) Placental HtrA3 is regulated by oxygen tension and serum levels are altered during early pregnancy in women destined to develop preeclampsia. J. Clin. Endocrinol. Metab. 96, 403–411 [DOI] [PubMed] [Google Scholar]
- 23. Dynon K., Heng S., Puryer M., Li Y., Walton K., Endo Y., Nie G. (2012) HtrA3 as an early marker for preeclampsia: specific monoclonal antibodies and sensitive high-throughput assays for serum screening. PLoS ONE 7, e45956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Inagaki A., Nishizawa H., Ota S., Suzuki M., Inuzuka H., Miyamura H., Sekiya T., Kurahashi H., Udagawa Y. (2012) Up-regulation of HtrA4 in the placentas of patients with severe pre-eclampsia. Placenta 33, 919–926 [DOI] [PubMed] [Google Scholar]
- 25. Uchida H., Maruyama T., Nishikawa-Uchida S., Oda H., Miyazaki K., Yamasaki A., Yoshimura Y. (2012) Studies using an in vitro model show evidence of involvement of epithelial-mesenchymal transition of human endometrial epithelial cells in human embryo implantation. J. Biol. Chem. 287, 4441–4450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gonzalez M., Neufeld J., Reimann K., Wittmann S., Samalecos A., Wolf A., Bamberger A. M., Gellersen B. (2011) Expansion of human trophoblastic spheroids is promoted by decidualized endometrial stromal cells and enhanced by heparin-binding epidermal growth factor-like growth factor and interleukin-1β. Mol. Hum. Reprod. 17, 421–433 [DOI] [PubMed] [Google Scholar]
- 27. Gellersen B., Wolf A., Kruse M., Schwenke M., Bamberger A. M. (2013) Human endometrial stromal cell-trophoblast interactions: mutual stimulation of chemotactic migration and promigratory roles of cell surface molecules CD82 and CEACAM1. Biol. Reprod. 88, 80. [DOI] [PubMed] [Google Scholar]