

Background: Ubiquitination of intracellular N termini lysines inhibits cleavage/stimulation of α,β,γ epithelial sodium channel (ENaC) extracellular domains.
Results: The N terminus of γ-ENaC attains secondary structure upon sensing membrane phospholipids. Basic residues promote ENaC cleavage.
Conclusion: Ubiquitination obscures residues mediating allosteric stimulatory N-terminal structural transition that promotes ENaC cleavage/stimulation.
Significance: This study presents a new mechanism of allosteric linkage between cytosolic signaling pathways and extracellular ENaC proteolysis.
Keywords: Allosteric Regulation, Electrophysiology, Ion Channel, Molecular Dynamics, Protein Conformation, Gamma ENaC, Structural Model
Abstract
The epithelial sodium channel (ENaC) is activated upon endoproteolytic cleavage of specific segments in the extracellular domains of the α- and γ-subunits. Cleavage is accomplished by intracellular proteases prior to membrane insertion and by surface-expressed or extracellular soluble proteases once ENaC resides at the cell surface. These cleavage events are partially regulated by intracellular signaling through an unknown allosteric mechanism. Here, using a combination of computational and experimental techniques, we show that the intracellular N terminus of γ-ENaC undergoes secondary structural transitions upon interaction with phosphoinositides. From ab initio folding simulations of the N termini in the presence and absence of phosphatidylinositol 4,5-bisphosphate (PIP2), we found that PIP2 increases α-helical propensity in the N terminus of γ-ENaC. Electrophysiology and mutation experiments revealed that a highly conserved cluster of lysines in the γ-ENaC N terminus regulates accessibility of extracellular cleavage sites in γ-ENaC. We also show that conditions that decrease PIP2 or enhance ubiquitination sharply limit access of the γ-ENaC extracellular domain to proteases. Further, the efficiency of allosteric control of ENaC proteolysis is dependent on Tyr370 in γ-ENaC. Our findings provide an allosteric mechanism for ENaC activation regulated by the N termini and sheds light on a potential general mechanism of channel and receptor activation.
Introduction
Amiloride-sensitive epithelial sodium channels (ENaC)3 are heteromultimeric glycoproteins mediating active sodium reabsorption in the kidney, colon, lung, and sweat glands in vertebrates (1). Coexpression of three homologous subunits (α, β, and γ) forms functional channels with unresolved stoichiometry (2). Each subunit of ENaC spans the plasma membrane twice, with more than 50% of the protein presenting itself to the extracellular milieu (3). ENaC is synthesized as an inactive precursor, the activation of which is contingent on cleavage of the extracellular domain at multiple sites by an array of intracellular, membrane-associated, and extracellular proteases (4). Among the three subunits, cleavage of γ-ENaC plays an important role in activation of nearly silent channels (5). Cleavage of the extracellular domains is said to render ENaC constitutively active; however, intracellular conditions and signaling that involves the N and C termini of the subunits modulates the open probability (Po) of active channels (6, 7). For instance, point mutations at a highly conserved glycine residue in the N termini of any of the three subunits dramatically decrease the Po via alterations in channel open and close times (8). In addition, ENaC-mediated Na+ conductance is regulated by internalization and proteasomal degradation following ubiquitination of the intracellular N termini of the ENaC subunits (9, 10).
Biosynthesis and expression of ENaC are strongly regulated by signaling pathways that influence ubiquitination of the ENaC N termini. Recent studies indicate that the balance of ubiquitination/deubiquitination of ENaC N termini is coupled to the efficiency of proteolysis of the extracellular domain (11–13). It is hypothesized that ubiquitination of the cytosolic N termini controls the accessibility of cleavage sites in the extracellular domain to channel-activating proteases through conformational changes that propagate along the length of the channel. Knight et al. (11) demonstrated that intracellular sodium regulates the proteolytic activation of ENaC possibly by altering accessibility of protease cleavage sites. Although these observations indicate that intracellular signaling or conditions can significantly influence extracellular cleavage and activation of ENaC, the molecular mechanism of such transmembrane allosteric regulation of ENaC is currently unknown.
In addition to ubiquitination, ENaC is regulated by anionic phospholipids (AP) localized to the inner leaflet of the plasma membrane (14, 15). Both phosphatidylinositol 4,5-bisphosphate (PIP2) and phosphatidylinositol 3,4,5-triphosphate (PIP3) modulate channel function and gating via direct interaction with the cytosolic termini, explaining regulation of ENaC by signaling pathways in addition to ubiquitination (e.g. stimulation by PI3K activation and inhibition by purinergic P2Y receptor signaling) (16–19). Electrophysiology and mutation analyses have revealed the influence, via interaction with PIP3, of a stretch of basic residues immediately distal to the second transmembrane region of γ-ENaC on channel function (20). Depletion of PIP2 levels either through manipulation of physiological signaling via membrane receptors or other pharmacological maneuvers results in a marked decrease in ENaC activity and channel Po (16, 21). Clusters of basic residues in the N termini of β- and γ-ENaC have been proposed as PIP2-binding sites including lysines that are ubiquitinated. Thus, it is conceivable that the basic residues in the cytosolic tails of ENaC “sense” major intracellular conditions, including PIP2 levels and ubiquitination, and control proteolysis of the extracellular domain through an allosteric regulatory mechanism. Such a sensor function necessitates a structural response of the cytosolic ENaC tails to intracellular events that acts through an allosteric signaling pathway linking the N termini to functionally critical extracellular sites.
Here, we used computational and experimental methods to investigate the importance of N-terminal lysine clusters (K-clusters) in an allosteric mechanism that changes the susceptibility of ENaC to proteolytic cleavage and activation in response to PIP2 levels and ubiquitination. Using fluorescence spectroscopy and anisotropy, we demonstrate direct interaction of the synthetic N terminus of γ-ENaC with PIP2 in vitro. Molecular dynamics simulations and circular dichroism spectroscopy have revealed that the N terminus of γ-ENaC gains α-helical propensity in the presence of PIP2. Using electrophysiology and mutation analyses, we demonstrate that the N-terminal K-clusters are critical for efficient cleavage of the extracellular domain of γ-ENaC and expression of amiloride-sensitive sodium currents (INa). We analyzed channel activation by chymotrypsin and found that elimination of charge provided by the K-clusters results in delayed activation of ENaC, suggesting that a lack of PIP2 binding alters the accessibility of the protease cleavage sites via conformational change. Based on our results, we propose an allosteric activation model where N-terminal PIP2 binding favors a conformational change in the extracellular domain of γ-ENaC that facilitates proteolytic activation.
EXPERIMENTAL PROCEDURES
Ab Initio Folding Simulations
Parallel replica exchange discrete molecular dynamics (DMD) simulations were performed to study the structural features of γNT and its mutant variants (22, 23). Simulations at a high temperature (0.7 DMD units, ∼350 K) were performed to generate random initial configurations. The temperature unit used in DMD is kcal/mol·kB (where kB is the Boltzmann constant) and 1 unit = 5.03 × 102 Kelvin. More information on the units of measurement used in DMD is reported elsewhere (22). To simulate the presence of PIP2, the head groups of four PIP2 molecules were constrained to be on the XY plane. Harmonic potential with a force constant of 0.1 units were applied to the C terminus of γNT to maintain the peptide at the center of the simulation box. That γNT is followed by the first transmembrane helix justifies the use of such constraints on the peptide. Appropriate repulsive potential was applied such that all amino acids in γNT are maintained on the same side of the PIP2 plane. This constraint was used to avoid formation of unphysical configurations, thereby retaining only meaningful structural states. Ab initio replica exchange folding simulations were performed for 250 ns with eight replicas with the temperature ranging from 0.5 (∼250 K) to 0.745 (∼375 K) DMD units at a step of 0.035 (∼17.5 K) DMD units. Thirty independent replicates, amounting to a total simulation time of 7.5 μs, were used to estimate the specific heat, radius of gyration, and secondary structural content of γNT and its mutant variants. Trajectories were analyzed using the weighted histogram analysis method and perl/shell scripts developed in house (24). Snapshots from replica exchange molecular dynamics simulations were collected at regular intervals and clustered based on pairwise root mean square deviation to obtain the representative structural models.
Structural Model of Rat γ-ENaC
Primary sequence of rat γ-ENaC was obtained from the National Center for Biotechnology Information protein sequence repository. The crystal structure of cASIC1 was used to generate a homology-based structural model of γ-ENaC (25). Amino acid sequence encoding the γ-subunit from 15 different species were aligned with respect to each other and to that of cASIC using ClustalW2 and manually edited to preserve conservation of cysteines in the extracellular domain (26). The γ-subunit shares a sequence identity of 27% with cASIC1, whereas the α- and β-subunits share 24 and 29% identity, respectively, making cASIC1 an acceptable template for modeling the ENaC subunits. Medusa, a comprehensive protein design toolkit, was used to perform model building (27). The structural model thus generated was refined using Chiron, an automated protein energy minimization methodology, to remove steric clashes and nonphysical interactions that emerge from homology modeling (28).
Materials
Chloroform stock solutions of 1,2-dioleoyl-3-sn-phosphatidylcholine (DOPC), 1,2-dioleoyl-3-sn-phosphatidylglycerol (DOPG), and the lyophilized powder of 1,2-dioctanoyl-sn-glycero-3-[phosphoinositol-4,5-biphosphate] (PIP2) were purchased from Avanti Polar Lipids (Birmingham, AL) and used without further purification. For the peptide synthesis and purification, Fmoc-protected amino acids, resin, and coupling agents were obtained from EMD Novabiochem (Billerica, MA). Solvents were purchased from Sigma-Aldrich.
Peptide Synthesis
The peptide with the sequence H-MAPGEKIKAK10IKKNLPVRGP20QAPTIKDLMH30WYCMNTNTHG40CRRIVVSRGR50LRRLL-OH was synthesized chemically on a Tribute automated peptide synthesizer (Protein Technologies, Inc., Tuscon, AZ) using standard Fmoc solid phase techniques. The crude peptide was purified by reverse phase HPLC (Hitachi, Pleasanton, CA). The purity of the final product was confirmed by MALDI-TOF (Applied Biosystems, Foster City, CA) mass spectrometry.
CD Spectroscopy
Circular dichroism spectra were measured using a ChiraScan spectropolarimeter (Applied Photophysics, Surrey, UK). The data were collected using 2-mm-path length cells at wavelengths from 260 to 185 nm with intervals of 0.2 nm. The lipid to peptide ratio was maintained at 100:1 for all measurements, where the peptide concentration was 10 μm. Stock solution of the peptide was made in phosphate buffer, and the required volume of peptide was added externally to the small unilamellar vesicles made with different lipid composition. The spectrum with small unilamellar vesicles (without any peptide) was used as background and was subtracted from peptide-containing vesicle spectra. All the spectra have been corrected using the correction methodology published earlier (29), and the presented data are the averages of at least three independent measurements.
Fluorescence Measurement
Fluorescence spectra and anisotropy were measured using a Jobin Yvon Fluorolog Tau3 (Edison, NJ) spectrofluorimeter, which was equipped with both excitation and emission polarizer. The sole tryptophan (Trp31) in the peptide was excited at 295 nm, and emission was monitored from 305 to 450 nm using a 10-mm-path length cuvette. Reported fluorescence emission maximum data are the averages of at least three independent measurements. Fluorescence anisotropy (r) was measured using the standard equation,
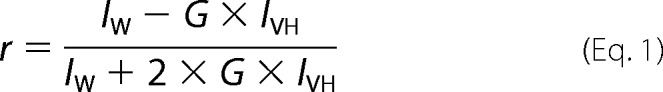 |
where G = IHV/IHH and IVV indicate vertically polarized excitation and vertically polarized emission, IVH indicates vertically polarized excitation and horizontally polarized emission, IHH indicates horizontally polarized excitation and horizontally polarized emission, and IHV indicates horizontally polarized excitation and vertically polarized emission.
Kinetics of ENaC Activation by Proteases
At the apical membrane, ENaC exists in the active and inactive states, and the total number of ENaC molecules (nT) is the sum of these two subpopulations,
where nI and nA are the numbers of inactive and active ENaC molecules, respectively. In vitro, ENaC is converted from inactive to active state upon addition of proteases like trypsin in less than 5 min. In the presence of trypsin, (inactive ENaC) → (active ENaC) can be described by the rate equation,
 |
where k is the reaction rate constant. Given that the total amount of ENaC (nT) is approximately constant in short time scales (<5 min), Equation 2 implies that dnA/dt = −dnI/dt, so that Equation 3 becomes
 |
Solving this differential equation, we get the following,
where nI0 is the concentration of inactive ENaC at time 0, which decays exponentially upon trypsin addition. The number of active ENaC molecules (nA) can be obtained from Equations 2 and 5 as follows,
where nA0 is the number of active ENaC molecules at time 0.
Having described the kinetics of ENaC activation by trypsin, we now need to understand how the number of active ENaC channels at the cell membrane is related to the electric current measured in whole cell voltage clamp experiments. The main ion channels expressed in oocytes are Na+, K+, Cl−, and Ca2+ channels; therefore the total current is the sum of currents associated with each of these ions.
In particular, the Na+ current (INa) is the sum of contributions from all individual ENaC channels, so that,
where nA is the number of active ENaC molecules, and INasingle is the current through a single active ENaC channel. Note that in writing Equation 8, we assumed that uncleaved ENaC is completely silent, so that only cleaved ENaC molecules contribute to the Na+ current.
We are interested in computing the rate of change in current (dI/dt) after trypsin addition. Given that all ion concentrations remain constant and that the membrane potential also remains constant (voltage clamped at −50 or −100 mV), the electrochemical gradients for K+, Cl−, and Ca2+ are constant during the experiments. Therefore, assuming that the permeability of the other ion channels are not affected by trypsin, the terms IK, ICl, and ICa in Equation 7 are constants, and we get the following,
 |
indicating that changes in total current after trypsin addition are solely due to changes in Na+ conduction.
Substituting Equation 8 into Equation 9, we get a relationship between the experimental electric current and the number of active ENaC channels, given by the following.
 |
From Equation 6, we have dnA/dt = knI0e−kt, so that Equation 10 becomes Equation 11.
 |
Upon integration, we get the following,
where Ibaseline is the current before trypsin addition (time t = 0), and Iplateau is the steady-state current after trypsin addition (limit t → ∞). These two currents are related by the following,
which means that the new steady-state current (Iplateau) is the sum of previous baseline current (Ibaseline) and the total current from activated ENaC (nI0INasingle).
Electrophysiology
Measurement of INa was performed as described previously (30). Four to six recordings for each condition from the same batch of oocytes were averaged to obtain average traces. An exponential fit was calculated for each trace using Equation 12. The time constant of activation (τ) was defined as
 |
One should note that the reaction rate constant k reflects the chemical affinity between reactants. Therefore, mutations in ENaC that affect its reactivity with trypsin are reflected in different values of k and τ. Finally, we must note that the reaction rate constant k is independent of the unitary conductance INasingle) (see Equations 12 and 13), so that any differences in the unitary conductance of ENaC (e.g. caused by mutations) do not affect the estimated time constant of activation τ.
Surface Labeling
Biotinylation was performed as described previously (30).
RESULTS
N Terminus of γ-ENaC Is α-Helical in Presence of PIP2
Binding of several membrane APs has been implicated in the regulation of ENaC, mostly involving the K-clusters in the N termini of β- and γ-ENaC. For example, direct phosphoinositide binding of short peptide fragments from the N terminus of different ENaC subunits has been demonstrated using lipid-protein overlay assays on PIP StripsTM (31). However, specific structural features (requirements and consequences) of interaction of ENaC N termini with APs have not been reported. To assess potential phosphoinositide-induced structural changes in ENaC, we performed DMD simulations of the N terminus of rat γ-ENaC (γNT) in the presence and absence of PIP2. We selected the γ-subunit because of its crucial role in ENaC activation by proteases (5). Analysis of the specific heat (Cv) of γNT in the presence and absence of PIP2 revealed that γNT is more stable in the presence of PIP2, as indicated by the shift in the peak corresponding to Cv of γNT+PIP2 (Fig. 1A). In addition, the radius of gyration of γNT is smaller in the presence of PIP2 than in its absence, indicating that γNT is more compact in the presence of PIP2. We also estimated the fraction of secondary structure assumed by γNT during simulations, in either the presence or the absence of PIP2, and found that the presence of PIP2 facilitates formation of secondary structure (Fig. 1, B and C). Our results suggest that γNT is more stable and compact and gains secondary structure in the presence of PIP2 (Fig. 1, A–C).
FIGURE 1.

γNT gains secondary structure in the presence of PIP2. A, comparison of structural/thermodynamic parameters of γNT in the presence and absence of PIP2. ENaC γNT is more stable (peak in Cv) and compact (radius of gyration (Rg)) and gains secondary structure (SS) in the presence of PIP2 (red versus black profile). Trajectories from all simulations were used to obtain the reported parameters (“Experimental Procedures”). B, representative structural model of γNT in the absence of PIP2. Lysines and arginines are shown as sticks and lines, respectively. Note that the NT does not present any major secondary structural features. C, representative structural model of γNT in the presence of PIP2. Lysines and arginines are represented as sticks and lines, respectively. PIP2 head groups are shown in ball and stick representation. Notice the preference for helical content in the representative model in the presence of PIP2. D, circular dichroism spectroscopy of γNT in DOPC lipid vesicles doped with DOPG or PIP2 to demonstrate the preference of γNT toward the presence of PIP2 over DOPG to form secondary structures. E, fluorescence anisotropy measurements of γNT peptide in the presence and absence of anioinic phospholipids. In the presence of anionic phospholipids, γNT undergoes a significant secondary structural rearrangement as shown by the emission profile of the NT peptide.
For biophysical characterization of PIP2 binding to γNT, we synthesized the first 55 amino acids of the N terminus of rat γ-ENaC (“Experimental Procedures”). We performed CD spectroscopy to determine the secondary structural features of synthetic γNT in aqueous solution and in the presence of membranes made with and without APs. Analysis of the mean molar ellipticity profile of γNT in aqueous buffer and in different types of lipid environments indicates that γNT is a highly unstructured peptide in aqueous buffer but gains a significant amount of secondary structure in the presence of negatively charged lipids, especially PIP2 (Fig. 1D). Small unilamellar vesicles better represent the membrane and are ideal for performing CD measurements of membrane proteins/peptides because of their relatively low scattering. Nonetheless, we have corrected all the CD spectra using a correction method developed recently (29). We measured the CD spectra of γNT in the presence of DOPC (zwitterionic) and a mixture of DOPC and PIP2 in a molar ratio of 9:1. To show that the effect of PIP2 is not completely due to its negative charge, we also measured the CD spectra of γNT in a 9:1 mixture of DOPC/DOPG. The double negative minima at 208 and 222 nm of the mean molar ellipticity profile indicate strong preference for α-helical content in the presence of PIP2 (Fig. 1D). γNT also acquired significant secondary structure in the presence of another negatively charged lipid, DOPG, but there was no significant effect of the zwitterionic lipid (DOPC) on secondary structure.
To further characterize the secondary structure gained by γNT in the presence of PIP2, we monitored the environment of the sole tryptophan (Trp31) present in the peptide by measuring Trp fluorescence as a function of lipid concentration. In fluorescence spectra, γNT showed an emission maximum at 357 nm in phosphate buffer, indicating that Trp31 experiences an extremely aqueous (polar) environment, which is in accordance with the unstructured nature of the peptide observed using CD and DMD simulations (Fig. 1). In addition, the change (blue shift) in Trp emission maxima in γNT follows a pattern similar to what we observed in CD measurements, PIP2 > DOPG ≫ DOPC (Fig. 1E). The blue shift in emission maxima confirms that γNT gains secondary structure (disorder to order transition) in the presence of APs generally and to a greater extent in the presence of PIP2 in particular. Furthermore, the emission profiles of γNT in the presence of DOPG and PIP2 indicate that the number of charges on the head group does not solely guide the AP-induced change in secondary structure. To confirm that the blue shift in emission maxima is not due to the incorporation of the peptide into the hydrophobic environment of the membrane/vesicle, we performed fluorescence anisotropy measurements. Tryptophan fluorescence anisotropy represents the ordering of the hydrophobic Trp environment. Fluorescence anisotropy of Trp31 did not change in the presence of any lipid, indicating that the peptide is not incorporated into the membrane.4 This result reiterates our observations that γNT acquires secondary structure in the presence of APs, with higher specificity for PIP2.
K-clusters in β- and γ-ENaC Strongly Influence ENaC Proteolytic Regulation
Previous studies have reported direct interaction with APs of the N terminus of β-ENaC (βNT) and γ-ENaC (γNT) (31). We therefore extended our computational analyses to study the possible structural changes induced by PIP2 in βNT. Our results indicate that the interaction with PIP2 improves stability of βNT with no significant change in predicted secondary structure content (Fig. 2, A–C). Moreover, the representative structure of βNT in the presence of PIP2 is similar to the one in its absence (Fig. 2D). Nonetheless, the βNT includes a cluster of conserved lysines (residues 4, 5, 9, 16, and 23 in rat ENaC) that are theoretically exposed to the same conditions as the K-cluster in γNT (residues 6, 8, 10, 12, 13, and 26 in rat ENaC). We therefore substituted the early K-cluster in βNT, as well as in γNT with glutamines (βNT-5K/Q and γNT-6K/Q) and made functional and biochemical assessments of rENaC with one or both mutagenized β- and γ-subunits by coexpressing all three subunits in oocytes.
FIGURE 2.

β-ENaC NT does not undergo significant structural transformation. A–C, the percentage of secondary structure (includes α helix, β strand, and turns) (A), radius of gyration (B), and specific heat (C) obtained by averaging values from different snapshots of the βNT during the course of simulation in the presence (red) and absence (black) of PIP2. D, overlay of representative structural models in the presence (red) and absence (gray) of PIP2, after clustering the snapshots from the simulations (“Experimental Procedures”).
WT ENaC expressed in oocytes produces a basal amiloride-sensitive sodium current (INa) that is further stimulated upon addition of an exogenous protease such as trypsin or chymotrypsin. Such a response to exogenous proteases indicates that ENaC can reside on the cell surface in a partially proteolytically activated state. However, coexpression of matriptase/channel-activating protease 3, a surface active protease, leads to increased basal INa that is not further activated by proteases, consistent with complete proteolytic activation (Fig. 3A) (30). ENaC generated by coexpression of WT α-subunits and mutagenized β- and γ-subunits was characterized by very low basal INa that was stimulated by trypsin, but not to the full extent seen with WT ENaC (Fig. 3A). Remarkably, coexpression of matriptase did not result in stimulation of mutant ENaC INa (Fig. 3A). Examination of γ-ENaC fragmentation in cell lysates revealed decreased intensity of the band associated with cleavage at the furin site (Fig. 3B), which is a prerequisite for further activation by matriptase (30). Diminished cleavage at the γ-ENaC furin site could explain the ineffectiveness of matriptase and possibly the incomplete recovery of INa by trypsin. However, a previous report alluded to the impact of βNT K-cluster on proper assembly and plasma membrane expression of ENaC (13). Therefore, we measured surface expression of ENaC by biotinylation and found that channels comprising the mutant β- and γ-subunits were expressed to the same levels as WT channels (Fig. 3B). In the presence of matriptase, ENaC in the surface pools appeared as a matriptase cleaved fragment only, whereas upon expression of the mutant without charged residues (βNT-5K/Q and γNT-6K/Q), some of the channels were uncleaved at the surface, whereas the others were cleaved at the furin site (Fig. 3B). The resistance of these mutants to complete activation by matriptase and exogenous trypsin suggests that expression of βNT-5K/Q and γNT-6K/Q dramatically influences accessibility of key proteolytic sites in the ENaC extracellular domain.
FIGURE 3.

Mutation of K-clusters in γNT significantly impairs ENaC proteolytic processing. A, ENaC containing β- and γ-subunits in which NT K-clusters were mutated to glutamines (β,γ-NT-K/Q) exhibit reduced INa and marked resistance to activation by coexpressed channel-activating protease 3. WT ENaC INa was fully stimulated by coexpressed channel-activating protease 3 (matriptase), whereas mutant ENaC INa was not stimulated at all. The results represent recordings from 24 oocytes per group combined from four separate batches of oocyte injections. Basal indicates steady-state INa before stimulation. Trypsin indicates INa following 3 min of perfusion with 2 μg/ml trypsin. B, cleavage of γENaC was followed upon mutation of NT K-clusters of β- and γ-subunits in the presence and absence of coexpressed matriptase (Mtp.). Cleavage of the γ-subunit was diminished in βNT-K/Q and γNT-K/Q ENaC. Shown are Western blots of lysates and surface-biotinylated protein (surface) from oocytes expressing WT ENaC or βNT-K/Q, γNT-K/Q ENaC (β,γNT-K/Q) ± matriptase. γ-ENaC was detected using V5 mAb (Invitrogen) that detects the C-terminal V5 tag. C, stimulation of INa by perfusion with 2 μg/ml trypsin. The activation time constant (τ) for activation of WT and mutant ENaC is calculated as described under “Experimental Procedures.” Inset, Tau for β,γ-NTK/Q ENaC (n = 5) is increased compared with WT (n = 5, p < 0.001). D, Tau calculated for WT ENaC (15) or ENaC containing β 5K/Q (6) or γ 6K/Q (14).
To further demonstrate the different behavior of mutant ENaC residing at the oocyte surface, we assayed the susceptibility of ENaC to proteolysis by measuring the rate of ENaC stimulation by exogenous proteases (5). This assay is based on the acute change in ENaC current when oocytes are exposed to soluble proteases that have been shown to activate ENaC through proteolytic cleavage (5, 32, 33). The cleavage event catalyzed by these soluble enzymes is the completion of an endoproteolytic process that severs γ-ENaC at or near its furin site and at sites within 20–40 residues downstream (30). Cleavage at the furin site and more distal residues is hypothesized to allow portions of the extracellular domain to attain conformations that promote conduction through the ENaC pore. We reasoned that if mutagenesis of ENaC N-terminal K-clusters reduced the accessibility of key γ-ENaC extracellular segments to cleavage by proteases, then the rate of ENaC activation by an exogenous protease would be slowed. Indeed, we found that ENaC with βNT-5K/Q and γNT-6K/Q have a much slower activation compared with WT channels, seen as differences in the time constant of activation after trypsin addition (τ = 129 and 41.2 s, respectively) (Fig. 3C). Similar results were obtained with exogenous chymotrypsin, indicating that such effects on ENaC activation are not specific to trypsin (data not shown). This observation, along with our biochemical results showing equivalence in expression of the mutants at the oocyte surface, indicates that NT lysines influence the susceptibility of ENaC extracellular domains to proteolytic cleavage (Fig. 3, B and C). We next distinguished the contributions of βNT and γNT to control of proteolytic activation of ENaC and found that mutagenesis of γNT exhibits a relatively stronger effect on τ than that of βNT (Fig. 3D). Therefore, we focused on the role of the γNT K-cluster in ENaC activation in the remainder of this study.
Balance between Ubiquitination and AP Binding of NT K-clusters Regulates Extracellular Proteolysis
Our data indicate that the lysines in the extreme N terminus of γ-ENaC are critical for regulation of ENaC activation. To further elucidate the role of individual lysines in the γNT K-cluster, we determined the propensity of each of these residues to interact with PIP2 in our simulations by enumerating the time points in which the lysines are in proximity (within 4 Å) to any of the PIP2 head groups (Fig. 4). We found that except for Lys26, all the other lysines interact with PIP2 for at least 10% (arbitrary cutoff) of the duration of the simulation. We retained the identity of this amino acid intact to determine the role of this residue in ENaC activation. We found that γNT-5K/Q has as profound an effect on ENaC activation as γNT-6K/Q (compare Fig. 5A with Fig. 3D). Therefore, we more thoroughly investigated the effects of γNT-5K/X (where X indicates Arg or Gln) on regulation of ENaC proteolytic activation. To ask whether the capability to bind APs by γNT K-clusters promoted accessibility of proteolytic sites, we substituted the K-cluster with arginines (γNT-5K/R), thereby preserving the basic charge requirement for AP sensing. Strikingly, the activation rate of γNT-5K/R is comparable with that of WT ENaC, suggesting that the charge and not the identity of the K-cluster in γNT is critical for regulation of ENaC activation (Fig. 5A). Acute activation of INa by applied proteases to oocytes expressing ENaC only or together with ubiquitin or ubiquitin-specific peptidase 17-like 2 (Usp17L2 or DUB3) revealed that the differences in ENaC basal INa, the extent of proteolytic activation, and Po under conditions of enhanced and reduced ubiquitination are strongly influenced because of altered access of extracellular proteolytic sites to activating proteases (13) (Fig. 5B). The effects of ubiquitinating conditions on basal and trypsin-stimulated INa were paralleled by differences we observed in the rate of trypsin stimulation of ENaC coexpressed with ubiquitin (τ increased from 30 to 50 s) or DUB3 (τ decreased to 20 ± 4 s) (Fig. 5B). The latter result establishes ubiquitination of NT K-clusters as an intracellular signal that controls susceptibility of ENaC extracellular domains to proteolysis.
FIGURE 4.
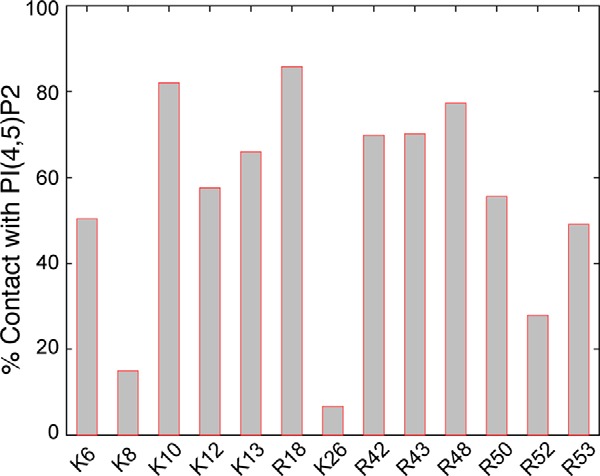
N-terminal lysines and arginines interact with PIP2 head groups. Terminal amines of all N-terminal positive charges except for Lys8 and Lys26 were found to be within 4 Å of any of the phosphate groups of the PIP2 head groups used in the simulations for at least 20% of the total duration (“Experimental Procedures”).
FIGURE 5.

γNT K-cluster is critical for regulation of ENaC proteolysis under ubiquitinating conditions. A, mutagenesis that eliminates any possibility of ubiquitination of the K-cluster and eliminates its basic charge (γNT-5K/Q) slows activation by chymotrypsin (increased Tau). However, mutation (γNT-5K/R) that preserves basic charge at the K-cluster has no effect on chymotrypsin stimulation. B, enhanced ubiquitinating conditions increased Tau (coexpressed ubiquitin (+UB, n = 14, p < 0.001)), whereas diminished ubiquitination conditions decreased Tau (coexpressed deubiquitinating enzyme 3 (+DUB3, n = 8, p < 0.01)) compared with WT ENaC alone (n = 14) stimulated by trypsin. C, deubiquitinating conditions increase the rate of WT ENaC stimulation (Tau decreased to 0.72 ± 0.04 of WT ENaC control without DUB3, n = 22, p < 0.001) but not in ENaC containing γNT 5 K/R (0.90 ± 0.09 of control without DUB3, n = 14, p = 0.20). In contrast, mutagenesis of potential ubiquitin targeted lysines in α-ENaC did not prevent stimulation of ENaC by DUB3 coexpression (Tau was 0.49 ± 0.03 of control without DUB3, n = 9, p < 0.001). D, deletion of residues 1–67 in α-ENaC caused a slight decrease in Tau (n = 21, p < 0.05), whereas deletion of residues 1–13 in the γNT to eliminate the K-cluster increased Tau (n = 25, p < 0.001).
We established that substitution of the γNT K-cluster has a more profound effect on the rate of ENaC activation compared with the βNT K-cluster (Fig. 3D). To understand the role of the αNT lysines in ENaC activation, we substituted the two lysines in α-ENaC with arginines. Ruffieux-Daidié and Staub (13) demonstrated that overexpression of a ubiquitin-specific protease (Usp2–45) has no effect on the cleavage of lysine-substituted ENaC variants. Our finding that the γNT K-cluster influences the rate of ENaC activation prompted us to hypothesize that the NT of γ-ENaC might also regulate the rate of ENaC activation upon coexpression of the ubiquitin protease DUB3. To test our hypothesis, we coexpressed WT, αNT-2K/R, or γNT-5K/R with DUB3 in oocytes and measured the rate constants of ENaC activation, after confirming that the same lysines in these subunits are ubiquitinated (data not shown). In agreement with published biochemical data for Usp2–45, we found that WT ENaC was activated faster in the presence of DUB3 than in its absence (Fig. 5C) (13). This effect is reflected in the increased basal INa and a concomitant decrease in the ratio of exogenously stimulated and basal INa of WT ENaC in the presence of DUB3, suggesting that the extracellular domain is primed for proteolytic stimulation because of deubiquitination of the cytosolic N termini (data not shown). To investigate the relative importance of arginines in α- and γ-NT in mediating such allosteric regulation of ENaC activation, we expressed αNT-2K/R,β,γ ENaC or α,β,γNT-5K/R ENaC in oocytes and followed their activation kinetics. Interestingly, in addition to featuring higher basal INa compared with WT ENaC, channels comprising αNT-2K/R were activated faster than WT in the presence of DUB3, suggesting a role for the γNT K-cluster in DUB3-mediated channel activation (Fig. 5C). On the other hand, coexpression of ENaC containing γNT-5K/R with DUB3 hardly had any effect on rate of channel activation, indicating that the γNT K-cluster plays a critical role in modulating kinetics of activation of ENaC (see “Discussion”) (Fig. 5C). To further implicate the γNT K-cluster in regulation of ENaC activation kinetics, we deleted the extreme N terminus of α-ENaC (Δ1–67) and γ-ENaC (Δ1–13) and expressed all three subunits with the mutant α- or γ-subunit in oocytes. We found that coexpression of γNTΔ1–13 dramatically decreases the rate of channel activation compared with WT ENaC (Fig. 5D). In contrast, coexpression of αNTΔ1–67 with WT β- and γ-ENaC resulted in faster channel activation, reiterating the role of the γ subunit in allosteric activation of ENaC (Fig. 5D).
Our results using Lys/Arg mutagenesis to link low ubquitination states of ENaC to enhanced susceptibility to proteolytic regulation is in broad agreement with the recent report by Ruffieux-Daidié and Staub (13). However, NT-K/Q mutagenesis sharply curtailed the susceptibility of ENaC to proteolytic regulation, contrary to the prediction that diminished ubiquitination of ENaC enhances proteolysis (Figs. 3D and 5A). To determine whether the basis for this discrepancy lay in the inability of γNT-K/Q to sense changes in AP levels, we examined the effects of P2Y2-R on ENaC proteolysis. P2Y2-R couples to phospholipase C, and purinergic signaling inhibits ENaC in part by depleting PIP2 (16, 21). ENaC-mediated sodium current in oocytes coexpressed with P2Y2-R was activated by trypsin with a time constant that was identical to control (ENaC alone) (Fig. 6). However, application of UTP to stimulate P2Y2-R resulted in reduced basal INa that was activated significantly more slowly than ENaC-mediated current in nonstimulated oocytes (Fig. 6). We have established earlier that γNT-5K/R has a time constant of activation similar to WT ENaC (Fig. 5A). To confirm that the positive charge on γNT K-cluster is required for response to APs, we coexpressed WT α- and β-subunits with γNT-5K/R or γNT-5K/Q ENaC with P2Y2-R and stimulated the receptor with UTP. Strikingly, γNT-5K/R responded acutely to UTP, whereas stimulation of P2Y2-R with UTP had no effect on the activation of γNT-5K/Q ENaC. Together, these results implicate the K-cluster in γNT in linking proteolytic susceptibility of ENaC to physiologic intracellular signaling events besides ubiquitination.
FIGURE 6.
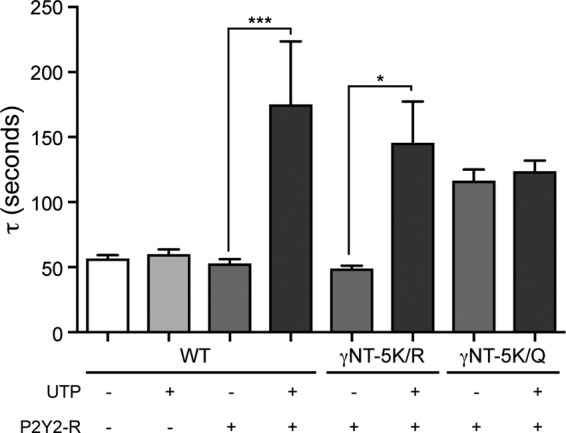
UTP stimulation of P2Y2-R slowed proteolytic stimulation of ENaC. UTP (10 μm) was included in the chymotrypsin containing perfusate. Tau was calculated as described in Fig. 2. UTP also slowed proteolytic activation when the K-cluster in the γNT was mutated to arginines (γNT-5K/R) to preserve basic charge. However, mutation of the γNT K-cluster to glutamines (γNT-5K/Q) caused slower stimulation of INa that was not affected by UTP in oocytes coexpressing P2Y2-R (n = 12–14 for each experimental group, one-way analysis of variance; *, p < 0.05; ***, p < 0.005)).
Tyr370 in the γ-Subunit Affects Allosteric Regulation of Proteolytic Activation of ENaC
Previous reports and our data support a hypothesis that γNT serves as a sensor of balance between inner leaflet AP levels and ubiquitination of ENaC. Because cleavage of extracellular sites is required for ENaC to be maximally active and because either lysines or arginines must be retained in the N terminus for efficient proteolysis, we believe that γNT regulates the extent of extracellular proteolysis in an allosteric manner. This mechanism requires long range communication between the γNT and extracellular proteolytic sites. We therefore investigated the putative “path” followed by such allosteric communication between the intracellular and extracellular sites. We used a graph theory-based computational approach to identify key residues involved in allosteric signal propagation from one region of a protein to another.5 We have successfully used this approach to identify key residues involved in allosteric signal propagation in the first nucleotide binding domain of the cystic fibrosis transmembrane conductance regulator (34).5 Recently, other groups have focused on extracellular events modulating gating of ENaC and related ion channels (35, 36). Based on a hypothesis put forth by Jasti et al. (25), validated by Li et al. (35), and based on the crystal structure of cASIC1, Shi et al. (36) studied the role of a loop near the extracellular membrane boundary in ENaC gating. By mutagenizing a stretch of amino acids forming this loop, Shi et al. (36) concluded that this loop has a role in modulating ENaC gating in response to external stimuli, as observed via altered sodium self-inhibition and response to shear stress. To gain structural perspective for the role of the extracellular loop in mediating allosteric communication between the NT and the proteolytic cleavage sites, we constructed a homology model of the rat γ-subunit using Medusa, based on the structure of cASIC1 (25, 27). In our structural model, similar to its equivalent residue in cASIC1, the side chain of Tyr370 (residue in rat γ-ENaC equivalent to Tyr375 in mouse γENaC) interacts with the residues in the extracellular boundaries of transmembrane helices 1 and 2 that form a pocket (Fig. 7A, inset). One of the walls of this pocket is lined by negatively charged amino acids, whereas the other is predominantly hydrophobic (Fig. 7A, inset). We investigated the role of Tyr370 in communicating the effects of intracellular signaling events to the extracellular protease cleavage sites by mutagenizing it to a lysine, expecting perturbation of interactions of this residue with the pocket. Expression of γY370K in oocytes resulted in decreased basal and trypsin-stimulated INa, an effect similar to γNT-K/Q variant, suggesting that Y370K exerts its effect via decreasing susceptibility of the extracellular domain to proteolysis (Fig. 7B). Because MTSET stimulation of ENaC harboring Y370K substitution was unaffected upon coexpression of WT α and γY370K ENaC with the MTSET-responsive βS518C ENaC, we conclude that folding and surface expression of ENaC is not affected by the Y370K substitution (Fig. 7B). That MTSET stimulation is independent of the effects of Y370K on activation of ENaC indicates that these two modifications affect ENaC function via mutually exclusive mechanisms. To exclude the possibility that the effect of Y370K on ENaC activation is driven solely by electrostatics, we replaced Tyr370 with an asparagine. Coexpression of γY370N with WT α and βS518C subunits in oocytes resulted in impaired ENaC activation, albeit to a lesser extent compared with the γY370K substitution (Fig. 7B).
FIGURE 7.
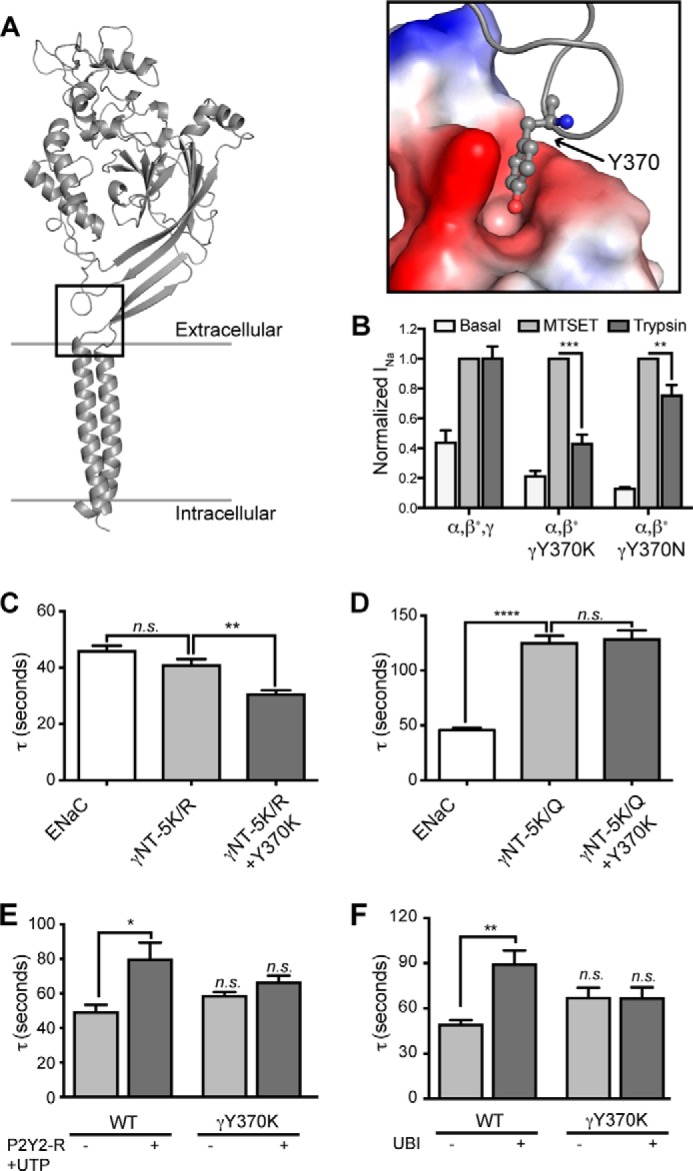
Tyr370 in γENaC mediates allosteric signal propagation required for channel activation in response to cytosolic signaling. A, structural model of rat γENaC constructed based on the crystal structure of cASIC1 (“Experimental Procedures”). Inset, surface representation of the residues in the pocket that forms a docking site for Tyr370. B, oocytes were injected with WT α- and γ-subunits and β-ENaC S518C, resulting in channels that are “locked” in the open state when exposed to the cysteine reactive MTSET. Other oocytes were injected identically except that γ-ENaC Y370K or Y370N replaced WT γ-ENaC. After basal current was recorded, oocytes were exposed to either MTSET (1 mm) or trypsin (20 μg/ml) for 5 min in the presence of amiloride. Amiloride was washed off to reveal the effect of MTSET or trypsin on INa. β-ENaC S518C is indicated by an asterisk on β in each group. C and D, oocytes were injected with WT subunits (ENaC) or with WT α-ENaC and β-ENaC and the indicated mutations of γ-ENaC. Tau was calculated as in Fig. 2. E, WT ENaC coexpressed with P2Y2-R and incubated for 5–20 min with 1 mm UTP were more slowly activated by perfusion with 4 mg/ml chymotrypsin (*, p < 0.05; one-way analysis of variance). UTP did not affect the chymotrypsin activation time constant of INa of ENaC containing γ-ENaCY370K and coexpressed P2Y2-R. F, Xenopus oocytes were injected with WT α- and β-ENaC subunits and either WT (ENaC) or Y370K γ-ENaC (γ370K) (0.3 ng/subunit/oocyte). Oocytes in each group were also injected with ubiquitin (1.0 ng/oocyte) (ENaC + UBI and γY370K + UBI). INa was recorded following washout of amiloride and during 5 min of perfusion with 4 mg/ml chymotrypsin, and the time constant of activation (τ) was calculated as described from four to nine traces performed for each group. **, different from ENaC, p < 0.01 by one-way analysis of variance. n.s., not significant.
To determine whether Try370 participates in propagation of an allosteric effect that stems from intracellular signaling events, we compared the effect of Y370K on γNT-K/R and K/Q substitutions (Fig. 7, C and D). We chose the Y370K substitution because of its stronger effect on ENaC activation compared with Y370N. γNT-K/R cannot be ubiquitinated, whereas it still senses APs, making it a suitable variant to study the effect of Y370K on ENaC proteolytic stimulation. Analysis of the rate of WT and mutant ENaC activation in oocytes revealed that Y370K substitution results in accelerated proteolytic processing of γNT-5K/R (compare Fig. 7C with Fig. 5D). Based on these observations, we speculate that Tyr370 in the γ subunit influences allosteric control upon ENaC activation in response to changes in PIP2 levels. To determine the effect of Y370K on AP sensing, we expressed WT or Y370K ENaC in oocytes and stimulated the coexpressed P2Y2-R with UTP in oocytes (Fig. 7E). Although the rate of WT ENaC activation decreased (larger τ) upon UTP stimulation, Y370K substitution in γENaC did not respond to UTP, suggesting that the mutant channels are not as dependent on APs and/or have diminished ability to sense APs as effectively as WT channels (Fig. 7E). Similarly, γY370K substitution minimized the ability of overexpression of ubiquitin in oocytes to slow the rate of ENaC activation (Fig. 7F). These results, in context of the recent studies elucidating the role in ENaC activation of residues in the vicinity of Tyr370 (Fig. 7A) (36, 37), provide further evidence that allosteric information from intracellular signaling events propagate via a pathway containing Tyr370 to modify exposure of the extracellular proteolytic sites.
DISCUSSION
Complete activation of ENaC is contingent upon cleavage of the extracellular domains of the α- and γ-subunits by serine proteases (4, 30, 38). Interestingly, the susceptibility of ENaC to proteolysis is linked to intracellular conditions. Knight et al. (11) reported that intracellular Na+ impaired extracellular ENaC proteolysis possibly by altering the accessibility of cleavage sites to proteases. A proposed mechanism for such regulation involves the ENaC N termini, which interact with the polypeptide components of diverse signaling pathways that tightly regulate surface expression and function of ENaC. Principal among these is ubiquitination of the N termini catalyzed by Nedd4-2 (neuronal precursor cell expressed, developmentally down-regulated 4-2) protein (9, 39). Although ubiquitination is a post-translational modification that directs protein recycling or localization, Staub and co-workers (9, 13) discovered that this modification uniquely regulates proteolytic activation of ENaC at the cell surface. Substitution of the N-terminal lysines targeted for ubiquitination to arginines not only results in enhanced surface expression of ENaC but also promotes robust proteolytic activation. ENaC-mediated INa is also regulated by phosphatidylinositides in the inner leaflet of the plasma membrane in a manner independent of channel trafficking to the cell surface (17, 19). The resulting stimulation places ENaC in a large group of ion channels whose activity is influenced by membrane-associated anionic phospholipids (40). This study, for the first time, provides a mechanistic framework for how basic charges in the γNT provided by clustered lysines enable critical ENaC signaling pathways to both positively and negatively regulate ENaC proteolysis.
Several studies have addressed the role of multiple highly conserved basic residues in the β- and γ-ENaC NT in the influence of APs on ENaC activity (15, 16, 19–21). These studies establish the rationale for the presence of an allosteric mechanism whereby the activation of ENaC via limited endoproteolysis is linked to intracellular conditions. Although ubiquitination is thought to affect channel dwell time at the surface, AP binding has been proposed to regulate gating and both processes affect trafficking of ENaC to the surface (14). Despite these indications of rich complexity in the roles of ENaC NT in receiving signaling inputs, the NT themselves have received scant attention in structural studies. Lack of structural insight into the roles of ENaC NT can partly be ascribed to their intrinsic dynamics: cytosolic termini of ASIC, an ENaC family member, had to be excised before crystallographic structure determination (25). In this study, we used a molecular dynamics-driven approach, as well as experimental strategies to determine the structural events underlying NT control of ENaC activity. We found that the γ-ENaC NT undergoes structural transition upon interaction with PIP2. This structural transition is critical for acute activation of ENaC because arresting the structural change by mutagenesis resulted in significant delay in activation following exposure to exogenous proteases.
The binding of PIP2 by proteins is broadly thought to be mediated either by specialized protein domains (e.g. pleckstrin homology domain) with a positively charged pocket of basic residues or by unstructured polypeptides with contiguous clusters of basic residues (e.g. myristoylated alanine-rich C kinase substrate (MARCKS) peptide) (40). Primary sequence analysis of the β- and γ-ENaC NT indicates the presence of charge clusters analogous to the MARCKS peptide with little or no similarity to any known pleckstrin homology domains. Secondary structure prediction using JPred revealed a propensity for the first 13 amino acids of γNT to form an α helix (41).5 Unlike potassium channels, ENaC does not feature a well structured PIP2 binding cytosolic domain. A recent structural study focusing on the calmodulin-binding region of MARCKS demonstrated the formation of a “fixed” conformation in the presence of calmodulin (Protein Data Bank code 1IWQ) (42). These observations prompted us to compare the structure of ENaC NT in the presence and absence of PIP2. We observed that PIP2 enhances secondary structural content in the γNT both in simulations and biophysical studies (Fig. 1). That the γNT is structurally more compact in the presence of PIP2 (Fig. 1, A–C) indicates that engagement of NT K-clusters in PIP2 sensing might further preclude these sites from ubiquitination. Furthermore, we observed that the presence of basic charges in the extremity of γNT is required for its structural compactness in the presence of PIP2. Although we focused on the lysines in this study because of their connection to ubiquitination, other groups have proposed that the arginines that are close to the first membrane-spanning helix in the β- and γ-ENaC NT might interact with PIP2 (16). Our simulations support this notion where the arginines in the C terminus of γNT are in contact with PIP2 for more than 50% of the duration of the simulations (Fig. 4). Interestingly, Lys8 and Lys26 make contact with PIP2 head groups for the least amount of time during the simulations. Based on this trend, we speculate that the conformational dynamics of γNT are such that the configurations in which Lys8 and Lys26 are not in contact with PIP2 are energetically favored to maintain contacts between the remaining lysines and the lipid head groups. Further studies are required to investigate the roles of the positive charges with lower predicted propensity to interact with PIP2 head groups in simulations. Although previous experimental studies report equal likelihood for the β- and γNT to interact with PIP2, in our simulations, only γNT underwent structural transformation in the presence of PIP2 (Fig. 1; compare Figs. 2 and 4). This difference could mean that additional factors are required for the βNT to interact with PIP2 at the plasma membrane. For instance, a recent study reported that the βNT is palmitoylated at Cys43 (43). Such modifications might place the N terminus of the β-subunit proximal to the plasma membrane. In our simulations, the N termini are not subject to any restraints, and hence the consequences of such post-translational modifications are not observed. The observation that the effect of palmitoylation of β-ENaC on channel gating is more profound compared with that of γ-ENaC further supports the notion that palmitoylation and PIP2 sensing are probably independent events in case of the γ-subunit (43). Thus, our findings support a mechanism in which AP sensing favors a distinct structural conformation of the γNT that in turn primes ENaC extracellular domains to proteolytic processing.
The proteolytic activation state of ENaC under a given condition is revealed by the acute increase in INa induced by treatment with a soluble protease. The ratio of stimulated to basal current indicates the initial extent of ENaC proteolysis and, within limits, reflects the properties of ENaC resident at the cell surface during the period of recording. Further, the rate of proteolytic stimulation of ENaC is influenced by the accessibility/conformation of remaining cleavage sites. In this study, we calculated the time constant of activation to quantify the allosteric control exerted over extracellular ENaC proteolytic stimulation by dynamic structural transitions of the NT in response to cytosolic signaling. We reasoned that if PIP2 binding and ubiquitination of the NT competed for the same lysines, the two signaling processes would have contrasting effects on ENaC proteolytic stimulation. Therefore, we measured the time constant of ENaC activation upon cleavage by chymotrypsin under the following conditions: βNT-K/Q, γNT-K/Q, γNT-K/R, and β,γNT-K/Q expressed in the presence of complementing WT subunits. Because expression of α,β,γNT-K/Q resulted in a significant decrease in rate (greater τ) of ENaC activation in oocytes, we inferred that elimination of charge with these substitutions disallowed a condition favorable for exposure of ENaC cleavage sites (Fig. 3D). Our molecular dynamics simulations of γNT-K/Q,R peptides in the presence and absence of PIP2 support this notion. Although the stability and predicted secondary structure content of γNT-K/Q were unaffected in the presence and absence of PIP2, we observed increased stability as well as secondary structure content of γNT-K/R (comparable with WT γNT) in the presence of PIP2 (Fig. 8). Thus, preserving charge on the γNT is critical for enhancing its secondary structural propensity in the presence of PIP2.
FIGURE 8.
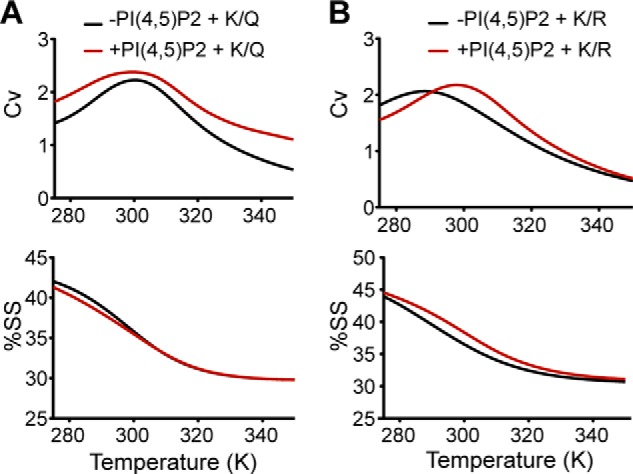
Positive charge in the γNT is required for stability and secondary structure in the presence of PIP2. Mutation of γNT K-cluster to glutamines does not show significant difference in stability or secondary structure in the presence or absence of PIP2 (A), whereas mutation to arginines increases stability and secondary structure content in the presence of PIP2 compared with in its absence (B) (compare with Fig. 1). SS, secondary structure.
The rate of ENaC activation by proteases is slowed when the K-cluster in the γNT is mutated to glutamines (Fig. 3D) or deleted (Fig. 5D), but not when its basic charge is preserved by K/R mutagenesis (Fig. 5A). Thus, through its positive charge, the γNT K-cluster mediates accessibility of extracellular cleavage sites. Furthermore, we found that P2Y2-R inhibition of ENaC proteolysis utilized basic charges at the residues occupied by the γNT K-cluster (Fig. 6). Because our simulations predict that basic residues at these positions mediate a structural transition in the γNT upon PIP2 binding, we conclude that PIP2 sensing at the γ-ENaC NT is a signal that increases the accessibility of extracellular ENaC cleavage sites. Because we also found that the γNT K-cluster contributed to the regulation of ENaC proteolysis under ubiquitinating conditions, we propose that ubiquitination of γ-ENaC inhibits ENaC proteolysis by eliminating the positive effect of PIP2 binding on cleavage site accessibility.
Recent studies have concluded that the extracellular domain might undergo allosteric structural transitions upon proteolysis (36, 37, 44, 45). However, the identities of amino acids involved in transmission of a signal responsible for such structural changes upon intracellular stimuli have yet to be mapped. Recent structural studies using crystallography and electron microscopy revealed that ATP binding in P2X receptors triggers conformational changes in the extracellular domain resulting in receptor activation and initiation of downstream signaling (46, 47). Considering that ENaC shares homology with P2X receptors, structural similarities in activation mechanisms could be drawn between the two families. However, the structure of ENaC remains to be solved. The result of our study is the identification of a putative path involving residues in γENaC that play a crucial role in signal propagation from the intracellular N termini to the extracellular protease cleavage sites. Consequently, the concept has emerged of such putative transmembrane paths for propagating regulatory signals in ENaC and related channels and receptors. Using this method and based on our homology model of rat γENaC, we identified Tyr370 as a potential hot spot in the putative allosteric pathway connecting the intracellular N termini with the extracellular protease cleavage sites. We found that mutagenesis of this site results in impaired cleavage and hence activation of ENaC (Fig. 6). Furthermore, substitution of this residue partially reverts the impaired proteolytic stimulation caused by P2Y2-R-mediated depletion of PIP2 levels (Fig. 7E). In conclusion, our results support a model where PIP2 sensing by the K-clusters in the N terminus of γENaC results in a structural transformation favoring formation of secondary structural elements (Fig. 9). Such structural changes trigger an allosteric response that is propagated via Tyr370 in γ-ENaC, culminating in a conformational change in the extracellular domain that promotes the proteolytic processing required for robust channel activation.
FIGURE 9.

Proposed model for allosteric activation of ENaC. ENaC subunits processed in the Golgi are cotrafficked with surface-active proteases to the plasma membrane, where they are further activated by membrane-bound and soluble proteases (bottom panel). Proteolytic activation of ENaC is reviewed in detail in Ref. 4. The prevailing model of allosteric activation of ENaC states that the intracellular N termini must be in a deubiquitinated form for the extracellular proteases to robustly activate the channel. We propose that in addition to the ubiquitination status of the N termini, a positive charge in the extreme N terminus of the γ-subunit is a prerequisite for allosteric signal propagation to the extracellular domains. Preserving the positive charge enhances the propensity of the γNT to form a structured, compact form that acts as a sensor for AP levels in the inner leaflet of the plasma membrane, and such sensing is a critical step in allosteric activation by proteolytic cleavage of the extracellular domain.
Acknowledgments
We thank Dr. Feng Ding and Dr. Srinivas Ramachandran for insightful discussions.
This work was supported, in whole or in part, by National Institutes of Health Program Project Grant P01 HL 110873-01. This work was also supported by National Science Foundation Career Grant 0846140 (to J. K.).
P. Kota, G. Buchner, H. Chakraborty, Y. L. Dang, H. He, G. J. M. Garcia, J. Kubelka, M. Gentzsch, M. J. Stutts, and N. V. Dokholyan, unpublished observations.
P. Kota, A. A. Aleksandrov, L. He, J. R. Riordan, and N. V. Dokholyan, manuscript in preparation.
- ENaC
- epithelial sodium channel
- AP
- anionic phospholipids
- cASIC1
- chicken acid-sensing ion channel 1
- DMD
- discrete molecular dynamics
- DOPC
- 1,2-dioleoyl-3-sn-phosphatidylcholine
- DOPG
- 1,2-dioleoyl-3-sn-phosphatidylglycerol
- DUB
- deubiquitinating enzyme
- K-cluster
- cluster of lysine residues at the N terminus of ENaC subunits
- MARCKS
- myristoylated alanine-rich C kinase substrate
- MTSET
- 2-(trimethylammonium)ethyl methanethiosulfonate
- NT
- N-terminal tail
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- Po
- open probability
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl.
REFERENCES
- 1. Garty H., Palmer L. G. (1997) Epithelial sodium channels: function, structure, and regulation. Physiol. Rev. 77, 359–396 [DOI] [PubMed] [Google Scholar]
- 2. Harris M., Garcia-Caballero A., Stutts M. J., Firsov D., Rossier B. C. (2008) Preferential assembly of epithelial sodium channel (ENaC) subunits in Xenopus oocytes: role of furin-mediated endogenous proteolysis. J. Biol. Chem. 283, 7455–7463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Snyder P. M., McDonald F. J., Stokes J. B., Welsh M. J. (1994) Membrane topology of the amiloride-sensitive epithelial sodium channel. J. Biol. Chem. 269, 24379–24383 [PubMed] [Google Scholar]
- 4. Rossier B. C., Stutts M. J. (2009) Activation of the epithelial sodium channel (ENaC) by serine proteases. Annu. Rev. Physiol. 71, 361–379 [DOI] [PubMed] [Google Scholar]
- 5. Diakov A., Bera K., Mokrushina M., Krueger B., Korbmacher C. (2008) Cleavage in the γ-subunit of the epithelial sodium channel (ENaC) plays an important role in the proteolytic activation of near-silent channels. J. Physiol. 586, 4587–4608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anantharam A., Tian Y., Palmer L. G. (2006) Open probability of the epithelial sodium channel is regulated by intracellular sodium. J. Physiol. 574, 333–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tong Q., Gamper N., Medina J. L., Shapiro M. S., Stockand J. D. (2004) Direct activation of the epithelial Na+ channel by phosphatidylinositol 3,4,5-trisphosphate and phosphatidylinositol 3,4-bisphosphate produced by phosphoinositide 3-OH kinase. J. Biol. Chem. 279, 22654–22663 [DOI] [PubMed] [Google Scholar]
- 8. Gründer S., Firsov D., Chang S. S., Jaeger N. F., Gautschi I., Schild L., Lifton R. P., Rossier B. C. (1997) A mutation causing pseudohypoaldosteronism type 1 identifies a conserved glycine that is involved in the gating of the epithelial sodium channel. EMBO J. 16, 899–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Staub O., Gautschi I., Ishikawa T., Breitschopf K., Ciechanover A., Schild L., Rotin D. (1997) Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 16, 6325–6336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou R., Patel S. V., Snyder P. M. (2007) Nedd4-2 catalyzes ubiquitination and degradation of cell surface ENaC. J. Biol. Chem. 282, 20207–20212 [DOI] [PubMed] [Google Scholar]
- 11. Knight K. K., Wentzlaff D. M., Snyder P. M. (2008) Intracellular sodium regulates proteolytic activation of the epithelial sodium channel. J. Biol. Chem. 283, 27477–27482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ruffieux-Daidié D., Poirot O., Boulkroun S., Verrey F., Kellenberger S., Staub O. (2008) Deubiquitylation regulates activation and proteolytic cleavage of ENaC. J. Am. Soc. Nephrol. 19, 2170–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ruffieux-Daidié D., Staub O. (2011) Intracellular ubiquitylation of the epithelial Na+ channel controls extracellular proteolytic channel activation via conformational change. J. Biol. Chem. 286, 2416–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ma H. P., Eaton D. C. (2005) Acute regulation of epithelial sodium channel by anionic phospholipids. J. Am. Soc. Nephrol. 16, 3182–3187 [DOI] [PubMed] [Google Scholar]
- 15. Pochynyuk O., Tong Q., Staruschenko A., Stockand J. D. (2007) Binding and direct activation of the epithelial Na+ channel (ENaC) by phosphatidylinositides. J. Physiol. 580, 365–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kunzelmann K., Bachhuber T., Regeer R., Markovich D., Sun J., Schreiber R. (2005) Purinergic inhibition of the epithelial Na+ transport via hydrolysis of PIP2. FASEB J. 19, 142–143 [DOI] [PubMed] [Google Scholar]
- 17. Ma H. P., Saxena S., Warnock D. G. (2002) Anionic phospholipids regulate native and expressed epithelial sodium channel (ENaC). J. Biol. Chem. 277, 7641–7644 [DOI] [PubMed] [Google Scholar]
- 18. Tong Q., Stockand J. D. (2005) Receptor tyrosine kinases mediate epithelial Na+ channel inhibition by epidermal growth factor. Am. J. Physiol. Renal Physiol. 288, F150–F161 [DOI] [PubMed] [Google Scholar]
- 19. Yue G., Malik B., Yue G., Eaton D. C. (2002) Phosphatidylinositol 4,5-bisphosphate (PIP2) stimulates epithelial sodium channel activity in A6 cells. J. Biol. Chem. 277, 11965–11969 [DOI] [PubMed] [Google Scholar]
- 20. Pochynyuk O., Staruschenko A., Tong Q., Medina J., Stockand J. D. (2005) Identification of a functional phosphatidylinositol 3,4,5-trisphosphate binding site in the epithelial Na+ channel. J. Biol. Chem. 280, 37565–37571 [DOI] [PubMed] [Google Scholar]
- 21. Pochynyuk O., Bugaj V., Vandewalle A., Stockand J. D. (2008) Purinergic control of apical plasma membrane PI(4,5)P2 levels sets ENaC activity in principal cells. Am. J. Physiol. Renal Physiol. 294, F38–F46 [DOI] [PubMed] [Google Scholar]
- 22. Ding F., Tsao D., Nie H., Dokholyan N. V. (2008) Ab initio folding of proteins with all-atom discrete molecular dynamics. Structure 16, 1010–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dokholyan N. V., Buldyrev S. V., Stanley H. E., Shakhnovich E. I. (1998) Discrete molecular dynamics studies of the folding of a protein-like model. Fold Des. 3, 577–587 [DOI] [PubMed] [Google Scholar]
- 24. Kumar S., Bouzida D., Swendsen R. H., Kollman P. A., Rosenberg J. M. (1992) The weighted histogram analysis method for free-energy calculations on biomolecules: I. the method. J. Comput. Chem. 13, 1011–1021 [Google Scholar]
- 25. Jasti J., Furukawa H., Gonzales E. B., Gouaux E. (2007) Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature 449, 316–323 [DOI] [PubMed] [Google Scholar]
- 26. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 27. Ding F., Dokholyan N. V. (2006) Emergence of protein fold families through rational design. PLoS Comput. Biol. 2, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ramachandran S., Kota P., Ding F., Dokholyan N. V. (2011) Automated minimization of steric clashes in protein structures. Proteins 79, 261–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chakraborty H., Lentz B. R. (2012) A simple method for correction of circular dichroism spectra obtained from membrane-containing samples. Biochemistry 51, 1005–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kota P., García-Caballero A., Dang H., Gentzsch M., Stutts M. J., Dokholyan N. V. (2012) Energetic and structural basis for activation of the epithelial sodium channel by matriptase. Biochemistry 51, 3460–3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang Z. R., Chou C. F., Wang J., Liang Y. Y., Ma H. P. (2010) Anionic phospholipids differentially regulate the epithelial sodium channel (ENaC) by interacting with α, β, and γ ENaC subunits. Pflugers Arch. 459, 377–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haerteis S., Krappitz M., Diakov A., Krappitz A., Rauh R., Korbmacher C. (2012) Plasmin and chymotrypsin have distinct preferences for channel activating cleavage sites in the γ subunit of the human epithelial sodium channel. J. Gen. Physiol. 140, 375–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harris M., Firsov D., Vuagniaux G., Stutts M. J., Rossier B. C. (2007) A novel neutrophil elastase inhibitor prevents elastase activation and surface cleavage of the epithelial sodium channel expressed in Xenopus laevis oocytes. J. Biol. Chem. 282, 58–64 [DOI] [PubMed] [Google Scholar]
- 34. Aleksandrov A. A., Kota P., Cui L., Jensen T., Alekseev A. E., Reyes S., He L., Gentzsch M., Aleksandrov L. A., Dokholyan N. V., Riordan J. R. (2012) Allosteric modulation balances thermodynamic stability and restores function of DeltaF508 CFTR. J. Mol. Biol. 419, 41–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li T., Yang Y., Canessa C. M. (2009) Interaction of the aromatics Tyr-72/Trp-288 in the interface of the extracellular and transmembrane domains is essential for proton gating of acid-sensing ion channels. J. Biol. Chem. 284, 4689–4694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shi S., Ghosh D. D., Okumura S., Carattino M. D., Kashlan O. B., Sheng S., Kleyman T. R. (2011) Base of the thumb domain modulates epithelial sodium channel gating. J. Biol. Chem. 286, 14753–14761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shi S., Carattino M. D., Kleyman T. R. (2012) Role of the wrist domain in the response of the epithelial sodium channel to external stimuli. J. Biol. Chem. 287, 44027–44035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Passero C. J., Mueller G. M., Myerburg M. M., Carattino M. D., Hughey R. P., Kleyman T. R. (2012) TMPRSS4-dependent activation of the epithelial sodium channel requires cleavage of the γ-subunit distal to the furin cleavage site. Am. J. Physiol. Renal Physiol. 302, F1–F8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Goulet C. C., Volk K. A., Adams C. M., Prince L. S., Stokes J. B., Snyder P. M. (1998) Inhibition of the epithelial Na+ channel by interaction of Nedd4 with a PY motif deleted in Liddle's syndrome. J. Biol. Chem. 273, 30012–30017 [DOI] [PubMed] [Google Scholar]
- 40. Suh B. C., Hille B. (2008) PIP2 is a necessary cofactor for ion channel function: how and why? Annu. Rev. Biophys. 37, 175–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cole C., Barber J. D., Barton G. J. (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36, W197–W201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yamauchi E., Nakatsu T., Matsubara M., Kato H., Taniguchi H. (2003) Crystal structure of a MARCKS peptide containing the calmodulin-binding domain in complex with Ca2+-calmodulin. Nat. Struct. Biol. 10, 226–231 [DOI] [PubMed] [Google Scholar]
- 43. Mueller G. M., Maarouf A. B., Kinlough C. L., Sheng N., Kashlan O. B., Okumura S., Luthy S., Kleyman T. R., Hughey R. P. (2010) Cys palmitoylation of the β subunit modulates gating of the epithelial sodium channel. J. Biol. Chem. 285, 30453–30462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kashlan O. B., Adelman J. L., Okumura S., Blobner B. M., Zuzek Z., Hughey R. P., Kleyman T. R., Grabe M. (2011) Constraint-based, homology model of the extracellular domain of the epithelial Na+ channel α subunit reveals a mechanism of channel activation by proteases. J. Biol. Chem. 286, 649–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Winarski K. L., Sheng N., Chen J., Kleyman T. R., Sheng S. (2010) Extracellular allosteric regulatory subdomain within the γ subunit of the epithelial Na+ channel. J. Biol. Chem. 285, 26088–26096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jiang R., Taly A., Lemoine D., Martz A., Cunrath O., Grutter T. (2012) Tightening of the ATP-binding sites induces the opening of P2X receptor channels. EMBO J. 31, 2134–2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roberts J. A., Allsopp R. C., El Ajouz S., Vial C., Schmid R., Young M. T., Evans R. J. (2012) Agonist binding evokes extensive conformational changes in the extracellular domain of the ATP-gated human P2X1 receptor ion channel. Proc. Natl. Acad. Sci. U.S.A. 109, 4663–4667 [DOI] [PMC free article] [PubMed] [Google Scholar]