

Background: E3 ubiquitin ligases facilitate destruction of other proteins.
Results: In frog egg extract, the DNA repair factor thymine DNA glycosylase (TDG) was destroyed during DNA replication and repair, dependent on the E3 ubiquitin ligase CRL4Cdt2.
Conclusion: TDG is a novel target of CRL4Cdt2.
Significance: We identified a novel form of TDG regulation that informs how cells regulate S phase and epigenetic inheritance.
Keywords: Base Excision Repair (BER), DNA Methylation, DNA Replication, E3 Ubiquitin Ligase, Xenopus, CRL4-Cdt2, Proteolysis, Thymine DNA Glycosylase (TDG)
Abstract
The E3 ubiquitin ligase CRL4Cdt2 targets proteins for destruction in S phase and after DNA damage by coupling ubiquitylation to DNA-bound proliferating cell nuclear antigen (PCNA). Coupling to PCNA involves a PCNA-interacting peptide (PIP) degron motif in the substrate that recruits CRL4Cdt2 while binding to PCNA. In vertebrates, CRL4Cdt2 promotes degradation of proteins whose presence in S phase is deleterious, including Cdt1, Set8, and p21. Here, we show that CRL4Cdt2 targets thymine DNA glycosylase (TDG), a base excision repair enzyme that is involved in DNA demethylation. TDG contains a conserved and nearly perfect match to the PIP degron consensus. TDG is ubiquitylated and destroyed in a PCNA-, Cdt2-, and PIP degron-dependent manner during DNA repair in Xenopus egg extract. The protein can also be destroyed during DNA replication in this system. During Xenopus development, TDG first accumulates during gastrulation, and its expression is down-regulated by CRL4Cdt2. Our results expand the group of vertebrate CRL4Cdt2 substrates to include a bona fide DNA repair enzyme.
Introduction
The E3 ubiquitin ligase CRL4Cdt2 is a master regulator of S phase progression whose activity is coupled to DNA replication or repair via the processivity factor PCNA3 (1, 2). Fewer than 10 CRL4Cdt2 substrates are known, but all have a role in DNA replication or cell cycle regulation (2). The best-characterized substrate is the replication licensing factor Cdt1 (3–6), which is required to recruit the MCM2-7 helicase to origins of replication in the G1 phase of the cell cycle. After cells enter S phase, CRL4Cdt2 marks Cdt1 for destruction, thereby preventing reinitiation from origins that have already fired. CRL4Cdt2 also targets Cdt1 for destruction in response to DNA damage, although the reasons are unclear. In vertebrates, CRL4Cdt2 also targets the histone methyltransferase Set8 (7, 8) and the cyclin-dependent kinase inhibitor p21 (9, 10). In both cases, destruction is thought to inhibit licensing in S phase. The destruction of p21 after DNA damage grants translesion DNA polymerases access to PCNA (11, 12). The smallest subunit of DNA polymerase δ was recently identified as the newest CRL4Cdt2 substrate in mammalian cells (13, 14). Zhang et al. (13) proposed that destruction converts DNA polymerase δ to a three-subunit enzyme, which is less error-prone, whereas Terai et al. (14) suggested that destruction is necessary for fork stalling following DNA damage. Other substrates of CRL4Cdt2 include the transcription factor E2f1 in flies (15) (to turn off the G1 expression program in S phase), the ribonucleotide reductase inhibitor Spd1 in fission yeast (16, 17) (to up-regulate dNTP synthesis in S phase and after DNA damage), and the translesion DNA polymerase POLH-1 in worms (18) (to avoid mutagenic translesion DNA synthesis).
For most CRL4Cdt2 substrates, it has been shown that ubiquitylation requires a PCNA-interacting peptide (PIP) degron. The degron comprises a canonical, 8-amino acid PIP box motif (Fig. 1A, purple residues) (2, 4, 19, 20), which confers binding to PCNA (21). Additionally, most CRL4Cdt2 substrates contain threonine and aspartate residues at positions 5 and 6 of the PIP box (“TD motif”), which confer high affinity binding of the substrate to PCNA. The degron also contains at least one basic residue 4 amino acids downstream of the PIP box (“B+4” residue), which is required for efficient recruitment of CRL4Cdt2 to the PCNA-substrate complex (22, 23). CRL4Cdt2 activity also requires residues on PCNA that probably make direct contact with Cdt2 (22, 24). Because canonical PCNA-binding proteins such as replicative DNA polymerases lack the TD motif and/or B+4 residues, they are not destroyed.
FIGURE 1.
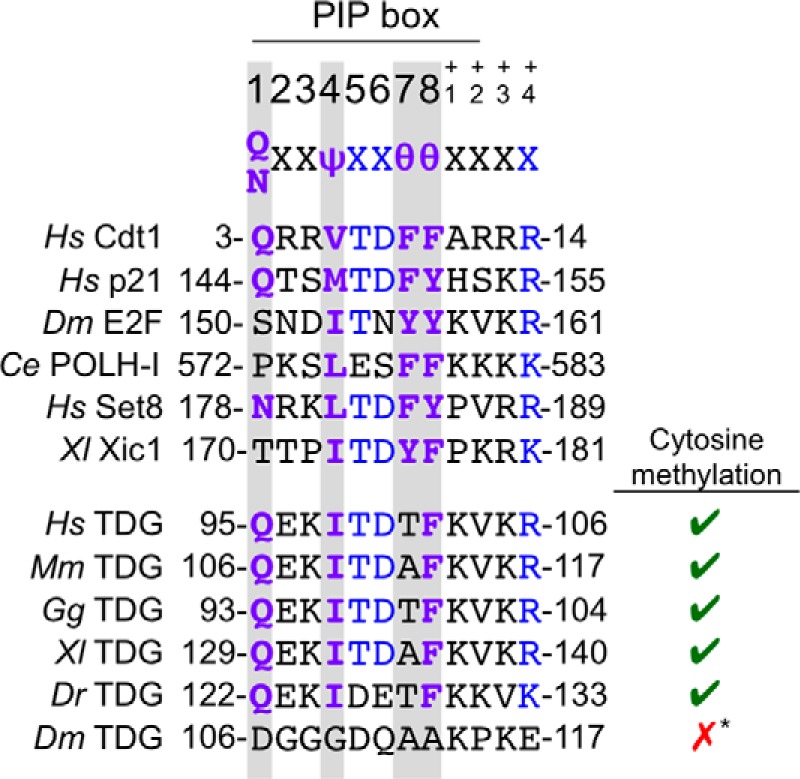
TDG contains a putative PIP degron. The PIP degrons of confirmed CRL4Cdt2 substrates are shown above the putative PIP degron of TDG in human, mouse, chicken, frog, and zebrafish. TDG of lower eukaryotes, including Drosophila, does not contain a PIP box in the same region of the protein or elsewhere. Ψ, any hydrophobic residue (I/L/V/M); θ, any aromatic residue (Y/F/W). Residues shown in purple are part of the conserved PIP box, and residues shown in blue additionally contribute to the “PIP degron.” The right-hand column indicates whether each animal regulates gene expression via cytosine methylation. *, although there is some evidence of cytosine methylation in Drosophila, recent whole-genome sequencing studies suggest that the genome remains primarily unmethylated and that there are no conserved patterns of DNA methylation (65). Additionally, Drosophila lack homologs of maintenance and de novo DNA methyltransferases that are typically present in organisms that use cytosine methylation for epigenetic regulation.
TDG is a glycosylase that acts in the first step of base excision repair (BER) to remove thymine, uracil, and certain modified cytosine residues when they are paired with guanine, generating an abasic (AP) site. Subsequent processing by AP endonuclease and other BER enzymes restores the AP site to cytosine. TDG is product-inhibited due to irreversible binding to AP sites, but TDG SUMOylation decreases its affinity for DNA, facilitating its removal from AP sites (25–27). Interestingly, TDG is eliminated from S phase cells by ubiquitin-mediated proteolysis (28). The S phase destruction of TDG might prevent the collision of DNA replication forks with TDG-AP site complexes that have not yet been disrupted by TDG SUMOylation (28). In possible agreement with this model, overexpression of TDG causes an accumulation of cells in S phase (28).
Recent evidence has shown that TDG functions in DNA demethylation and thus participates in the epigenetic regulation of gene expression (29). In animals, Tet proteins serially oxidize 5-methylcytosine to 5-hydroxylmethylcytosine, 5-formylcytosine, and 5-carboxymethylcytosine. TDG removes 5-formylcytosine and 5-carboxymethylcytosine from DNA via the conventional BER pathway, leading to cytosine demethylation (30–39). Additionally, TDG and other BER proteins cooperate with AID and APOBEC1, deaminases that process 5-methylcytosine to thymine, which TDG and BER convert to cytosine (40–43). Mice lacking TDG die at embryonic day 11.5, probably due to a failure to establish and maintain epigenetic gene expression programs during cell and tissue differentiation (42, 44).
Here, we show that TDG is a CRL4Cdt2 substrate with a highly conserved PIP degron. When damaged DNA was incubated in Xenopus egg extract, TDG was targeted for destruction dependent on CRL4Cdt2, PCNA, and the TDG PIP degron. Interestingly, the PIP degron of TDG deviates from the consensus sequence by one amino acid, and changing the degron to the consensus dramatically enhanced the efficiency of TDG destruction. The addition of non-degradable TDG to egg extract did not affect S phase progression, but this might be due to an inhibitor of TDG activity in the extract. During frog development, TDG protein accumulated late in gastrulation, and its abundance was down-regulated by CRL4Cdt2, probably as a result of S phase destruction. However, expression of non-degradable TDG in frog embryos had no detectable effects on S phase progression or development. Our results identify TDG as the first bona fide DNA repair protein targeted by CRL4Cdt2 and suggest that TDG might be inhibited in S phase by multiple redundant mechanisms.
EXPERIMENTAL PROCEDURES
Xenopus Egg Extract
High-speed supernatant (HSS) of Xenopus egg extract and nucleoplasmic extract (NPE) were prepared as described previously (45). Before use, NPE was diluted by 40–60% with egg lysis buffer (ELB; 250 mm sucrose, 2.5 mm MgCl2, 50 mm KCl, 10 mm HEPES, pH 7.7). All extracts were supplemented with an energy regeneration mix (2 mm ATP, 20 mm phosphocreatine, and 5 μg/ml creatine kinase). HSS was also supplemented with nocodazole (3–5 μg/ml), and diluted NPE was supplemented with an additional 10 mm DTT. All experiments in Xenopus egg extract were performed in a 2:1 mixture of diluted NPE to HSS, unless otherwise noted.
For damage-dependent TDG destruction assays, NPE and HSS were mixed together prior to the start of the experiment. After the addition of recombinant TDG (see below), methyl methanesulfonate (MMS)-damaged DNA was added to trigger DNA repair and CRL4Cdt2 function. MMS-damaged plasmid or linear DNA was generated as described previously (46). Unless otherwise indicated, MMS-damaged plasmid was used at a final concentration of 10 ng/μl in egg extract to trigger TDG destruction. For all destruction assays, TDG was added to egg extract at a concentration of 1 ng/μl (20 nm). Linear, MMS-damaged DNA was coupled to beads, and beads were recovered as described previously (22). In experiments examining DNA-bound TDG, methyl ubiquitin (2 mg/ml; Boston Biochem) was included to allow monoubiquitylation on multiple sites while blocking polyubiquitin chain formation and subsequent proteolysis. Where indicated, CRL4Cdt2 function was inhibited with a previously described p21 peptide (47) at a final concentration of 200 μm. MG132 (1 mm; Boston Biochem) was used where indicated to inhibit the proteasome.
For DNA replication assays and replication-dependent TDG destruction assays, DNA replication in egg extract (HSS/NPE) was monitored as described previously (45). Briefly, plasmid DNA was first incubated in HSS, which facilitates DNA licensing, and then diluted NPE (40–60% with ELB) was added to trigger replication initiation. DNA replication was measured via the incorporation of radiolabeled dATP into high molecular weight DNA, as measured by gel electrophoresis and autoradiography (45). TDG was added with the diluted NPE at the start of replication. Where indicated, licensing was inhibited with 400 nm geminin (48) added to HSS.
Embryo Microinjections
Eggs were fertilized according to published protocols (49) and stored in 0.1× MMR containing 50 μg/ml gentamycin. Embryos were staged according to the Nieuwkoop and Faber anatomical stages (50, 51). 20 pg of TDG mRNA (10 nl at 2 ng/μl in water) was microinjected into each cell of dejellied stage 2 embryos in 0.1× MMR + 5% Ficoll containing gentamycin (50 μg/ml) at 18 °C. Embryos were allowed to heal in 0.1× MMR + 5% Ficoll containing gentamycin for 1–2 h at 18 °C and then stored in 0.1× MMR containing gentamycin at 14 °C until they reached N-F stage 7, after which development was monitored at 23 °C.
Frozen embryos were lysed via homogenization with a pipette tip and light vortexing in embryonic lysis buffer (250 mm sucrose, 1% Nonidet P-40, 10 mm EDTA, 25 mm HEPES, pH 7.5, supplemented with a Roche Applied Science Complete protease inhibitor tablet; 15 μl/embryo). Yolk proteins were removed via centrifugation at 10,000 × g, 4 °C for 10 min. The soluble fraction was recleared of yolk proteins with a second spin. In a typical experiment, 30–40 embryos were injected with WT or ΔPIP mRNA, and at each developmental time point, two embryos were frozen to prepare extract for blotting or mRNA extraction. By the end of the experiment, there were at least five embryos left. In five repetitions of the experiment, 98 of 108 WT-injected, 137 of 148 ΔPIP-injected, and 135 of 149 uninjected embryos showed no phenotype through at least stage 14.
Quantitative PCR
RNA was purified from Xenopus embryos using the RNAqueous® total RNA isolation kit (Invitrogen), following the manufacturer's instructions. Five embryos were pooled per time point, lysed in 300 μl of lysis/binding solution, and cleared by centrifugation at 21,130 × g at 4 °C for 30 min prior to RNA purification. cDNA were synthesized using the iScriptTM cDNA synthesis kit (Bio-Rad), and quantitative PCR was performed using the iTaqTM Universal SYBR Green Supermix (Bio-Rad) on a 7900HT fast real-time PCR system (Applied Biosystems), following the manufacturer's instructions. Primers for TDG were designed and validated (5′-GTTACAATTCTGCCCTTGGA-3′, 5′-TGCCCATTAAACTCTGCATT-3′). All samples were normalized to Hprt1 (5′-TGGGAGGTCACCATATTG-3′, 5′-CTTGTCACTGTTGCGGTT-3′).
Plasmid Construction, Mutagenesis, and Protein Purification
The TDG gene was amplified from cDNA generated from unfertilized eggs using primers 5′-GCTAAGGATCCATGGAGGCCCAGGACCCAAGC-3′ and 5′-TTTTTCTCGAGTCAAGCGTTGCTGCCTCCTTGC-3′ (Operon/Eurofins Genomics). The TDG gene was inserted into a modified pET28 vector that contains a Prescission Protease (GE Healthcare) cleavage site for expression in bacteria or into a pCS2+ vector for mRNA production at BamHI and XhoI (New England Biolabs) sites in the respective vector. TDG point mutants were generated with a QuikChange II site-directed mutagenesis kit (Agilent).
To prepare mRNA for microinjections, pCS2+TDG plasmid was linearized with a NotI (New England Biolabs) overnight digest and purified using a PCR purification kit (Qiagen). mRNA was prepared using the mMessage mMachine Sp6 kit (Ambion) and purified using an RNeasy minikit (Qiagen). Purified mRNA was ethanol-precipitated and diluted in RNase-free water.
To obtain native, functional TDG, expression of His-tagged TDG was induced in Arctic Express cells (Agilent) with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside for 24 h at 15 °C, and TDG was purified in batch. Cells were lysed via sonication in lysis buffer (750 mm NaCl, 20% glycerol, 10 mm β-mercaptoethanol, 10 mm imidazole, 50 mm sodium phosphate, pH 8.0) containing protease inhibitors (0.5 mm PMSF (Amresco), 1 mm benzamidine (Sigma), 10 μg/ml aprotinin, and 10 μg/ml leupeptin) and cleared via centrifugation at 30,000 × g. Soluble His-tagged TDG was bound to nickel-nitrilotriacetic acid beads (Qiagen) for 1 h and washed twice with lysis buffer containing 20 mm imidazole and once with lysis buffer containing 50 mm imidazole (aprotinin and leupeptin were omitted from these wash buffers). To separate the Cpn60/10 chaperonin protein from TDG (52), an additional 2-h wash was performed in an ATP-based chaperonin removal buffer (375 mm NaCl, 20% glycerol, 10 mm β-mercaptoethanol, 10 mm imidazole, 10 mm MgCl2, 150 mm KCl, 5 mm ATP, and 50 mm sodium phosphate, pH 7.6) modified from Joseph et al. (52). Finally, the beads were washed again twice with lysis buffer containing 20 mm imidazole and once with lysis buffer containing 50 mm imidazole before eluting in lysis buffer containing 400 mm imidazole. 10 units of Prescission protease was added to the native protein eluate, which was then dialyzed overnight in 50 mm NaCl, 50 mm Tris, 10% glycerol, 10 mm β-mercaptoethanol, and 1 mm EDTA, pH 8. TDG was used at a final concentration of 1 ng/μl in extract for destruction assays or at 15 ng/μl to measure its activity in extract.
To obtain TDG for antibody production, TDG was expressed in BL21 (DE3) cells via induction with 0.25 mm isopropyl 1-thio-β-d-galactopyranoside at 37 °C for 3 h. Cells were lysed in 7 m urea, 10 m sodium phosphate, and 10 mm Tris, pH 8.0, cleared via centrifugation at 30,000 × g, and bound to nickel-nitrilotriacetic acid beads for 1 h. Beads were washed with 8 m urea, 100 mm sodium phosphate, 10 mm Tris, pH 6.3. His-TDG was eluted directly into 2× SDS-PAGE loading buffer (20% glycerol, 6.1% SDS, 125 mm Tris-HCl, pH 6.8, 0.01% bromphenol blue) and separated on an SDS-polyacrylamide gel. Protein was stained with Gelcode Blue (Thermo Scientific) and electroeluted into SDS-PAGE running buffer (0.1% SDS, 250 mm glycine, 25 mm Tris, pH 8.3). The resulting purified TDG was >99% pure as measured by Coomassie staining.
Immunological Methods
Denatured His-TDG was sent to Pocono Rabbit Farm and Laboratory (Canadensis, PA) for antibody production in two rabbits. Serum from one rabbit (number 197) was used for all immunoblotting at a concentration of 1:5000–1:2500. Serum from the second rabbit (number 198) was used for immunoprecipitation. The Msh2 antiserum was raised against N-terminally His-tagged full-length Xenopus Msh2 protein, expressed in and purified from E. coli, and the Msh6 serum was raised against a peptide corresponding to the C-terminal 17 amino acids of xMsh6 (CNGSPEGLALHKRLKLLQ).
Antibodies to Xenopus Cdt1 (53), Cdt2 (3), Orc2 (54), MCM7 (55), Rcc1 (56), and replication protein A (55) were described previously. Commercial antibodies were used to blot for PCNA (Santa Cruz Biotechnology, Inc.). Where indicated, band density was quantified using ImageJ software and normalizing to the density of an appropriate loading control.
Immunodepletions were performed using a 3:1 ratio of serum to protein A-Sepharose Fastflow resin (Amersham Biosciences). Antibody-bound resin was used at a 1:5 ratio to egg extract. Immunodepletions were performed separately in HSS and diluted NPE (40–60% in ELB) before the two extracts were combined or otherwise used in experiments. To deplete Cdt2, either one or two 1-h rounds were performed, both leading to the same results shown. To deplete MutSα, Msh2 and Msh6 antiserum was first combined in a 1:1 ratio and then treated as a single serum to deplete HSS in two 1-h rounds.
To immunoprecipitate TDG from extract, 1 μl crude serum (rabbit number 198) was added to 10 μl of Protein A-Sepharose Fastflow resin. The beads were washed five times with PBS and two times with ELB containing 500 mm NaCl. 13 μl of HSS/NPE extract was diluted 2-fold in ELB and incubated with the beads for 1 h at 4 °C. Beads were recovered and washed five times in ELB containing 0.1% Triton X-100 and eluted directly into 2× sample buffer.
TDG Activity Assay
Base release assays were used to measure TDG activity on duplex oligonucleotide templates containing a single G-T mispair (30, 36, 57). 29-mer double-stranded oligonucleotides (Operon/Eurofins Genomics) were prepared by end labeling the strand containing the base to be hydrolyzed by TDG (5′-CCGCTGAGGGATATNGAATTCCTGCAGGC-3′, where N represents C or T) with [γ-32P]ATP and annealing it to an unlabeled complementary strand (5′-GCCTGCAGGAATTCGATATCCCTCAGCGG-3′) by heating the mixture to 85 °C and allowing it to cool slowly to 23 °C. Polynucleotide kinase (New England Biolabs) was used to attach the radiolabeled phosphate in polynucleotide kinase buffer (10 mm MgCl2, 5 mm DTT, 70 mm Tris, pH 7.6). The DNA strands were annealed in the same buffer. Free label was removed with G-50 Probequant columns (GE Healthcare). 90-mer double-stranded oligonucleotides (5′-CTCCGTATTGCGAGCTCCATTGACTCGGCCCGAACTCCCTGGTGCCATAACGAATTNGCGGCGGTCAGTCGTCAACTGCTTGTGCACCGC-3′, where N represents C or T; reverse complement 5′-GCGGTGCACAAGCAGTTGACGACTGACCGCCGCGAATTCGTTATGGCACCAGGGAGTTCGGGCCGAGTCAATGGAGCTCGCAATACGGAG-3′) were prepared by the same method. All three 90-mer oligonucleotides were synthesized by Integrated DNA Technologies with a 3′ biotin modification, which did not affect TDG activity toward the substrates (data not shown).
To test TDG mutants for activity, TDG was incubated with double-stranded 29-mer in activity buffer (20 mm NaCl, 0.5 mm EDTA, 1 mm DTT, 50 mm Tris, pH 7.5, and 10 μg/μl aprotinin and leupeptin, along with trace amounts of β-mercaptoethanol and aprotinin and leupeptin contributed by the protein itself). Activity assays were incubated for 40 min at 23 °C. TDG-generated abasic sites were cleaved with the addition of 90 mm NaOH and heating to 95 °C for 3 min. Equimolar acetic acid was added to neutralize the reaction, and samples were diluted in formamide loading dye (Ambion). Samples were reheated to 75 °C and loaded immediately on a small urea-polyacrylamide gel (7 m urea, 0.8× glycerol-tolerant buffer (U.S. Biochemical Co.), 15% polyacrylamide (from a 40% Rapid Gel XL concentrate; U.S. Biochemical Co.), and ammonium persulfate and TEMED for polymerization) that had been prerun for 20 min at 200 V in 0.8× glycerol-tolerant buffer. Gels were run at 200 V until the size range of interest reached the middle of the gel. TDG activity was measured by a change in the size of the end-labeled DNA strand, as visualized by PhosphorImager analysis. Analysis of the TDG activity on the 90-mer substrate was performed by the same protocol, except that samples were reheated to 85 °C prior to gel electrophoresis, and gels were run at 30 watts.
RESULTS
Identification of a Putative PIP Degron in the TDG N Terminus
Given that TDG levels drop in S phase due to proteasome-dependent destruction (28), we looked for and found a close match to the PIP degron consensus in human TDG (Fig. 1). The putative TDG degron contains a PIP box whose only deviation from the consensus is that it lacks the first of two aromatic residues. In addition, it contains the B+4 residue and TD motif characteristic of PIP degrons (Fig. 1). The putative PIP degron is conserved in human, mouse, chicken, and frog TDG. In zebrafish, the PIP box and B+4 residue are conserved, but the TD motif is not. In flies, the PIP degron is absent. Interestingly, the PIP degron is only present in organisms that undergo robust cytosine methylation of DNA (Fig. 1).
Proteasome- and Cdt2-dependent TDG Destruction during DNA Repair in Xenopus Egg Extract
Xenopus egg extracts recapitulate CRL4Cdt2-dependent proteolysis during DNA replication and repair (3, 7, 53). To study the latter process, DNA that has been damaged with MMS is added to egg extract. Extract-mediated excision repair of this damage involves a PCNA-dependent gap filling step, which activates CRL4Cdt2-dependent destruction of endogenous and exogenous PIP degron substrates. We raised an antibody against Xenopus TDG, which revealed that the concentration of endogenous TDG in unfertilized egg extracts is very low (Fig. 2) (see below). We therefore purified Xenopus TDG (Fig. 3A) and used a base release assay (Fig. 3B) to verify that WT recombinant TDG and several mutant TDGs used throughout this study are active (Fig. 3C). When we added TDG to egg extract, within 1 min, a slow mobility form of the protein appeared (Fig. 4A; S-TDG), which we showed corresponds to SUMOylated TDG (see below). As shown in Fig. 4B, recombinant TDG was efficiently destroyed in the presence of MMS-damaged plasmid but not in the presence of undamaged plasmid. Destruction was impaired by MG132, an inhibitor of the proteasome (Fig. 4C), and by immunodepletion of Cdt2, the putative substrate receptor for CRL4Cdt2 (Fig. 4D). These results suggest that CRL4Cdt2 can cause the destruction of recombinant TDG in response to DNA damage.
FIGURE 2.
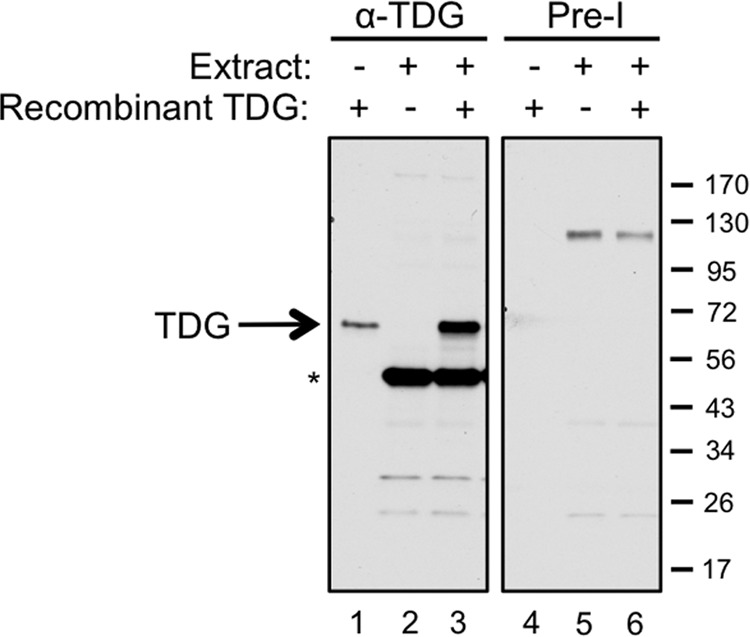
Characterization of Xenopus TDG antibody. TDG antiserum (left) or the matched preimmune serum (right) was used for Western blotting to probe 0.6 ng of recombinant TDG (lanes 1 and 4), 0.6 μl of a 2:1 mixture of NPE (diluted by 50% with ELB) plus HSS (lanes 2 and 5), and 0.6 ng of TDG combined with 0.6 μl of the NPE/HSS mixture (lanes 3 and 6). The band running at 50 kDa is a nonspecific cross-reacting band, as evidenced by the fact that it is not recognized by antiserum from a second rabbit that efficiently recognizes TDG.
FIGURE 3.

Characterization of Xenopus TDG protein. A, native TDG from E. coli was purified and analyzed by SDS-PAGE and Coomassie Blue staining. B, schematic of the base release assay used to monitor TDG activity. Double-stranded DNA (29 bp) containing a site-specific G-T mispair is 5′-radiolabeled on the DNA strand containing the thymine of the mispair. TDG activity generates an abasic site, and subsequent treatment with sodium hydroxide and heat leads to hydrolysis of the backbone at the abasic site. TDG activity is measured as the appearance of a 14-nt product. As a control, the oligonucleotide was treated with EcoRI, which cleaves adjacent to the G-T mispair in the fully complementary duplex to generate a 12-nt product. C, recombinant wild type TDG and all TDG mutants used in this work, except the catalytically inactive form (CI), are active in the base release assay diagramed in B. Detailed descriptions of these mutants can be found upon their first experimental usage in Figs. 5 and 6. The concentration of TDG in all reactions was 100 nm, and the DNA concentration was 50 nm. Strand cleavage was measured using a 15% polyacrylamide gel.
FIGURE 4.
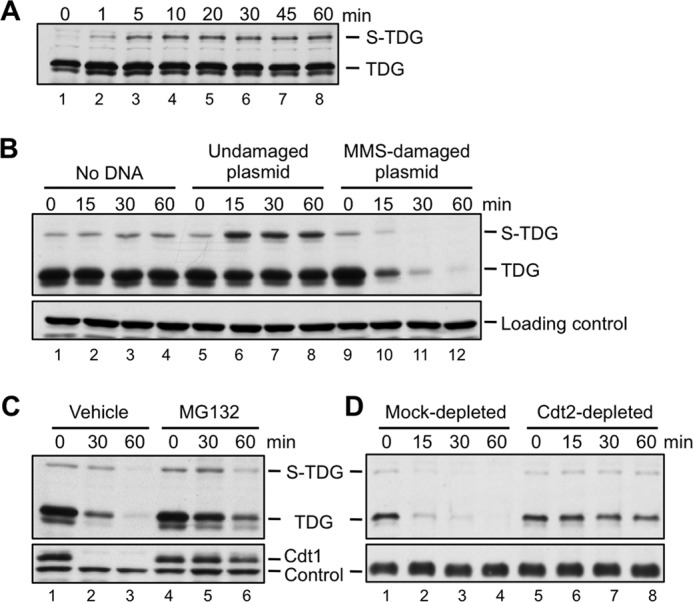
TDG is destroyed in Xenopus egg extract during DNA repair. A, recombinant TDG is rapidly modified to SUMOylated TDG (S-TDG) upon the addition to HSS. TDG (1 ng/μl) was added to HSS in the absence of added DNA, and the reaction was stopped at the indicated times and blotted for TDG. B, TDG is destroyed during DNA repair. For all destruction assays, egg extract was supplemented with TDG (1 ng/μl) and then, to start the reaction, with MMS-damaged plasmid (10 ng/μl), unless stated otherwise. For this reason, S-TDG is visible at the 0 min time point. TDG levels in extract were monitored over time by Western blotting with anti-TDG antibody. C, TDG destruction depends on the proteasome. TDG destruction was measured as in Fig. 4B in the presence of DMSO (vehicle) or 1 mm MG132, a proteasome inhibitor. D, depletion of Cdt2 stabilizes TDG. Egg extract was mock-depleted with preimmune serum or immunodepleted with anti-Cdt2 antiserum, and TDG destruction was measured as in B. For B and D, an extract-specific, non-TDG band detected by the TDG antibody was used as a loading control. For C, an extract-specific, non-Cdt1 band detected by the Cdt1 antibody was used as a loading control.
The Role of SUMOylation in TDG Destruction
A detailed time course revealed that the destruction of recombinant TDG was considerably slower than that of endogenous Cdt1 (Fig. 5, A (lanes 1–7) and B (navy blue and green graphs)). Because SUMOylation decreases the affinity of TDG for DNA, we suspected that SUMOylation might protect TDG from destruction by limiting its access to DNA. To test this, we mutated the conserved SUMOylation site, Lys-364, to alanine, yielding TDGΔSUMO. Upon the addition to extract, TDGΔSUMO migrated as a single species (Fig. 5A, lane 8), confirming that the slow mobility form (S-TDG) represented SUMOylated TDG. As expected, TDGΔSUMO bound to chromatin more efficiently than TDGWT (Fig. 5C, compare lanes 3 and 4). However, TDGWT and TDGΔSUMO were destroyed with virtually identical kinetics (Fig. 5, A and B, compare navy blue and orange traces). Destruction of TDGΔSUMO also depended on damaged DNA, the proteasome, and Cdt2 (Fig. 5, D–F). Interestingly, the small fraction of SUMOylated TDG disappeared at a slower rate than unmodified TDG (Fig. 5, A (lanes 1–7) and B (compare dotted and dashed blue lines)). A possible explanation of these results is that SUMOylation inhibits TDG destruction but that S-TDG is in rapid exchange with unmodified TDG, such that the overall kinetics of TDG destruction are unaffected by SUMOylation.
FIGURE 5.

TDGΔSUMO is regulated similarly to TDGWT. A, TDG is destroyed at a slow rate compared with Cdt1, and SUMOylation does not affect the total TDG destruction rate. Destruction of TDGWT or TDGΔSUMO was triggered with 5 ng/μl MMS-damaged plasmid and measured as in Fig. 4B. B, quantification of the rate of destruction of Cdt1 and different forms of TDG in A. C, mutation of the SUMOylation site (K364A) eliminates modification of TDG to a higher molecular weight and increases the affinity of TDG for DNA. TDGWT or TDGΔSUMO was added to extract that was subsequently added to linear, MMS-damaged DNA immobilized on magnetic beads. Methyl ubiquitin was included to allow monoubiquitylation on multiple sites but to prevent polyubiquitylation and destruction. Loading control in total extract was as in Fig. 4B; loading control for DNA-bound fraction was Orc2. D, TDGΔSUMO is destroyed during DNA repair. Buffer, MMS-damaged plasmid, or undamaged plasmid was added to egg extract, and TDG levels were measured as in Fig. 4B. E, TDGΔSUMO destruction depends on the proteasome. TDGΔSUMO destruction was measured as in Fig. 4B in the presence of DMSO (vehicle) or 1 mm MG132. F, depletion of Cdt2 stabilizes TDGΔSUMO. Egg extract was mock-depleted with preimmune serum or immunodepleted with anti-Cdt2 antiserum, and TDGΔSUMO destruction was measured as in Fig. 4B. Loading controls for A and E were as in Fig. 4C. Loading controls for D and F were as in Fig. 4B. Error bars, S.D.
The PIP Degron of TDG Is Required for TDG Destruction
To characterize the nature of the TDG degron, we first added a competitor peptide comprising the p21 PIP degron to egg extract, which we previously showed inhibited Cdt1 destruction (47). The wild type p21 peptide stabilized TDG, whereas a mutated control peptide lacking the PIP box did not (Fig. 6A). We observed the same result with TDGΔSUMO (data not shown). Mutation of the core PIP box residues in TDG (TDGΔPIP; Fig. 6B) abolished TDG destruction (Fig. 6C, lanes 4–6). Mutation of the B+4 residue in TDG (TDGΔB+4; Fig. 6B) strongly inhibited but did not abolish destruction (Fig. 6C, lanes 7–9), whereas mutation of B+3 and B+4 together (TDGΔB+3/4; Fig. 6B) almost completely stabilized TDG (Fig. 6C, lanes 10–12). These mutations had the same effect on TDGΔSUMO as on TDG (Fig. 6D). In summary, the destruction of TDG requires both a PIP box and positively charged residues downstream of the PIP box.
FIGURE 6.

TDG destruction depends on its PIP degron. A, a p21 PIP box peptide prevents the destruction of TDGWT. TDG destruction was measured as in Fig. 4B in extract supplemented with 200 μm WT or p21 peptide whose PIP box was mutated (ΔPIP). B, TDG PIP degron mutations used in this study. ΔPIP, Q128A/I131A/F135A; ΔB+4, R139A; ΔB+3/4, K138A/R139A; PIP*, A134F. C, mutation of the TDG PIP box or downstream basic residues stabilizes TDGWT. Destruction of TDGWT, TDGΔPIP, TDGΔB+4, and TDGΔB+3/4 was measured as in Fig. 4B. D, mutation of the TDG PIP box or downstream basic residues stabilizes TDGΔSUMO. The experiment was performed identically to that in Fig. 6C except that TDGΔSUMO replaced TDGWT. Loading control for A was as in Fig. 4C. Loading controls for C and D were as in Fig. 4B.
The TDG PIP Degron Is Imperfect
The PIP degron of TDG lacks one of the two conserved aromatic residues normally found in PIP box proteins (Fig. 1) (21). We postulated that this deviation from the PIP consensus might explain the slower destruction of TDG compared with Cdt1 (Fig. 5, A and B). To test this idea, we mutated alanine 134 to phenylalanine (A134F), yielding TDGPIP* (Fig. 6B). TDGPIP* was destroyed much faster than TDGWT, with kinetics approaching those of Cdt1 (Fig. 7, A and B). The same PIP box mutation also promoted more rapid destruction in the context of TDGΔSUMO (data not shown). Interestingly, human, mouse, chicken, zebrafish, and frog TDG all lack the first aromatic residue of the PIP box (Fig. 1), suggesting that suboptimal destruction of TDG benefits cellular or organismal fitness.
FIGURE 7.
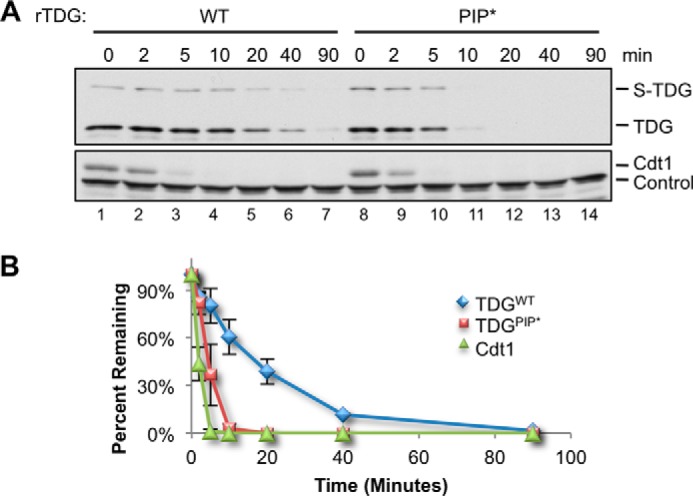
Improving the TDG PIP box results in faster destruction. A, MMS-damaged plasmid was added to egg extract at a final concentration of 5 ng/μl, and destruction of TDGWT and TDGPIP* was measured as in Fig. 4B. Loading control was as in Fig. 4C. B, quantification of A. Error bars, S.D.
TDG Is Ubiquitylated on Chromatin
Because TDGΔSUMO binds more efficiently to DNA than TDGWT (Fig. 5D), we used the former to look for evidence of CRL4Cdt2-dependent ubiquitylation, which normally occurs on chromatin (47, 53). To monitor the chromatin-bound fraction, MMS-damaged DNA was immobilized on magnetic beads and added to egg extract containing methyl-ubiquitin, which prevents polyubiquitin chain formation but allows monoubiquitylation on multiple sites. Upon recovery of the immobilized DNA, we detected ubiquitylated TDGΔSUMO (Fig. 8A, lane 1, second panel). Mutation of the PIP box eliminated detectable TDGΔSUMO ubiquitylation (Fig. 8A, lane 2, second panel), and mutation of the B+4 residue greatly decreased ubiquitylation (Fig. 8A, lane 3, third panel), whereas additional mutation of the B+3 residue eliminated ubiquitylation (Fig. 8A, lane 4, third panel). Conversely, improving the TDG PIP box via addition of the second aromatic residue dramatically enhanced TDGΔSUMO ubiquitylation on DNA (Fig. 8A, lane 5, second panel), such that ubiquitylation could even be detected in total extract (Fig. 8A, lane 5, first panel). This enhanced ubiquitylation was abolished by the p21 PIP box competitor peptide (Fig. 8B, lane 4). As expected, Cdt2 depletion inhibited TDG ubiquitylation (Fig. 8C). Together, our results show that TDG is ubiquitylated on chromatin in a PIP degron-, PCNA-, and Cdt2-dependent manner.
FIGURE 8.
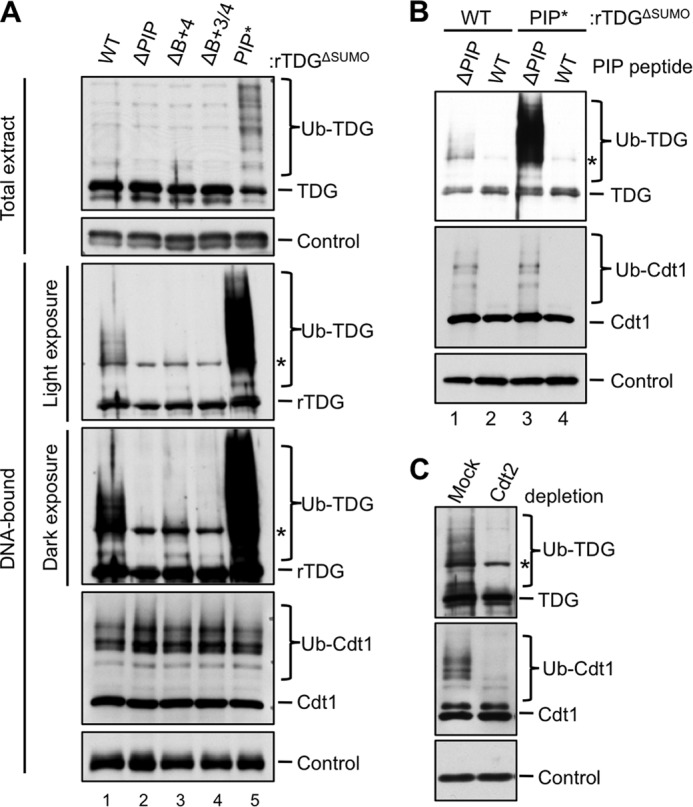
TDG is ubiquitylated on chromatin in a PIP box-, PCNA-, and Cdt2-dependent manner. A, the effect of different TDG mutations on its ubiquitylation. The indicated TDG proteins (described in the legend to Fig. 6B) were added to egg extract containing methyl ubiquitin (2 mg/ml) before the addition of 50 ng/μl MMS-damaged linear DNA immobilized on magnetic beads. The beads were recovered, and associated TDG was monitored via immunoblotting with anti-TDG antibodies. TDG in total extract is also shown. B, the addition of a PIP box competitor peptide abolishes ubiquitylation of both TDGΔSUMO and TDGΔSUMO/PIP*. p21 peptide was added to egg extract as in Fig. 6A, and TDG ubiquitylation was monitored as in Fig. 8A. C, depletion of Cdt2 eliminates ubiquitylation of DNA-bound TDGΔSUMO. Extract was mock-depleted or immunodepleted of Cdt2 and supplemented with TDG, methyl ubiquitin, and linear DNA. TDG ubiquitylation was monitored as in Fig. 8A. In all panels, the asterisk indicates two overlapping bands: specific signal (ubiquitylated TDG) and a nonspecific signal. The loading controls for each panel are as follows: Orc2 (A, DNA-bound), nonspecific band detected by TDG antibody (A, Total extract), RPA-14 (B), and PCNA (C).
Endogenous TDG Is Destroyed after DNA Damage
We next asked whether endogenous TDG is destroyed after DNA damage. As shown in Fig. 9, lanes 3 and 4, endogenous TDG levels declined after the addition of damaged DNA. Due to the low levels of TDG in Xenopus egg extract, the effect was more evident when TDG was immunoprecipitated prior to Western blotting (Fig. 9, lanes 9 and 10). The reduction in TDG levels was abolished by the p21 PIP box peptide (Fig. 9, compare lanes 10 and 12), showing that destruction of the endogenous protein is linked to PCNA. We conclude that, like recombinant TDG, endogenous TDG is targeted by CRL4Cdt2 during DNA repair.
FIGURE 9.
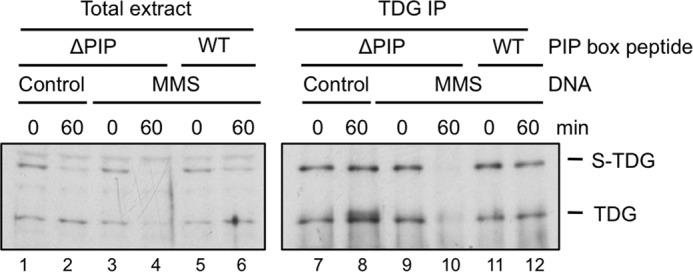
Endogenous TDG is destroyed in a PCNA-dependent manner. Egg extract was supplemented with undamaged or MMS-damaged DNA and p21 control (ΔPIP) peptide or p21 competitor (WT) peptide as indicated. Total extract (lanes 1–6) or TDG immunoprecipitates (lanes 7–12) were blotted with TDG antibody.
TDG Destruction during S Phase
TDG is destroyed during S phase in human cells (28). We therefore sought to recapitulate replication-coupled destruction in egg extract. To this end, we licensed plasmid DNA in HSS before adding recombinant TDG and NPE, which promotes replication initiation and a single round of DNA replication (54). Under these conditions, TDGWT was stable, whereas Cdt1 was rapidly destroyed (Fig. 10A, lanes 1–4). In contrast, TDGPIP* was destroyed efficiently in the replicating extract, but not when replication was blocked with geminin, an inhibitor of licensing (Fig. 10, A and B). We observed similar results with TDGΔSUMO (Fig. 10, C and D). Thus, TDG containing an improved PIP degron is destroyed during replication in Xenopus egg extract, arguing that CRL4Cdt2 can target the protein during S phase.
FIGURE 10.

TDG containing an improved PIP box is destroyed during replication in Xenopus egg extract. A, plasmid was incubated in HSS containing buffer or geminin, followed by NPE supplemented with TDG, and TDG levels were monitored by Western blotting (0 min time corresponds to the moment of NPE addition). B, quantification of replication in the reactions described in A. C, identical to A except that TDGΔSUMO replaces TDGWT. D, identical to B except that TDGΔSUMO replaces TDGWT. Loading controls for A and C were as in Fig. 4C.
TDG Is Not Active in Xenopus Egg Extract
We next asked whether CRL4Cdt2-dependent TDG destruction is required for S phase progression when exogenous TDG is added to egg extract. For example, TDG proteolysis might prevent the irreversible binding of TDG to abasic sites and thereby avoid replication fork stalling. In this scenario, TDG destruction and SUMOylation could represent redundant mechanisms to prevent such a replication fork block. To test this model, we measured replication of a plasmid containing a G-T mispair in the presence of TDG lacking the PIP degron. To eliminate regulation by SUMOylation, we also mutated the TDG SUMOylation site. However, neither TDGΔSUMO nor TDGΔSUMO/ΔPIP significantly impaired replication of the G-T plasmid in egg extract (data not shown). These proteins also failed to induce detectable nascent strand pausing at the G-T mispair (data not shown).
These negative results led us to address whether TDG is active in Xenopus egg extract. To test this, we added a duplex oligonucleotide containing a G-T mispair to extract supplemented with TDG. After 30 min, the DNA was recovered and treated with alkali, which cleaves the DNA backbone at abasic sites (Fig. 11A). Under these conditions, no cleavage of the DNA was detected (Fig. 11B, lane 4). In contrast, TDG was highly active in buffer (Fig. 11B, lane 3). Importantly, when we recovered the DNA from the extract and subsequently treated it with TDG in buffer, cleavage occurred, demonstrating that the G-T mispair had not been repaired in the extract before it could be cleaved by TDG (Fig. 11B, lane 5). We conclude that TDG is not active in Xenopus egg extract. Importantly, G-U mispairs were rapidly repaired in egg extract by endogenous uracil DNA glycosylase activity (data not shown), demonstrating that the extract does not inhibit all DNA glycosylases.
FIGURE 11.

TDG is inactive in Xenopus egg extract. A, schematic depicting TDG activity assay. Left, TDG action on a G-T mispair-containing duplex oligonucleotide labeled on the bottom strand leads to an abasic site whose cleavage by NaOH yields a 56-nt radiolabeled product. Right, when the same oligonucleotide as in the left scheme but lacking the G-T mispair is cut with EcoRI, a radiolabeled, 52-nucleotide product is generated. B, the G-T mispair-containing duplex oligonucleotide depicted in A (left) was incubated with 300 nm TDGWT in the presence of buffer (lane 3) or HSS (lane 4). In lane 1, TDG was excluded from the reaction. In lane 5, the oligonucleotide was incubated with HSS and subsequently isolated and incubated with buffer and TDG. Glycosylase activity was measured as depicted in A (left). In lane 2, fully complementary oligonucleotide was digested with EcoRI, yielding a 52-nt species. C, HSS was immunodepleted of MutSα with antibodies to Msh2 and Msh6 or mock-depleted by incubation with preimmune serum, and different amounts of the resulting extracts were blotted for Msh2 and Msh6. 100% equals 1 μl of extract. D, MutSα-depleted extract is unable to support TDG activity. A G-T-containing oligonucleotide was incubated with TDG in ELB (lane 1), HSS depleted of MutSα (lane 3), or mock-depleted extract (lane 2). Activity was measured as described in A (left).
We previously showed that efficient DNA replication in egg extract requires a threshold concentration of DNA (45). We reasoned there might be a similar DNA threshold for TDG activity. However, the addition of plasmid carrier DNA did not stimulate TDG activity on the G-T oligonucleotide (data not shown). We further postulated that MutSα, which recognizes DNA mismatches, might compete with TDG for binding to the G-T base pair. However, immunodepletion of Msh2 and Msh6, the two proteins that comprise MutSα, from egg extract (Fig. 11C) had no effect on TDG activity (Fig. 11D). We also failed to observe TDG activity on oligonucleotides containing G-carboxy-C base pairs in extract (data not shown). Future work will be required to determine how TDG is inhibited in Xenopus egg extract (see “Discussion”).
Developmental Regulation of TDG
The lack of TDG activity in egg extract made it difficult to determine the function of TDG destruction in this setting. We therefore asked whether TDG is destroyed during frog development and, if so, what happens when this process is disrupted. We first determined the pattern of endogenous TDG expression. To this end, frog eggs were fertilized in vitro, and TDG expression was monitored at different stages of development by Western blotting. Fig. 12A shows that TDG expression was extremely low during early embryonic development (lanes 3–7), consistent with the difficulty of detecting TDG in egg extract (Fig. 2, lane 2). During gastrulation (Nieuwkoop and Faber (NF) stages 10–12), TDG became detectable, and its levels continued to increase drastically during neurulation (represented by NF stages 14, 16, and 20) (Fig. 12A). In contrast, mRNA levels increased less than 5-fold during this period (Fig. 12B), suggesting that TDG is regulated post-transcriptionally during Xenopus laevis development.
FIGURE 12.

TDG expression is regulated during X. laevis development. A, TDG is not detectable until mid-gastrulation. X. laevis eggs were fertilized in vitro, and embryos were harvested at the indicated NF stage. TDG was detected in embryo lysates via immunoblotting. PCNA is shown as a loading control. B, TDG mRNA levels increase ∼5-fold after stage 10 (66). Pax6 mRNA, which is not substantially expressed until after the mid-blastula transition, is shown for comparison (66). Error bars, S.D. from multiple published experiments. C, TDGΔPIP accumulates earlier than TDGWT. Stage 2 embryos were microinjected with 20 pg of the indicated TDG mRNA, and the TDG protein level was analyzed at the indicated stages via Western blotting. Uninjected control is included. PCNA was used as a loading control. D, after microinjection as in C, TDG mRNA levels were compared in stage 7 embryos. E, embryos were microinjected as in C and photographed at the indicated stages of development. In five repetitions of the experiment, 98 of 108 WT-injected, 137 of 148 ΔPIP-injected, and 135 of 149 uninjected embryos showed no phenotype through at least stage 14.
We next microinjected both cells of NF stage 2 X. laevis embryos with TDGWT or TDGΔPIP mRNA and monitored TDG expression and embryonic development. TDGΔPIP levels were higher than TDGWT levels from the blastula stages (NF stage 7–9) until at least late gastrulation (stage 12) (Fig. 12C, compare lanes 7–11 and 13–17). By the early tail bud stages (represented by NF stage 25), their concentrations became more similar (Fig. 12C, lanes 12 and 18). We confirmed by quantitative PCR that injected embryos contained approximately the same level of TDGWT and TDGΔPIP mRNA (Fig. 12D). These data suggest that TDG is normally down-regulated in a PIP degron-dependent manner from the blastula until at least the late gastrula stages of development. Because there should be no significant damage-dependent destruction during unperturbed development, these results imply that TDG is normally subject to replication-dependent destruction by CRL4Cdt2 in the developing embryo.
Remarkably, embryos expressing TDGΔPIP exhibited no detectable developmental defects through the tadpole stage (Fig. 12E and data not shown). Expression of much higher levels of TDGΔPIP did cause developmental abnormalities, but these were often also observed with TDGWT (data not shown) and therefore probably reflected the effects of unphysiologically high TDG expression rather than a specific failure to destroy TDG in S phase. It is possible that S phase TDG proteolysis is required for later development, but injected mRNAs only persist in embryos for approximately 2 days (58), making this question difficult to address.
DISCUSSION
In recent years, the DNA glycosylase TDG has emerged as a key component of the enzymatic cascade that leads to DNA demethylation. Several forms of TDG regulation have been discovered: 1) SUMOylation facilitates TDG dissociation from abasic sites on DNA (59); 2) both acetylation and phosphorylation regulate the access of TDG to DNA (60, 61); and 3) S phase-specific proteolysis eliminates TDG during DNA replication (28). Here, we show that TDG is also destroyed after DNA damage, and our results indicate that replication and damage-dependent destruction of TDG both involve the E3 ubiquitin ligase CRL4Cdt2.
We found that during the repair of MMS-damaged DNA in Xenopus egg extract, recombinant TDG destruction depended on Cdt2, PCNA, the proteasome, and a PIP degron. Moreover, as seen for other CRL4Cdt2 substrates, TDG was ubiquitylated on chromatin. In the same setting, endogenous TDG was destroyed in a PCNA-dependent manner. Together, these results demonstrate that TDG is a CRL4Cdt2 target after DNA damage. Although we were not able to detect destruction of endogenous or recombinant TDGWT during chromosomal replication in egg extract, TDG carrying an enhanced PIP degron (TDGPIP*) was destroyed efficiently during replication. TDGPIP* was also destroyed more efficiently than TDGWT in response to DNA damage. Thus, TDG contains a suboptimal PIP degron that is sufficient for damage-dependent destruction but not replication-dependent destruction in egg extract.
Importantly, our data suggest that during early frog development, TDG is destroyed in S phase. We infer this from the fact that TDGΔPIP accumulated to higher levels than TDGWT when we microinjected embryos with the corresponding mRNAs. It appears that in our in vitro extract experiments, the trigger for TDG destruction after DNA damage is more robust than during replication. However, in vivo, the replication-dependent signal suffices to promote TDG destruction. Together, our data support the conclusion that TDG is a CRL4Cdt2 substrate during DNA repair and replication. Similarly, TDG is targeted by CRL4Cdt2 in S phase and after DNA damage in mammalian cells (67).
The PIP degron of TDG lacks one of the two aromatic residues seen in most PIP boxes. This feature, which makes TDG a suboptimal CRL4Cdt2 substrate, is conserved among TDG orthologs. We speculate that this suboptimal PIP degron helps establish a hierarchy among CRL4Cdt2 substrates such that those with optimal degrons (e.g. Cdt1) are destroyed first as cells enter S phase. The discovery of a bona fide CRL4Cdt2 substrate lacking the first aromatic residue of the PIP box relaxes the sequence requirements for CRL4Cdt2 substrates and thus expands the potential universe of such substrates.
Why is TDG targeted by CRL4Cdt2? Most CRL4Cdt2 substrates are toxic for S phase progression (2). It is possible that TDG competes with the MMR pathway for the correction of G-T mispairs generated in the process of replication and thereby causes mutagenesis (because MMR but not TDG can distinguish nascent from parental strands). Additionally, irreversible binding of TDG to abasic sites might block replication fork progression. In possible agreement with these ideas, endogenous TDG was not expressed during the early embryonic cleavage divisions when S phase comprises most of the cell cycle. To directly test for replication inhibition, we incubated a plasmid containing a G-T mispair in Xenopus egg extract in the presence of non-degradable TDG that also lacked the SUMOylation site. Although we observed no defects in DNA replication, the results were inconclusive because TDG did not act on its cognate substrates in egg extract (Fig. 11). We speculate that egg extracts contain an inhibitor that acts redundantly with TDG proteolysis to ensure that no TDG activity is present during early embryogenesis. To elucidate the mechanistic consequences of TDG expression in S phase, it will be important to find conditions that allow its activity in egg extract.
An alternative proposal is that TDG expression in S phase interferes with epigenetic inheritance. For example, when heavily methylated heterochromatin decondenses during replication fork passage, TDG might trigger aberrant DNA demethylation at sites that are typically inaccessible to TDG. In general agreement with such an idea, the PIP degron is only present in animals that exhibit cytosine DNA methylation (Fig. 1A) (59).
CRL4Cdt2-dependent TDG destruction might be important to regulate DNA methylation levels during development. Notably, total methyl-CpG levels in frogs remain fairly constant through the midblastula transition (62). Then total methyl-CpG levels drop during gastrulation, suggesting that the rate of new methylation decreases or active demethylation increases (62). Consistent with the latter model, we found that endogenous TDG protein was not detectable from fertilization through early gastrulation (until NF stage 10), and then it increased sharply during gastrulation. Moreover, the mRNA of Tet3, which oxidizes methylated cytosine in preparation for excision by TDG, also increases during early gastrulation (63). Thus, the drop in methylation seen during gastrulation might be due, in whole or in part, to collaborative DNA demethylation by Tet3 and TDG. Similarly, regulated transcription of developmental genes begins at the early gastrula transition (between NF stages 10 and 10.5) in Xenopus (64). Therefore, TDG- and Tet-dependent demethylation in the gastrula embryo might help to activate developmentally regulated genes that act in lineage commitment, as seen in mammals (42, 44).
In embryos injected with TDGΔPIP mRNA, TDG protein accumulated prematurely, during the blastula stages (NF stages 7–9). This observation indicates that CRL4Cdt2 can attenuate TDG expression before gastrulation. Total TDG levels might be particularly sensitive to CRL4Cdt2 regulation during this time, because most cells in the embryo are still undergoing DNA replication and cell division. It is possible that the down-regulation of total TDG by CRL4Cdt2 during the blastula and early gastrulation stages helps prevent premature promoter demethylation and activation of certain developmentally regulated genes. Although non-degradable TDG expression did not visibly perturb frog development up to the tadpole stage, this might be due to the absence of Tet3 protein before gastrulation or due to redundant inhibitory mechanisms that restrict TDG activity during early development, as we observed in unfertilized egg extracts.
In conclusion, we provide compelling evidence that TDG is a bona fide new substrate of CRL4Cdt2. Given the conserved nature of the PIP degron in TDG, TDG destruction almost certainly serves an important function in cell physiology or development. To identify this function, replacement of the endogenous TDG gene with TDGΔPIP in cells and animals may ultimately be required.
Acknowledgments
We thank Primo Schär for encouragement, helpful discussions, and comments on the manuscript and Anindya Dutta for communicating results prior to publication. We thank Marc Kirschner for use of the embryo injection facility (supported by National Institutes of Health Grant GM26875).
This work was supported, in whole or in part, by National Institutes of Health Grant GM80676 (to J. C. W.).
- PCNA
- proliferating cell nuclear antigen
- AP site
- abasic (apurinic/apyrimidinic) site
- TEMED
- N,N,N′,N′-tetramethylethylenediamine
- NF
- Nieuwkoop and Faber
- PIP
- PCNA-interacting peptide
- BER
- base excision repair
- TDG
- thymine DNA glycosylase
- HSS
- high-speed supernatant
- NPE
- nucleoplasmic extract
- ELB
- egg lysis buffer
- MMS
- methyl methanesulfonate
- SUMO
- small ubiquitin-like modifier
- nt
- nucleotide.
REFERENCES
- 1. Abbas T., Dutta A. (2011) CRL4Cdt2: master coordinator of cell cycle progression and genome stability. Cell Cycle 10, 241–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Havens C. G., Walter J. C. (2011) Mechanism of CRL4Cdt2, a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 25, 1568–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jin J., Arias E. E., Chen J., Harper J. W., Walter J. C. (2006) A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell 23, 709–721 [DOI] [PubMed] [Google Scholar]
- 4. Higa L. A., Banks D., Wu M., Kobayashi R., Sun H., Zhang H. (2006) L2DTL/CDT2 interacts with the CUL4/DDB1 complex and PCNA and regulates CDT1 proteolysis in response to DNA damage. Cell Cycle 5, 1675–1680 [DOI] [PubMed] [Google Scholar]
- 5. Ralph E., Boye E., Kearsey S. E. (2006) DNA damage induces Cdt1 proteolysis in fission yeast through a pathway dependent on Cdt2 and Ddb1. EMBO Rep. 7, 1134–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sansam C. L., Shepard J. L., Lai K., Ianari A., Danielian P. S., Amsterdam A., Hopkins N., Lees J. A. (2006) DTL/CDT2 is essential for both CDT1 regulation and the early G2/M checkpoint. Genes Dev. 20, 3117–3129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Centore R. C., Havens C. G., Manning A. L., Li J.-M., Flynn R. L., Tse A., Jin J., Dyson N. J., Walter J. C., Zou L. (2010) CRL4Cdt2-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol. Cell 40, 22–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abbas T., Shibata E., Park J., Jha S., Karnani N., Dutta A. (2010) CRL4Cdt2 regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol. Cell 40, 9–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abbas T., Sivaprasad U., Terai K., Amador V., Pagano M., Dutta A. (2008) PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 22, 2496–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim Y., Starostina N. G., Kipreos E. T. (2008) The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 22, 2507–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mansilla S. F., Soria G., Vallerga M. B., Habif M., Martínez-López W., Prives C., Gottifredi V. (2013) UV-triggered p21 degradation facilitates damaged-DNA replication and preserves genomic stability. Nucleic Acids Res. 41, 6942–6951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tsanov N., Kermi C., Coulombe P., Van der Laan S., Hodroj D., Maiorano D. (2014) PIP degron proteins, substrates of CRL4Cdt2, and not PIP boxes, interfere with DNA polymerase η and κ focus formation on UV damage. Nucleic Acids Res. 42, 3692–3706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang S., Zhao H., Darzynkiewicz Z., Zhou P., Zhang Z., Lee E. Y. C., Lee M. Y. W. T. (2013) A novel function of CRL4Cdt2: regulation of the subunit structure of DNA polymerase δ in response to DNA damage and during the S phase. J. Biol. Chem. 288, 29550–29561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Terai K., Shibata E., Abbas T., Dutta A. (2013) Degradation of p12 subunit by CRL4Cdt2 E3 ligase inhibits fork progression after DNA damage. J. Biol. Chem. 288, 30509–30514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shibutani S. T., de la Cruz A. F. A., Tran V., Turbyfill W. J., 3rd, Reis T., Edgar B. A., Duronio R. J. (2008) Intrinsic negative cell cycle regulation provided by PIP box- and Cul4Cdt2-mediated destruction of E2F1 during S phase. Dev. Cell 15, 890–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu C., Powell K. A., Mundt K., Wu L., Carr A. M., Caspari T. (2003) Cop9/signalosome subunits and Pcu4 regulate ribonucleotide reductase by both checkpoint-dependent and -independent mechanisms. Genes Dev. 17, 1130–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Salguero I., Guarino E., Shepherd M. E. A., Deegan T. D., Havens C. G., MacNeill S. A., Walter J. C., Kearsey S. E. (2012) Ribonucleotide reductase activity is coupled to DNA synthesis via proliferating cell nuclear antigen. Curr. Biol. 22, 720–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim D. H., Budhavarapu V. N., Herrera C. R., Nam H. W., Kim Y. S., Yew P. R. (2010) The CRL4Cdt2 ubiquitin ligase mediates the proteolysis of cyclin-dependent kinase inhibitor Xic1 through a direct association with PCNA. Mol. Cell. Biol. 30, 4120–4133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Senga T., Sivaprasad U., Zhu W., Park J. H., Arias E. E., Walter J. C., Dutta A. (2006) PCNA is a cofactor for Cdt1 degradation by CUL4/DDB1-mediated N-terminal ubiquitination. J. Biol. Chem. 281, 6246–6252 [DOI] [PubMed] [Google Scholar]
- 20. Nishitani H. (2001) The human licensing factor for DNA replication Cdt1 accumulates in G1 and is destabilized after initiation of S-phase. J. Biol. Chem. 276, 44905–44911 [DOI] [PubMed] [Google Scholar]
- 21. Moldovan G.-L., Pfander B., Jentsch S. (2007) PCNA, the maestro of the replication fork. Cell 129, 665–679 [DOI] [PubMed] [Google Scholar]
- 22. Havens C. G., Walter J. C. (2009) Docking of a specialized PIP box onto chromatin-bound PCNA creates a degron for the ubiquitin ligase CRL4Cdt2. Mol. Cell 35, 93–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Michishita M., Morimoto A., Ishii T., Komori H., Shiomi Y., Higuchi Y., Nishitani H. (2011) Positively charged residues located downstream of PIP box, together with TD amino acids within PIP box, are important for CRL4Cdt2-mediated proteolysis. Genes Cells 16, 12–22 [DOI] [PubMed] [Google Scholar]
- 24. Havens C. G., Shobnam N., Guarino E., Centore R. C., Zou L., Kearsey S. E., Walter J. C. (2012) Direct role for proliferating cell nuclear antigen in substrate recognition by the E3 ubiquitin ligase CRL4Cdt2. J. Biol. Chem. 287, 11410–11421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hardeland U., Steinacher R., Jiricny J., Schär P. (2002) Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J. 21, 1456–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smet-Nocca C., Wieruszeski J.-M., Léger H., Eilebrecht S., Benecke A. (2011) SUMO-1 regulates the conformational dynamics of thymine-DNA glycosylase regulatory domain and competes with its DNA binding activity. BMC Biochem. 12, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baba D., Maita N., Jee J.-G., Uchimura Y., Saitoh H., Sugasawa K., Hanaoka F., Tochio H., Hiroaki H., Shirakawa M. (2005) Crystal structure of thymine DNA glycosylase conjugated to SUMO-1. Nature 435, 979–982 [DOI] [PubMed] [Google Scholar]
- 28. Hardeland U., Kunz C., Focke F., Szadkowski M., Schär P. (2007) Cell cycle regulation as a mechanism for functional separation of the apparently redundant uracil DNA glycosylases TDG and UNG2. Nucleic Acids Res. 35, 3859–3867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu H., Zhang Y. (2014) Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 156, 45–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. He Y. F., Li B. Z., Li Z., Liu P., Wang Y., Tang Q., Ding J., Jia Y., Chen Z., Li L., Sun Y., Li X., Dai Q., Song C. X., Zhang K., He C., Xu G. L. (2011) Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ito S., Shen L., Dai Q., Wu S. C., Collins L. B., Swenberg J. A., He C., Zhang Y. (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo J. U., Su Y., Zhong C., Ming G.-L., Song H. (2011) Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145, 423–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wossidlo M., Nakamura T., Lepikhov K., Marques C. J., Zakhartchenko V., Boiani M., Arand J., Nakano T., Reik W., Walter J. O. R. (2011) 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat. Commun. 2, 241–248 [DOI] [PubMed] [Google Scholar]
- 34. Hashimoto H., Liu Y., Upadhyay A. K., Chang Y., Howerton S. B., Vertino P. M., Zhang X., Cheng X. (2012) Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 40, 4841–4849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang L., Lu X., Lu J., Liang H., Dai Q., Xu G.-L., Luo C., Jiang H., He C. (2012) Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat. Chem. Biol. 8, 328–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maiti A., Drohat A. C. (2011) Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J. Biol. Chem. 286, 35334–35338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Iqbal K., Jin S.-G., Pfeifer G. P., Szabó P. E. (2011) Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc. Natl. Acad. Sci. U.S.A. 108, 3642–3647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Inoue A., Shen L., Dai Q., He C., Zhang Y. (2011) Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res. 21, 1670–1676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Inoue A., Zhang Y. (2011) Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science 334, 194–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Morgan H. D., Dean W., Coker H. A., Reik W., Petersen-Mahrt S. K. (2004) Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J. Biol. Chem. 279, 52353–52360 [DOI] [PubMed] [Google Scholar]
- 41. Rai K., Huggins I. J., James S. R., Karpf A. R., Jones D. A., Cairns B. R. (2008) DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and Gadd45. Cell 135, 1201–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cortellino S., Xu J., Sannai M., Moore R., Caretti E., Cigliano A., Le Coz M., Devarajan K., Wessels A., Soprano D., Abramowitz L. K., Bartolomei M. S., Rambow F., Bassi M. R., Bruno T., Fanciulli M., Renner C., Klein-Szanto A. J., Matsumoto Y., Kobi D., Davidson I., Alberti C., Larue L., Bellacosa A. (2011) Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 146, 67–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Popp C., Dean W., Feng S., Cokus S. J., Andrews S., Pellegrini M., Jacobsen S. E., Reik W. (2010) Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 463, 1101–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cortázar D., Kunz C., Selfridge J., Lettieri T., Saito Y., MacDougall E., Wirz A., Schuermann D., Jacobs A. L., Siegrist F., Steinacher R., Jiricny J., Bird A., Schär P. (2011) Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 470, 419–423 [DOI] [PubMed] [Google Scholar]
- 45. Lebofsky R., Takahashi T., Walter J. C. (2009) DNA replication in nucleus-free Xenopus egg extracts. Methods Mol. Biol. 521, 229–252 [DOI] [PubMed] [Google Scholar]
- 46. Stokes M. P. (2003) DNA damage-induced replication arrest in Xenopus egg extracts. J. Cell Biol. 163, 245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Arias E. E., Walter J. C. (2006) PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat. Cell Biol. 8, 84–90 [DOI] [PubMed] [Google Scholar]
- 48. Wohlschlegel J. A. (2000) Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science 290, 2309–2312 [DOI] [PubMed] [Google Scholar]
- 49. Sive H. L., Grainger R. M., Harland R. M. (2010) Early Development of Xenopus laevis: A Laboratory Manual, 1st Ed., pp. 99–100, Cold Spring Harbor Press, Cold Spring Harbor, NY [Google Scholar]
- 50. Bowes J. B., Snyder K. A., Segerdell E., Gibb R., Jarabek C., Noumen E., Pollet N., Vize P. D. (2008) Xenbase: a Xenopus biology and genomics resource. Nucleic Acids Res. 36, D761–D767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Faber J., Nieuwkoop P. D. (1994) Normal Table of Xenopus laevis (Daudin) (Faber J., Nieuwkoop P. D., eds) Garland Science, New York [Google Scholar]
- 52. Joseph R. E., Andreotti A. H. (2008) Bacterial expression and purification of interleukin-2 tyrosine kinase: single step separation of the chaperonin impurity. Protein Expr. Purif. 60, 194–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Arias E. E., Walter J. C. (2005) Replication-dependent destruction of Cdt1 limits DNA replication to a single round per cell cycle in Xenopus egg extracts. Genes Dev. 19, 114–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Walter J., Sun L., Newport J. (1998) Regulated chromosomal DNA replication in the absence of a nucleus. Mol. Cell 1, 519–529 [DOI] [PubMed] [Google Scholar]
- 55. Walter J., Newport J. (2000) Initiation of eukaryotic DNA replication: origin unwinding and sequential chromatin association of Cdc45, RPA, and DNA polymerase α. Mol. Cell 5, 617–627 [DOI] [PubMed] [Google Scholar]
- 56. Dasso M., Newport J. W. (1990) Completion of DNA replication is monitored by a feedback system that controls the initiation of mitosis in vitro: studies in Xenopus. Cell 61, 811–823 [DOI] [PubMed] [Google Scholar]
- 57. Hardeland U., Bentele M., Jiricny J., Schär P. (2000) Separating substrate recognition from base hydrolysis in human thymine DNA glycosylase by mutational analysis. J. Biol. Chem. 275, 33449–33456 [DOI] [PubMed] [Google Scholar]
- 58. Blitz I. L., Andelfinger G., Horb M. E. (2006) Germ layers to organs: using Xenopus to study “later” development. Semin. Cell Dev. Biol. 17, 133–145 [DOI] [PubMed] [Google Scholar]
- 59. Cortázar D., Kunz C., Saito Y., Steinacher R., Schär P. (2007) The enigmatic thymine DNA glycosylase. DNA Repair 6, 489–504 [DOI] [PubMed] [Google Scholar]
- 60. Tini M., Benecke A., Um S.-J., Torchia J., Evans R. M., Chambon P. (2002) Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol. Cell 9, 265–277 [DOI] [PubMed] [Google Scholar]
- 61. Mohan R. D., Litchfield D. W., Torchia J., Tini M. (2010) Opposing regulatory roles of phosphorylation and acetylation in DNA mispair processing by thymine DNA glycosylase. Nucleic Acids Res. 38, 1135–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stancheva I., El-Maarri O., Walter J., Niveleau A., Meehan R. R. (2002) DNA methylation at promoter regions regulates the timing of gene activation in Xenopus laevis embryos. Dev. Biol. 243, 155–165 [DOI] [PubMed] [Google Scholar]
- 63. Xu Y., Xu C., Kato A., Tempel W., Abreu J. G., Bian C., Hu Y., Hu D., Zhao B., Cerovina T., Diao J., Wu F., He H. H., Cui Q., Clark E., Ma C., Barbara A., Veenstra G. J. C., Xu G., Kaiser U. B., Liu X. S., Sugrue S. P., He X., Min J., Kato Y., Shi Y. G. (2012) Tet3 CXXC domain and dioxygenase activity cooperatively regulate key genes for Xenopus eye and neural development. Cell 151, 1200–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Veenstra G. J., Destrée O. H., Wolffe A. P. (1999) Translation of maternal TATA-binding protein mRNA potentiates basal but not activated transcription in Xenopus embryos at the midblastula transition. Mol. Cell. Biol. 19, 7972–7982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Raddatz G., Guzzardo P. M., Olova N., Fantappié M. R., Rampp M., Schaefer M., Reik W., Hannon G. J., Lyko F. (2013) Dnmt2-dependent methylomes lack defined DNA methylation patterns. Proc. Natl. Acad. Sci. U.S.A. 110, 8627–8631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yanai I., Peshkin L., Jorgensen P., Kirschner M. W. (2011) Mapping gene expression in two Xenopus species: evolutionary constraints and developmental flexibility. Dev. Cell 20, 483–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shibata E., Dar A., Dutta A. (2014) CRL4Cdt2 E3 ubiquitin ligase and proliferating cell nuclear antigen (PCNA) cooperate to degrade thymine DNA glycosylase in S phase. J. Biol. Chem. 289, 23056–23064 [DOI] [PMC free article] [PubMed] [Google Scholar]