

Background: TDG is degraded in S phase via the proteasomal pathway.
Results: CRL4Cdt2 ubiquitinates and targets TDG associated with PCNA for degradation.
Conclusion: CRL4Cdt2-dependent degradation of TDG is important for preventing toxicity from excess TDG.
Significance: This is the first report demonstrating that TDG is regulated by CRL4Cdt2.
Keywords: DNA Damage Response, E3 Ubiquitin Ligase, Proliferating Cell Nuclear Antigen (PCNA), Protein Degradation, Ubiquitin, Thymine-DNA Glycosylase (TDG)
Abstract
Thymine DNA glycosylase (TDG) is an essential enzyme playing multiple roles in base excision repair, transcription regulation, and DNA demethylation. TDG mediates the cytotoxicity of the anti-cancer chemotherapeutic drug 5-fluorouracil (5-FU) by prolonging S phase, generating DNA strand breaks, and inducing DNA damage signaling. During S phase of the cell cycle, TDG is degraded via the proteasomal pathway. Here we show that CRL4Cdt2 E3 ubiquitin ligase promotes ubiquitination and proteasomal degradation of TDG in S phase in a reaction that is dependent on the interaction of TDG with proliferating cell nuclear antigen (PCNA). siRNA-mediated depletion of PCNA or components of CRL4Cdt2, specifically cullin4A/B or substrate adaptor Cdt2, stabilizes TDG in human cells. Mutations in the PCNA-interacting peptide (PIP) motif of TDG that disrupt the interaction of TDG with PCNA or change critical basic residues essential for the action of the PIP degron prevent the ubiquitination and degradation of TDG. Thus physical interaction of TDG with PCNA through the PIP degron is required for targeting TDG to the CRL4Cdt2 E3 ubiquitin ligase complex. Compared with forced expression of wild type TDG, CRL4Cdt2- resistant TDG (ΔPIP) slows cell proliferation and slightly increases the toxicity of 5-FU. Thus, CRL4Cdt2-dependent degradation of TDG occurs in S phase because of the requirement for TDG to interact with chromatin-loaded PCNA, and this degradation is important for preventing toxicity from excess TDG.
Introduction
TDG3 is one of the four uracil DNA glycosylase enzymes (UNG2, SMUG1, TDG, and MBD4) involved in the base excision repair pathway (1–4). Like other uracil DNA glycosylases, TDG has a broad range of substrate specificity and actively processes DNA lesions arising from oxidation, alkylation, and deamination of cytosine, 5-methyl-cytosine, and thymine. Although TDG is known to remove T from G-T lesions, it removes U from G-U lesions more efficiently (3). Unlike other glycosylases, TDG remains tightly bound to the abasic site left after removal of the mismatched base. The dissociation of TDG from its bound abasic site is the rate-limiting step of the glycosylase reaction. The SUMOylation at lysine 330 (by SUMO-1 or SUMO-2/3) and/or direct interaction with APE1 endonuclease increases the dissociation rate of TDG from AP sites, giving the downstream enzymes access to the lesion (5). Very recently, TDG and TET enzymes have been implicated in epigenetic regulation by carrying out a cascade of enzymatic reactions leading to DNA demethylation in mammalian cells (6). The methylcytosine is converted to 5-hydroxymethylcytosine and further to 5-formylcytosine and 5-carboxylcytosine by TET enzymes. TDG specifically recognizes 5-formylcytosine and 5-carboxylcytosine paired with a G residue to excise the modified cytosine residue from DNA, leaving an abasic site. APE1 excises the abasic residue to allow a DNA polymerase to insert a cytosine across from the G and thus complete the demethylation of methylcytosine. Thus, TDG is intimately involved in the epigenetic regulation of gene expression.
In a range of human cancers, such as colorectal, gastrointestinal, breast, and ovarian, 5-FU (uracil analog) is used as the first line of treatment (7, 8). 5-FU inhibits thymidylate synthase enzyme and gets incorporated into the DNA and RNA. The excision of 5-FU by TDG mediates the cytotoxicity of the drug by creating an excess of abasic sites, prolonging S phase, and inducing DNA damage-induced checkpoint pathways (9).
TDG localizes to gene promoters, where it interacts with a number of transcription factors and transcription coactivators, including estrogen receptor, retinoic acid receptor, and p300/CBP, supporting a direct role of TDG in transcriptional regulation (10–13). TDG knock-out in mice is embryonically lethal, and the lethal phenotype is associated with aberrant promoter methylation and imbalanced histone modifications (13, 14).
Cullin ring finger ligase (CRL) ubiquitin ligases are a large family of enzymes based on one of seven cullins (Cul1, Cul2, Cul3, Cul4A, Cul4B, Cul5, and Cul7). The C-terminal “cullin homology domain” of the cullin associates with a ring finger protein that helps recruit an E2, whereas the N-terminal domain of the Cullin interacts through a bridge protein to a substrate-adaptor protein that recruits substrates to the cullin (15). In CRL4 ubiquitin ligases, Cul4A or -B associates with RBX1 through the cullin homology domain and with DDB1 at the other end. The DDB1 protein interacts with a large family of DDB1-cullin-associated factors (DCAFs) that directly bind to substrates (16). The CRL4Cdt2 ubiquitin ligase uses Cdt2 as the DCAF and has been implicated in the polyubiquitination and degradation of several substrates involved in S phase regulation, in a reaction that is dependent on PCNA (17, 18). The substrates include the DNA replication initiation factor Cdt1, the cell cycle inhibitor p21, the histone H4 Lys-20 methyltransferase Set8, Cdt2 itself, cyclin-dependent kinase inhibitor Xic1, and the fourth subunit of the replicative DNA polymerase δ, PolD4 or p12 (17–26). The target substrates of the CRL4Cdt2 E3 ubiquitin ligase complex are known to have a PIP degron comprising a consensus PCNA-interacting protein motif (PIP motif) with additional sequence requirements (17, 18, 27, 28). The conventional PIP motif is present on many proteins that interact with PCNA (proliferating cell nuclear antigen), and such interaction does not usually lead to recognition of the substrate by CRL4Cdt2. The PIP degron is a specialized PIP motif that usually has a Thr and Asp at positions 5 and 6 of the PIP motif, which increases interaction with PCNA, and a positively charged amino acid 4 amino acids downstream of the PIP box (position +4) that is not essential for interaction with PCNA but important for recognition by CRL4Cdt2. The PIP degron on a substrate protein is essential for the binding of the substrate to chromatin-bound PCNA and its recognition by CRL4Cdt2 for polyubiquitination on the way to degradation of the substrate by the proteasome.
The expression of TDG is tightly regulated during the cell cycle. In human cells, TDG accumulates during G2-M and G1 phases and is degraded during S phase by proteasomes (29). In this study, we demonstrate that the CRL4Cdt2 E3 ubiquitin ligase complex ubiquitinates and degrades TDG in a PCNA- and PIP degron-dependent manner. Because the association of PCNA with chromatin increases in S phase, the requirement that the substrate interact with PCNA before recognition by CRL4Cdt2 makes this an S phase-specific pathway of protein degradation. We show that physical interaction of TDG with PCNA, and the substrate specificity factor Cdt2, are necessary for CRL4Cdt2 to ubiquitinate and target TDG for degradation. An excess of wild type TDG slows down cell proliferation, as does CRL4Cdt2-resistant TDG(ΔPIP), indicating that the active degradation of TDG by this pathway is critical for keeping cellular levels of the protein below toxic levels.
EXPERIMENTAL PROCEDURES
Cell Lines, Plasmids, and Transfection
Human cervical carcinoma HeLa cells and human embryonic kidney 293T cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. To generate constitutive TDG-expressing vector, TDG cDNA was cloned into pMSCV-FLAG or pFEM vector. Doxycycline-inducible TDG-expressing lentiviral vector was generated by inserting TDG cDNA into FUW-tetO vector. To construct the bacterial TDG expression vector, TDG cDNA was cloned into pGEX vector. All of the constructs were generated using In-Fusion HD Plus (Clontech). TDG(ΔPIP) and TDG(ΔKR) were generated by site-directed mutagenesis. siRNA transfection was performed using Lipofectamine-RNAiMAX (Invitrogen) according to the manufacturer's instruction. siRNA targeting sequences are siGL2 against luciferase (CGTACGCGGAATACTTCGA), siCdt2 (GAATTATACTGCTTATCGA), siPCNA (GATCGAGGATGAAGAAGGA), siCul4A (GACAATCCGAATCAGTACC), and siCul4B (AGATAAGGTTGACCATATA). MG132 was purchased from Boston Biochem. MLN4924 was provided by Millennium Pharmaceuticals. The remaining reagents were purchased from Sigma-Aldrich.
Immunoprecipitations and Western Blot Analysis
For immunoprecipitations, cells were treated with 2 mm HU for 12 h and then with HU and 40 μm MG132 for an additional 6 h before lysing in lysis buffer (50 mm Tris-HCl (pH8.0), 10% glycerol, 150 mm NaCl, 1 mm EDTA, 0.1% Triton X-100, 1 mm DTT, protease inhibitors) by incubating on ice for 30 min followed by centrifugation at 15,000 rpm for 10 min. The supernatant recovered was immunoprecipitated with antibodies against PCNA or FLAG. Bound proteins were resolved on SDS-polyacrylamide gel, transferred to nitrocellulose membrane, and probed with specific antibodies. For Western blotting, cells were lysed in IPH buffer (50 mm Tris-HCl (pH8.0), 150 mm NaCl, 5 mm EDTA, 0.5% Nonidet P-40, 1 mm DTT, 10% glycerol, 10 mm NaF, 2 mm Na3VO4, protease inhibitor) at 4 °C for 30 min, and lysates were cleared by centrifugation or directly lysed in SDS sample buffer. Antibodies against TDG (Proteintech), FLAG (Sigma-Aldrich), β-actin, tubulin, HSP90, PCNA, Cul4, and GST (Santa Cruz Biotechnology, Inc.) were purchased. Antibodies against Cdt2, Myc (9E10), and HA (12CA5) have been described previously (22).
In Vivo Ubiquitination Assay
For the in vivo ubiquitination assay, 293T cells transiently transfected with HA-ubiquitin and Myc-TDG, with or without FLAG-Cdt2, were treated with 40 μm MG132 for 1 h before harvest. Cells were harvested in denaturing ubiquitination buffer (50 mm Tris-Cl (pH 8.0), 5 mm DTT, and 1% SDS) and immediately boiled for 10 min at 95 °C, followed by cooling on ice for 10 min. The lysates were sonicated, and supernatants were recovered after centrifugation at 15,000 rpm for 20 min. The supernatants were diluted with 9 volumes of buffer containing 50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 5% glycerol, 0.4% Nonidet P-40, and protease inhibitors, and Myc-TDG was immunoprecipitated by anti-Myc antibodies. Ubiquitinated TDG protein in the immunoprecipitate was detected by SDS-PAGE and immunoblotting with anti-HA antibody (30).
In Vitro Ubiquitination Assay
In vitro ubiquitination of TDG was conducted as described previously (22) with a minor modification. 293T cells were transiently transfected with Myc-TDG-expressing plasmid. Immunoprecipitate with anti-Myc antibody was eluted with Myc peptide and used as a substrate for the assay.
GST Pull-down Assay
GST, GST-TDG(WT), GST-TDG(ΔPIP), and GST-TDG(ΔKR) were purified from Escherichia coli under native conditions. The pull-down assay was carried out by incubating glutathione beads coupled with GST or GST-TDG proteins with recombinant, bacterially produced PCNA in pull-down buffer (25 mm Tris-Cl (pH 7.5), 100 mm NaCl, 1 mm DTT, 5% glycerol, 0.01% Nonidet P-40, protease inhibitors) for 2 h at 4 °C. The beads were washed three times with wash buffer (25 mm Tris-Cl (pH 7.5), 150 mm NaCl, 0.01% Nonidet P-40) and boiled in 2× SDS sample buffer. The samples were analyzed by Western blotting using anti-GST and anti-PCNA antibodies.
MTT Assay
Cells were seeded at a density of 1000/well in 96-well plates. The MTT assay was performed with CellTiter 96® Non-Radioactive Cell Proliferation Assay (Promega) according to the manufacturer's instruction.
Immunostaining
For PCNA and TDG staining, HeLa cells were fixed with ice cold methanol for 5 min. Cells were then stained as described previously (19).
Cell Cycle Analysis
Cells were trypsinized and fixed with 70% ethanol. Fixed cells were stained with 50 μg/ml propidium iodide and 50 μg/ml RNase A in PBS and analyzed by FACSCalibur flow cytometer (BD Biosciences). The graphs in Fig. 4C show the change in S phase after expression of the indicated forms of TDG, relative to the same cells where TDG was not induced by doxycycline: ((percentage of cells in S phase in doxycycline − percentage of cells in S phase in the absence of doxycycline)/percentage of cells in S phase in the absence of doxycycline) × 100%.
FIGURE 4.

TDG overexpression decreases cell proliferation, increases S phase population, and increases DNA breaks. A, Western blot analysis of doxycycline-induced TDG expression in stably transfected HeLa cell lines. B, top, doxycycline-induced TDG overexpression reduces cell growth. Cell growth was assessed by an MTT assay. Bottom, TDG(ΔKR) is not as toxic as TDG(ΔPIP). Cells grown in the absence of doxycycline still express some TDG from leaky expression (Western blot). C, TDG-overexpressing cells accumulate in S phase. Cells were treated with 50 ng/ml of doxycycline for 3 days to induce TDG(WT), TDG(ΔPIP), or TDG(ΔKR). Cell cycle profile was analyzed by flow cytometry of propidium iodide-stained cells. Left, changes in the percentage of S phase cells induced by doxycycline, relative to cells not treated with doxycycline, are plotted (mean ± S.D. (error bars) of >3 measurements). Right, percentage of cells in each phase of the cell cycle in three independent experiments. D, increase of TDG increases DNA breaks. Cells (transfected with TDG(WT) or mock-transfected) were treated with 100 ng/ml doxycycline for 3 days. DNA breaks were analyzed by an alkaline comet assay. A box and whiskers plot (median and interquartile range) shows the yield of strand breaks as evaluated from the tail moment (n > 100 cells).
Comet Assay
The alkaline comet assay was performed with CometAssay (Trevigen) according to the manufacturer's instruction. The captured images were analyzed by OpenComet and ImageJ software.
RESULTS
TDG Is Degraded in S Phase by a Proteasome-dependent Pathway
Treating HeLa cells with MG132 or MLN4924 to block proteasomal or cullin ubiquitin ligase activities, respectively, increased basal TDG protein levels (Fig. 1A). MLN4924 is an inhibitor of Nedd8-activating enzyme and inhibits the action of all cullins by preventing their neddylation (31). We confirmed by immunofluorescence that the amount of TDG protein is selectively diminished in S phase cells that can be distinguished by the appearance of foci of PCNA in the nucleus (Fig. 1B, top). Treating cells with the proteasome inhibitor MG132 results in the accumulation of TDG in the cells containing S phase-specific PCNA foci (Fig. 1B, bottom). This suggested that cullin-mediated ubiquitylation and proteasomes are involved in the degradation of TDG in S phase.
FIGURE 1.
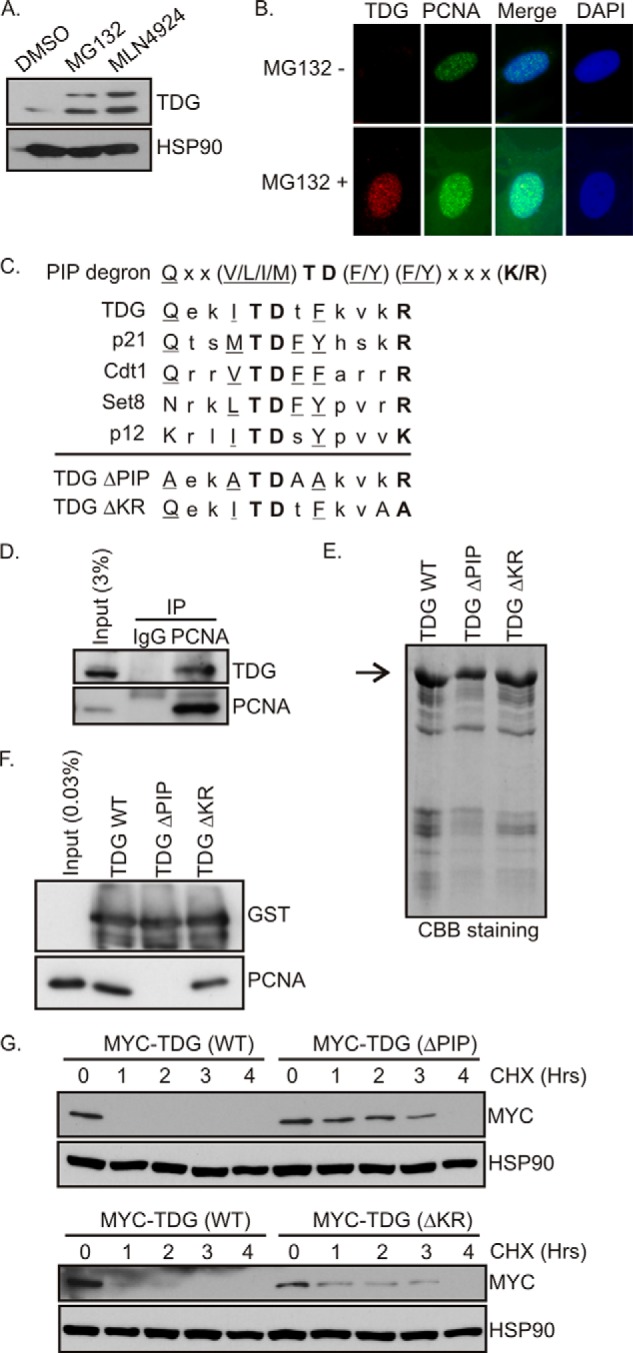
TDG interacts with PCNA. A, MG132 and MLN4924 stabilize TDG. HeLa cells (asynchronous) were treated with DMSO, MG132 (20 μm), or MLN4924 (5 μm) for 6 h. The cell lysates were probed with the indicated antibodies. B, immunofluorescence of TDG and PCNA. TDG staining is absent in the cells with S phase-specific PCNA foci unless cells are treated with MG132. C, PIP degron of CRL4 Cdt2 substrates. Conserved residues in the canonical PIP box are underlined. Conserved residues in the PIP degron are in boldface type. The mutations made in TDG(ΔPIP) and TDG(ΔKR) are shown. D, TDG interacts with PCNA. Cell extracts from HeLa cells treated with 40 μm MG132 with or without 2 mm HU were immunoprecipitated (IP) with anti-PCNA or control IgG antibodies, and the precipitates were immunoblotted with anti-TDG and anti-PCNA antibodies. E, purification of recombinant proteins. Coomassie Brilliant Blue staining of gel showing the purification of GST-TDG(WT), GST-TDG(ΔPIP), and GST-TDG(ΔKR). F, in vitro GST pull-down assay. Purified PCNA was incubated either with GST-TDG(WT) or its mutants immobilized on glutathione beads. After washing, proteins bound to beads were resolved on 10% SDS gel, and the blot was probed with anti-PCNA and GST antibodies. G, mutations in the PIP degron increase half-life of TDG. 293T cells were transfected with Myc-TDG(WT), Myc-TDG(ΔPIP), or Myc-TDG(ΔKR). After 48 h of transfection, cells were treated with cycloheximide (50 μg/ml) and harvested at different time points as indicated. The blots were probed with anti-Myc antibodies.
TDG Interacts with PCNA through a PIP Degron
A consensus PIP degron (from amino acid 95 to 106) was identified in TDG (GenBankTM, NP 003202.3) using ClustalW analysis (Fig. 1C). Besides the residues typical of all PIP motifs, the sequence had the characteristic TD and basic residue that marks PIP degrons and is present on several targets of CRL4Cdt2.
To investigate whether TDG physically interacted with PCNA, we treated cells with MG132 to prevent the degradation of any TDG and immunoprecipitated PCNA. Immunoprecipitating endogenous PCNA co-precipitated endogenous TDG from these lysates (Fig. 1D). To confirm the role of the PIP degron in mediating the interaction of TDG with PCNA, we made point mutations in the PIP degron (Fig. 1C) of GST-tagged recombinant TDG protein (Fig. 1E) for in vitro pull-down assays. Upon incubation with purified recombinant PCNA, GST-TDG(WT) pulled down PCNA, whereas mutations in the PIP box (TDG(ΔPIP)) disrupted the interaction (Fig. 1F). Mutations in the KR amino acids in the PIP degron (TDG(ΔKR)) did not affect the interaction of TDG with PCNA (Fig. 1F), consistent with the previous observation that these residues are not essential for stable interaction with PCNA, although they are essential for degradation.
To investigate the role of the PIP degron in TDG degradation, we compared the half-life of exogenously expressed TDG(WT), TDG(ΔPIP), and TDG(ΔKR) after blocking new protein synthesis with cycloheximide. The PIP degron mutants, TDG(ΔPIP) or TDG(ΔKR) are more stable than TDG(WT) (Fig. 1G). These results demonstrate that association of TDG with PCNA through the PIP degron is important for the turnover of TDG in cells.
The PIP Degron of TDG Is Required for Its Degradation in S Phase
To elucidate whether the PIP degron of TDG is needed for its destruction during S phase, cells expressing wild type FLAG-TDG(WT) or PIP degron mutants, FLAG-TDG(ΔPIP) and FLAG-TDG(ΔKR), were treated with HU to arrest cells in S phase. Compared with wild type TDG, which was decreased in HU, the PIP degron mutants were not decreased in HU-arrested cells (Fig. 2A).
FIGURE 2.
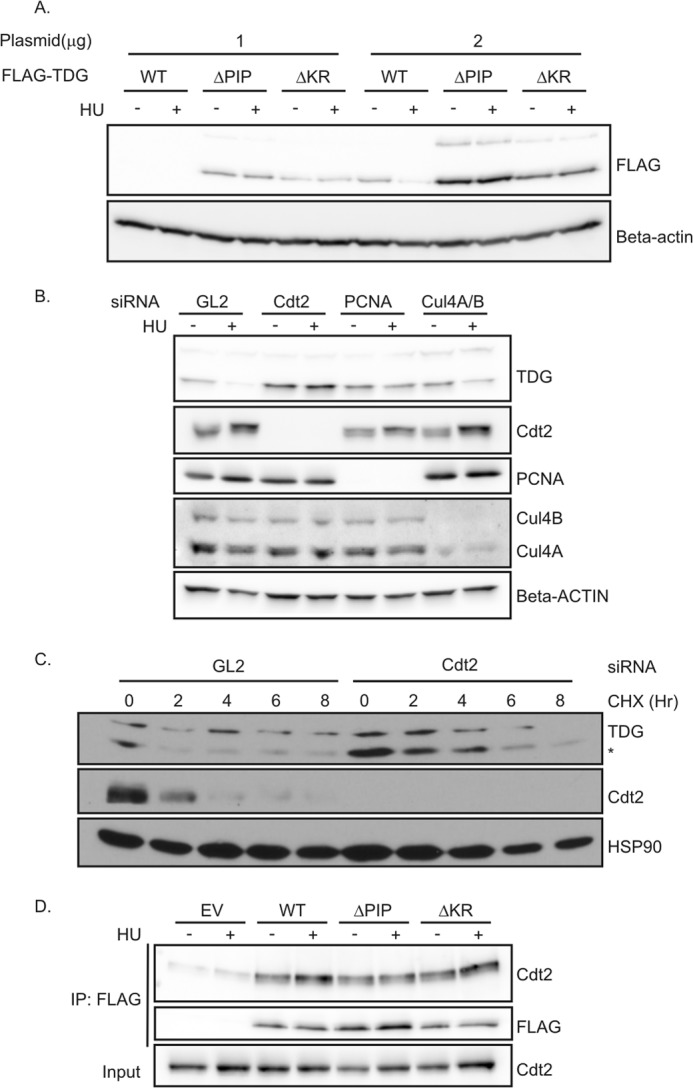
CRL4Cdt2 is required for the degradation of TDG. A, TDG mutants were not degraded after HU treatment. 293T cells in 6-well plates were transiently transfected with 1 or 2 μg of TDG(WT)-, TDG(ΔPIP)-, or TDG(ΔKR)-expressing plasmids for 24 h, followed by treatment with 1.5 mm HU for 24 h. Western blot analyses were performed using the indicated antibodies. B, HeLa cells were treated with the indicated siRNA. siRNA against luciferase (GL2) was used as a negative control. After 48 h, 1.5 mm HU was added for 24 h. Cell lysates were subjected to immunoblotting with anti-TDG and anti-β-actin antibodies. C, knockdown of Cdt2 increased the half-life of TDG. HeLa cells were treated with GL2 siRNA or siRNA against Cdt2 for 60 h, followed by protein synthesis inhibitor cycloheximide treatment (100 μg/ml) for the indicated times. The whole cell lysate was probed with antibodies against indicated proteins. *, lower band is TDG. D, TDG(WT) and mutants co-immunoprecipitate with Cdt2. Nuclear fraction of 293T cells transiently expressing the indicated forms of FLAG-TDG were immunoprecipitated (IP) with anti-FLAG. Association was analyzed by Western blot using anti-Cdt2 and anti-FLAG antibodies. EV, empty vector.
The Down-regulation of TDG in S Phase Requires CRL4Cdt2
CRL4Cdt2 is known to degrade its substrates during S phase of the cell cycle in cooperation with PCNA (19, 21–25). Having established above that the PIP degron is responsible for TDG destruction in S phase, we wanted to address whether CRL4Cdt2 and PCNA are involved in TDG degradation. We treated asynchronously cycling cells with siRNAs to knock down individual components of the CRL4Cdt2 complex or PCNA and then arrested them in S phase with HU. Compared with cells transfected with negative control luciferase GL2 siRNA, cells transfected with siRNAs targeting Cdt2, Cul4A/B, or PCNA expressed elevated levels of TDG protein in HU-arrested cells (Fig. 2B).
We next examined whether Cdt2 is required for the turnover of TDG in cells asynchronously cycling through the cell cycle. Depleting Cdt2 increased the basal level of TDG and increased the half-life of TDG from <2 to 4 h, as measured after blocking new protein synthesis with cycloheximide (Fig. 2C).
It has been suggested that the interaction of a substrate with Cdt2 is dependent on the interaction of the substrate with PCNA and on the basic residue of the PIP degron (28). To examine whether a substrate of CRL4Cdt2 had to be associated with PCNA or required the basic residue at +4, before it could be recognized by the E3 ligase, we examined the interaction of TDG WT and mutants with Cdt2 by immunoprecipitation (Fig. 2D). Cdt2 was co-immunoprecipitated equally well by WT and by the ΔPIP or ΔKR mutants of TDG, an observation that will be discussed later. These results demonstrate that CRL4Cdt2 E3 ubiquitin ligase is required to degrade TDG in a PCNA- and PIP degron-dependent manner.
TDG Is Polyubiquitinated by CRL4Cdt2 E3 Ubiquitin Ligase in a PIP Degron-dependent Manner
To demonstrate the role of CRL4Cdt2 in ubiquitination of TDG, we measured the ubiquitination of Myc-tagged TDG in vivo after overexpressing FLAG-Cdt2. The ubiquitination was followed by co-transfecting HA-ubiquitin and then carrying out an immunoblot for HA-ubiquitin on Myc-TDG immunoprecipitates. Myc-TDG was polyubiquitinated when transfected by itself, but overexpression of FLAG-Cdt2 significantly increased the polyubiquitination (Fig. 3A). Thus, Cdt2 may be rate-limiting for the polyubiquitination of TDG.
FIGURE 3.
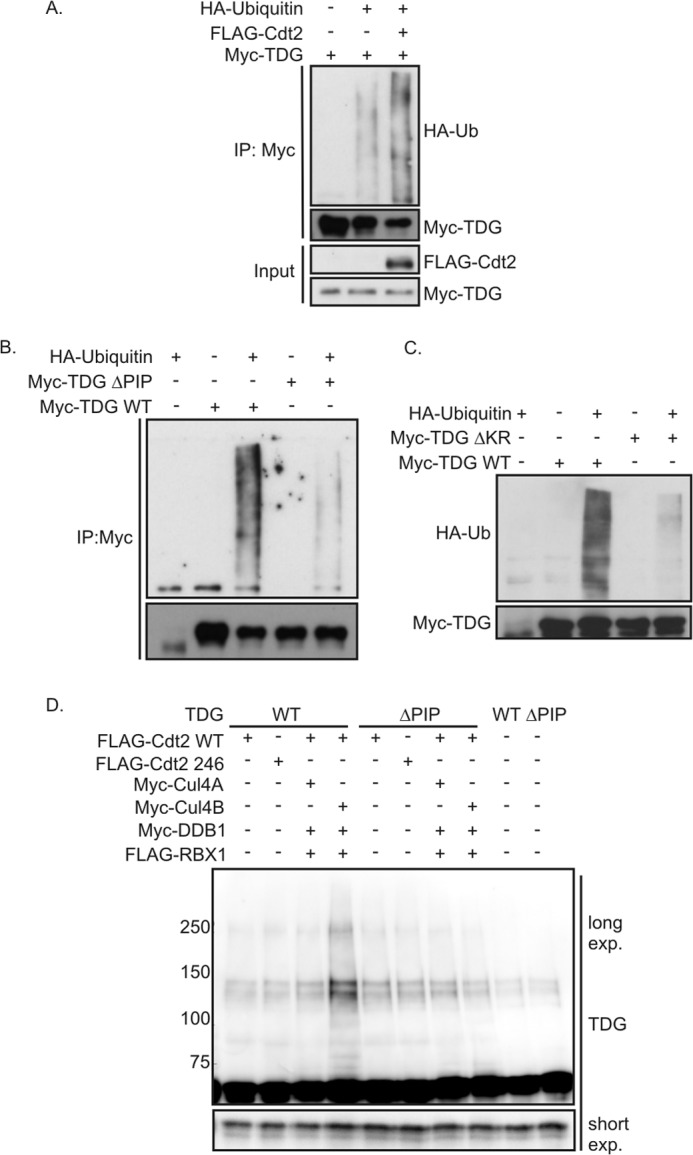
CRL4Cdt2 ubiquitinates TDG. A, in vivo ubiquitination assay of TDG. Myc-TDG was transfected into 293T cells with or without FLAG-Cdt2 and HA-ubiquitin. Cells were lysed under denaturing conditions and subjected to immunoprecipitation (IP) with anti-Myc and processed for immune blotting with the indicated antibodies. B and C, in vivo ubiquitination assay. Myc TDG WT or mutants were transfected into 293T cells with or without HA-ubiquitin. Cells were lysed under denaturing conditions and subjected to immune precipitation with anti-Myc and processed for immune blotting with the indicated antibodies. The exposure time is longer than in A to better see the polyubiquitination carried out by endogenous Cdt2. D, in vitro ubiquitination assay. Myc TDG was immunopurified from 293T cells to use as substrate. CRL4Cdt2 complex created by transiently overexpressing FLAG-Cdt2, FLAG-RBX1, Myc-DDB1 with Myc-Cul4A, or Myc-Cul4B was immunopurified with anti-FLAG from 293T cells and used as E3 ligase. Anti-TDG Western blot was performed on the reaction products to detect high molecular weight forms of TDG.
We next asked in a similar assay, but without co-transfection of FLAG-Cdt2, whether the PIP degron of TDG is essential for polyubiquitination in vivo. The polyubiquitination of PIP degron mutants, Myc-TDG(ΔPIP) or Myc-TDG(ΔKR), was decreased compared with that of Myc-TDG(WT) (Fig. 3, B and C).
The polyubiquitination of TDG seen in the figures above could have been indirect and downstream of CRL4Cdt2, although it is unlikely that a different E3 ubiquitin ligase will be as dependent as CRL4Cdt2 on the presence of a PIP degron in the substrate. To test whether TDG is directly polyubiquitinated by CRL4Cdt2, we reconstituted the polyubiquitination reaction in vitro. Components of CRL4Cdt2 E3 ubiquitin ligase complex were co-expressed in cells and immunopurified by anti-FLAG antibody and eluted with the FLAG epitope peptide. Cdt2R246A, a mutant that cannot interact with DDB1, will not co-purify with the whole E3 ligase complex and so is used as a negative control. The substrate, Myc-TDG (WT or ΔPIP), was expressed in a different culture of cells and purified by anti-Myc antibody and eluted with Myc epitope peptide. After in vitro incubation, polyubiquitination of TDG is detected by immunoblotting the reaction mix with anti-TDG antibody and looking for higher molecular weight bands of TDG (Fig. 3D). Despite the presence of a background doublet of around 140 kDa, which increased a little in all lanes upon incubation even with partial Cdt2 complexes, the full E3 ligase clearly stimulated the polyubiquitination of wild type TDG but not the PCNA binding-deficient ΔPIP mutant (Fig. 3D, lane 4 versus lane 8). Overall, these results suggest that PCNA-bound TDG is directly polyubiquitinated by the CRL4Cdt2 E3 ubiquitin ligase complex.
CRL4Cdt2-resistant TDG Slows Cell Proliferation More than Its Wild Type Counterpart
To understand why TDG needs to be degraded by CRL4Cdt2, we overexpressed WT and mutant TDG in various cell lines. Constitutive stable overexpression of untagged WT TDG, however, slowed cell proliferation at very early passages and made it difficult to obtain cells with overexpressed TDG (data not shown) (29). Thus, we stably transfected TDG(WT) and TDG(ΔPIP) in HeLa cells under a doxycycline-inducible promoter (Fig. 4A). At 0 and 10 ng/ml doxycycline, the TDG(ΔPIP) protein was present at a higher concentration compared with TDG(WT), as would be expected for the stable mutant, but the differences were not apparent at higher concentrations of 20 and 50 ng/ml doxycycline. At these higher concentrations, both forms of TDG induced markers of DNA damage and of checkpoint activation, such as phospho-H2AX (or γH2AX) and phospho-Chk1 (Fig. 4A).
At even 0 ng/ml doxycycline, there was enough leaky expression of TDG for TDG(ΔPIP) to significantly inhibit cell proliferation compared with TDG(WT) and TDG(ΔKR) (Fig. 4B, both panels). At the highest dose of doxycycline, cell proliferation was slowed even by cells expressing TDG(WT), although TDG(ΔPIP) inhibited cell proliferation even further (Fig. 4B, top). TDG(ΔKR) was surprisingly not as toxic as TDG(WT) (Fig. 4B, bottom), suggesting that the KR sequence itself is required for some function of TDG responsible for its toxicity. These results suggest that a higher than normal level of wild type TDG was sufficient to slow cell proliferation and that the CRL4Cdt2-mediated degradation of TDG is important to keep the level of TDG below this toxicity threshold.
Because TDG is degraded by CRL4Cdt2 in S phase, we tested whether the effect of elevated TDG was particularly deleterious to cells in S phase or later in the cell cycle. Propidium iodide-stained FACS analysis of these cells showed a significant increase in the S phase population when 50 ng/ml doxycycline was added even to cells expressing TDG(WT) (Fig. 4C). The S phase accumulation was the same with TDG(ΔPIP), which suppressed cell proliferation further, suggesting that an increase in TDG may be mildly toxic to passage of cells in S phase, but this is not the major explanation for the toxic effect of elevated TDG. TDG(ΔKR) caused significantly less S phase accumulation (Fig. 4C), consistent with the notion that the KR mutations affect some function of TDG that diminishes its toxicity.
Some DNA glycosylases, such as 3-methyladenine DNA glycosylase II (AlkA) from E. coli and MPG from humans removes normal bases from undamaged DNA (32). The resulting abasic sites could be excised by endonucleases leading to DNA breaks in the cell. To explore whether TDG overexpression is toxic because it induces more DNA breaks in cells, we conducted alkaline comet assays to quantitate the amount of single strand and double strand DNA breaks when TDG(WT) is induced by doxycycline. Compared with mock-transfected cells, TDG(WT)-overexpressing cells showed a significant increase in DNA breaks (Fig. 4D), suggesting that the toxicity seen when TDG levels exceed a particular threshold are mostly from the DNA repair activity of TDG rather than the demethylating activity of TDG. This is consistent with the reported low levels of hydroxymethylcytosine (33) (and thus low levels of 5-formylcytosine and 5-carboxylcytosine) in HeLa cells, suggesting that any excess TDG is unlikely to have a huge epigenetic change in such cells.
CRL4Cdt2-resistant TDG Enhances DNA Damage and Promotes Sensitivity of Cells to 5-FU Treatment
5-FU is known to arrest cells in G1/S phase in a TDG-dependent manner by eliciting Chk1 phosphorylation. We wondered whether an elevated level of TDG would trigger excessive DNA damage in 5-FU-treated cells. Cells stably expressing FLAG-TDG(WT), FLAG-TDG(ΔPIP), or FLAG-TDG(ΔKR) were incubated with 5-FU for 20 h (Fig. 5A). Although FLAG-TDG(WT) was significantly more abundant than wild type endogenous TDG, it was degraded in 5-FU-treated cells. TDG(ΔPIP) or TDG(ΔKR), however, was stable in the continued presence of 5-FU. Parenthetically, TDG is also decreased after DNA damage induced by other agents, such as camptothecin (inhibitor of DNA topoisomerase I) or doxorubicin (inhibitor of topoisomerase II) (data not shown).
FIGURE 5.
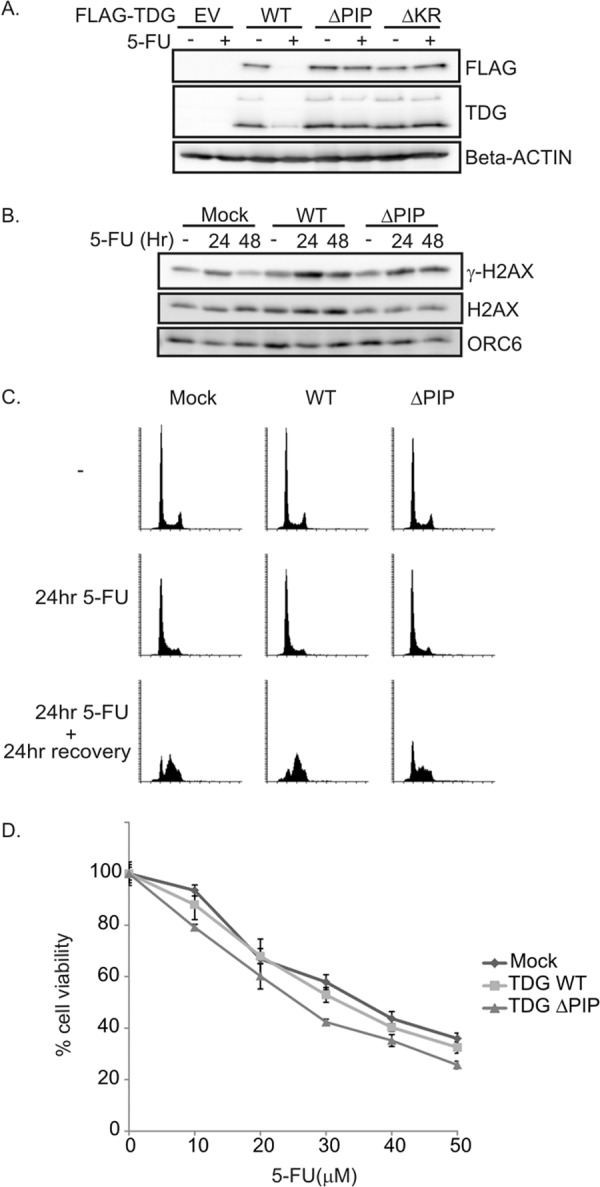
TDG(ΔPIP)-expressing cells are slightly sensitive to 5-FU. A, TDG mutants were not degraded upon DNA damage with 5-FU treatment. HeLa cell lines expressing TDG(WT) or mutants were exposed to 5-FU for 20 h. Western blot analyses were carried out using the indicated antibodies. B, delayed repair of DNA damage induced by 5-FU in HeLa cells expressing TDG(WT) or TDG (ΔPIP). Cells were exposed to 20 μm 5-FU for 24 h followed by a 24-h recovery period. P-H2AX (Ser-139) and H2AX levels were analyzed by Western blot. ORC6 was used as a loading control. C, TDG(ΔPIP) expression slows down resumption of S phase after G1/S arrest induced by 5-FU treatment. HeLa cells expressing TDG(WT) or TDG(ΔPIP) were exposed to 20 μm 5-FU for 24 h, followed by a 24-h recovery period. Cell cycle profile was analyzed by flow cytometry of propidium iodide-stained cells. D, HeLa cells expressing TDG(WT) or TDG(ΔPIP) were cultured with the indicated concentrations of 5-FU for 24 h, and cell viability was assessed on day 5 with the MTT assay. The relative cell viability was expressed as a percentage relative to the untreated cells. Error bars, S.D.
To test the impact of TDG overexpression upon exposure to 5-FU, cells expressing TDG(WT) or TDG(ΔPIP) were incubated with 5-FU for 24 h followed by 24 h of recovery. Because the difference in expression of TDG(WT) and TDG(ΔPIP) was evident at 0 nm doxycycline (Fig. 4A), the following experiments were done without any doxycycline. γH2AX, a marker of DNA damage, was induced at 24 h upon 5-FU exposure but decreased to normal levels in the mock-transfected cells after 24 h of recovery without 5-FU (Fig. 5B). In contrast, γ-H2AX induced by 5-FU treatment was not decreased even after 24 h of recovery in cells overexpressing TDG(WT) or TDG(ΔPIP) (Fig. 5B). Thus, although TDG(WT) is degraded during 5-FU exposure (Fig. 5A), the overexpression of TDG(WT) during the recovery period is deleterious for the quick repair of the DNA damage during the next 24 h. The stable TDG(ΔPIP) was not degraded during 5-FU treatment and, of course, was also overexpressed during the recovery period and thus predictably delayed the quick repair of DNA damage.
To examine the cell cycle progression during the recovery from 5-FU treatment in TDG-overexpressing cells, cells were incubated with 5-FU for 24 h and allowed to recover for 24 h without 5-FU, and then the cell cycle profile was analyzed. 5-FU caused the cells to accumulate in G1 or early S to the same extent regardless of whether wild-type or stable TDG was overexpressed (Fig. 5C). However, following release from 5-FU, more of the control cells (mock-transfected) or cells expressing TDG(WT) progressed through S phase than cells expressing TDG(ΔPIP), where more cells remained at the G1/S boundary (Fig. 5C). Although γH2AX levels were elevated in cells overexpressing TDG(WT), those cells had no difficulty in resuming S phase once 5-FU was removed. These results indicate that TDG(ΔPIP)-expressing cells have difficulty recovering from the 5-FU and resuming S phase when the drug is removed but that this delay is not explained by the delay in repair of DNA damage.
Next we asked whether the initial delay in repair of damage or delay in re-entering a normal cell cycle makes TDG(ΔPIP)-expressing cells more susceptible to 5-FU in the longer term. MTT assays were used to measure the viability of cells expressing TDG(WT) or TDG(ΔPIP) 5 days after treatment with different doses of 5-FU for 24 h. Although the basal level of TDG(WT) was much higher than endogenous TDG and there was an initial delay in the repair of DNA damage, cells overexpressing TDG(WT) were not more sensitive to 5-FU than control cells with empty vector (Fig. 5D). In contrast, the cells overexpressing TDG(ΔPIP) were slightly more sensitive to 5-FU. The difference between TDG(ΔPIP) and TDG(WT) suggests that the initial delay in resuming S phase when 5-FU was removed in the former contributed more to the longer term toxicity than the delay in repairing the DNA damage. In keeping with the suggestion that the KR residues of TDG are required for a function of TDG independent of direction to CRL4Cdt2, TDG(ΔKR) did not make the cells more susceptible to 5-FU (data not shown).
DISCUSSION
CRL4Cdt2 E3 ubiquitin ligase complex regulates the cell cycle by degrading key replication initiation proteins such as Cdt1 to ensure a single round of replication licensing per cell cycle. Additionally, the enzyme complex degrades CDK inhibitor p21 as cells recover from DNA damage and degrades Set8 histone methyltransferase for progression from G2 to M phase of the cell cycle. CRL4Cdt2 is also involved in the degradation of several substrates after DNA damage: Cdt1, p21, Set8, Pold4, etc. (19, 21–25). It is believed that the DNA repair processes that follow DNA damage lead to the recruitment of PCNA to chromatin, and this triggers the polyubiquitination of PIP degron-containing substrates by CRL4Cdt2. In this study, we report that TDG is a CRL4Cdt2 substrate for polyubiquitination during S phase, consistent with the previous observation of low levels of TDG in S phase and its accumulation during G2/M-G1 phases (29). Like other substrates of CRL4Cdt2 (e.g. Cdt1 and Set8), TDG is degraded in a PCNA-dependent manner. We found that TDG contains a conserved PIP degron, through which it comes in association with chromatin-bound PCNA so as to present itself as a substrate for ubiquitination activity of the CRL4Cdt2 E3 ligase complex. Disruption of the interaction between TDG and PCNA or decrease of CRL4Cdt2 enzyme complex prevents TDG ubiquitination and degradation.
The mechanism by which PCNA-substrate interaction and the basic residue at +4 of the PIP degron contribute to polyubiquitination of a substrate by CRL4Cdt2 is not completely clear. Although it has been suggested that the interaction of a substrate with Cdt2 is dependent on the interaction of the substrate with PCNA, we had noted that p21 with a deletion of its PIP box still co-immunoprecipitated with components of CRL4Cdt2 (22). In addition, the basic residue at +4 has been suggested to be essential for interaction between substrate and Cdt2 (28). It is thus surprising that the ΔPIP or ΔKR mutants of TDG co-immunoprecipitated Cdt2 as well as wild-type TDG (Fig. 2D). The interaction of TDG(ΔPIP) with Cdt2 is similar to the interaction of p21 ΔPIP with CRL4Cdt2 and suggests that physical association of some substrates and Cdt2 can occur in the absence of an associated PCNA. Similarly, the basic residue in the PIP degron is dispensable for the co-immunoprecipitation of TDG(ΔKR) with Cdt2. However, the absolute requirement of the PIP degron, of the basic residue in the PIP degron, and of PCNA for the degradation of CRL4Cdt2 substrates also suggests that simple co-immunoprecipitation of a substrate (TDG or p21) with CRL4Cdt2 does not indicate a productive interaction that can lead to polyubiquitination. Most likely, the various components mentioned (PCNA, PIP degron, and the basic residue of the PIP degron) are required for more than simply promoting the association of Cdt2 with its substrate.
The degradation-resistant TDG(ΔPIP) decreases cell proliferation. Stable TDG (or simply overexpressed TDG(WT)) increases the S phase population by about 25%, although UNG2, another DNA glycosylase, is more important for base excision during S phase (29). Most likely, increase of TDG through overexpression or stabilization increases the pool of glycosylases that can recognize a mismatched T (or U) in S phase, so more glycosylases bind to mismatched T-G pairs for deglycosylation and thus slow down S phase progression. The increased glycosylase activity and attendant AP endonuclease activity probably explain why a global increase in TDG or prevention of the degradation of TDG in S phase slows cell proliferation, increases the S phase population, and increases γH2AX and DNA breaks.
TDG is involved in epigenetic regulation of the genome by playing a role in DNA demethylation. Homozygous knockout of TDG results in an embryonic lethal phenotype, indicating that essential functions other than base mismatch repair are associated with TDG and are required in early stages of embryonic development. DNA methyltransferase DNMT1 methylates cytosine bases in S phase in a replication-coupled manner. The methylation (methylcytosine) is predominant at CpG islands. In collaboration with TET dioxygenase enzymes, TDG is involved in 5-methyl cytosine demethylation and regulates the gene expression. We explored whether degradation of TDG by CRL4Cdt2 complex in S phase is a mechanism to prevent the initiation of antagonistic DNA demethylation by TET and TDG enzymatic reaction cascade in S phase. However, overexpressing FLAG-TDG (WT or ΔPIP) did not significantly change gene expression, as measured by microarray hybridization, compared with control cells (data not shown), suggesting that TET is the rate-limiting enzyme in CpG demethylation.
The chemotherapeutic drug 5-FU is used in therapy of colorectal, gastrointestinal, breast, and ovarian cancers. TDG mediates DNA-directed toxicity and DNA damage response in response to 5-FU. Inactivation of TDG leads to cellular resistance to 5-FU, although TDG(WT) is mostly degraded after a mere 24 h of exposure to 5-FU (Fig. 5A). On the other hand, we show that forced expression of degradation-resistant TDG (ΔPIP) slightly sensitizes cells to 5-FU. The increase in sensitivity was not remarkable and may indicate that there is already enough TDG in the cell to produce maximal toxicity in response to the doses of 5-FU used. Any further increase of TDG levels does not increase the toxicity of 5-FU significantly. Similar results are seen in two other cell lines, NIH3T3 and HEK293T. Two points are worth exploring to test whether the sensitization to 5-FU is of clinical utility. (a) will multiple cycles of 5-FU treatment and recovery further increase the 5-FU sensitivity of cells expressing stable TDG? (b) Will the stabilization of TDG by MLN4924 sensitize cancer cells to 5-FU?
Walter and co-workers (34) have independently discovered that Xenopus TDG is degraded by CRL4Cdt2 in Xenopus egg extracts in a reaction dependent on PCNA and the PIP degron of TDG. The conservation of this S phase- and DNA damage-specific degradation pathway in at least two vertebrates suggests that TDG degradation by this pathway is useful for the cell. Although excess TDG is toxic, stable TDG is not significantly more toxic than overexpressed TDG(WT) when measuring cell proliferation or S phase progression. We suggest, therefore, that although this is a cell cycle stage-specific degradation, the main function of the pathway is to keep the TDG levels below a toxicity threshold and not specifically an S phase function. This is thus different from many of the other CRL4Cdt2 substrates like p21, Cdt1, Set8, or PolD4, whose degradation in S phase or following DNA damage is important for regulation of DNA replication or DNA damage repair.
Acknowledgments
We thank members of the Dutta laboratory for helpful discussions, Dr. J. C. Walter (Harvard University) for communicating results prior to publication, and Dr. A. Bellacosa (University of Pennsylvania) for valuable discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 CA60499 and CA166054 (to A. D.).
- TDG
- thymine DNA glycosylase
- TET
- ten-eleven translocation
- 5-FU
- 5-fluorouracil
- CBP
- cAMP-response element-binding protein-binding protein
- CRL
- cullin ring finger ligase
- PIP
- PCNA-interacting peptide
- PCNA
- proliferating cell nuclear antigen
- HU
- hydroxyurea
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- SUMO
- small ubiquitin-like modifier
- DCAF
- DDB1-cullin-associated factor.
REFERENCES
- 1. Hendrich B., Hardeland U., Ng H. H., Jiricny J., Bird A. (1999) The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature 401, 301–304 [DOI] [PubMed] [Google Scholar]
- 2. Haushalter K. A., Todd Stukenberg M. W., Kirschner M. W., Verdine G. L. (1999) Identification of a new uracil-DNA glycosylase family by expression cloning using synthetic inhibitors. Curr. Biol. 9, 174–185 [DOI] [PubMed] [Google Scholar]
- 3. Hardeland U., Bentele M., Lettieri T., Steinacher R., Jiricny J., Schar P. (2001) Thymine DNA glycosylase. Prog. Nucleic Acids Res. Mol. Biol. 68, 235–253 [DOI] [PubMed] [Google Scholar]
- 4. Slupphaug G., Eftedal I., Kavli B., Bharati S., Helle N. M., Haug T., Levine D. W., Krokan H. E. (1995) Properties of a recombinant human uracil-DNA glycosylase from the UNG gene and evidence that UNG encodes the major uracil-DNA glycosylase. Biochemistry 34, 128–138 [DOI] [PubMed] [Google Scholar]
- 5. Hardeland U., Steinacher R., Jiricny J., Schär P. (2002) Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J. 21, 1456–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. He Y. F., Li B. Z., Li Z., Liu P., Wang Y., Tang Q., Ding J., Jia Y., Chen Z., Li L., Sun Y., Li X., Dai Q., Song C. X., Zhang K., He C., Xu G. L. (2011) Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Longley D. B., Harkin D. P., Johnston P. G. (2003) 5-Fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer 3, 330–338 [DOI] [PubMed] [Google Scholar]
- 8. Heidelberger C., Chaudhuri N. K., Danneberg P., Mooren D., Griesbach L., Duschinsky R., Schnitzer R. J., Pleven E., Scheiner J. (1957) Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature 179, 663–666 [DOI] [PubMed] [Google Scholar]
- 9. Kunz C., Focke F., Saito Y., Schuermann D., Lettieri T., Selfridge J., Schär P. (2009) Base excision by thymine DNA glycosylase mediates DNA-directed cytotoxicity of 5-fluorouracil. PLoS Biol. 7, e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tini M., Benecke A., Um S. J., Torchia J., Evans R. M., Chambon P. (2002) Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol. Cell. 9, 265–277 [DOI] [PubMed] [Google Scholar]
- 11. Um S., Harbers M., Benecke A., Pierrat B., Losson R., Chambon P. (1998) Retinoic acid receptors interact physically and functionally with the T:G mismatch-specific thymine-DNA glycosylase. J. Biol. Chem. 273, 20728–20736 [DOI] [PubMed] [Google Scholar]
- 12. Chen D., Lucey M. J., Phoenix F., Lopez-Garcia J., Hart S. M., Losson R., Buluwela L., Coombes R. C., Chambon P., Schär P., Ali S. (2003) T:G mismatch-specific thymine-DNA glycosylase potentiates transcription of estrogen-regulated genes through direct interaction with estrogen receptor α. J. Biol. Chem. 278, 38586–38592 [DOI] [PubMed] [Google Scholar]
- 13. Cortellino S., Xu J., Sannai M., Moore R., Caretti E., Cigliano A., Le Coz M., Devarajan K., Wessels A., Soprano D., Abramowitz L. K., Bartolomei M. S., Rambow F., Bassi M. R., Bruno T., Fanciulli M., Renner C., Klein-Szanto A. J., Matsumoto Y., Kobi D., Davidson I., Alberti C., Larue L., Bellacosa A. (2011) Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 146, 67–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cortázar D., Kunz C., Selfridge J., Lettieri T., Saito Y., MacDougall E., Wirz A., Schuermann D., Jacobs A. L., Siegrist F., Steinacher R., Jiricny J., Bird A., Schär P. (2011) Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 470, 419–423 [DOI] [PubMed] [Google Scholar]
- 15. Petroski M. D., Deshaies R. J. (2005) Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6, 9–20 [DOI] [PubMed] [Google Scholar]
- 16. Jackson S., Xiong Y. (2009) CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem. Sci. 34, 562–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arias E. E., Walter J. C. (2006) PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat. Cell Biol. 8, 84–90 [DOI] [PubMed] [Google Scholar]
- 18. Senga T., Sivaprasad U., Zhu W., Park J. H., Arias E. E., Walter J. C., Dutta A. (2006) PCNA is a cofactor for Cdt1 degradation by CUL4/DDB1-mediated N-terminal ubiquitination. J. Biol. Chem. 281, 6246–6252 [DOI] [PubMed] [Google Scholar]
- 19. Abbas T., Shibata E., Park J., Jha S., Karnani N., Dutta A. (2010) CRL4Cdt2 regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol. Cell. 40, 9–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Abbas T., Mueller A. C., Shibata E., Keaton M., Rossi M., Dutta A. (2013) CRL1-FBXO11 promotes Cdt2 ubiquitylation and degradation and regulates Pr-Set7/Set8-mediated cellular migration. Mol. Cell. 49, 1147–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Terai K., Shibata E., Abbas T., Dutta A. (2013) Degradation of p12 subunit by CRL4Cdt2 E3 ligase inhibits fork progression after DNA damage. J. Biol. Chem. 288, 30509–30514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Abbas T., Sivaprasad U., Terai K., Amador V., Pagano M., Dutta A. (2008) PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 22, 2496–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jin J., Arias E. E., Chen J., Harper J. W., Walter J. C. (2006) A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell. 23, 709–721 [DOI] [PubMed] [Google Scholar]
- 24. Oda H., Hübner M. R., Beck D. B., Vermeulen M., Hurwitz J., Spector D. L., Reinberg D. (2010) Regulation of the histone H4 monomethylase PR-Set7 by CRL4Cdt2-mediated PCNA-dependent degradation during DNA damage. Mol. Cell. 40, 364–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Centore R. C., Havens C. G., Manning A. L., Li J. M., Flynn R. L., Tse A., Jin J., Dyson N. J., Walter J. C., Zou L. (2010) CRL4Cdt2-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol. Cell. 40, 22–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim D. H., Budhavarapu V. N., Herrera C. R., Nam H. W., Kim Y. S., Yew P. R. (2010) The CRL4Cdt2 ubiquitin ligase mediates the proteolysis of cyclin-dependent kinase inhibitor Xic1 through a direct association with PCNA. Mol. Cell. Biol. 30, 4120–4133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Michishita M., Morimoto A., Ishii T., Komori H., Shiomi Y., Higuchi Y., Nishitani H. (2011) Positively charged residues located downstream of PIP box, together with TD amino acids within PIP box, are important for CRL4Cdt2-mediated proteolysis. Genes Cells 16, 12–22 [DOI] [PubMed] [Google Scholar]
- 28. Havens C. G., Walter J. C. (2009) Docking of a specialized PIP Box onto chromatin-bound PCNA creates a degron for the ubiquitin ligase CRL4Cdt2. Mol. Cell. 35, 93–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hardeland U., Kunz C., Focke F., Szadkowski M., Schär P. (2007) Cell cycle regulation as a mechanism for functional separation of the apparently redundant uracil DNA glycosylases TDG and UNG2. Nucleic Acids Res. 35, 3859–3867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dar A., Shibata E., Dutta A. (2013) Deubiquitination of Tip60 by USP7 determines the activity of the p53-dependent apoptotic pathway. Mol. Cell. Biol. 33, 3309–3320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Soucy T. A., Smith P. G., Milhollen M. A., Berger A. J., Gavin J. M., Adhikari S., Brownell J. E., Burke K. E., Cardin D. P., Critchley S., Cullis C. A., Doucette A., Garnsey J. J., Gaulin J. L., Gershman R. E., Lublinsky A. R., McDonald A., Mizutani H., Narayanan U., Olhava E. J., Peluso S., Rezaei M., Sintchak M. D., Talreja T., Thomas M. P., Traore T., Vyskocil S., Weatherhead G. S., Yu J., Zhang J., Dick L. R., Claiborne C. F., Rolfe M., Bolen J. B., Langston S. P. (2009) An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736 [DOI] [PubMed] [Google Scholar]
- 32. Berdal K. G., Johansen R. F., Seeberg E. (1998) Release of normal bases from intact DNA by a native DNA repair enzyme. EMBO J. 17, 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Song C. X., Szulwach K. E., Fu Y., Dai Q., Yi C., Li X., Li Y., Chen C. H., Zhang W., Jian X., Wang J., Zhang L., Looney T. J., Zhang B., Godley L. A., Hicks L. M., Lahn B. T., Jin P., He C. (2011) Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 29, 68–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Slenn T. J., Morris B., Havens C. G., Freeman R. M., Jr., Takahashi T. S., Walter J. C. (2014) Thymine DNA glycosylase is a CRL4Cdt2 substrate. J. Biol. Chem. 289, 23043–23055 [DOI] [PMC free article] [PubMed] [Google Scholar]