

Background: Mechanisms by which β-2-adrenoreceptor agonists effect bronchorelaxation remain unestablished.
Results: Direct inhibition of PKA via molecular approaches reversed β-agonist-mediated antagonism of procontractile signaling and relaxation of contracted airway smooth muscle (ASM) despite augmenting intracellular cAMP.
Conclusion: PKA is the primary mechanism by which β-agonists relax ASM.
Significance: PKA-dependent signaling and functions should guide the development of bronchodilator drugs.
Keywords: Adrenergic Receptor, Asthma, G Protein-coupled Receptor (GPCR), Protein Kinase A (PKA), Smooth Muscle
Abstract
Inhaled β-agonists are effective at reversing bronchoconstriction in asthma, but the mechanism by which they exert this effect is unclear and controversial. PKA is the historically accepted effector, although this assumption is made on the basis of associative and not direct evidence. Recent studies have asserted that exchange protein activated by cAMP (Epac), not PKA, mediates the relaxation of airway smooth muscle (ASM) observed with β-agonist treatment. This study aims to clarify the role of PKA in the prorelaxant effects of β-agonists on ASM. Inhibition of PKA activity via expression of the PKI and RevAB peptides results in increased β-agonist-mediated cAMP release, abolishes the inhibitory effect of isoproterenol on histamine-induced intracellular calcium flux, and significantly attenuates histamine-stimulated MLC-20 phosphorylation. Analyses of ASM cell and tissue contraction demonstrate that PKA inhibition eliminates most, if not all, β-agonist-mediated relaxation of contracted smooth muscle. Conversely, Epac knockdown had no effect on the regulation of contraction or procontractile signaling by isoproterenol. These findings suggest that PKA, not Epac, is the predominant and physiologically relevant effector through which β-agonists exert their relaxant effects.
Introduction
β-2-Adrenoreceptor (β2AR)4 agonists (β-agonists) are a cornerstone of asthma therapy, serving as both a means of long term disease control and the most effective therapy for reversing the acute bronchoconstriction associated with an asthma attack. Despite clear therapeutic benefits, β-agonist use has several disadvantages that have been the focus of continued scrutiny since their introduction nearly 50 years ago. Chronic use of β-agonist has been shown to result in a waning of effectiveness (1–3), including decreased efficacy in bronchoprotection and deterioration of asthma control (4, 5), events that have long been attributed to desensitization of the β2AR (6). Long term clinical trials and meta analyses have also raised concerns regarding long-acting β-agonists and severe exacerbation of asthma symptoms, possibly leading to an increase in asthma-related deaths among users (7–10). Although the validity of this effect is controversial (11), these findings recently led the Food and Drug Administration to add a black box warning on long-acting β-agonists and to implement more stringent guidelines for their clinical use (reviewed in Ref. 12). A mechanistic understanding of these adverse effects, as well as the need for improved or alternative bronchodilatory agents, represent key problems in the fields of asthma and chronic obstructive pulmonary disease (13).
Canonical signaling paradigms indicate that PKA is the primary effector molecule of G protein-coupled receptors signaling via Gαs, such as the β2AR activated by β-agonists. This notion has been accepted historically in the field of airway biology despite a lack of direct evidence. Many of the conclusions regarding PKA actions, in airway smooth muscle (ASM) as well as many other cell types, are made on the basis of associative data typically derived from studies in which agonists of Gs-coupled G protein-coupled receptors and other agents (e.g. forskolin) known to stimulate PKA activity yield the same outcome (e.g. relaxation of contracted ASM). The general lethality of genetic strategies for inhibiting PKA activity in vivo, nonspecific properties of most pharmacologic PKA inhibitors, and difficulty in characterizing intracellular PKA activity and confirming its inhibition, have contributed to the difficulty in establishing a mechanistic role for PKA in signaling and functional outcomes in primary cell types (14–16). To date, the most compelling data implicating PKA in β-agonist-mediated regulation of contraction is limited to studies of heat shock protein 20 (HSP-20) (17) and K+ channel regulation (18, 19). Currently, no published evidence exists directly linking PKA with the relaxant effects of β-agonists on ASM.
Challenging the long-held dogma, recent studies have implicated the exchange protein activated by cAMP (Epac) as a potentially important cAMP effector in smooth muscle cells. Published findings include the capability of Epac to mediate relaxation (20, 21), inhibit mitogen-stimulated growth (22), and modulate phenotype switching in ASM (23, 24). However, to date, studies have not established Epac as a critical or physiologically relevant effector of β-agonist-mediated relaxation in human ASM. Further progress related to therapeutic targeting of the β2AR or its downstream prorelaxant signaling is critically dependent on resolving the question of which effector (PKA, Epac, or another) comprises the required or dominant mechanism of action for these agents.
Having previously determined the unreliability of many commonly used pharmacological tools for inhibiting PKA activity in ASM cells (14, 15), we developed molecular approaches for accomplishing this task. The PKA inhibitor peptide PKI or the mutant regulatory PKA subunit RevAB (which does not bind cAMP) can be expressed as a GFP chimera utilizing viral infection of cultured cells or tissue. Expression of these proteins has already been implemented successfully to study the role of PKA in cytokine regulation of β2AR signaling (15, 16) as well as the requirement for PKA in mediating the antimitogenic properties of multiple agents, including β-agonists (25). Using these tools, this study sought to clarify the role of PKA as a cAMP effector mediating the relaxant effect of β-agonists on ASM.
EXPERIMENTAL PROCEDURES
Materials
Antibodies against vasodilator-stimulated phosphoprotein (VASP) were from BD Biosciences. Antibodies against phospho-p42/p44 and phospho-MLC-20 were from Cell Signaling Technology (Beverly, MA). Antibodies against phospho-HSP-20 was from Abcam (Cambridge, MA). Antibodies against β-actin were from Sigma. IRDye 680 or 800 secondary antibodies were from Invitrogen. All other materials were obtained from Sigma or from sources identified previously (16, 26).
Constructs
The open reading frames of PKI (from pcDNA3-PKI, provided by Tung Chan, Thomas Jefferson University) and RevAB (provided as pcDNA3-RevAB by G. Stanley McKnight, University of Washington) were PCR-cloned into the HindIII/SalI sites of pEGFP-N1 to generate pEGFPN1-PKI and pEGFPN1-RevAB, encoding the GFP chimeras PKI-GFP and RevAB-GFP. The HindIII/NotI fragments of pEGFPN1-PKI and pEGFPN1-RevAB, containing sequences encoding PKI-GFP and RevAB-GFP, respectively, were each subcloned into the retroviral vector pLNCX2 (Invitrogen) as well as the lentiviral vector pLenti (Invitrogen).
Cell Culture
Human ASM (HASM) cultures were established from human tracheae and primary bronchi as described previously (10). Third- to fifth-passage cells, stably selected after retroviral infection as described below, were plated at a density of 104 cells/cm2 in either 24-well (cAMP radioimmunoassay) or 12-well plates (immunoblotting) and maintained in Ham's F-12 medium supplemented with 10% FBS. 24 h prior to stimulation, cells were arrested in serum-free Ham's F-12 medium supplemented with 0.1% BSA.
Retroviral Expression of PKA Inhibitory Peptides
Stable expression of GFP, PKI-GFP, and RevAB-GFP was achieved by retroviral infection as described previously (15). Briefly, a retrovirus for the expression of each was produced by cotransfecting GP2-293 cells with the pVSV-G vector (encoding the pantropic vesicular stomatitis Indiana virus G protein envelope protein) and either pLNCX2-GFP, pLNCX2-PKI-GFP, or pLNCX2-RevAB-GFP using FuGENE HD (Promega, Madison, WI). 48 h after transfection, supernatants were harvested, filter-sterilized, and used to infect early (second or third) passage human ASM cultures. Cultures were selected to homogeneity (typically >95% GFP-positive, as demonstrated in Ref. 15) with 250 μg/ml G418. Stable lines expressing GFP exhibited properties similar to those of uninfected naive cells with respect to mitogen-stimulated DNA synthesis and cell proliferation, as reported previously (16).
Epac Isoform Knockdown
Human ASM cultures were transfected with Dharmacon SMARTpool oligonucleotides targeting Epac 1 using Dharmafect reagent as in previous studies (27). 24 h later, cells were plated into 6-well (real-time quantitative PCR analysis), 12-well (immunoblot analysis), or 96-well (magnetic twisting cytometry, see below) plates and maintained in culture for another 72 h until time-specific experiments were performed. Epac isoform knockdown was assessed by real-time PCR quantification of mRNA abundance (ΔΔCt values as in Ref. 27), which was determined to be a more reliable indication of magnitude of knockdown given that the immunoblot analysis of Epac isoform protein suggested that expression was either very low (Epac1) or undetectable (Epac 2) in ASM cultures (not shown).
Immunoblotting
Cultured HASM cells stably expressing GFP, PKI-GFP, and RevAB-GFP were grown to near confluence in 12-well plates, and growth was arrested for 24 h in serum-free Ham's F-12 medium with 0.1% BSA as described above. The medium was changed to plain Ham's F-12 4 h prior to stimulation. Cells were stimulated with vehicle, 50 nm and 1 μm isoproterenol (ISO), 100 μm 8-pCPT-2′-O-Me-cAMP, or 100 μm forskolin (FSK) in the absence or presence of 1 μm histamine (HIST) for 5 min. Cells were lysed as reported previously (15), sonicated briefly, electrophoresed on 10% or 4–20% SDS-polyacrylamide gels, transferred to nitrocellulose membranes, and subsequently probed with the indicated primary and secondary antibodies conjugated with infrared fluorophores (28).
Cellular cAMP Accumulation
HASM cells stably expressing GFP, PKI-GFP, or RevAB-GFP were grown to near confluence in 24-well plates as above and stimulated in PBS containing 0.1% dimethyl sulfoxide, vehicle, 50 nm and 1 μm ISO, or 100 μm FSK for 10 min. Stimulation was terminated by aspiration of the medium and quenching with 100% cold ethanol, after which cAMP was isolated and quantified by radioimmunoassay as reported previously (14).
Single-cell Ca2+ Mobilization
HASM cells stably expressing GFP or PKI-GFP were washed and loaded with 5 μm Fura-2/AM for 30 min at 37 °C. The cells were washed and maintained in Hanks' balanced salt solution containing 10 mm HEPES, 11 mm glucose, 2.5 mm CaCl2, and 1.2 mm MgCl2 (pH 7.4). The coverslips were mounted onto an open slide chamber and [Ca2+]i flux was assessed using a dual excitation fluorescence photomultiplier system (Metafluor, Molecular Devices, Sunnyvale, CA) as described previously (29). The fluorescence intensities were converted into absolute calcium concentration using a calibration curve derived from maximum (ionophore) and minimum (EGTA) calcium flux in these cells according to the software. The cells were stimulated with 10 μm HIST in Hanks' balanced salt solution to determine the agonist-induced increase in [Ca2+]i, followed by washing with plain Hanks' balanced salt solution. The cells were then stimulated with 10 μm HIST ± 5-min pretreatment with 1 μm ISO. The net calcium response was calculated by subtracting the basal from peak [Ca2+]i on agonist stimulation. Calcium response data are reported as ratio of the second HIST response to the first (S2/S1). Experiments were repeated using HASM cells obtained from different donors.
Magnetic Twisting Cytometry (MTC)
Dynamic changes in cytoskeletal stiffness in response to HIST and ISO were measured as an indicator of contraction and relaxation of isolated human ASM cells using MTC. Although the effects on cytoskeleton assembly/disassembly independent of pharmacomechanical coupling-mediated cross-bridge cycling and cell contraction per se can promote changes in cell stiffness measured by MTC, the technique has proven to be a valuable tool for the assessment of regulation of ASM contraction (30). In brief, an arginylglycylaspartic acid-coated ferrimagnetic microbead functionalized to the cytoskeleton through cell surface integrin receptors was magnetized horizontally (parallel to the surface on which cells were plated) with a brief, 1000-G pulse and twisted in a vertically aligned homogenous magnetic field (20 G) that was varying sinusoidally in time. Measurements were performed at a single frequency of 0.75 Hz. The sinusoidal twisting magnetic field causes a rotation and a pivoting displacement of the bead. As the bead moves, the cell develops internal stresses that resist bead motion (31). Lateral bead displacements in response to the resulting oscillatory torque were detected optically (in spatial resolution of ∼5 nm), and the ratio of specific torque-to-bead displacements was computed and expressed as the cell stiffness in units of pascals per nanometer.
For each individual ASM cell, stiffness was measured for a duration of 300 s. Baseline stiffness was measured for the first 0–60 s, and changes in cell stiffness in response to HIST and ISO (alone or following contraction with HIST) were measured continuously. For each cell, stiffness was normalized to its baseline stiffness before agonist stimulation.
Lentiviral Expression of Peptides
The lentivirus for the expression of each peptide was produced by cotransfecting HEK 293T cells with lentivirus packaging mix (Applied Biological Materials, Richmond, BC, Canada) and either pLenti-GFP or pLenti-PKI-GFP using LentiFectin (Applied Biological Materials) as described by the manufacturer. The medium was changed after 24 h, and virus was collected at 48 and 72 h. These collections were pooled and concentrated using 100,000 kDa molecular mass cutoff cellulose filters (Millipore, Billerica, MA).
Ex Vivo ASM Tension Development
Tracheae were excised from C57BL6 mice after euthanasia by CO2 inhalation and cleaned of surrounding connective tissue. Rings were isolated and washed in Hanks' balanced salt solution containing amphotericin, penicillin, and streptomycin before being treated with 0.05% trypsin for 5 min. Rings were washed and plated in 24-well plates containing DMEM with antibiotics. Concentrated lentivirus (pLenti) encoding GFP or PKI-GFP was added to each well along with 10 μg/ml Polybrene. Rings were incubated with virus for 48 h. Prepared rings were mounted in a multiwire myograph (ADInstruments, Colorado Springs, CO) in Krebs-Henseleit solution (pH 7.40–7.45) maintained at 37 °C with 5% CO2 and 95% O2, with frequent changing of the solution. The chambers were mounted onto the Myo-Interface (model 610 m) and connected to the transducer and PowerLab (ADInstruments) for data transferring and recording. Chart5 software for Windows (ADInstruments) was used to collect and analyze the data. A basal tension of 0.5 g was set, and the tracheal rings were stimulated with 10 μm methacholine (MCh) for 5 min. Rings were then washed, stimulated with 60 mm KCl, washed, and again stimulated with 10 μm MCh and allowed to contract for 10 min. Stimulation of the rings with 10 μm MCh produces ∼80% maximum tension in the rings, as determined previously (32). After steady-state tension was achieved, rings were treated in a dose-dependent manner with ISO (10 nm to 10 μm), and tension was recorded. The net contractile responses were determined by subtracting the basal tension from that of the peak tension on agonist stimulation, and the change in the tension was normalized to the change in tension in response to KCl. Relaxation responses were calculated as percentage change in MCh-induced tension on stimulation with different concentrations of ISO.
Statistical Analysis
Data are presented as mean ± S.E. values from n experiments, in which each experiment was performed using a different culture derived from a unique donor. Individual data points from a single experiment were calculated as the mean value from three replicate observations for cAMP accumulation assays. For immunoblot analyses, band intensities representing signals from secondary antibody conjugated with infrared fluorophores were visualized and quantified directly using the Odyssey infrared imaging system (Li-Cor, Lincoln, NE). These values were normalized to values determined for β-actin and compared among stimuli and experimental groups. Statistically significant differences among groups were assessed by either analysis of variance with Fisher's protected least significant difference post hoc analysis (GraphPad Prism 6.0; GraphPad Software, La Jolla, CA) or by Student's t test for paired samples, with values of p < 0.05 sufficient to reject the null hypothesis.
RESULTS
To examine the role of PKA in β-agonist-mediated relaxation of the airway, HASM cultures stably expressing GFP, the PKA-inhibiting peptide PKI-GFP, or the mutant regulatory subunit RevAB-GFP were generated as described under “Experimental Procedures.” Expression was verified by visualization of green fluorescence (not shown) and via immunoblotting (Fig. 1A). Inhibition of PKA activity is easily confirmed by immunoblotting for total VASP, which undergoes phosphorylation by PKA at Ser-157 to cause a mobility shift (46 to 50 kDa). This shift correlates with the efficacy of β-agonists in promoting intracellular cAMP (33) and has been used previously as a marker for PKA activity (15, 25, 34). Compared with control cells, PKI-GFP and RevAB-GFP HASM cells stimulated with ISO (50 nm or 1 μm) or FSK (100 μm) exhibited significantly less VASP shift (stimulated with FSK, 23 ± 2.3 and 21 ± 2.4% versus 77 ± 3.0% for GFP), indicating greatly attenuated PKA activity.
FIGURE 1.

β-Agonist regulation of VASP and phospho-HSP-20 and the effect of PKA inhibition. GFP-, PKI-GFP-, or RevAB-expressing HASM cells were plated into 12-well plates and stimulated with vehicle (Bas), ISO (50 nm or 1 μm) or FSK (100 μm) ± HIST (1 μm) as described under “Experimental Procedures.” After 5 min of stimulation, cell lysates were harvested, and expression levels of various proteins were assessed by immunoblotting. Representative blots (A) are shown. Band intensities for VASP phosphorylation and phospho-HSP-20 were quantified as described under “Experimental Procedures,” adjusted for quantified β-actin levels and presented in graph form in B and C, respectively. VASP bands present as either a 46- or 50-kDa species, the latter representing VASP in which Ser-157 is phosphorylated by PKA. Expression of GFP in all three lines was confirmed by Western blot using anti-GFP antibody. HSP-20 phosphorylation has also been shown previously to be PKA-dependent. Quantified phospho-HSP20 was normalized to the GFP + FSK value. Data are mean ± S.E. †, p < 0.0001 PKI/RevAB versus GFP.
Phosphorylation of HSP-20 in ASM has been demonstrated previously to be directly dependent on PKA activity (17). As expected, stimulation of PKI-GFP and RevAB-GFP HASM with ISO resulted in virtually no phosphorylation of HSP-20, whereas, in GFP cells, the protein undergoes significant phosphorylation (Fig. 1A). Quantified data for VASP phosphorylation and HSP-20 phosphorylation from multiple experiments are shown in the bar graph (Fig. 1B). Collectively these data indicate a strong inhibition of PKA activity in PKI- and RevAB-expressing cells.
We then investigated the role of PKA in β-agonist-mediated inhibition of signaling events induced by the contractile agonist HIST. There was no significant difference among lines in the ability of HIST to induce p42/p44 (ERK1/2) phosphorylation (Fig. 2). However, HIST-induced ERK1/2 phosphorylation was attenuated by addition of ISO or FSK in control (GFP) cells but not in PKI- or RevAB-expressing cells.
FIGURE 2.
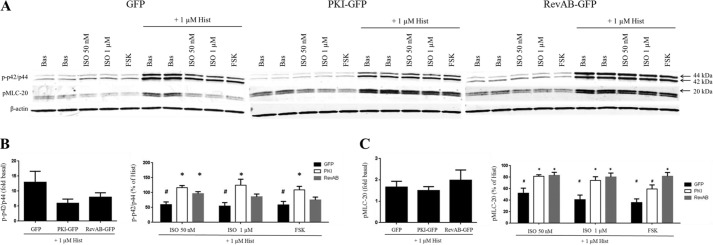
β-Agonist regulation of phospho-p42/p44 and phospho-MLC-20 levels and the effect of PKA inhibition. Cultures of GFP-, PKI-GFP-, or RevAB-expressing HASM cells were plated into 12-well plates and stimulated with vehicle (Bas), ISO (50 nm or 1 μm), or FSK (100 μm) ± HIST (1 μm) according to the experiments in Fig. 1. Representative blots in A, derived from the same representative experiment depicted in Fig. 1A, depict the regulation of phospho-p42/p44 and phospho-MLC-20 levels by the indicated agonists. Band intensities for phospho-p42/p44 and phospho-MLC-20 were quantified as the ratio of HIST-stimulated to basal (left panel) and as the percent of HIST alone (right panel) and are presented in graph form in B and C, respectively. A, VASP bands present as either a 46- or 50-kDa species, the latter representing VASP in which Ser-157 is phosphorylated by PKA. Expression of GFP in all three lines was confirmed by Western blot analysis using anti-GFP antibody. HSP-20 phosphorylation has also been shown previously to be PKA-dependent. Quantified phospho-HSP20 was normalized to the GFP + FSK value. B and C, phospho-p42/p44 (B) and phospho-MLC-20 (C) levels were quantified as the ratio of HIST-stimulated to basal (left panel) and as the percent of HIST alone (right panel). The HIST alone value (not shown) was set to 100%. Data are mean ± S.E. *, p < 0.05, PKI/RevAB versus GFP; #, p < 0.05 versus HIST alone (n = 5).
The phosphorylation state of MLC-20 is one mechanism by which contraction of ASM cells is regulated (35). There was no difference among lines in the ability of HIST to stimulate MLC-20 phosphorylation (Fig. 2). In GFP cells, addition of ISO or FSK significantly reduced HIST-stimulated MLC-20 phosphorylation. ISO had no significant effect on phospho-MLC-20 levels when PKA activity was inhibited.
Next we investigated the role of PKA activity in second messenger generation in HASM (Fig. 3). We demonstrated previously that, under conditions of intracellular phosphodiesterase inhibition, PKA inhibition via PKI-GFP, or RevAB-GFP expression resulted in significantly increased ISO- mediated cAMP accumulation. Because our analyses of procontractile signaling and contraction in this study are conducted in the absence of pharmacological phosphodiesterase inhibition, regulation of β-agonist-stimulated cAMP accumulation in HASM was assessed under the same conditions. As shown in Fig. 3, ISO-mediated cAMP accumulation was significantly greater in PKI-GFP or RevAB-GFP expressing cells (relative to GFP cells). This was true for both absolute (ISO-stimulated) cAMP values and those normalized to FSK-stimulated values.
FIGURE 3.

Effect of PKI-GFP or RevAB-GFP expression on β-agonist-mediated cAMP accumulation. GFP-, PKI-GFP-, or RevAB-GFP-expressing HASM cells were plated into 24-well plates and stimulated with vehicle (Bas), ISO (50 nm or 1 μm), or FSK (100 μm). Global cAMP accumulation was measured after 10-min stimulation and plotted as raw values (picomoles/well) (A) or percent of the FSK-stimulated value (B). Data are mean ± S.E. *, p < 0.05 PKI/RevAB versus GFP (n = 6).
The ability of ISO to attenuate HIST-stimulated calcium release under conditions of PKA inhibition was also determined (Fig. 4). Representative traces of the intracellular calcium mobilization assessed for cultures expressing GFP or PKI-GFP are presented in Fig. 4, A–D, with mean calcium response data reported as the ratio of the second HIST response (following a 5-min incubation in the presence or absence of 1 μm ISO) to the first (S2/S1) in Fig. 4E. No differences were observed in HIST-stimulated, single-cell calcium release between GFP- and PKI-GFP-expressing cells. ISO pretreatment significantly reduced HIST-stimulated calcium flux in GFP cells (79 ± 8.0% versus 37 ± 13% with pretreatment). However, no effect of ISO pretreatment was seen in PKI-expressing cells.
FIGURE 4.

Effect of PKI-GFP and RevAB-GFP expression on β-agonist inhibition of HIST-stimulated Ca2+ mobilization. HASM cultures expressing GFP or PKI-GFP were loaded with Fura-2/AM. Ca2+ mobilization was assessed in response to stimulation with HIST (10 μm). Cells were then washed and restimulated with HIST ± ISO (1 μm) pretreatment. A—D, representative traces for GFP-expressing (A and B) and PKI-GFP-expressing (C and D) cells assessing the effect of prior vehicle (A and C) or ISO (B and D) treatment. The ratio of second histamine response to the first (S2/S1) was calculated and compared between groups. Data are mean ± S.E. *, p < 0.05 versus HIST alone; #, p < 0.05 versus matched GFP (GFP HIST, n = 82; PKI HIST, n = 93; GFP ISO + HIST, n = 187; PKI ISO + HIST, n = 170).
To link the observed changes in signaling to functional outcomes, the effect of PKA inhibition on contraction of cultured HASM cells was investigated using MTC (30). MTC utilizes ferrimagnetic microbeads bound to the cytoskeleton of ASM cells, and changes in cell stiffness elicited by either ASM contraction or relaxation can be measured as the pivoting displacement of beads. Treatment with ISO significantly reduced the stiffness of GFP-expressing HASM from the baseline, whereas no effect was observed with PKI or RevAB expression (Fig. 5A). All three lines exhibited increased stiffness in response to addition of HIST, although the PKI and RevAB lines exhibited a slightly lesser response than control cells (Fig. 5B). Addition of ISO reduced stiffness in GFP cells precontracted with HIST but failed to alter the stiffness of PKI- or RevAB-expressing cells under these conditions (Fig. 5C).
FIGURE 5.
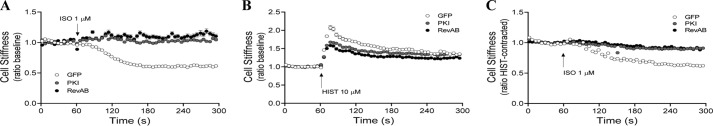
Effects of PKA inhibition on β-agonist-induced changes in cell stiffness. HASM cultures stably expressing GFP, PKI-GFP, or RevAB-GFP were assessed for changes in cell stiffness in response to HIST and ISO, measured using magnetic twisting cytometry as described under “Experimental Procedures.” A, time-dependent response to ISO (1 μm) from baseline. B, response to HIST (10 μm) from baseline. C, ability of ISO (1 μm) to alter stiffness in cells prestimulated with HIST (10 μm). †, p < 0.0001 PKI/RevAB versus GFP. Differences in mean values for baseline stiffness were less than 20% among the GFP, PKI-GFP, and RevAB-GFP groups. Data points represent mean ± S.E. of ≥ 86 cells/experimental group.
To complement our cell-based assay of contraction, we assessed the contractile regulation of ASM tissue using murine tracheal rings. Rings were isolated and infected with lentivirus encoding GFP or PKI-GFP as described under “Experimental Procedures.” Expression was evidenced throughout the tissue by direct visualization of GFP (Fig. 6A) and confirmed by Western blotting of whole tissue lysates (Fig. 6B). There was no significant difference in the ability of MCh (1 μm) to contract GFP versus PKI-GFP rings (Fig. 6C). Rings were precontracted with MCh and then treated with increasing doses of ISO. A significantly lesser relaxation was observed for PKI-GFP rings than for their matched GFP counterparts at each concentration of β-agonist (Fig. 6D).
FIGURE 6.
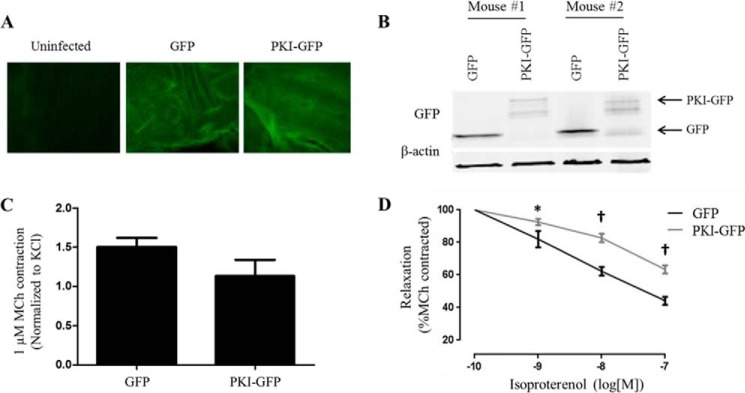
Effect of PKA inhibition on the ability of β-agonist to relax precontracted murine tracheal rings. Tracheal rings were isolated from C57BL6 mice and infected for 48 h with lentivirus encoding GFP or PKI-GFP. A, GFP images of control, GFP-, or PKI-GFP-infected murine tracheal rings. The control ring was maintained in culture for 48 h but not infected with virus. B, representative immunoblot for GFP expression in lysates from GFP- and PKI-GFP-infected murine tracheal rings. C, KCl-normalized contraction of rings stimulated with MCh (1 μm). D, tracheal rings were precontracted with MCh (1 μm), and then changes in contractile force were measured in response to treatment with increasing doses of ISO. *, p < 0.05; †, p < 0.0001 versus GFP (GFP, n = 9; PKI, n = 12).
Lastly, in an attempt to discern some role of Epac in mediating the anticontractile effect of β-agonist on ASM, we knocked down Epac 1 (71% as determined by quantitative PCR) and assessed the regulation of histamine-induced signaling (Fig. 7) and contraction (Fig. 8). Knockdown had no apparent effect on any outcome. Moreover, stimulation of cells with 100 μm 8-pCPT-2′-O-Me-cAMP, unlike stimulation with 1 μm ISO, had no effect on histamine-induced MLC-20 phosphorylation or contraction. Experiments assessing the effect of Epac 2 siRNA or combined Epac1 and 2 siRNA revealed a similar lack of effect on ISO-mediated inhibition of histamine-stimulated, procontractile signaling and contraction (data not shown).
FIGURE 7.

Effect of Epac knockdown on β-agonist regulation of VASP, phospho-HSP-20, phospho-p42/p44, and phospho-MLC-20 levels. 96 h after transfection of HASM cells with either scrambled (Scr) siRNA or siRNA targeting Epac1, cells plated into 12-well plates were stimulated with vehicle (Bas), ISO (1 μm), 8-pCPT-2′-O-Me-cAMP (8CPT, 100 μm), or FSK (100 μm) ± HIST (1 μm) as described under “Experimental Procedures.” After 5 min of stimulation, cell lysates were harvested, and expression levels of VASP, phospho-HSP-20, phospho-p42/p44 and phospho-MLC-20, and β-actin were assessed by immunoblotting. Blots are representative of three experiments providing similar results.
FIGURE 8.
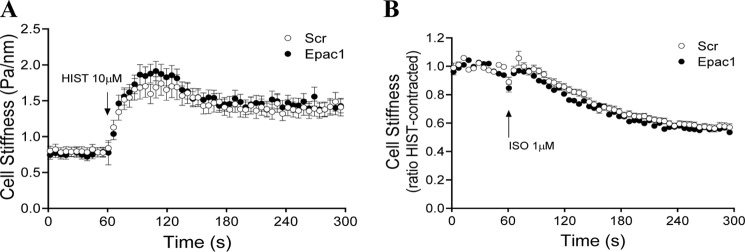
Effects of Epac knockdown on β-agonist-induced changes in cell stiffness. HASM cells were subjected to Epac1 knockdown as in Fig. 7. Experiments were assessed for changes in cell stiffness in response to HIST and ISO using magnetic twisting cytometry as described under “Experimental Procedures.” Scr, scrambled.
DISCUSSION
Although the effects of β-agonists have traditionally been attributed to activation of PKA, recent studies have demonstrated that β2AR signaling can initiate numerous other downstream events. cAMP-dependent but PKA-independent activation of Epac by the β2AR has been demonstrated in many cell types (reviewed in Ref. 36). Other mechanisms of G protein-independent signaling by β2ARs, including that mediated by arrestins, have been described in various systems (37). The combination of these findings with the lack of direct evidence tying PKA activity to the action of β-agonists challenges the validity of many long-held assumptions regarding the physiological relevance of PKA in effecting the functional changes mediated by β-agonists.
Recent studies have reported a role for cAMP-dependent but PKA-independent activation of Epac in mediating the bronchorelaxant effects of β-agonists. In guinea pig tracheae, the activity of the β-agonist isoprenaline has been proposed to be PKA-independent because treatment with the PKA inhibiting cAMP analog 8-(4-chlorophenylthio)adenosine-cAMP, Rp-isomer (Rp-8-cP-cAMPs) failed to alter the ability of isoprenaline to antagonize contraction (38). In both guinea pig ASM cultures and human telomerase reverse transcriptase-immortalized human ASM cells, Epac activation by the selective analog 8-pCPT-2′-O-Me-cAMP completely inhibited the MCh-induced phosphorylation of MLC-20 (20), reportedly via inhibition of RhoA and activation of Rac1 (20, 21). Finally, selective activation of Epac with 8-pCPT-2′-O-Me-cAMP produced dose-dependent relaxation of MCh-contracted guinea pig tracheal rings that was not affected by pretreatment with the PKA inhibitory analog Rp-8-cPT-cAMPs (20).
Although these studies suggest the capability of specific Epac activation to relax various forms of ASM, the non-physiological nature and questionable specificity of these selective cAMP analogues raise concerns over the meaning of these findings. The development of analogues was necessary simply because PKA and Epac activation by cAMP cannot be uncoupled in a physiological setting. Elevation of endogenous cAMP results in activation of both effectors, although PKA is activated at lower concentrations of cAMP than Epac (half-maximal concentration for activation (AC50); PKAI AC50, 0.09 μm; PKAII AC50, 0.08 μm; Epac1 AC50, 45 μm) (39). Several commonly used analogues, including 8-pCPT-2′-O-Me-cAMP, are potent phosphodiesterase inhibitors and/or substrates (40, 41), raising the concern that interaction of the analogues with phosphodiesterases may alter the metabolism of endogenous cAMP and, therefore, artificially elevate or prolong its intracellular concentration (41, 42). The ability of these agents to specifically activate one effector is also questionable because at least one commonly used “selective” analog (8-bromo-cAMP) has been shown to activate both PKA and Epac at normal concentrations (40). Finally, PKA is capable of activating Rap1 in an Epac-independent manner (43), and, therefore, Rap1-dependent effects attributed to Epac may, in fact, be a product of PKA activity. Although cAMP analogues are currently the best available tools to study the role of Epac in various systems, these issues suggest that findings made using these agents regarding the capability of Epac to exert the described effects may not be helpful in understanding the role of Epac in mediating physiological, cAMP-dependent actions. Consistent with this notion, but in contrast to Zieba et al. (21), we failed to observe any effect of selective Epac activation, with 8-pCPT-2′-O-Me-cAMP, on phospho-MLC-20 regulation (Fig. 7).
In this study, we attempted to circumvent the limitations of current techniques to clarify the question of whether or not the functional effects of β-agonists on ASM are truly PKA-independent. We previously utilized retroviral expression of PKI and RevAB to demonstrate the role of PKA in regulating proinflammatory cytokine expression and functional consequences (15, 16) as well as the requirement for PKA in effecting the antimitogenic properties of β-agonists (25). This study extends our findings to the role of PKA in β-agonist-mediated relaxation of the airway. Expression of the PKI and RevAB peptides inhibited a significant portion of PKA activity within the cell, as indicated by a marked attenuation in VASP shift and HSP-20 phosphorylation (Fig. 1A). PKA inhibition resulted in increased β-agonist-mediated cAMP release, which not only demonstrated the ability of PKA to regulate β2AR responsiveness in ASM but also demonstrated that the ability of PKA inhibition to reverse ISO-mediated inhibition of contraction and procontractile signaling occurred in the context of increased intracellular cAMP levels.
Such PKA inhibitory effects included near abolishment of the inhibitory effect of ISO on intracellular calcium flux and significantly attenuated HIST-stimulated MLC-20 phosphorylation. Functional measurements using both HASM cultures and murine tracheal rings also indicated that ISO-mediated relaxation of contracted smooth muscle is highly dependent on PKA activity (Figs. 4 and 5).
Despite the highly significant level of PKA inhibition achieved with PKI and RevAB expression, some residual PKA activity was present in the cultures. We attribute this to either expression levels that were stoichiometrically insufficient to inhibit all PKA activity or the failure to express the inhibitory peptides in 100% of the cells despite rigorous antibiotic selection of cultures. Despite the retention of some PKA activity, the functional consequences of PKI or RevAB expression in ASM cells were profound, causing an almost complete reversal of β-agonist-mediated inhibition of procontractile signaling events and elimination of β-agonist-mediated inhibition of ASM cell contraction assessed by MTC.
Analysis of ASM tissue contractile regulation demonstrates a significant but incomplete reversal of the relaxant effect of β-agonist when PKA is inhibited (Fig. 5). The failure to reverse completely can be explained by several possibilities, including the limitations of infecting intact tissue. Although we have successfully demonstrated the ability to express GFP and PKI-GFP in murine tracheal rings via lentiviral infection, the extent to which PKA activity is inhibited is unclear. Successful infection of, and PKA inhibition in, all ASM cells within the tissue may be confounded by tissue thickness and by the presence of other cell types, such as the epithelia. Despite these modest limitations, it remains clear that, in both cultured HASM cells and murine tracheal rings, PKA activity is the predominant effector of prorelaxant signaling and functional effects of β-agonists.
Another possible explanation for the residual relaxant effect of isoproterenol on PKI-expressing rings involves cAMP-dependent activation of protein kinase G (PKG). PKG activation by various agents has been shown to relax smooth muscle (44, 45), and cAMP-dependent activation of PKG has been reported in vascular smooth muscle (46, 47). Although to date we have failed to procure evidence that cAMP can activate PKG in ASM (Ref. 17 and data not shown), we cannot eliminate a cAMP/PKG-dependent mechanism effecting relaxation in this study.
As discussed, recent studies have suggested that Epac plays an important role in several functional characteristics of ASM, including β-agonist-mediated relaxation (20, 22–24). However, given that our results demonstrate that PKA inhibition in ASM results in increased cAMP generation following β-agonist treatment, cAMP-dependent Epac activity would be expected to increase under these conditions. Although Epac levels in ASM appear to be extremely low (Epac1 is not consistently detected in all cultures of ASM, and Epac2 is not detectable), we performed siRNA-mediated knockdown of Epac1 in an attempt to discern any role for Epac. Knockdown failed to affect the ability of isoproterenol to either inhibit MLC-20 phosphorylation or reverse ASM contraction, as evidenced by MTC. Moreover, we were unable to detect an effect of the Epac selective activator 8-pCPT-2′-O-Me-c AMP on histamine-induced contraction or procontractile signaling. Therefore, although the data presented here do not necessarily exclude the capability of Epac to relax ASM, they do suggest that any role of Epac in mediating the relaxant effect of β-agonists is minimal at best.
Although β-agonists are critical to the management of asthma symptoms, issues with long term efficacy and adverse events associated with current formulations leave considerable room for improvement. A mechanistic understanding of both the beneficial and problematic effects of β-agonists is essential for the development of better or alternative bronchodilator therapies. Our findings that PKA activity is the predominant mechanism by which β-agonist-mediated signaling and ASM relaxation occur provide clear direction for the design of new therapeutic agents for asthma.
This work was supported, in whole or in part, by National Institutes of Health Grants HL58506 (to R. B. P.), AG041265 and HL087560 (to D. A. D.), and HL114471 (to R. B. P., R. A. P., and S. S. A.).
- β2AR
- β-2-adrenoreceptor
- ASM
- airway smooth muscle
- Epac
- exchange protein activated by cAMP
- VASP
- vasodilator-stimulated phosphoprotein
- HASM
- human airway smooth muscle
- ISO
- isoproterenol
- 8-pCPT-2′-O-Me-cAMP
- 8-(4-chlorophenylthio)-2′-O-methyladenosine-cAMP
- FSK
- forskolin
- HIST
- histamine
- MTC
- magnetic twisting cytometry
- MCh
- methacholine
- Veh
- vehicle
- PKG
- protein kinase G
- Rp-8-cPT-cAMPs
- 8-(4-chlorophenylthio)adenosine-cAMP Rp-isomer.
REFERENCES
- 1. Newnham D. M., McDevitt D. G., Lipworth B. J. (1994) Bronchodilator subsensitivity after chronic dosing with eformoterol in patients with asthma. The American Journal of Medicine 97, 29–37 [DOI] [PubMed] [Google Scholar]
- 2. Newnham D. M., Grove A., McDevitt D. G., Lipworth B. J. (1995) Subsensitivity of bronchodilator and systemic β 2 adrenoceptor responses after regular twice daily treatment with eformoterol dry powder in asthmatic patients. Thorax 50, 497–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grove A., Lipworth B. J. (1995) Bronchodilator subsensitivity to salbutamol after twice daily salmeterol in asthmatic patients. Lancet 346, 201–206 [DOI] [PubMed] [Google Scholar]
- 4. Sears M. R. (2002) Adverse effects of b-agonists. J. Allergy Clin. Immunol. 110, S322–S328 [DOI] [PubMed] [Google Scholar]
- 5. Salpeter S. R., Buckley N. S., Ormiston T. M., Salpeter E. E. (2006) Meta-analysis: effect of long-acting b-Agonists on severe asthma exacerbations and asthma-related deaths. Annals Intern. Med. 144, 904–912 [DOI] [PubMed] [Google Scholar]
- 6. Deshpande D. A., Penn R. B. (2006) Targeting G protein-coupled receptor signaling in asthma. Cell Signal. 18, 2105–2120 [DOI] [PubMed] [Google Scholar]
- 7. Stolley P. D., Schinnar R. (1978) Association between asthma mortality and isoproterenol aerosols: a review. Prev. Med. 7, 519–538 [DOI] [PubMed] [Google Scholar]
- 8. Spitzer W. O., Suissa S., Ernst P., Horwitz R. I., Habbick B., Cockcroft D., Boivin J. F., McNutt M., Buist A. S., Rebuck A. S. (1992) The use of b-agonists and the risk of death and near death from asthma. N. Engl. J. Med. 326, 501–506 [DOI] [PubMed] [Google Scholar]
- 9. Pearce N., Beasley R., Crane J., Burgess C., Jackson R. (1995) End of the New Zealand asthma mortality epidemic. Lancet 345, 41–44 [DOI] [PubMed] [Google Scholar]
- 10. Nelson H. S., Weiss S. T., Bleecker E. R., Yancey S. W., Dorinsky P. M. (2006) The salmeterol multicenter asthma research trial: a comparison of usual pharmacotherapy for asthma or usual pharmacotherapy plus salmeterol. Chest 129, 15–26 [DOI] [PubMed] [Google Scholar]
- 11. Taylor D. R. (2009) The b-agonist saga and its clinical relevance: on and on it goes. Am. J. Respir. Crit. Care Med. 179, 976–978 [DOI] [PubMed] [Google Scholar]
- 12. Ortega V. E., Peters S. P. (2010) β-2 adrenergic agonists: focus on safety and benefits versus risks. Curr. Opin. Pharmacol. 10, 246–253 [DOI] [PubMed] [Google Scholar]
- 13. Walker J. K., Penn R. B., Hanania N. A., Dickey B. F., Bond R. A. (2011) New perspectives regarding b2-adrenoceptor ligands in the treatment of asthma. Br. J. Pharmacol. 163, 18–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Penn R. B., Parent J. L., Pronin A. N., Panettieri R. A., Jr., Benovic J. L. (1999) Pharmacological inhibition of protein kinases in intact cells: antagonism of β adrenergic receptor ligand binding by H-89 reveals limitations of usefulness. J. Pharmacol. Exp. Ther. 288, 428–437 [PubMed] [Google Scholar]
- 15. Guo M., Pascual R. M., Wang S., Fontana M. F., Valancius C. A., Panettieri R. A., Jr., Tilley S. L., Penn R. B. (2005) Cytokines regulate β-2-adrenergic receptor responsiveness in airway smooth muscle via multiple PKA- and EP2 receptor-dependent mechanisms. Biochemistry 44, 13771–13782 [DOI] [PubMed] [Google Scholar]
- 16. Misior A. M., Yan H., Pascual R. M., Deshpande D. A., Panettieri R. A., Penn R. B. (2008) Mitogenic effects of cytokines on smooth muscle are critically dependent on protein kinase A and are unmasked by steroids and cyclooxygenase inhibitors. Mol. Pharmacol. 73, 566–574 [DOI] [PubMed] [Google Scholar]
- 17. Komalavilas P., Penn R. B., Flynn C. R., Thresher J., Lopes L. B., Furnish E. J., Guo M., Pallero M. A., Murphy-Ullrich J. E., Brophy C. M. (2007) The small heat shock-related protein, HSP20, is a cAMP-dependent protein kinase substrate that is involved in airway smooth muscle relaxation. Am. J. Physiol. Lung Cell Mol. Physiol. 294, L69–L78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Z. W., Kotlikoff M. I. (1996) Activation of KCa channels in airway smooth muscle cells by endogenous protein kinase A. Am. J. Physiol. 271, L100–L105 [DOI] [PubMed] [Google Scholar]
- 19. Kume H., Hall I. P., Washabau R. J., Takagi K., Kotlikoff M. I. (1994) b-Adrenergic agonists regulate KCa channels in airway smooth muscle by cAMP-dependent and -independent mechanisms. J. Clin. Invest. 93, 371–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roscioni S. S., Maarsingh H., Elzinga C. R., Schuur J., Menzen M., Halayko A. J., Meurs H., Schmidt M. (2011) Epac as a novel effector of airway smooth muscle relaxation. J. Cell. Mol. Med. 15, 1551–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zieba B. J., Artamonov M. V., Jin L., Momotani K., Ho R., Franke A. S., Neppl R. L., Stevenson A. S., Khromov A. S., Chrzanowska-Wodnicka M., Somlyo A. V. (2011) The cAMP-responsive Rap1 guanine nucleotide exchange factor, Epac, induces smooth muscle relaxation by down-regulation of RhoA activity. J. Biol. Chem. 286, 16681–16692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kassel K. M., Wyatt T. A., Panettieri R. A., Jr., Toews M. L. (2008) Inhibition of human airway smooth muscle cell proliferation by β 2-adrenergic receptors and cAMP is PKA independent: evidence for EPAC involvement. Am. J. Physiol. Lung Cell Mol. Physiol. 294, L131–L138 [DOI] [PubMed] [Google Scholar]
- 23. Roscioni S. S., Prins A. G., Elzinga C. R., Menzen M. H., Dekkers B. G., Halayko A. J., Meurs H., Maarsingh H., Schmidt M. (2011) Protein kinase A and the exchange protein directly activated by cAMP (Epac) modulate phenotype plasticity in human airway smooth muscle. Br. J. Pharmacol. 164, 958–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roscioni S. S., Dekkers B. G., Prins A. G., Menzen M. H., Meurs H., Schmidt M., Maarsingh H. (2011) cAMP inhibits modulation of airway smooth muscle phenotype via the exchange protein activated by cAMP (Epac) and protein kinase A. Br. J. Pharmacol. 162, 193–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yan H., Deshpande D. A., Misior A. M., Miles M. C., Saxena H., Riemer E. C., Pascual R. M., Panettieri R. A., Penn R. B. (2011) Anti-mitogenic effects of β-agonists and PGE2 on airway smooth muscle are PKA dependent. FASEB J. 25, 389–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kong K. C., Billington C. K., Gandhi U., Panettieri R. A., Jr., Penn R. B. (2006) Cooperative mitogenic signaling by G protein-coupled receptors and growth factors is dependent on Gq11. FASEB J. 20, 1558–1560 [DOI] [PubMed] [Google Scholar]
- 27. Saxena H., Deshpande D. A., Tiegs B. C., Yan H., Battafarano R. J., Burrows W. M., Damera G., Panettieri R. A., Dubose T. D., Jr., An S. S., Penn R. B. (2012) The GPCR OGR1 (GPR68) mediates diverse signalling and contraction of airway smooth muscle in response to small reductions in extracellular pH. Br. J. Pharmacol. 166, 981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Billington C. K., Kong K. C., Bhattacharyya R., Wedegaertner P. B., Panettieri R. A., Jr., Chan T. O., Penn R. B. (2005) Cooperative regulation of p70S6 kinase by receptor tyrosine kinases and G protein-coupled receptors augments airway smooth muscle growth. Biochemistry 44, 14595–14605 [DOI] [PubMed] [Google Scholar]
- 29. Naik S., Billington C. K., Pascual R. M., Deshpande D. A., Stefano F. P., Kohout T. A., Eckman D. M., Benovic J. L., Penn R. B. (2005) Regulation of cysteinyl leukotriene type 1 receptor internalization and signaling. J. Biol. Chem. 280, 8722–8732 [DOI] [PubMed] [Google Scholar]
- 30. An S. S., Fabry B., Trepat X., Wang N., Fredberg J. J. (2006) Do biophysical properties of the airway smooth muscle in culture predict airway hyperresponsiveness? Am. J. Respir. Cell Mol. Biol. 35, 55–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fabry B., Maksym G. N., Butler J. P., Glogauer M., Navajas D., Fredberg J. J. (2001) Scaling the microrheology of living cells. Phys. Rev. Lett. 87, 148102. [DOI] [PubMed] [Google Scholar]
- 32. Gavett S. H., Wills-Karp M. (1993) Elevated lung G protein levels and muscarinic receptor affinity in a mouse model of airway hyperreactivity. Am. J. Physiol. Lung Cell. Mol. Physiol. 265, L493–L500 [DOI] [PubMed] [Google Scholar]
- 33. Freyer A. M., Billington C. K., Penn R. B., Hall I. P. (2004) Extracellular matrix modulates β2-adrenergic receptor signalling in human airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 31, 440–445 [DOI] [PubMed] [Google Scholar]
- 34. Butt E., Abel K., Krieger M., Palm D., Hoppe V., Hoppe J., Walter U. (1994) cAMP- and cGMP-dependent protein kinase phosphorylation sites of the focal adhesion vasodilator-stimulated phosphoprotein (VASP) in vitro and in intact human platelets. J. Biol. Chem. 269, 14509–14517 [PubMed] [Google Scholar]
- 35. Billington C. K., Penn R. B. (2003) Signaling and regulation of G protein-coupled receptors in airway smooth muscle. Respir. Res. 4, 2. [PMC free article] [PubMed] [Google Scholar]
- 36. Grandoch M., Roscioni S. S., Schmidt M. (2010) The role of Epac proteins, novel cAMP mediators, in the regulation of immune, lung and neuronal function. Br. J. Pharmacol. 159, 265–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Penn R. B. (2008) Embracing emerging paradigms of G protein-coupled receptor agonism and signaling to address airway smooth muscle pathobiology in asthma. Naunyn Schmiedebergs Arch. Pharmacol. 378, 149–169 [DOI] [PubMed] [Google Scholar]
- 38. Spicuzza L., Belvisi M. G., Birrell M. A., Barnes P. J., Hele D. J., Giembycz M. A. (2001) Evidence that the anti-spasmogenic effect of the β-adrenoceptor agonist, isoprenaline, on guinea-pig trachealis is not mediated by cyclic AMP-dependent protein kinase. Br. J. Pharmacol. 133, 1201–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rehmann H., Schwede F., Døskeland S. O., Wittinghofer A., Bos J. L. (2003) Ligand-mediated activation of the cAMP-responsive guanine nucleotide exchange factor Epac. J. Biol. Chem. 278, 38548–38556 [DOI] [PubMed] [Google Scholar]
- 40. Poppe H., Rybalkin S. D., Rehmann H., Hinds T. R., Tang X. B., Christensen A. E., Schwede F., Genieser H. G., Bos J. L., Doskeland S. O., Beavo J. A., Butt E. (2008) Cyclic nucleotide analogs as probes of signaling pathways. Nat. Methods 5, 277–278 [DOI] [PubMed] [Google Scholar]
- 41. Holz G. G., Chepurny O. G., Schwede F. (2008) Epac-selective cAMP analogs: new tools with which to evaluate the signal transduction properties of cAMP-regulated guanine nucleotide exchange factors. Cell. Signal. 20, 10–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Laxman S., Riechers A., Sadilek M., Schwede F., Beavo J. A. (2006) Hydrolysis products of cAMP analogs cause transformation of Trypanosoma brucei from slender to stumpy-like forms. Proc. Natl. Acad. Sci. 103, 19194–19199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Altschuler D. L., Peterson S. N., Ostrowski M. C., Lapetina E. G. (1995) Cyclic AMP-dependent activation of Rap1b. J. Biol. Chem. 270, 10373–10376 [DOI] [PubMed] [Google Scholar]
- 44. Perez-Zoghbi J. F., Bai Y., Sanderson M. J. (2010) Nitric oxide induces airway smooth muscle cell relaxation by decreasing the frequency of agonist-induced Ca2+ oscillations. J. Gen. Physiol. 135, 247–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Carvajal J. A., Germain A. M., Huidobro-Toro J. P., Weiner C. P. (2000) Molecular mechanism of cGMP-mediated smooth muscle relaxation. J. Cell Physiol. 184, 409–420 [DOI] [PubMed] [Google Scholar]
- 46. White R. E., Kryman J. P., El-Mowafy A. M., Han G., Carrier G. O. (2000) cAMP-dependent vasodilators cross-activate the cGMP-dependent protein kinase to stimulate BKCa channel activity in coronary artery smooth muscle cells. Circ. Res. 86, 897–905 [DOI] [PubMed] [Google Scholar]
- 47. Jiang H., Colbran J. L., Francis S. H., Corbin J. D. (1992) Direct evidence for cross-activation of cGMP-dependent protein kinase by cAMP in pig coronary arteries. J. Biol. Chem. 267, 1015–1019 [PubMed] [Google Scholar]