

Background: IL-10 inhibits the production of inflammatory mediators in TLR signaling.
Results: β-arrestin 2 interacts with p38 and promotes p38 activation and IL-10 production.
Conclusion: The results provide the first evidence for a mechanism of β-arrestin 2-mediated IL-10 response in a p38-dependent mechanism in TLR4 signaling.
Significance: The data indicate new therapeutic approaches to treatment of inflammatory diseases.
Keywords: Cell Signaling, Inflammation, p38, Protein Kinase, Toll-like Receptor (TLR), IL-10, β-Arrestin 2, Endotoxic Shock
Abstract
The control of IL-10 production in Toll-like receptor (TLR) signals remains to be elucidated. Here, we report that β-arrestin 2 positively regulates TLR-triggered IL-10 production in a p38 mitogen-activated protein kinase (MAPK)-dependent mechanism. In vitro studies with cells including peritoneal macrophages and HEK293/TLR4 cells have demonstrated that β-arrestin 2 forms complexes with p38 and facilitates p38 activation after lipopolysaccharide (LPS) stimulation. Deficiency of β-arrestin 2 and inhibition of p38 MAPK activity both ameliorate TLR4-stimulated IL-10 response. Additionally, in vivo experiments show that mice lacking β-arrestin 2 produce less amount of IL-10, and are more susceptible to LPS-induced septic shock which is further enhanced by blocking IL-10 signal. These results reveal a novel mechanism by which β-arrestin 2 negatively regulates TLR4-mediated inflammatory reactions.
Introduction
Toll-like receptors (TLRs)4 are widely described key regulators involved in both initiation of innate resistance and the development of adaptive immune responses against infectious pathogens (1). Engagement of ligands to respective TLRs initiates series of intracellular signaling events ultimately culminating in the activation of nuclear factor-κB (NF-κB) or mitogen-activated protein kinase (MAPK) cascades and thereby transcription of inflammatory genes (2).
Triggered TLRs appropriately orchestrate the duration and intensity of inflammatory reactions upon pathogenic organisms encountered. For example, the release of pro-inflammatory mediators is increased immediately after lipopolysaccharide (LPS) challenge but subsequently down-modulated by a battery of anti-inflammatory cytokines such as interleukin-10 (IL-10) (3, 4). IL-10, initially designated a cytokine synthesis inhibitory factor, inhibits the synthesis of proinflammatory cytokines in response to TLR stimulation (5). The immunoregulatory action of IL-10 is also shown by its suppressive effect on the development of experimental autoimmune optic neuritis and experimental autoimmune encephalitis (6, 7). In vivo studies have suggested that mice deficient in the production of IL-10 become more sensitive to endotoxic shock, while pre-treatment of IL-10 suppresses endotoxemia and improves survival rate (8). Thus, the balanced inflammatory responses effectively rely on an appropriate regulation of IL-10 synthesis. However, little is known concerning the mechanisms of regulating IL-10 production by TLR signals.
Multifunctional signaling molecules β-arrestins (β-arrestin 1 and β-arrestin 2) are ubiquitously expressed cytosolic proteins, which are originally appreciated attributable to the modulation of receptor endocytosis and attenuation of G protein-coupled receptor (GPCR) signaling through binding to phosphorylated GPCRs (9). In addition to these established functions, β-arrestins increasingly represent an active line of investigation where β-arrestins bind with various target molecules and thus modulate a wide range of biological processes (9, 10). Accumulating evidence has shown that β-arrestins are functionally involved in the regulation of immune responses by modulating various signaling pathways (11). It has been implicated that β-arrestin 2 is a negative regulator of TLR-mediated inflammatory reactions (12). β-Arrestin 2 interacts with TRAF6, a major adaptor required for initiation of most TLR/IL-1R signaling events, and prevents autoubiquitination of TRAF6 and subsequent activation of NF-κB and AP-1 (12). Additionally, β-arrestin 2 also exerts the ability of binding with IκB α directly, preventing the phosphorylation and degradation of IκB α, and consequently inhibiting NF-κB activation (13). However, there is still a limited understanding about the regulatory effect of β-arrestin 2 on the anti-inflammatory products of TLR signaling.
The affinity between β-arrestin 2 and MAPKs has been displayed in numerous cases of GPCR signaling (14–16). β-arrestin 2 scaffolds MAPK components such as the MAP kinases c-Jun-N-terminal kinase (JNK) and ERK, leading to phosphorylation, activation and accumulation of MAPKs in defined cellular compartments (17). MAPKs are important mediators of TLR signaling pathways. In an attempt to define the mechanisms by which β-arrestin 2 modules TLR-mediated immune regulation, we focus on investigation of β-arrestin 2 to TLR-activated MAPK pathways. The results of our present study illustrate the molecular mechanism for the orchestration of TLR-triggered immune responses by β-arrestin 2. Importantly, p38 MAPK is identified as a key partner of β-arrestin 2 in the integration of IL-10 expression upon TLR stimulation.
EXPERIMENTAL PROCEDURES
Mice
β-arrestin 2 knock-out (KO) mice on a C57BL/6 background were generously provided by Dr. Robert Lefkowitz, Duke University Medical Center. TLR4 KO mice on a C57BL/6 background and wild type (WT) C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME). All mice were maintained in the Division of Laboratory Animal Resources at East Tennessee State University (ETSU), a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC). All animal studies were approved by the ETSU Committee on Animal Care.
Cells and Cell Cultures
WT, β-arrestin 1 KO, β-arrestin 2 KO, and double β-arrestin 1/2 KO (DKO) mouse embryonic fibroblasts (MEFs) were generously provided by Dr. Robert Lefkowitz, Duke University Medical Center. MKK3 KO MEFs were generously provided by Dr. Bing Su, Yale University School of Medicine. HEK293 cells and HEK293 cells stably transfected with TLR4 (HEK293/TLR4) were kindly provided by Dr. Evelyn A. Kurt-Jones of the University of Massachusetts Medical School. THP-1 cells were purchased form ATCC (American Type Culture Collection, USA). All cells were cultured in DMEM containing 10% FBS, 100 units/ml penicillin, and 100 mg/ml streptomycin. For isolation of peritoneal macrophages, mice were administered with 2 ml of 4% thioglycollate by intraperitoneal (intraperitoneal) injection. Three days later, peritoneal macrophages were harvested and washed with PBS. Cells were resuspended in DMEM containing 10% FBS and incubated overnight at 37 °C in a humidified incubator containing 5% CO2. Nonadherent cells were removed by washing with PBS and adherent macrophages were stimulated with LPS.
Reagents
Anti-phospho-MKK3/6, Anti-ASK1, anti-phospho-JNK, anti-JNK, anti-phospho-ERK, anti-ERK, anti-phospho-p38, anti-p38, and anti-GAPDH antibodies were purchased from Cell Signaling Technology (Beverly, MA). Anti-β-arrestin 2 and anti-GFP antibodies were obtained from Abcam (Cambridge, MA). Anti-FLAG antibody was purchased from Sigma-Aldrich. Anti-phospho-ASK1 and anti-MKK3/6 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). TLR4 ligand LPS, TLR2 ligand peptidoglycan (PGN), and Morphine were purchased from Sigma. Murine TLR9 ligand CpG ODN 1826 and control ODN were obtained from InvivoGen (San Diego, CA). TNF-α, anti-IL-10 antibody, and recombinant mouse IL-10 were purchased from R&D systems (Minneapolis, MN). P38 inhibitor SB203580, ERK inhibitor PD98059 and JNK inhibitor SP600125 were obtained from Alexis (San Diego, CA).
Transfection
β-Arrestin 2 full-length GFP vector, p38 plasmid and their control vectors were generous gifts from Dr. Gang Pei, Shanghai Institutes for Biological Sciences, China. HEK293 cells, HEK293/TLR4 cells, β-arrestin 1/2 DKO MEFs, and MKK3 KO MEFs were seeded in culture plates 24 h before transfection. Transfection was performed using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA).
Measurement of Cytokine Production
Peritoneal macrophages and MKK3 KO MEFs were cultured in 12-well plates and stimulated with LPS. Then culture supernatants were collected. For serum cytokine measurement, blood was collected from all experimental and control mice 3 h after LPS treatment. Samples were allowed to clot for 2 h at room temperature before centrifugation for 20 min at 2000 × g. Serum was removed and stored at −20 °C for subsequent assay. The amount of cytokines in the supernatants and serum was examined by using Quantikine Mouse ELISA kits (R&D Systems).
Western Blot
Cells were lysed in ice-cold lysis buffer containing 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, 1 mm phenylmethyl sulfonyl fluoride. After centrifugation, all extracts were resolved by 10% SDS-PAGE and analyzed by immunoblot. The membranes were probed with appropriate primary antibodies. The blot was exposed to the SuperSignal West Dura Extended Duration substrate (Pierce Biotechnology, Rockford, IL). The signals were quantified by scanning densitometry using a Bio-Image Analysis System (Bio-Rad).
Confocal Microscopy
MEFs and peritoneal macrophages were seeded on glass coverslips and stimulated with LPS for the desired times. After fixation with 4% paraformaldehyde, cells were exposed to a permeabilization solution (0.1% Triton X-100, 0.1% sodium citrate) for 10 min and incubated with appropriated primary antibodies at 4 °C overnight. After washing with PBS, cells were labeled with secondary antibodies conjugated with FITC (Abcam) or Alexa Fluor® 594 (Abcam). Confocal mages were obtained with a Leica TCS SP2 confocal microscope system (Leica Microsystems Inc., Bannockburn, IL) with dual line switching excitation (488 nm and 543 nm) and emission (500–560 nm for FITC, 580–670 nm for Alexa Fluor® 594) filter sets.
Co-Immunoprecipitation (Co-IP)
HEK293/TLR4 and HEK293cells were transfected with β-arrestin 2 full-length GFP vectors or empty vector for 48 h. THP-1 cells were co-transfected with β-arrestin 2 full-length GFP vectors and p38 plasmids for 48 h. The cells were treated with LPS, TNF-α or CpG-ODN for the indicated time periods. Then Co-IP was performed with Pierce® Co-Immunoprecipitation Kit according to the manufacturer's protocol. Briefly, cells were incubated with IP Lysis/Wash Buffer on ice for 5 min to obtain cell lysates. Anti-GFP antibody was immobilized using AminoLink Plus coupling resin. The antibody-coupled resin was incubated with cell lysates overnight at 4 °C. Protein complexes bound to the antibody were then eluted with elution buffer and analyzed by Western blot as described before.
Luciferase Reporter Assay
A 1010 bp hIL-10 promoter fragment was subcloned into the XhoI polylinker site of pGL3B luciferase reporter plasmid as described (18). Transfection of HEK293/TLR4 cells and HEK293 cells with the above plasmid was performed using Lipofectamine 2000 reagent. Briefly,10 μg of the target plasmid and 5 μg of pSV-β-galactosidase internal control vector (Promega) were incubated with 10 μl of Lipofectamine reagent in 200 μl of OPTI-MEM reduced serum medium (Invitrogen) for 45 min. These complexes were added to each well containing cells and culture medium in 6-well plates for 24 h. The cells were stimulated with 1 μg/ml LPS. 24 h later, cells were lysed in 150 μl of luciferase lysis reagent/well. After 15 min of incubation at room temperature, lysates were centrifuged at 7200 × g for 15 s, and the supernatant solutions were kept for the following assay. Luciferase assay substrates (100 μl) were mixed with 30 μl of cell lysate, and luciferase activity was measured by a 1250 Luminometer (Bio-Orbit, Finland).
Quantitative Real-time RT-PCR
Total RNA was isolated from cells and tissues using a RNeasy Plus Mini Kit (Qiagen Sciences) according to the manufacturer's instructions. One microgram of RNA from each sample was used for reverse transcription and synthesis of cDNA using a Reaction ReadyTM first strand cDNA synthesis kit (SABiosciences, Frederick, MD). PCR was performed using RT2 real-timeTM SYBR Green Fluorescien PCR Master Mix (SABiosciences). GAPDH expression was used as internal control. The primer sequences used were as follows: IL-10 forward 5′-TGC TAA CCG ACT CCT TAA TGC AGG AC-3′, IL-10 reverse 5′-CCT TGA TTT CTG GGC CAT GCT TCT C-3′, GAPDH forward 5′-TGA CCA CAG TCC ATG CCA TC-3′, GAPDH reverse 5′-GAT GGG GGT TAC ACA GGC AG-3′.
Endotoxin Shock
For endotoxicity studies, WT and β-arrestin 2 KO mice received an intraperitoneal injection of LPS (20 mg/kg body weight). At 3 h after the injection, the liver and spleen were harvested. Relative mRNA in liver and spleen was determined by quantitative real-time RT-PCR. Blood was collected, and serum IL-10 was examined by ELISA. For endotoxic shock, mice were challenged with LPS (10 mg/kg body weight, intraperitoneal) and monitored for survival for up to 48 h after injection.
Statistical Analyses
Student's t test was used to determine the significance of the differences between groups. Survival curves were generated using the Kaplan-Meier method, and the significance of difference was calculated by the log-tank test. A value of p < 0.05 was considered statistically significant.
RESULTS
β-Arrestin 2 Deficiency Impairs IL-10 Production in Response to LPS Stimulation
To investigate the role of β-arrestin 2 in TLR4-triggered inflammatory signaling, we isolated peritoneal macrophages from WT and β-arrestin 2 KO or TLR4 KO mice and examined multiple cytokine production by these macrophage stimulated with LPS. The results of ELISA showed that minimal cytokine production was detected from TLR4-deficient macropahges (data not shown), indicating that TLR4 is specifically required for cytokine response to LPS. LPS significantly increased the level of anti-inflammatory cytokine IL-10 in WT macrophages, whereas less induction of IL-10 was observed in macrophages lacking β-arrestin 2. By contrast, the amount of LPS-induced IL-4 secretion was comparable between these two cell types (Fig. 1). The unique control of IL-10 production by β-arrestin 2 in TLR4 signaling was also confirmed in microglia cells isolated from WT and β-arrestin 2 KO mice (data not shown). These results indicate a potential role of β-arrestin 2 in the regulation of TLR4-stimulated IL-10 production.
FIGURE 1.
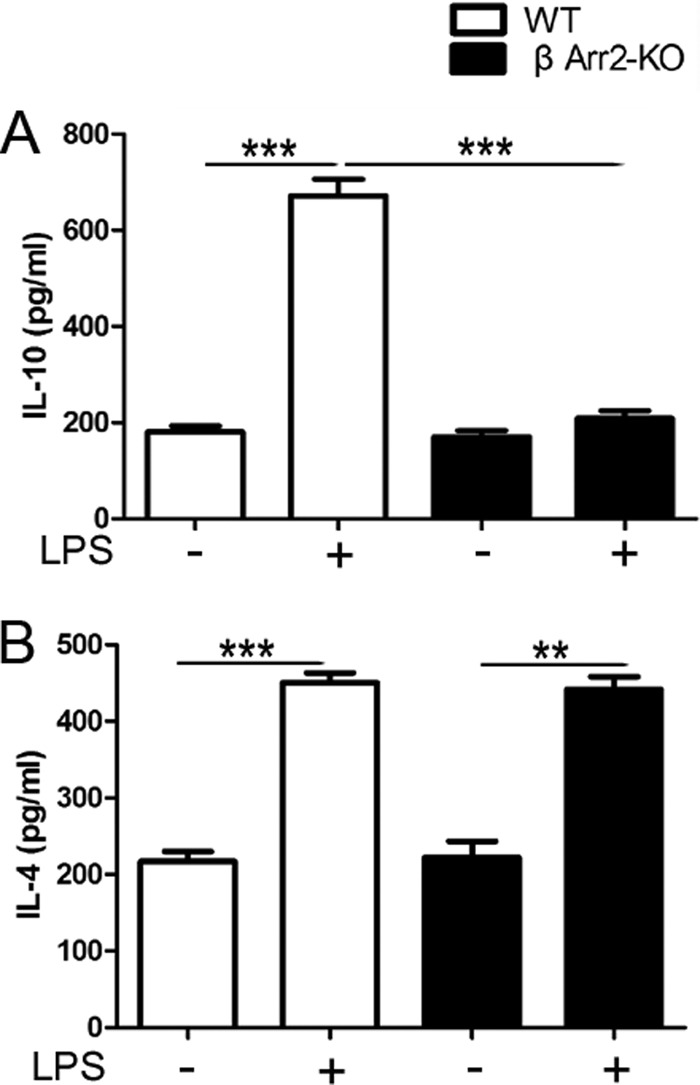
β-Arrestin 2 is required for IL-10 induction by TLR4 stimulation. Peritoneal macrophages were isolated from wild type (WT) and β-arrestin 2 knock-out (KO) mice (n = 5 per group) and stimulated in vitro with LPS (10 μg/ml) for 6 h. Cytokine production in the supernatants, anti-inflammatory cytokines IL-10 (A) and IL-4 (B) were assessed by ELISA. Data are expressed as means ± S.D. **, p < 0.01; ***, p < 0.001 compared with indicated groups.
Enhancement of IL-10 Promoter Activity by β-Arrestin 2
To obtain more evidence that supports the importance of β-arrestin 2 in LPS-induced IL-10 production, HEK293 and HEK293/TLR4 were cotransfected with hIL-10-luciferase reporter plasmid and either β-Arrestin 2 full-length GFP vector or control vector. The transfected cells were treated with 1 μg/ml LPS. As shown in Fig. 2, there were no significant changes of the luciferase activity in LPS-stimulated HEK293 cells with or without overexpression of β-arrestin 2, compared with untreated cells. In contrast, LPS stimulation induced a remarkable enhancement of hIL-10 promoter activity in HEK293/TLR4 cells, which is consistent with the finding that LPS treatment resulted in a significant increase of IL-10 synthesis in macrophages. More importantly, β-arrestin 2 overexpression strikingly enhanced this action of LPS. These results supports that β-arrestin 2 plays a positive role in LPS-induced IL-10 response, combined with the observation of disturbed IL-10 expression by β-arrestin 2 deletion as in Fig. 1.
FIGURE 2.
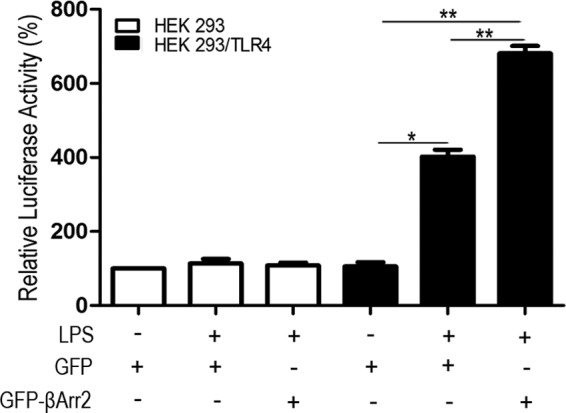
β-arrestin 2 facilitates TLR4-triggered IL-10 transcription. HEK293 and HEK293/TLR4 cells were transfected with hIL-10-luciferase reporter plasmid and either GFP-β-arrestin 2 full-length vector or control vector. The transfected cells were analyzed for luciferase activity after treatment with 1 μg/ml LPS for 2 h. Data are means ± S.D. from three independent experiments. *, p < 0.05; **, p < 0.01 compared with indicated groups.
TLR4-triggered P38 Activation Requires β-Arrestin 2 Recruitment
MAPKs activation may be mediated in a β-arrestin-dependent manner (19, 20). To investigate the mechanisms responsible for the β-arrestin 2-regulated IL-10 response to LPS, we first evaluated the role of β-arrestin 2 in activation of MAPK downstream of TLR4 signaling. Here, MEF cells were employed and then we tested the expression of TLR4 on them. Importantly, both WT and β-arrestin 2 KO MEFs expressed high levels of TLR4 protein, comparable to that in HEK/TLR4 cells (data not shown). Fig. 3A shows that in WT MEFs, all MAPKs were rapidly activated within 15 min after LPS stimulation. Interestingly, LPS-enhanced JNK and ERK1/2 activity was both attenuated by β-arrestin 2 deletion, whereas the dynamic of p38 activation by LPS was completely abolished in β-arrestin 2-deficient cells. Our findings reveal an indispensable role of β-arrestin 2 in the regulation of p38 MAPK in TLR4-triggered signaling.
FIGURE 3.

p38 is activated through a β-arrestin 2-dependent mechanism in TLR4 signaling. A, WT, β-arrestin 2 KO, and β-arrestin 1 KO MEFs were stimulated with LPS (20 μg/ml) for the indicated times. Levels of phospho-p38, phospho-JNK and phospho-ERK were examined by immunoblotting with specific antibodies. B, β-arrestin 1/2 DKO MEFs were transfected with β-arrestin 2 full-length construct or control vector, and analyzed for the expression of phospho-p38 by Western blot following stimulation with LPS (20 μg/ml) for 15 min. Results are representatives of three independent experiments.
β-arrestin 1 and β-arrestin 2 share ∼70% similar sequence and repetitive functions. In some case, they play distinct roles in the regulation of innate immune responses (21). Therefore, we measured p38 cascade response to LPS in β-arrestin 1-deficient MEFs, and found that the activation of p38 MAPK as well as JNK and ERK1/2 was effectively induced with a kinetic similar to that in WT MEFs following LPS stimulation (Fig. 3A). This result suggests that β-arrestin 1 seems to function redundantly in LPS-elicited p38 activation. To further confirm that LPS induces p38 MAPK activation through a β-arrestin 2-dependent mechanism, β-arrestin 1/2 double KO MEFs were cultured and re-transfected with β-arrestin 2 full-length vector prior to LPS treatment. As shown in Fig. 3B, LPS-stimulated phosphorylation of p38 was restored by the presence of β-arrestin 2.
Regulation of P38 MAPK Pathway by β-Arrestin 2 Is Typical of TLR2/4 Signaling Pathway
To determine whether the β-arrestin 2 dependent p38 activation is specific to TLR4 signal, we assessed TLR2 and TLR9-triggered p38 pathway in the same cell lines. Impaired activation of p38 MAPK following treatment with TLR2 ligand PGN was also observed in the setting of β-arrestin 2 depletion, whereas TLR9 ligand CpG-ODN-induced p38 phosphorylation appeared to be comparable between WT and β-arrestin 2 KO MEFs (Fig. 4A). It is possible that cell surface TLRs and intracellular TLRs use mechanistically distinct adaptors to provoke p38 cascade. To exclude the possibility that defective β-arrestin 2 was sufficient to disturb p38 activation in regardless of TLR triggering, we examined the impact of β-arrestin 2 deficiency on cytokine receptor signaling (TNF-α) or the GPCR signaling (morphine)-activated p38 cascade under similar conditions. Interestingly, β-arrestin 2 deficiency did not ruin p38 activation after treatment of either TNF-α or morphine (Fig. 4B). Taken together (from Figs. 3 and 4), β-arrestin 2-regulated p38 cascade seems a unique pattern within cell surface TLR2/4 signaling.
FIGURE 4.

β-Arrestin 2-regulated p38 activity is typical of TLR2/TLR4 signaling. A, WT and β-arrestin 2 KO MEFs were stimulated with 10 μg/ml PGN (TLR2 ligand) or 2 μm CPG (TLR9 ligand) for the indicated times. The level of total and phospho-p38 was determined by immunoblotting. B, WT and β-arrestin 2 KO MEFs were stimulated with 40 ng/ml TNF-α or 20 μm morphine for the indicated times. The expression of total and phospho-p38 was examined as in A.
P38 Activation Is Facilitated through Interaction with β-Arrestin 2
To dissect which constituent is the direct target of β-arrestin 2 required for the increased activity of p38 in response to LPS, we examined ASK1 and MEK3/6, the upstream activators of p38. As shown in Fig. 5A, identical kinetics of ASK1 and MEK3/6 activation in a time-dependent manner stimulated by LPS was observed between WT and β-arrestin 2 KO MEFs, indicating that β-arrestin 2-mediated p38 activation is dispensable for ASK1/MEK3/6 signaling. We verified this notion in peritoneal macrophages isolated from WT and β-arrestin 2 KO mice. P38 MAPK was activated in WT macrophages after treatment of LPS, which was abolished by β-arrestin 2 deficiency (Fig. 5B). Intriguingly, there were no significant changes in the levels of phosphorylated ASK1 or MEK3/6 both in WT and β-arrestin 2 KO macrophages following LPS stimulation. These results inspire us to reasonably speculate that β-arrestin 2 is more likely to associate with p38 MAPK directly.
FIGURE 5.

β-Arrestin 2 dynamically interacts with p38 after TLR4 triggering. A, WT and β-arrestin 2 KO MEFs were stimulated with LPS (20 μg/ml) for the indicated times. Levels of total and phospho-ASK1, total and phospho-MEK3/6 were determined by immunoblotting. B, peritoneal macrophages isolated from WT and β-arrestin 2 KO mice (n = 5 per group) were treated with 10 μg/ml LPS for the indicated times. The expression of total and phospho-p38, total and phospho-ASK1, total and phospho-MEK3/6 was examined as in A. C, WT MEFs and peritoneal macrophages were stimulated with LPS at the dose of 20 μg/ml and 10 μg/ml, respectively, for the indicated times. p38 (green) and β-arrestin 2 (red) distribution were viewed using confocal microscopy. Bar, 15 μm. D, control vector or β-arrestin 2 plasmid-transfected HEK293/TLR4 cells were cultured in the absence or presence of LPS (1 μg/ml) for the indicated times. Cell extracts were immunoprecipitated with anti-GFP antibody and then analyzed together with cell lysates by immunoblotting with indicated primary antibodies. IP and IB denote immunoprecipitation and immunoblotting, respectively. E, THP-1 cells, co-transfected with β-arrestin 2 plasmid and p38 plasmid, were cultured with LPS (1 μg/ml for 5 min), TNF-α (40 ng/ml for 5 min), or CpG-ODN (2 μm for 30 min). Interaction of β-arrestin 2 and p38 were determined as in (D). All of the results are representatives of three independent experiments.
To evaluate this possibility, we used confocal immunofluorescence microscopy to monitor β-arrestin 2 and p38 distribution in peritoneal macrophages and MEFs. Both p38 (green) and β-arrestin2 (red) were mainly located in the cytoplasm (Fig. 5C). Before LPS stimulation, no significant colocalization was detected between these two proteins. After 5 min of LPS treatment, the merging of β-arrestin 2 and p38 signals was found. However, the colocalizaion disappeared again after 15 min of LPS stimulation. These data suggest that there is a rapid and transient interaction between β-arrestin 2 and p38 in response to LPS. Whether the interaction is dependent on p38 phosphorylation or not will be further investigated in our future work.
The potential interaction was further assessed by Co-IP assay. HEK293/TLR4 cells were transfected with β-arrestin 2 full-length plasmid. After stimulation with LPS, p38 immunoprecipitated together with β-arrestin 2 is in a time dependent manner with a peak at 5 min (Fig. 5D). By contrast, the activation of p38 reached a peak at 15 min of LPS stimulation, implicating that the interaction between p38 and β-arrestin 2 facilitate p38 MAPK activity. Taken together, a transient and dynamic regulatory mechanism controlled by β-arrestin 2 participates in LPS-triggered p38 activation.
Fig. 4 has shown that deficiency of β-arrestin 2 failed to disturb p38 activation upon CpG-ODN or TNF-α stimulation. We speculated that there might be no interaction of p38 and β-arrestin 2 in these situations. As expected, p38 was not co-immunoprecipitated with β-arrestin 2 after stimulation of CpG-ODN or TNF-α as shown by Co-IP analysis (Fig. 5E). The lack of interaction was further confirmed by ICC (data not shown).
Optimal Induction of IL-10 by LPS Requires P38 MAPK Activity
MAPKs, including ERK and p38 MAPK, play a role in the regulation of IL-10 production in different systems such as TLR-triggered signals (22, 23). Since β-arrestin 2-deficient significantly disturbed both p38 activation and IL-10 production following LPS stimulation (Figs. 1 and 3), we next defined whether p38 MAPK activity is required for the induction of IL-10 by LPS. The dose of pharmacological inhibitor used thereafter did not affect cell viability (data not shown). Fig. 6A shows that cells treated with different doses of p38 inhibitor SB203580 produced less IL-10 than those untreated cells after stimulation of LPS. In contrast, neither ERK nor JNK inhibitor prevented the induction of IL-10 by LPS. To confirm the role of p38 pathway in IL-10 production, we also assessed IL-10 expression in MKK3 KO MEFs. Notably, a remarkably decreased production of IL-10 stimulated by LPS occurred in the cells lacking MKK3 compared with WT cells (Fig. 6B). Consistently, MKK3 KO MEFs overexpressing p38 significantly enhanced IL-10 release following LPS treatment (Fig. 6C).
FIGURE 6.
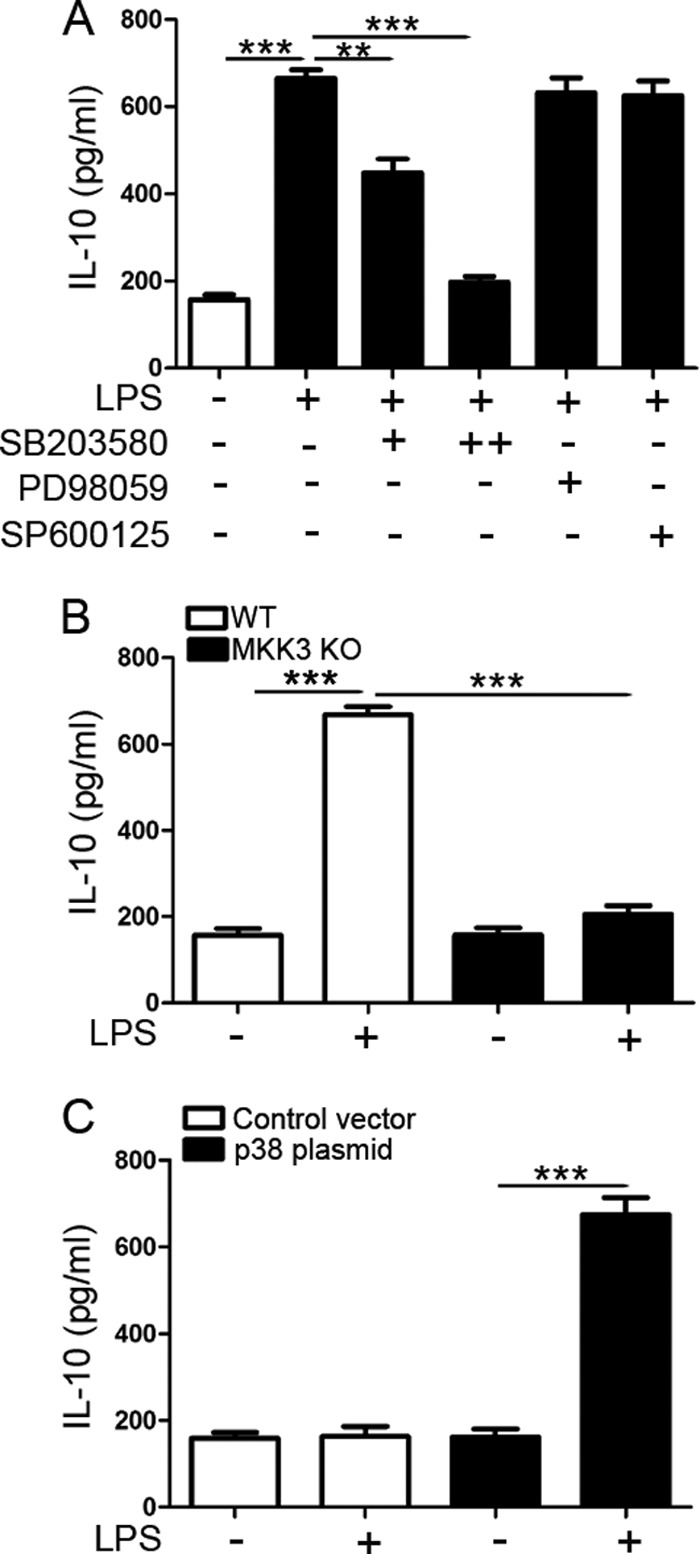
IL-10 induction by LPS requires p38 MAPK. A, WT peritoneal macrophages were stimulated with LPS (10 μg/ml) for 6 h in the absence of presence of the p38 inhibitor SB203580 (10 and 20 μm), the ERK inhibitor PD98059 (10 μm) and the JNK inhibitor SP600125 (10 μm). IL-10 level in cell-free supernatants was assessed by ELISA. For SB203580, + and ++ denote the lower dose (10 μm) and the higher dose (20 μm), respectively. B, WT and MKK3 KO MEFs were stimulated with LPS (20 μg/ml) for 2 h. IL-10 production in cell-free supernatants was measured as in A. C, control vector or p38 plasmid-transfected MKK3 KO MEFs were stimulated with LPS (20 μg/ml) for 2 h. Production of IL-10 in cell-free supernatants was determined as in A. Data are means ± S.D. from three independent experiments; **, p < 0.01; ***, p < 0.001 compared with indicated groups.
Mice Deficient in β-Arrestin 2 Exert Less IL-10 Production and Enhanced Inflammatory Status upon LPS Stimulation
To gain insight into the physiological function of β-arrestin 2 in TLR4 signaling-triggered IL-10 response in vivo, WT and β-arrestin 2 KO mice were injected intraperitoneally with LPS. We found that β-arrestin 2 KO mice produced less IL-10 mRNA in the livers and spleens and less IL-10 protein in the serum than WT mice after LPS administration (Fig. 7, A and B), further supporting a pivotal role of β-arrestin 2 in TLR4-mediated IL-10 induction.
FIGURE 7.

β-Arrestin 2 KO mice exhibit impaired IL-10 response to LPS and are more susceptible to LPS-induced septic shock. A, WT and β-arrestin 2 KO mice (n = 5 per group) were injected with LPS (20 mg/kg, intraperitoneal). After 3 h injection, the mouse liver and spleen were harvested. Level of IL-10 mRNA was determined by real-time PCR. B, WT and β-arrestin 2 KO mice (n = 5 per group) were administered with LPS (20 mg/kg, intraperitoneal) for 3 h. Serum was collected and IL-10 production was assessed by ELISA. C, pretreated WT and β-arrestin 2 KO mice (n = 20–24 per group) with anti-IL-10 antibody (20 mg/kg, i.p) or isotype control IgG for 2 h were injected with LPS (10 mg/kg, intraperitoneal) and then monitored for survival for up to 48 h. D, pretreated β-arrestin 2 KO mice (n = 20–24 per group) with recombinant mouse IL-10 (3.0 μg/body) or 0.1% bovine serum albumin in phosphate-buffered saline (PBS, pH 7.0) for 2 h were injected with LPS (10 mg/kg, intraperitoneal) and then monitored for survival for up to 48 h. Data in A and B are means ± S.D.; *, p < 0.05; **, p < 0.01 compared with indicated groups.
To assess the in vivo involvement of β-arrestin 2 and IL-10 in TLR4 signaling, we administered WT and β-arrestin 2 KO mice with or without anti-IL-10 antibody (20 mg/kg, intraperitoneal) 1 h prior to the injection of LPS (10 mg/kg, intraperitoneal) and monitored their survival for 48 h. Forty-five percent of β-arrestin 2 KO mice died from LPS-induced endotoxic shock within 32 h, compared with only 9.5% of the WT group (Fig. 7C). Interestingly, the mice with anti-IL-10 antibody administration, especially β-arrestin 2 KO mice, were more susceptible to endotoxin shock than the mice without anti-IL-10 antibody injection (Fig. 7C).
To further evaluate the protective role of IL-10 in β-arrestin 2 KO mice, the mice were subcutaneously injected with recombinant mouse IL-10 (3.0 μg/body) at 2 h before LPS (10 mg/kg) injection. The results of survival rates revealed that IL-10 supplementation significantly alleviated the susceptibility of β-arrestin 2 KO mice to LPS-induced endotoxic shock (Fig. 7D). Taken together, these results suggest that β-arrestin 2 and IL-10 function as in vivo negative regulators of TLR4 signaling.
DISCUSSION
In this study, we have demonstrated that β-arrestin 2 plays a prominent role in the negative regulation of TLR4-triggered inflammatory responses by regulating p38 MAPK function and subsequent production of anti-inflammatory cytokine IL-10. Following TLR4 stimulation, β-arrestin 2 forms complexes with p38 kinase and thereby facilitates the activity of p38. β-arrestin 2 is also required for induction of IL-10 by TLR4 signaling, and p38 is a key partner of β-arrestin 2 among this process (Fig. 8). The role of β-arrestin 2 in regulating inflammatory reactions is significant, as depletion of β-arrestin 2 results in less IL-10 release after LPS stimulation. Our results reveal a new mechanism by which β-arrestin 2 orchestrates TLR4-mediated immune responses.
FIGURE 8.

Schematic diagram depicting that β-arrestin 2/p38 interaction mediates IL-10 expression in TLR4 signaling. Following TLR4 stimulation, β-arrestin 2 (β-Arr2) forms complexes with p38 and subsequently promotes p38 activation, resulting in increased IL-10 expression.
Accumulating evidence indicates that β-arrestin 2 has the ability to modulate immune functions through multiple mechanisms. For instance, β-arrestin 2 inhibits CXCR2-mediated neutrophil recruitment in the dorsal air-pouch model and the cutaneous wound-healing model (24). Another prior study suggests that β-arrestin 2 participates in the regulation of inflammatory responses during the development of allergic asthma (25). β-Arrestin 2 also exerts inhibitory effects on TLR signaling pathways. After activation of TLR4, β-arrestin 2 directly interact with TRAF6, prevents autoubiquitylation of TRAF6 and thus blocks the activation of NF-κB and AP-1, crucial regulators of cytokine production upon TLR stimulation (12). In the current study, a new mechanism was revealed for β-arrestin 2 in the resolution of TLR-mediated inflammatory responses.
IL-10 is a potent anti-inflammatory cytokine with significant roles in restraining immune responses and preventing excessive inflammatory response-induced host damage (26). Our study also supports its strong anti-inflammatory role in LPS-induced septic shock, as blocking IL-10 signal with anti-IL-10 antibody decreased the survival rate compared with untreated mice. A recent study presents a positive correlation between β-arrestin 2 expression and IL-10 level in patients with cryptococcal meningitis (27). Here, experiments using β-arrestin 2 KO macrophages showed a critical role for β-arrestin 2 in IL-10 response to TLR4 stimulation. In contrast, the expression of the other anti-inflammatory cytokine IL-4 was not affected by β-arrestin 2 deletion. Collectively, IL-10 is exclusively modulated by β-arrestin 2 among the anti-inflammatory cytokines assessed in this study. The relationship between β-arrestin 2 and IL-10 was also supported by the finding of enhanced IL-10 promoter luciferase activity observed in the β-arrestin 2-overexpressing cells. IL-10 gene expression is positively regulated by multiple transcriptional factors (e.g. Sp1, CREB, and AP-1) and there are specific binding sites for a given transcriptional factor in the IL-10 promoter region (28). Thus, which of them play a major role in the enhancement of IL-10 expression by β-arrestin 2 remains to be elucidated. More importantly, in vivo study presented here provide evidence that β-arrestin 2 and IL-10 are critical negative regulators of TLR4-induced inflammatory response, as β-arrestin 2 KO mice produced less IL-10, and deletion of β-arrestin 2 as well as blockage of IL-10 signal both enhanced the susceptibility to LPS-induced endotoxic shock. As expected, supplementation of IL-10 reversed the susceptibility of β-arrestin 2 KO mice to LPS stimulation. Additionally, Fig. 7C shows that IL-10 is not solely controlled by β-arrestin 2. In fact, the expression of IL-10 is regulated by a wide range of mechanisms (23). Thus, in our system, other regulatory mechanisms might also be involved in the fine tuning of IL-10 expression. Taken together, these data highlight the notion that β-arrestin 2 negatively regulates TLR4-mediated inflammatory responses by facilitating IL-10 response to TLR4 triggering. Some studies have demonstrated that LPS-treated mice die sooner when also treated with d-galactosamine (12). d-Galactosamine sensitizes mice to LPS treatment (12, 29). In comparison, when the mice were treated with LPS alone, they survive longer as we saw in our studies (30, 31). Thus, survival time of mice in this endotoxic shock model varies depending on d-galactosamine treatment.
Apart from its role in the negative regulation of GPCR signaling, β-arrestin 2 also functions as a molecular scaffold for many pathways such as MAPK modules (32, 33). It has been demonstrated that β-arrestin 2 forms complexes with the ERK cascade, including ERK2, after angiotensin stimulation, and the interaction enhances cRaf-1 and MEK-dependent cell signaling to ERK (16). The β-arrestin 2 scaffold for JNK3 activation by binding to the unique N-terminal region of JNK3 was also described in a previous study (34). Although β-arrestin 2 is required for p38 activation under some conditions (35, 36), whether there is an association between β-arrestin 2 and either p38 itself or its upstream kinases is unknown. MAPKs are central downstream mediators of TLR signals. In this study, we first evaluated LPS-induced MAPK activation. As expected, all three MAPK members (i.e. JNK, ERK, and p38) were rapidly activated after LPS stimulation. Intriguingly, unlike ERK and JNK activation, p38 activation in response to LPS was completely prevented by a deficiency of β-arrestin 2. In contrast, β-arrestin 1 deletion failed to block p38 phosphorylation, suggesting that β-arrestin 1 is not required for LPS regulation of p38. Thus, optimal LPS activation of p38 MAPK preferentially involves β-arrestin 2 isoform, despite both β-arrestin isotypes have been described to be involved in the negative regulation of TLR-triggered immune responses (12). One interpretation of this discrepancy is that the predominant mechanism used by a given arrestin to activate MAPK cascades varies between cell types and is influenced by certain cellular contexts. β-arrestin 2-dependent activation of p38 was also discovered in TLR2 signal. Curiously, WT and β-arrestin 2 KO cells presented identical activation of p38 in response to CpG-ODN, TNF-α or morphine, indicating that requirement of β-arrestin 2 for p38 cascade activation is not a general apparatus of major cell surface receptors. Additionally, β-arrestin 2-mediated p38 phosphorylation was independent of ASK1 and MEK3/6, the upstream activators of p38, which has been shown both in MEFs and macrophages in the present study. A plausible explanation for this phenomenon is the existence of the interaction between p38 and β-arrestin 2. Importantly, after LPS stimulation, the complex of p38 and β-arrestin 2 was detected both by confocal microscopy and Co-IP techniques, suggesting that β-arrestin 2 promotes p38 activation through binding with p38 in our system. To the best of our knowledge, this is the first report to demonstrate the association between p38 and β-arrestin 2. Whether p38 binds directly or indirectly to β-arrestin 2 will be explored in our future work.
In the current study, we observed a significant activation of all three MAPKs after LPS treatment, consistent with their roles in mediating TLR signals (37). MAPKs play a key role in the regulation of IL-10 expression (22, 29, 38–40). Then we attempted to elucidate whether they mediated IL-10 induction by LPS. Our studies suggest a critical role of p38 in the IL-10 response to TLR4 activation, since inhibition of p38 with a chemical inhibitor in macropahges or knock-out of its upstream activator in MEFs both blocked LPS-stimulated IL-10 expression. Conversely, p38 overexpression restored IL-10 response to LPS in MEK3 KO cells, further confirming the strong regulatory action of p38 in IL-10 synthesis. However, IL-10 produced by JNK inhibitor or ERK inhibitor-treated macrophages after LPS stimulation was comparable to that by non-treated macrophages. These observations indicated that the induction of IL-10 by TLR4 activation is p38- but not JNK- or ERK-dependent. The unique requirement of p38 for IL-10 production is also discovered in BV-2 cells following G protein-coupled adenosine receptor activation, and activation of CREB is demonstrated to be involved in the process of p38-mediated induction of IL-10 (22). Collectively, our studies reveal a model whereby β-arrestin 2 augments IL-10 production by promoting p38 activation.
In conclusion we have shown that β-arrestin 2 is a key regulator in the restraint of TLR4-mediated inflammatory reactions by forming complexes with p38 MAPK and thereby facilitating IL-10 release. For inflammatory diseases, determination of β-arrestin 2 expression level and further investigations may reveal a novel role of β-arrestin 2 in the pathogenesis and develop novel therapeutic strategies for the treatment.
Acknowledgments
We thank Dr. Robert Lefkowitz for WT, β-arrestin 1 KO, β-arrestin 2 KO, and β-arrestin 1/2 DKO MEF cells as well as breeding pairs of β-arrestin 2 KO mice, Dr. Bing Su for MKK3 KO MEF cells, and Dr. Evelyn A. Kurt-Jones for HEK293/TLR4 and HEK293 cells. We would also like to thank Dr. Gang Pei for β-arrestin 2 full-length GFP vector, p38 plasmid and their control vectors.
This work was supported in part by Research Grants NIDA020120 and NIGM094740 from the National Institutes of Health (to D. Y.). This work was also supported in part by the National Natural Science Foundation of China (81370433) (to Z. L.). This work was also supported in part by the Fundamental Research Funds for the Central Universities of China (121020) (to D. H.).
- TLR
- Toll-like receptor
- MAPK
- mitogen-activated protein kinase
- qRT-PCR
- quantitative RT-PCR
- TUNEL
- terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling
- LPS
- lipopolysaccharide
- GPCR
- G protein-coupled receptor.
REFERENCES
- 1. Krishnan J., Selvarajoo K., Tsuchiya M., Lee G., Choi S. (2007) Toll-like receptor signal transduction. Exp. Mol. Med. 39, 421–438 [DOI] [PubMed] [Google Scholar]
- 2. Kawai T., Akira S. (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384 [DOI] [PubMed] [Google Scholar]
- 3. Fiorentino D. F., Zlotnik A., Mosmann T. R., Howard M., O'Garra A. (1991) IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 147, 3815–3822 [PubMed] [Google Scholar]
- 4. Naiki Y., Michelsen K. S., Zhang W., Chen S., Doherty T. M., Arditi M. (2005) Transforming growth factor-β differentially inhibits MyD88-dependent, but not TRAM- and TRIF-dependent, lipopolysaccharide-induced TLR4 signaling. J. Biol. Chem. 280, 5491–5495 [DOI] [PubMed] [Google Scholar]
- 5. Murray P. J., Smale S. T. (2012) Restraint of inflammatory signaling by interdependent strata of negative regulatory pathways. Nat. Immunol. 13, 916–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matsuda R., Kezuka T., Nishiyama C., Usui Y., Matsunaga Y., Okunuki Y., Yamakawa N., Ogawa H., Okumura K., Goto H. (2012) Interleukin-10 gene-transfected mature dendritic cells suppress murine experimental autoimmune optic neuritis. Invest. Ophthalmol. Vis. Sci. 53, 7235–7245 [DOI] [PubMed] [Google Scholar]
- 7. Dai H., Ciric B., Zhang G. X., Rostami A. (2012) Interleukin-10 plays a crucial role in suppression of experimental autoimmune encephalomyelitis by Bowman-Birk inhibitor. J. Neuroimmunol. 245, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berg D. J., Kühn R., Rajewsky K., Müller W., Menon S., Davidson N., Grünig G., Rennick D. (1995) Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartzman reaction but not endotoxin tolerance. J. Clin. Invest. 96, 2339–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shukla A. K., Xiao K., Lefkowitz R. J. (2011) Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem. Sci. 36, 457–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shenoy S. K., Lefkowitz R. J. (2011) β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 32, 521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vroon A., Heijnen C. J., Kavelaars A. (2006) GRKs and arrestins: regulators of migration and inflammation. J. Leukoc. Biol. 80, 1214–1221 [DOI] [PubMed] [Google Scholar]
- 12. Wang Y., Tang Y., Teng L., Wu Y., Zhao X., Pei G. (2006) Association of β-arrestin and TRAF6 negatively regulates Toll-like receptor-interleukin 1 receptor signaling. Nat. Immunol. 7, 139–147 [DOI] [PubMed] [Google Scholar]
- 13. Gao H., Sun Y., Wu Y., Luan B., Wang Y., Qu B., Pei G. (2004) Identification of β-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-κB pathways. Mol. Cell 14, 303–317 [DOI] [PubMed] [Google Scholar]
- 14. McDonald P. H., Chow C. W., Miller W. E., Laporte S. A., Field M. E., Lin F. T., Davis R. J., Lefkowitz R. J. (2000) β-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science 290, 1574–1577 [DOI] [PubMed] [Google Scholar]
- 15. DeWire S. M., Ahn S., Lefkowitz R. J., Shenoy S. K. (2007) β-arrestins and cell signaling. Annu. Rev. Physiol. 69, 483–510 [DOI] [PubMed] [Google Scholar]
- 16. Luttrell L. M., Roudabush F. L., Choy E. W., Miller W. E., Field M. E., Pierce K. L., Lefkowitz R. J. (2001) Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proc. Natl. Acad. Sci. U. S. A. 98, 2449–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kovacs J. J., Hara M. R., Davenport C. L., Kim J., Lefkowitz R. J. (2009) Arrestin development: emerging roles for β-arrestins in developmental signaling pathways. Dev. Cell 17, 443–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ma W., Lim W., Gee K., Aucoin S., Nandan D., Kozlowski M., Diaz-Mitoma F., Kumar A. (2001) The p38 mitogen-activated kinase pathway regulates the human interleukin-10 promoter via the activation of Sp1 transcription factor in lipopolysaccharide-stimulated human macrophages. J. Biol. Chem. 276, 13664–13674 [DOI] [PubMed] [Google Scholar]
- 19. Décaillot F. M., Kazmi M. A., Lin Y., Ray-Saha S., Sakmar T. P., Sachdev P. (2011) CXCR7/CXCR4 heterodimer constitutively recruits β-arrestin to enhance cell migration. J. Biol. Chem. 286, 32188–32197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xie L., Qiao X., Wu Y., Tang J. (2011) β-Arrestin1 mediates the endocytosis and functions of macrophage migration inhibitory factor. PLoS One 6, e16428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seregin S. S., Appledorn D. M., Patial S., Bujold M., Nance W., Godbehere S., Parameswaran N., Amalfitano A. (2010) β-Arrestins modulate Adenovirus-vector-induced innate immune responses: differential regulation by β-arrestin-1 and β-arrestin-2. Virus Res. 147, 123–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koscso B., Csoka B., Selmeczy Z., Himer L., Pacher P., Virag L., Hasko G. (2012) Adenosine augments IL-10 production by microglial cells through an A2B adenosine receptor-mediated process. J. Immunol. 188, 445–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saraiva M., O'Garra A. (2010) The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 10, 170–181 [DOI] [PubMed] [Google Scholar]
- 24. Su Y., Raghuwanshi S. K., Yu Y., Nanney L. B., Richardson R. M., Richmond A. (2005) Altered CXCR2 signaling in β-arrestin-2-deficient mouse models. J. Immunol. 175, 5396–5402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Walker J. K., Fong A. M., Lawson B. L., Savov J. D., Patel D. D., Schwartz D. A., Lefkowitz R. J. (2003) β-arrestin-2 regulates the development of allergic asthma. J. Clin. Invest. 112, 566–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Banchereau J., Pascual V., O'Garra A. (2012) From IL-2 to IL-37: the expanding spectrum of anti-inflammatory cytokines. Nat. Immunol. 13, 925–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xia R., Hu Z., Sun Y., Chen S., Gu M., Zhou Y., Han Z., Zhong R., Deng A., Wen H. (2010) Overexpression of β-arrestin 2 in peripheral blood mononuclear cells of patients with cryptococcal meningitis. J. Interferon Cytokine Res. 30, 155–162 [DOI] [PubMed] [Google Scholar]
- 28. Mosser D. M., Zhang X. (2008) Interleukin-10: new perspectives on an old cytokine. Immunol. Rev. 226, 205–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hofmann S. R., Morbach H., Schwarz T., Rösen-Wolff A., Girschick H. J., Hedrich C. M. (2012) Attenuated TLR4/MAPK signaling in monocytes from patients with CRMO results in impaired IL-10 expression. Clin. Immunol. 145, 69–76 [DOI] [PubMed] [Google Scholar]
- 30. Trahey M., Weissman I. L. (1999) Cyclophilin C-associated protein: A normal secreted glycoprotein that down-modulates endotoxin and proinflammatory responses in vivo. Proc. Natl. Acad. Sci. U.S.A. 96, 3006–3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Porter K. J., Gonipeta B., Parvataneni S., Appledorn D. M., Patial S., Sharma D., Gangur V., Amalfitano A., Parameswaran N. (2010) Regulation of lipopolysaccharide-induced inflammatory response and endotoxemia by β-arrestins. J. Cell Physiol. 225, 406–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lefkowitz R. J., Rajagopal K., Whalen E. J. (2006) New roles for β-arrestins in cell signaling: not just for seven-transmembrane receptors. Mol. Cell 24, 643–652 [DOI] [PubMed] [Google Scholar]
- 33. DeFea K. A. (2011) β-arrestins as regulators of signal termination and transduction: how do they determine what to scaffold? Cell. Signal. 23, 621–629 [DOI] [PubMed] [Google Scholar]
- 34. Guo C., Whitmarsh A. J. (2008) The β-arrestin-2 scaffold protein promotes c-Jun N-terminal kinase-3 activation by binding to its nonconserved N terminus. J. Biol. Chem. 283, 15903–15911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morris G. E., Nelson C. P., Brighton P. J., Standen N. B., Challiss R. A., Willets J. M. (2012) Arrestins 2 and 3 differentially regulate ETA and P2Y2 receptor-mediated cell signaling and migration in arterial smooth muscle. Am. J. Physiol. Cell Physiol. 302, C723–C734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bruchas M. R., Macey T. A., Lowe J. D., Chavkin C. (2006) Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J. Biol. Chem. 281, 18081–18089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brown J., Wang H., Hajishengallis G. N., Martin M. (2011) TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J. Dent. Res. 90, 417–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Komori H., Watanabe H., Shuto T., Kodama A., Maeda H., Watanabe K., Kai H., Otagiri M., Maruyama T. (2012) α(1)-Acid glycoprotein up-regulates CD163 via TLR4/CD14 protein pathway: possible protection against hemolysis-induced oxidative stress. J. Biol. Chem. 287, 30688–30700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ley S., Weigert A., Weichand B., Henke N., Mille-Baker B., Janssen R. A., Brüne B. (2013) The role of TRKA signaling in IL-10 production by apoptotic tumor cell-activated macrophages. Oncogene 32, 631–640 [DOI] [PubMed] [Google Scholar]
- 40. Katholnig K., Kaltenecker C. C., Hayakawa H., Rosner M., Lassnig C., Zlabinger G. J., Gaestel M., Müller M., Hengstschläger M., Hörl W. H., Park J. M., Säemann M. D., Weichhart T. (2013) P38α Senses Environmental Stress To Control Innate Immune Responses via Mechanistic Target of Rapamycin. J. Immunol. 190, 1519–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]