

Background: The R21C mutation in cardiac troponin I (cTnI) prevents PKA-mediated phosphorylation of serines 23 and 24 of cTnI in vivo.
Results: Myofilament function is uncoupled from the intracellular [Ca2+] and delays muscle relaxation.
Conclusion: Long term ablation of cTnI phosphorylation leads to hypertrophy, diastolic dysfunction, and dysautonomia in mice.
Significance: Restoration of phosphorylated cTnI may prevent hypertrophic cardiomyopathy and diastolic dysfunction.
Keywords: Calcium, Cardiac Hypertrophy, Cardiac Muscle, Mouse, Troponin, R21C, Cardiac Troponin I, Heart Rate Variability, Hypertrophic Cardiomyopathy, Knock-in Mice
Abstract
The cardiac troponin I (cTnI) R21C (cTnI-R21C) mutation has been linked to hypertrophic cardiomyopathy and renders cTnI incapable of phosphorylation by PKA in vivo. Echocardiographic imaging of homozygous knock-in mice expressing the cTnI-R21C mutation shows that they develop hypertrophy after 12 months of age and have abnormal diastolic function that is characterized by longer filling times and impaired relaxation. Electrocardiographic analyses show that older R21C mice have elevated heart rates and reduced cardiovagal tone. Cardiac myocytes isolated from older R21C mice demonstrate that in the presence of isoproterenol, significant delays in Ca2+ decay and sarcomere relaxation occur that are not present at 6 months of age. Although isoproterenol and stepwise increases in stimulation frequency accelerate Ca2+-transient and sarcomere shortening kinetics in R21C myocytes from older mice, they are unable to attain the corresponding WT values. When R21C myocytes from older mice are treated with isoproterenol, evidence of excitation-contraction uncoupling is indicated by an elevation in diastolic calcium that is frequency-dissociated and not coupled to shorter diastolic sarcomere lengths. Myocytes from older mice have smaller Ca2+ transient amplitudes (2.3-fold) that are associated with reductions (2.9-fold) in sarcoplasmic reticulum Ca2+ content. This abnormal Ca2+ handling within the cell may be attributed to a reduction (2.4-fold) in calsequestrin expression in conjunction with an up-regulation (1.5-fold) of Na+-Ca2+ exchanger. Incubation of permeabilized cardiac fibers from R21C mice with PKA confirmed that the mutation prevents facilitation of mechanical relaxation. Altogether, these results indicate that the inability to enhance myofilament relaxation through cTnI phosphorylation predisposes the heart to abnormal diastolic function, reduced accessibility of cardiac reserves, dysautonomia, and hypertrophy.
Introduction
Inherited as an autosomal dominant disease, familial hypertrophic cardiomyopathy (HCM)2 is the most common genetic disorder of the heart (1). This clinical syndrome is characterized by an increase in left ventricular mass, diastolic dysfunction, and dysautonomia and carries a high incidence of sudden cardiac death (2). In many HCM cases, cardiac contractile dysfunction is attributed to inherited sarcomeric gene mutations, which can clinically present with variable penetrance, even within the same family pedigree (3, 4). Within the context of the sarcomere, it is well established that mutations in cardiac troponin (cTn) alter the Ca2+ sensitivity of cardiac muscle contraction (5, 6) and lead to excitation-contraction uncoupling, enhanced myofilament calcium buffering, and adaptations to intracellular calcium handling (7–10). Although HCM is characterized as a clinical syndrome, it is unclear how several HCM-linked mutations that yield different molecular and cellular phenotypes can manifest into a clinical population with abnormal diastolic function.
The kinetics of cardiac muscle activation and relaxation are intricately regulated by the handling of calcium in the cell and can be modified in a beat to beat fashion (11). During diastole, when cytosolic calcium is low, the role of cardiac troponin I (cTnI) within the cTn complex is to prevent the generation of force by inhibiting the interaction between the thick and thin filaments. Conversely, this inhibition is relieved when cytosolic calcium rises and binds to cardiac troponin C (cTnC) within the cTn complex (12). It is widely accepted that the activation of protein kinase A (PKA), mediated by β-adrenergic receptor (β-AR) stimulation, is responsible for the positive inotropic, chronotropic, and lusitropic effects on cardiac muscle dynamics (13). Among the several PKA targets, the phosphorylation of phospholamban (PLB) is predominantly responsible for the chronotropic responses (14), whereas the phosphorylation of myosin-binding protein C (MyBP-C) (15, 16) and cTnI are responsible for the lusitropic effects (17–20). Hence, phosphorylated cTnI serves to directly increase the rate of Ca2+ dissociation from cTnC and, together with phosphorylated MyBP-C, decreases the Ca2+ sensitivity of force generation (21–24).
Previously, we generated a homozygous knock-in (KI) mouse model that has an R21C amino acid substitution in cTnI (cTnI-R21C) that is associated with HCM and sudden cardiac death in humans (25, 26). Similar to human HCM patients, the mice developed a remarkable degree of hypertrophy and fibrosis (26). In a series of biochemical and proteomic assays, we demonstrated that cardiac fibers isolated from the homozygous mice were incapable of becoming phosphorylated in vivo and in vitro (by PKA) at serines 23 and 24 of cTnI due to the R21C mutation disrupting the PKA consensus sequence. Permeabilized cardiac fibers prepared from the R21C KI mice and WT littermates had comparable Ca2+ dependences of tension. However, unlike the WT, the R21C fibers were unable to decrease the Ca2+ sensitivity of force generation after incubation with PKA. These experiments confirmed an initial report that showed that recombinant cTnI-R21C blunts the PKA-mediated myofilament Ca2+ desensitization when reconstituted in skinned cardiac fibers (27).
Based on the above, we rationalized that one mechanism by which HCM can manifest is through impairing the phosphorylation of cTnI and, consequently, blunting its lusitropic effects. In the present study, we show that the homozygous R21C KI mice develop HCM late by recapitulating the aberrant morphologic, hemodynamic, and electrocardiographic properties accompanying the human disease phenotype. We begin to explore the cardiac autonomic regulation of R21C KI mice hearts and reveal some of the molecular and cellular events that are triggered in response to an impaired β-agonist-induced myofilament relaxation. The results herein demonstrate that the phosphorylation of cTnI by PKA is critical in maintaining normal cardiac diastolic function, preventing hypertrophy and, to some extent, adequate heart rate modulation.
EXPERIMENTAL PROCEDURES
Animal Models
All protocols and experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Miami and the Florida State University following the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication 85-234, revised 1996). We studied mice that have a homozygous KI mutation in the TNNI3 gene (encoding cardiac troponin I) which substitutes an arginine at residue 21 for cysteine (R21C) and completely prevents PKA-mediated phosphorylation of Ser-23/24 of cTnI (26). The R21C KI mice were generated by implanting recombinant embryonic stem cells (Tc1) within C57Bl/6 blastocysts. Male progeny with high color coat chimerism were bred to C57Bl/6 females. Heterozygous mice were then bred with transgenic CMV-Cre mice to remove the neo cassette that was flanked by two loxP sites. R21C KI homozygotes were generated through the breeding of heterozygous mice that had their neo cassette excised (26). Two age groups of mice were used in our investigation and are categorized as follows: young (∼6 months old) and old (between 12 and 15 months old).
Echocardiography (ECHO)
Young and old male R21C and WT littermate mice (n = 9, for each genotype and age group, totaling 36 mice) were lightly anesthetized with isoflurane (2%) and prepared for ECHO to assess the electrocardiographic and hemodynamic changes induced by the KI mutation. M-mode imaging (Vevo 2100, VisualSonics) of the parasternal short axis view was used to evaluate systolic and diastolic dimensions as well as posterior wall thickness. M-mode imaging was also used to calculate fractional shortening and ejection fraction. A four-chamber view was used to acquire mitral valve flow parameters. Pulsed wave spectral Doppler was used to measure mitral valve inflow, including its two main waves, early (E wave) and late atrial (A wave), from which deceleration time and isovolumetric relaxation time were derived. Mitral valve inflow parameters were used as an index of diastolic function (28).
Heart Rate Variability (HRV); Experimental Design and Data Acquisition and Analysis
Ten old and weight-matched male R21C and WT littermate mice were used in this experiment. ECGs were recorded non-invasively in conscious mice using the ECGenie system recording platform (Mouse Specifics, Boston, MA). We evaluated cardiac autonomic modulation before and after β-AR stimulation with an intraperitoneal injection of isoproterenol (ISO; 2.5 μg/g; Sigma) dissolved in 0.1 ml. ECGs were recorded within 5 min prior to each injection and between 1 and 3 min after each injection to capture the peak of the response to the drug (29). Mice were gently removed from their cages and positioned on the ECG recording platform. Three gel-coated ECG electrodes (Red Dot; 3M, St. Paul, MN) are embedded in the floor of the platform and spaced (3 cm) to provide contact between the electrodes and the animal's paws. This configuration provides an ECG wave similar to an Eithenoven lead I. After a 30-min acclimation or heart rate (HR) equilibration period, ECG signals were recorded for 2–3 s (25–39 QRS complexes) while the mice passively established contact between the underside of its paws and the electrodes. Data acquisition was carried out using a proprietary Mouse Acquisition Module (Mouse Specifics). The signals are digitized with a 16-bit pre-a sampling rate of 2500 Hz. Analysis of individual ECGs was performed using the EzCG Analysis Software Package (Mouse Specifics). Body temperatures were measured using an infrared thermometer to verify consistency before and after taking ECG readings. The HRV was accessed following established guidelines (30, 31) through the time domains total power, percentage of adjacent R-R intervals that differ by a length of time exceeding the experimental threshold of 6 ms similar to pNN50 in humans (pNN6), and root mean square of successive R-R differences. These are considered markers of cardiac parasympathetic (vagal) modulation. The frequency domains, high frequency component (HF; 1.5–4.0 Hz; vagal modulation), and low frequency component (LF; 0.4–1.5 Hz; vagal-sympathetic interplay and baroreflex function) (32) of the HRV parameters were calculated by means of fast Fourier transformation.
Isolated Myocyte Preparation
Cardiomyocytes (CMs) were isolated from young and old R21C and WT mice (n = 3 for each genotype and age group). Mice were first anesthetized with isoflurane and then injected intravenously with propranolol (1 mg/kg body weight; Calbiochem). After 30 min, they were given another exposure to isoflurane before cervical dislocation (26). Hearts were excised, and CMs were isolated by enzymatic retrograde perfusion through the aorta as described previously (33). CMs were incubated with Fura-2/AM (Invitrogen), followed by continuous superperfusion with Tyrode's solution containing 1.8 mmol/liter Ca2+. Sarcomere length (SL) and intracellular Ca2+ ([Ca2+]i) transients were recorded with an IonOptix iCCD camera outfitted with the IonOptix Hyper SwitchTM dual-excitation filter (IonOptix LLC, Milton, MA). Fura-2 was excited at 340 and 380 nm, and the ratio of the emissions at 510 nm (i.e. Fura-2 ratio) represents the amount of [Ca2+]i. SL and [Ca2+]i transients were measured in CMs paced from 0.5 to 4 Hz as described previously (34). To estimate diastolic SL relaxation and Ca2+ removal, the time constant of relaxation, t½ was used. To assess the response to β-AR stimulation, 100 nmol/liter ISO was added to the above Tyrode's perfusion solution for 10 min before acquiring the data.
Caffeine-induced Sarcoplasmic Reticulum (SR) Ca2+ Content Release
Following the pacing protocol, electrical stimulation was stopped, and the superfusing solution was switched to a Na+- and Ca2+-free solution to avoid Ca2+ fluxes through the sarcolemma. Then a rapid pulse of 20 mm caffeine was applied in order to assess the SR Ca2+ content as described previously (35). The in vivo calibration was performed by superfusing a free Ca2+ solution containing 10 μmol/liter ionomycin (Sigma) until a minimal value of fluorescence (Rmin) was reached. Then the myocytes were superfused with Tyrode's solution containing ionomycin and a saturating [Ca2+] (5 mm CaCl2) until a maximal value of fluorescence (Rmax) was reached. [Ca2+]i was calculated using calibration Equation 1,
where the Kd (dissociation constant), Rmin, and Rmax were measured experimentally in adult myocytes and were 224 nmol/liter, 0.179, and 0.788, respectively. The Ca2+-free and -bound scaling factors, Sf2 and Sb2, are 2059 and 1264, respectively, and were derived from calibration as described previously (36). SR Ca2+ content was calculated considering that the SR represents 3.5% and the cytosol represents 65% of the myocyte's volume and fit to Equation 2, as described previously (37),
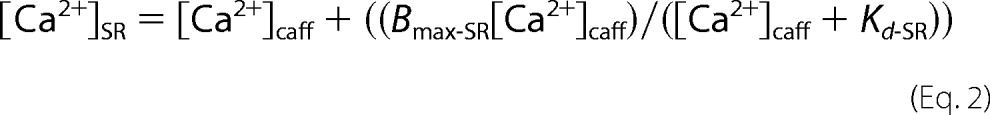 |
where [Ca2+]SR is the SR Ca2+ content, [Ca2+]caff is the SR Ca2+ released by caffeine, and Bmax-SR and Kd-SR are the usual Michaelis parameters for intra-SR Ca2+ binding.
Measurement of Cardiac Muscle Relaxation; Diazo-2 Flash Photolysis
Young WT and R21C mice were sacrificed as above after a propranolol injection. Skinned cardiac fibers (∼0.15 mm in diameter) isolated from WT and R21C mouse hearts were mounted onto a force transducer system (38). The fibers were chemically skinned with 1.0% Triton X-100 in pCa 8 (containing 10−8 mol/liter free [Ca2+]) solution for 30 min in order to remove membranes and organelles. Triton X-100 was removed by successive washes in pCa 8.0 solution prior to measuring the maximal force in pCa 4 (containing 10−4 mol/liter free [Ca2+]) contracting solution. To assess the effect of PKA-mediated phosphorylation on muscle relaxation, fibers were incubated with 0.5 units/μl of the catalytic subunit of PKA (Sigma) for 30 min in pCa 8 relaxing solution as described previously (26). Thereafter, fibers were incubated in a solution containing (mmol/liter) 2 Diazo-2 (Molecular Probes), 0.5 CaCl2, 60 TES (pH 7.0), 5 MgATP2−, 1 MgPr2, 10 creatine phosphate, along with 15 units/ml creatine phosphokinase in the cuvette surrounding the fiber (22). At the ratio of total added Ca2+ to Diazo-2 given above, the resulting average initial force was ∼80% of the initial force seen in the pCa 4 contracting solution. The fibers were flashed with a UV light to convert the Diazo-2 from a low to a high affinity Ca2+ chelator, which rapidly induces muscle relaxation (39). Each measurement was normalized to the maximal force in pCa 4, and the data were fit to Equation 3,
where Y is the quantity (i.e. relative force) at time Xi, A1 and A2 are the percentage amplitudes for both phases of the curve, and B1 and B2 are the corresponding rate constants (22). All solutions were made as described previously using the pCa Calculator program (40).
Immunoblotting
Immunoblot analyses were performed on total proteins isolated from young and old WT and R21C hearts (n = 3–4 for each genotype and age group) using the following antibodies: β1-AR (Abcam, 3442); calsequestrin (CSQ; Millipore, 06-382); GAPDH (Santa Cruz Biotechnology, 25778); phospho-MyBP-C_Ser-282 (MyBP-CSer-282; Enzo, ALX-215-067); Na+-Ca2+ exchanger (NCX1; SWANT, R3F1); total PLB (Millipore, 05-205); phospho-PLB_Ser-16 (PLBSer-16; Millipore, 07-052); phospho-PLB_Thr-17 (PLBThr-17; Badrilla Ltd., A010-13); and sarcoplasmic reticulum Ca2+ transport ATPase (SERCA2a; Pierce, MA3-919). GAPDH was used as a loading control, and all values were normalized to the WT control. Frozen ventricles were ground in liquid nitrogen, and an aliquot was thawed in 10% trichloroacetic acid containing 10 mmol/liter DTT. Precipitated protein was washed free of acid with three 5-min washes in ethyl ether and resuspended by vigorous agitation in urea sample buffer (8 mol/liter urea, 20 mmol/liter Tris base, 23 mmol/liter glycine, 0.2 mmol/liter EDTA, 10 mmol/liter DTT), using an orbital shaker (IKA Vibrax VXR) set at 1400 rpm for 30 min at room temperature. Complete denaturation and solubilization was achieved by the addition of urea crystals and prolonged agitation. Protein samples were centrifuged at 10,000 × g for 2 min, and protein concentrations in supernatant fractions were measured by a Bradford assay. For all proteins except NCX1, 2–40 μg were subjected to SDS-PAGE after boiling in Laemmli buffer and transferred to PVDF (Immobilon-FL, Millipore) or nitrocellulose (Protran, Whatman) and blotted by standard procedures. For NCX1 immunoblotting, solubilized samples were diluted into 2× Laemmli buffer and subjected to SDS-PAGE without boiling. The amount of protein loaded was optimized empirically for each antibody to ensure that density measurements were proportional to the amount of protein. Protein concentrations were determined by a Bradford assay using BSA as a control. Immunoblot band density was quantified by ImageQuant software.
Statistical Analysis
All values are presented as mean ± S.E. Error bars represent S.E. Statistical significance for ECHO, immunoblot, and SL transient data was determined by Student's t test. Significance for calcium transient, SR Ca2+ content, and flash photolysis data was determined by one-way analysis of variance using the Fisher LSD post hoc analysis. Significance for the HRV data was determined by a 2 × 2 ((R21C × WT) × (before −ISO × after +ISO)) repeat measures analysis of variance. When significant main effects or interactions were detected by repeated measures analysis of variance, a paired test was applied as a post hoc test.
RESULTS
Echocardiographic Features Associated with HCM in Old R21C KI Mice
Table 1 summarizes the baseline hemodynamic parameters measured by ECHO from anesthetized WT and R21C mice. At 6 months of age, the mitral velocities (E and A) from the R21C hearts were higher than the WT values (p < 0.05; Table 1). However, the mitral E/A ratio is comparable with that of the WT hearts. The young R21C mice do not present with left ventricle (LV) hypertrophy or other markers of diastolic dysfunction. Beyond 12 months of age, the older R21C mice have undergone morphological and hemodynamic changes associated with HCM. A comparison of the LV internal diameters of older WT and R21C mice in Fig. 1 reveals the marked changes in cardiac morphology associated with HCM.
TABLE 1.
Summary of echocardiographic parameters in WT and R21C mice
Data are mean ± S.E. and are derived from nine male R21C and nine WT littermates from each age group. HR, heart rate; SV, stroke volume; CO, cardiac output; ET, aortic ejection time; LV, left ventricular; EF, ejection fraction; FS, fractional shortening; M, mitral; E, passive LV filling peak velocity; A, atrial contraction flow peak velocity; EDV, end-diastolic volume; ESV, end-systolic volume; IVRT, isovolumetric relaxation time; IVCT, isovolumetric contraction time; PWd, posterior wall thickness at diastole; PWs, posterior wall thickness at systole. *, p < 0.05 versus WT. †, p < 0.01 versus WT.
| Parameter | Young (6 months) |
Old (12 months) |
||
|---|---|---|---|---|
| WT | R21C | WT | R21C | |
| HR (bpm) | 458 ± 9 | 450 ± 10 | 430 ± 9 | 476 ± 6† |
| R-R interval (ms) | 131 ± 2 | 131 ± 3 | 140 ± 3 | 126 ± 2† |
| SV (μl/beat) | 40.5 ± 3.5 | 41.0 ± 1.7 | 40.3 ± 3.2 | 28.5 ± 4.0* |
| CO (ml/min) | 18.6 ± 1.8 | 18.5 ± 0.9 | 17.5 ± 1.5 | 13.7 ± 1.9 |
| ET (ms) | 48.0 ± 1.9 | 50.0 ± 1.9 | 47.0 ± 3.1 | 37.9 ± 2.7* |
| %ETa | 29.9 ± 1.2 | 28.1 ± 0.8 | 33.7 ± 1.8 | 30.1 ± 2.3 |
| LV mass (mg) | 116.1 ± 4.8 | 115.1 ± 5.2 | 152.3 ± 11.8 | 218 ± 20.1* |
| EF (%) | 71.9 ± 3.0 | 70.7 ± 4.0 | 62.1 ± 2.6 | 65.6 ± 2.1 |
| FS (%)a | 41.3 ± 2.8 | 40 ± 2.3 | 33.5 ± 1.8 | 35.7 ± 1.6 |
| M E (cm/s) | 60 ± 3 | 70 ± 3* | 50 ± 4 | 45 ± 3 |
| M A (cm/s) | 41 ± 3 | 54 ± 4† | 31 ± 3 | 45 ± 4* |
| M E/A | 1.5 ± 0.2 | 1.3 ± 0.1 | 1.8 ± 0.3 | 1.0 ± 0.1* |
| E deceleration time (ms) | 12.4 ± 0.7 | 11.9 ± 0.4 | 13.7 ± 0.7 | 14.9 ± 0.3 |
| %E deceleration timea | 10.8 ± 0.6 | 12.9 ± 0.4 | 9.7 ± 0.5 | 11.9 ± 0.3† |
| EDV (μl) | 63.4 ± 7.3 | 58.5 ± 7.4 | 66.7 ± 7.6 | 44.2 ± 6.9* |
| ESV (μl) | 24.8 ± 4.5 | 26.4 ± 2.7 | 26.5 ± 4.8 | 15.7 ± 3.11 |
| IVRT (ms) | 24.1 ± 1.7 | 23 ± 1.7 | 22.4 ± 1.7 | 22.4 ± 1.7 |
| %IVRTa | 17.5 ± 1.1 | 17.1 ± 1.4 | 15.9 ± 1.1 | 17.8 ± 1.4 |
| IVCT (ms) | 18.1 ± 1.8 | 19.3 ± 1.6 | 22.2 ± 3.8 | 16.9 ± 1.7 |
| %IVCTa | 14.0 ± 1.1 | 14.1 ± 1.2 | 16.1 ± 2.9 | 13.3 ± 1.2 |
| PWd (mm) | 1.1 ± 0.1 | 0.9 ± 0.2 | 0.9 ± 0.1 | 1.6 ± 0.2* |
| PWs (mm) | 1.3 ± 0.1 | 1.3 ± 0.1 | 1.2 ± 0.1 | 1.9 ± 0.2* |
a %, denotes the temporal parameter was normalized to the R-R interval and expressed as percentage of the cardiac cycle.
FIGURE 1.

Representative live ECHO images of old (12 months) WT and R21C mice at diastole and systole. A and C, long axis parasternal view of left internal ventricular diameters during diastole in WT (A) and R21C mice (C). B and D, left internal ventricular diameters during systole in WT (B) and R21C mice (D).
The older R21C mice have a significantly shorter R-R interval (Δ = −14 ms; p < 0.01 versus WT; Table 1) and an increased HR (Δ = +46 beats/min; p < 0.001 versus WT; Table 1). Although the stroke and end diastolic volumes are smaller (p < 0.05 versus WT; Table 1) in the older R21C hearts, cardiac output (CO) is maintained. This suggests that the older R21C mice increase their HR in order to compensate for deficiencies in stroke volume and CO. The LV mass of the older R21C hearts is ∼1.43 times larger than that of the WT (p < 0.05; Table 1) and is accompanied by significant increases in posterior wall thickness during diastole and systole (p < 0.05 versus WT; Table 1). Because the HRs were significantly different among the genotypes, the temporal parameters (deceleration time, isovolumetric relaxation time, isovolumetric contraction time, and aortic ejection time) were normalized to the R-R interval and expressed as a percentage of the cardiac cycle as described previously (41, 42). Markers of HCM and diastolic dysfunction (coinciding with Stage I in humans) were present in the older R21C mice, as depicted by a significant reduction in the mitral E/A ratio (Δ = −0.8; p < 0.05 versus WT; Table 1) and by prolonging the normalized deceleration time by ∼20% (p < 0.01 versus WT; Table 1). No significant changes in the ejection fraction, fractional shortening, and normalized aortic ejection time existed (Table 1), suggesting preserved systolic function in the older R21C mice.
Electrocardiographic Features Associated with Dysautonomia in Older Conscious R21C Mice
We investigated whether abnormal cardiac autonomic modulation associated with HCM in humans is present in the older R21C mice. The ECGs were recorded and evaluated in conscious mice using markers of HRV. In the absence of ISO, the R21C mice have elevated HRs (Δ = +65 beats/min; p < 0.01 versus WT −ISO; Fig. 2A and Table 2). Notably, although ISO evokes a significant increase in the WT HRs (p < 0.05 versus WT −ISO; Fig. 2A), the HRs of older R21C mice do not increase in response to ISO, suggesting the existence of sympathetic hyperactivity and/or low vagal tone. In this context, we also observed that markers of vagal tone are reduced in the R21C mice, as shown by a lower root mean square of successive R-R differences (p < 0.01 versus WT −ISO; Fig. 2B and Table 2) and pNN6 (p < 0.05 versus WT −ISO; Table 2). After administering ISO to both groups, the WT markers of vagal tone decreased (p < 0.05 versus WT −ISO; Table 2) and were no longer statistically different from the values obtained from R21C mice. This suggests that the older R21C mice have impaired cardiovagal modulation and some degree of dysautonomia. Evaluation of other HRV parameters reveals that the total power and LF of older R21C mice are significantly smaller than the WT mice (p < 0.01 versus WT −ISO; Fig. 2, C and D, respectively); however, these effects become insignificant when both groups are exposed to ISO (Fig. 2, C and D). This suggests the existence of an aberrant heart rate modulation, such that the baroreflex function of the R21C mice is reduced. Altogether, these results indicate that long term ablation of cTnI phosphorylation leads to dysautonomia, increased sympathetic activity, and decreased vagal tone.
FIGURE 2.

Parameters of cardiac autonomic modulation before (gray) and after (black) ISO injections in older (12 months) conscious WT and R21C mice. A, HR; B, root mean square of successive R-R differences (RMSSD); C, total power (TP); D, LF. Data are given as mean ± S.E. Measurements were taken from 10 WT and 10 R21C mice. *, p < 0.05; †, p < 0.01 versus WT −ISO; ‡, p < 0.05; §, p < 0.01 versus WT −ISO. Error bars, S.E.
TABLE 2.
Parameters of cardiac autonomic modulation before and after isoproterenol (ISO) administration in older conscious WT and R21C mice
Data are given as mean ± S.E. and are derived from 10 male R21C and 10 WT littermate mice (12 months old). HR, heart rate; RMSSD, root mean square of successive R-R differences; pNN6 percentage of adjacent R-R intervals that differ by a length of time exceeding the experimental threshold (6 ms); TP, total power; HF, high frequency component of the heart rate variability; LF, low frequency component of the heart rate variability. *, p < 0.05 versus WT −ISO; †, p < 0.01 versus WT −ISO; ‡, p < 0.05 versus WT −ISO; §, p < 0.01 versus WT −ISO.
| Parameter | −ISO |
+ISOa |
||
|---|---|---|---|---|
| WT | R21C | WT | R21C | |
| HR (bpm) | 698 ± 16 | 763 ± 13† | 770 ± 12‡ | 767 ± 7 |
| RMSSD (ms) | 3.9 ± 1.3 | 0.9 ± 0.2* | 0.68 ± 0.1‡ | 0.8 ± 0.1 |
| pNN6 (ms) | 8.1 ± 0.9 | 2.9 ± 0.7† | 2.9 ± 1.1‡ | 2.1 ± 1.0 |
| TP (ms) | 2.1 ± 0.4 | 0.2 ± 0.1† | 1.0 ± 0.5‡ | 0.2 ± 0.1 |
| HF (ms2) | 1.97 ± 0.50 | 0.10 ± 0.06* | 0.33 ± 0.08‡ | 0.05 ± 0.07 |
| LF (ms2) | 4.05 ± 0.60 | 0.10 ± 0.09† | 0.12 ± 0.09§ | 0.10 ± 0.09 |
a Intraperitoneal injection of isoproterenol (2.5 μg/g).
Delayed Sarcomere Shortening Kinetics in Myocytes Isolated from Older R21C KI Mice
In order to further understand the cellular basis for the diastolic dysfunction in R21C mice, we examined whether these effects could arise from intramyocyte signaling independent of autonomic modulation. Table 3 shows that the contractile transient data measured in myocytes isolated from young R21C mice is not significantly different than the WT mice. However, Fig. 3 shows that beyond 12 months of age, R21C myocyte sarcomere shortening and relaxation are clearly affected. Fig. 3A illustrates the effects of the cTnI-R21C mutation on slowing sarcomere shortening and relaxation kinetics at 1 Hz. Under basal conditions (−ISO) and in the presence of 100 nmol/liter ISO, the t½ for SL relaxation is prolonged in the older R21C CMs at every pacing frequency (p < 0.05 versus WT; Fig. 3, A and B, respectively). In the presence and absence of ISO, the cTnI-R21C mutation also prolongs the time to peak (TTP) for SL shortening in myocytes paced from 0.5 to 3 Hz (p < 0.05 versus WT; Fig. 3, C and D, respectively). Faster frequencies and the addition of ISO have positive chronotropic and lusitropic effects on sarcomere shortening and relaxation kinetics in both groups. However, the R21C myocytes are unable to attain the WT ISO-mediated t½ SL relaxation times (Fig. 3C). Although the kinetics of sarcomere shortening and relaxation are slower in CMs isolated from the older R21C mice, the diastolic and systolic SLs are not different from those in the WT (see Table 4 for a summary).
TABLE 3.
Summary of Ca2+ and contractile transient parameters measured from cardiomyocytes isolated from young WT and R21C KI mice
*, p < 0.05 versus WT; †, p < 0.01 versus WT.
| Parameter | WTa |
R21Ca |
||||||
|---|---|---|---|---|---|---|---|---|
| 0.5 Hz | 2.0 Hz | 3.0 Hz | 4.0 Hz | 0.5 Hz | 2.0 Hz | 3.0 Hz | 4.0 Hz | |
| −ISOb | ||||||||
| [Ca2+]i decay (t½) (ms)c | 124 ± 12 | 89 ± 4 | 79 ± 5 | 64 ± 2 | 122 ± 4 | 86 ± 1 | 76 ± 2 | 66 ± 2 |
| Diastolic [Ca2+]i (Fura-2 ratio)d | 0.49 ± 0.02 | 0.51 ± 0.02 | 0.52 ± 0.02 | 0.53 ± 0.02 | 0.52 ± 0.01 | 0.53 ± 0.01 | 0.53 ± 0.001 | 0.54 ± 0.01 |
| Ca2+ amplitude (%)e | 19.9 ± 2.8 | 18.6 ± 2.7 | 19.4 ± 2.5 | 20.0 ± 2.5 | 14.2 ± 1.2* | 15.5 ± 1.2 | 16.2 ± 1.23 | 16.5 ± 1.0 |
| SL relaxation (t½) (ms)c | 58 ± 6 | 48 ± 4 | 46 ± 4 | 39 ± 3 | 64 ± 3 | 48 ± 2 | 45 ± 2 | 43 ± 2 |
| Diastolic SL (μm) | 1.75 ± 0.02 | 1.71 ± 0.02 | 1.69 ± 0.02 | 1.68 ± 0.03 | 1.73 ± 0.01 | 1.71 ± 0.01 | 1.69 ± 0.01 | 1.67 ± 0.02 |
| SL amplitude (%)e | 12.4 ± 0.8 | 10.6 ± 0.6 | 10.8 ± 0.6 | 9.9 ± 0.6 | 11.8 ± 0.8 | 10.9 ± 0.7 | 9.8 ± 0.9 | 9.2 ± 9 |
| +ISOb | ||||||||
| [Ca2+]a decay (t½) (ms)c | 72 ± 3 | 57 ± 2 | 55 ± 1 | 52 ± 2 | 76 ± 2 | 58 ± 1 | 56 ± 1 | 53 ± 1 |
| Diastolic [Ca2+]i (Fura-2 ratio)d | 0.53 ± 0.01 | 0.57 ± 0.02 | 0.57 ± 0.02 | 0.58 ± 0.02 | 0.51 ± 0.01 | 0.52 ± 0.01† | 0.52 ± 0.01* | 0.54 ± 0.02* |
| Ca2+ amplitude (%)e | 26.2 ± 3.5 | 27.0 ± 3.6 | 28.8 ± 4.1 | 28.6 ± 3.8 | 16.0 ± 1.2† | 16.2 ± 0.9† | 16.8 ± 1.1† | 18.3 ± 0.9† |
| SL relaxation (t½) (ms)c | 48 ± 4 | 41 ± 2 | 41 ± 3 | 41 ± 3 | 50 ± 2 | 41 ± 2 | 39 ± 1 | 37 ± 1 |
| Diastolic SL (μm) | 1.76 ± 0.02 | 1.72 ± 0.02 | 1.71 ± 0.02 | 1.67 ± 0.02 | 1.76 ± 0.01 | 1.73 ± 0.02 | 1.71 ± 0.02 | 1.69 ± 0.02 |
| SL amplitude (%)e | 15.7 ± 1.0 | 15.1 ± 0.9 | 15.2 ± 1.1 | 15.0 ± 1.0 | 15.6 ± 0.9 | 15.2 ± 0.9 | 14.8 ± 0.9 | 14.1 ± 0.9 |
a Values are given as mean ± S.E. (measurements were taken from 3 WT and 3 R21C KI mice (6 months old); 3–4 cells/heart were studied).
b +ISO and −ISO, presence and absence of 100 nmol/liter isoproterenol, respectively.
c t½, time it takes for the Ca2+ and SL amplitudes to decrease by 50%.
d Fura-2 was excited at 340 and 380 nm, and the ratio of their emissions at 510 nm (i.e. Fura-2 ratio) represents the [Ca2+]i.
e Ca2+ and SL amplitude represents the percentage increase of [Ca2+]i, and SL shortening that occurs during systole and is calculated using the equation, (|x − y|/y) × 100; where x is the peak systolic Fura-2 ratio (or SL), and y is the diastolic Fura-2 ratio (or SL).
FIGURE 3.

The effects of ISO and higher stimulation frequencies on sarcomere length shortening and relaxation kinetics in cardiomyocytes isolated from old WT and R21C mice. A, representative SL shortening transients recorded from WT (black) and R21C (gray) cardiomyocytes paced at 1 Hz in the absence (solid lines) and presence (dashed lines) of 100 nmol/liter ISO. B and C, time to half SL relaxation (t½) as a function of stimulation frequency in the absence (B) and presence (C) of 100 nmol/liter ISO. D and E, TTP SL shortening as a function of stimulation frequency in the absence (D) and presence (E) of ISO. t½, time it takes the SL transient amplitude to decrease 50%. Data are given as mean ± S.E. Measurements were taken from three WT and three R21C mice; 4–6 cells/heart were recorded and analyzed. The data are summarized in Table 4. *, p < 0.05; †, p < 0.01 versus WT. Error bars, S.E.
TABLE 4.
Summary of Ca2+ and contractile transient parameters measured from cardiomyocytes isolated from old WT and R21C KI mice
*, p < 0.05 versus WT; †, p < 0.01 versus WT.
| Parameter | WTa |
R21Ca |
||||||
|---|---|---|---|---|---|---|---|---|
| 0.5 Hz | 2.0 Hz | 3.0 Hz | 4.0 Hz | 0.5 Hz | 2.0 Hz | 3.0 Hz | 4.0 Hz | |
| −ISOb | ||||||||
| [Ca2+]i decay (t½) (ms)c | 175 ± 4 | 139 ± 3 | 126 ± 2 | 119 ± 2 | 177 ± 6 | 142 ± 4 | 129 ± 4 | 121 ± 3 |
| Diastolic [Ca2+]i (Fura-2 ratio)d | 0.51 ± 0.01 | 0.51 ± 0.01 | 0.52 ± 0.01 | 0.52 ± 0.01 | 0.54 ± 0.02 | 0.54 ± 0.02 | 0.54 ± 0.02 | 0.55 ± 0.03 |
| Time to peak [Ca2+]i (ms)e | 51.6 ± 3.1 | 45.7 ± 2.6 | 44.5 ± 2.3 | 44.7 ± 2.4 | 62.0 ± 2.6* | 53.8 ± 2.3* | 49.2 ± 2.9 | 49.1 ± 2.4 |
| Systolic [Ca2+]i (Fura-2 ratio)d | 0.58 ± 0.01 | 0.58 ± 0.02 | 0.58 ± 0.02 | 0.58 ± 0.02 | 0.60 ± 0.02 | 0.60 ± 0.02 | 0.60 ± 0.02 | 0.60 ± 0.02 |
| Ca2+ amplitude (%)f | 14.2 ± 1.4 | 14.4 ± 1.3 | 13.8 ± 1.4 | 13.9 ± 1.4 | 8.8 ± 0.5† | 9.7 ± 0.1† | 9.9 ± 1.0* | 9.3 ± 0.9* |
| SL relaxation (t½) (ms)c | 51.0 ± 3.4 | 41.0 ± 2.3 | 36.8 ± 2.2 | 32.4 ± 1.8 | 64.0 ± 3.3* | 52.4 ± 3.2† | 44.8 ± 2.2* | 38.7 ± 2.4* |
| Diastolic SL (μm) | 1.76 ± 0.01 | 1.75 ± 0.01 | 1.74 ± 0.02 | 1.74 ± 0.02 | 1.75 ± 0.03 | 1.73 ± 0.01 | 1.72 ± 0.01 | 1.72 ± 0.02 |
| Time to peak shortening (ms)e | 99.7 ± 4.7 | 83.0 ± 2.8 | 76.9 ± 2.9 | 73.1 ± 2.3 | 116.6 ± 4.7* | 92.8 ± 3.4* | 86.6 ± 3.3* | 79.2 ± 2.7 |
| SL amplitude (%)f | 10.8 ± 1.0 | 10.5 ± 0.8 | 10.3 ± 0.8 | 10.0 ± 0.7 | 9.9 ± 0.61 | 9.2 ± 1.0 | 8.9 ± 1.0 | 9.3 ± 1.0 |
| +ISOb | ||||||||
| [Ca2+]i decay (t½) (ms)c | 123 ± 2 | 104 ± 2 | 98 ± 2 | 96 ± 1 | 139 ± 3† | 113 ± 2† | 106 ± 2* | 103 ± 2* |
| Diastolic [Ca2+]i (Fura-2 ratio)d | 0.46 ± 0.01 | 0.47 ± 0.01 | 0.48 ± 0.01 | 0.49 ± 0.01 | 0.53 ± 0.02† | 0.53 ± 0.02† | 0.53 ± 0.02* | 0.53 ± 0.02* |
| Time to peak [Ca2+]i (ms)e | 34.9 ± 2.5 | 30.6 ± 2.0 | 30.6 ± 2.2 | 30.1 ± 2.3 | 46.4 ± 4.2* | 41.1 ± 2.7† | 38.7 ± 2.5* | 37.4 ± 2.7 |
| Systolic [Ca2+]i (Fura-2 ratio)d | 0.62 ± 0.02 | 0.63 ± 0.02 | 0.64 ± 0.03 | 0.66 ± 0.03 | 0.60 ± 0.02 | 0.59 ± 0.02 | 0.59 ± 0.02 | 0.60 ± 0.02 |
| Ca2+ amplitude (%)f | 31.2 ± 4.0 | 29.3 ± 3.6 | 29.7 ± 3.6 | 28.9 ± 3.3 | 12.6 ± 0.9† | 12.8 ± 0.8† | 13.5 ± 0.9† | 14.0 ± 0.9† |
| SL relaxation (t½) (ms)c | 43.2 ± 2.7 | 36.5 ± 2.6 | 33.8 ± 2.3 | 31.6 ± 2.3 | 52.7 ± 3.3* | 45.0 ± 2.6* | 42.6 ± 1.7† | 41.1 ± 1.9† |
| Diastolic SL (μm) | 1.75 ± 0.01 | 1.73 ± 0.01 | 1.72 ± 0.01 | 1.69 ± 0.02 | 1.74 ± 0.01 | 1.72 ± 0.01 | 1.70 ± 0.01 | 1.67 ± 0.01 |
| Time to peak shortening (ms)e | 82.3 ± 3.4 | 71.7 ± 1.8 | 67.9 ± 1.5 | 67.8 ± 1.9 | 94.2 ± 4.1* | 81.1 ± 2.5† | 76.7 ± 2.2† | 72.6 ± 2.0 |
| SL amplitude (%)f | 14.5 ± 1.0 | 13.9 ± 0.9 | 14.6 ± 0.9 | 14.7 ± 0.9 | 14.9 ± 0.8 | 13.8 ± 0.6 | 13.8 ± 0.7 | 13.7 ± 0.6 |
a Values are given as mean ± S.E. (measurements were taken from 3 WT and 3 R21C KI mice (15 months old); 4–6 cells/heart were studied).
b +ISO and −ISO, presence and absence of 100 nmol/liter isoproterenol, respectively.
c t½, time it takes for the Ca2+ and SL amplitudes to decrease by 50%.
d Fura-2 was excited at 340 and 380 nm, and the ratio of their emissions at 510 nm (i.e. Fura-2 ratio) represents the [Ca2+]i.
e Time to peak, time it takes for the [Ca2+]i and SL at diastole to reach the peak [Ca2+]i and SL shortening at the end of systole.
f Ca2+ and SL amplitude, percentage increase of [Ca2+]i and SL shortening that occurs during systole and is calculated using the equation, (|x − y|/y) × 100, where x is the peak systolic Fura-2 ratio (or SL), and y is the diastolic Fura-2 ratio (or SL).
Altered Ca2+ Transient Parameters in Myocytes Isolated from Old R21C KI Mice
We next investigated whether the slower sarcomere shortening and relengthening kinetics in CMs isolated from older R21C mice are associated with changes in the Ca2+ transient. Fig. 4 shows several Ca2+ transient parameters recorded from Fura-2-loaded WT and R21C CMs (old) that were electrically stimulated from 0.5 to 4 Hz in the presence and absence of ISO. Fig. 4A illustrates the effects of the cTnI-R21C mutation on slowing [Ca2+]i decay times as well as maintaining higher diastolic levels of Ca2+ in the presence of ISO at 1 Hz. Under basal conditions (−ISO), R21C CMs do not elicit significant changes in the t½ of [Ca2+]i decay in comparison with the WT when paced from 0.5 to 4 Hz (Fig. 4B). This is opposite to the effect seen in Fig. 3B, where the t½ for SL relaxation in myocytes from older R21C mice was prolonged in the absence of ISO. These results suggest that the cTnI-R21C mutation leads to the uncoupling of SL relaxation from the [Ca2+]i. TTP [Ca2+]i is also prolonged in the older R21C CMs when paced from 0.5 to 2 Hz (p < 0.05 versus WT −ISO; Fig. 4C), and ISO administration significantly accelerates this parameter (p < 0.05 versus R21C −ISO). Although the addition of ISO and higher pacing frequencies endow positive chronotropic and lusitropic effects on Ca2+ transient kinetics in both groups, the myocytes isolated from the older R21C mice are unable to attain the WT ISO-mediated t½ and TTP [Ca2+]i values (p < 0.05 versus WT +ISO; Fig. 4, B and C, respectively).
FIGURE 4.

The effects of ISO and higher stimulation frequencies on [Ca2+]i transient parameters in cardiomyocytes isolated from old WT and R21C mice. A, representative [Ca2+]i transients recorded from WT (black) and R21C (gray) cardiomyocytes paced at 1 Hz in the absence (solid lines) and presence (dashed lines) of 100 nmol/liter ISO. In B–E, the following parameters are plotted as a function of stimulation frequency in the absence (solid lines) and presence (dashed lines) of ISO. B, t½ of [Ca2+]i decay; C, TTP systolic [Ca2+]i; D, diastolic [Ca2+]i; E, Ca2+ amplitude. t½, time it takes the [Ca2+]i transient amplitude to decrease 50%. Data are given as mean ± S.E. Measurements were taken from three WT and three R21C mice; 4–6 cells/heart were recorded and analyzed. The data are summarized in Table 4. *, p < 0.05 versus WT −ISO; §, p < 0.05 versus R21C −ISO; †, p < 0.05 versus WT +ISO; ‡, p < 0.05 versus WT −ISO. Error bars, S.E.
Myocytes isolated from older R21C mice have significantly higher diastolic Ca2+ levels that are frequency-dissociated in the presence of ISO (p < 0.05 versus WT +ISO; Fig. 4D). This is in contrast to the stepwise frequency-dependent elevations in diastolic [Ca2+]i that occur in the WT controls (Fig. 4D). Moreover, the elevated diastolic [Ca2+]i is not coupled to significantly shorter diastolic SLs in the R21C CMs (Table 4). In the presence of ISO, the WT Ca2+ amplitudes increase ∼30% above the baseline (i.e. diastole) when paced from 0.5 to 4 Hz, whereas the R21C Ca2+ amplitudes are significantly reduced to ∼13% above the baseline (p < 0.05 versus WT +ISO; Fig. 4E). The peak systolic [Ca2+]i (i.e. Fura-2 ratio) is not significantly different between the WT and R21C CMs in the absence or presence of ISO (0.5–4 Hz; Table 4). Altogether, these results suggest that by uncoupling mechanical relaxation from the Ca2+ transient, the R21C CMs have adapted to compensate for the increase in myofilament Ca2+ sensitivity that would predominate during periods of PKA activation (summarized in Table 4).
Altered Ca2+ Transient Parameters in Myocytes Isolated from Young R21C KI Mice
The myocytes isolated from young R21C mice do not have any significant changes to the Ca2+ transient kinetics when ISO is absent (Table 3). However, when the young R21C myocytes are exposed to ISO, the Ca2+ amplitudes are ∼40% smaller than the WT controls at every pacing frequency (p < 0.05 versus WT +ISO; Table 3). When young WT myocytes are exposed to higher stimulation frequencies (i.e. 2–4 Hz) and ISO, frequency-dependent increases in diastolic [Ca2+]i occur (Table 3). However, these frequency-dependent elevations in diastolic [Ca2+]i are absent in the young R21C myocytes when stimulated from 0.5 to 3.0 Hz, which leads to significant reductions in the diastolic [Ca2+]i at higher pacing frequencies (p < 0.05 versus WT +ISO; Table 3).
cTnI-R21C Impairs the Rate of Muscle Relaxation in Skinned Cardiac Fibers
To further assess the effects of the cTnI-R21C mutation on cardiac lusitropy, we examined whether the mutant myofilaments could potentially impair the rate of cardiac muscle relaxation independent of intracellular Ca2+-handling proteins and sinks. Fig. 5 shows the effect of PKA phosphorylation on muscle relaxation in skinned cardiac fibers from WT and R21C mice. After rapid exposure to a UV pulse, the tension declined rapidly and reached a new steady state that was due to the Ca2+ chelation. Fig. 5A shows that the average rate of relaxation for WT fibers increased from a t½ of 40.88 ± 2.52 ms to 27.57 ± 1.89 ms (n = 13 and 12, respectively; p < 0.01) upon incubating the fiber with PKA. The t½ values for R21C fibers before and after PKA treatment are not statistically different from each other (37.99 ± 3.55 and 35.55 ± 2.51, respectively (Fig. 5B); n = 16 and 22, respectively); nor are they both different from the WT fibers not treated with PKA. Altogether, these results indicate that, independent of intracellular Ca2+-handling proteins and sinks, a higher myofilament Ca2+ sensitivity arising from the inability to phosphorylate cTnI will not facilitate cardiac muscle relaxation.
FIGURE 5.

The effect of PKA on the rate of muscle relaxation in skinned cardiac fibers from WT (A) and R21C (B) mice. The rates of muscle relaxation in the absence (solid lines) and presence (dashed lines) of PKA phosphorylation were normalized to the initial steady-state force before the UV flash (as described under “Experimental Procedures”). The muscle relaxed as Ca2+ was chelated by Diazo-2 that was converted to its high Ca2+ affinity form. A, the average rate of relaxation for WT fibers increased from t½ of 40.88 ± 2.52 ms to 27.57 ± 1.89 ms (n = 13 and 12, respectively; p < 0.01) after incubating the fiber with PKA. B, the t½ values for R21C fibers before and after PKA treatment are not statistically different from each other (37.99 ± 3.55 and 35.55 ± 2.51, n = 16 and 22, respectively); nor are they significantly different from those of WT fibers in the absence of PKA.
cTnI-R21C Affects the Expression and Phosphorylation of Ca2+-handling Proteins
We performed immunoblots to determine whether short and long term ablation of cTnI phosphorylation in R21C mice alters the expression of several intracellular Ca2+-handling proteins. Fig. 6A shows that the older R21C hearts decrease the CSQ expression by 42% (p < 0.01 versus WT) in conjunction with a significant increase (∼1.5-fold) in the amount of NCX1 expression (p < 0.01 versus WT; Fig. 6A). Taken together, the ratio of NCX1/CSQ is 3 times higher in the older R21C hearts when compared with the WT controls (p < 0.01 versus WT; data not shown). No detectable changes occur in the expression of PLB, SERCA2a, and β1-AR in the older R21C hearts. However, the older R21C mice have a ∼50% reduction in the amount of phosphorylated PLBThr-17 (p < 0.05 versus WT; Fig. 6B) that is not accompanied by changes in the phosphorylation of PLBSer-16 or MyBP-CSer-282 (Fig. 6B). Similar immunoblotting experiments performed on young R21C and WT hearts did not show any significant differences in the expression or phosphorylation of any of the above mentioned cellular Ca2+-handling proteins (Fig. 6).
FIGURE 6.
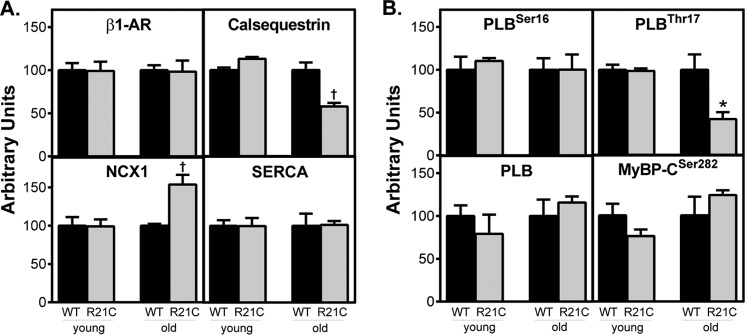
Quantification of β-adrenergic receptor and selected Ca2+-handling proteins from young (6 months) and old (15 months) WT and R21C heart homogenates. Expression of proteins relative to GAPDH was quantified by immunoblot analyses. The amount of each protein in R21C (gray) mouse heart homogenates was normalized to the WT (black) mean values. A, comparison of β1-AR, CSQ, NCX1, and SERCA2a expression. B, comparison of phosphorylated phospholamban (PLBSer-16 and PLBSer-17), total phospholamban (PLB), and phosphorylated MyBP-CSer-282. Data are given as mean ± S.E. (n = 3–4 hearts/genotype and age group). *, p < 0.05; †, p < 0.01 versus WT. Error bars, S.E.
SR Ca2+ Load Is Reduced in Myocytes Isolated from Old R21C KI Mice
Isolated CMs from young and old mice were loaded with Fura-2 and rapidly switched to a 0 Na+, 0 Ca2+ solution containing 20 mmol/liter caffeine to assess the cell's SR Ca2+ content. Fig. 7 shows that the cTnI-R21C mutation impacts the SR Ca2+ buffering capacity of myocytes from old R21C KI mice. When ISO is present, the R21C CMs increase their SR Ca2+ content 1.3-fold (p < 0.05 versus WT +ISO; Fig. 7), whereas the WT CMs increase it 3-fold (p < 0.05 versus WT −ISO; Fig. 7). Moreover, no statistical difference in the SR Ca2+ content was observed before and after the ISO addition to the older R21C CMs (Fig. 7). When the SR Ca2+ content of myocytes from young R21C and WT mice was assessed (±ISO), no significant differences were observed between the two genotypes (data not shown).
FIGURE 7.

The effects of the cTnI-R21C mutation on SR Ca2+ release in the presence and absence of ISO. Shown is functional SR Ca2+ content as measured by the Ca2+ transient amplitude during 20 mmol/liter caffeine-induced contractures in cardiomyocytes isolated from old (15 months) WT and R21C mice. Black and gray vertical bars represent the presence and absence of 100 nmol/liter isoproterenol, respectively, in the myocyte perfusion bath. *, p < 0.05 versus WT +ISO; §, p < 0.01 versus WT −ISO. Error bars, S.E.
DISCUSSION
It is well established that mutations associated with cardiomyopathies affect the cardiac sarcomere, hemodynamics, and electrical conductivity of the heart (43, 44). Efforts to identify unifying mechanistic pathways among the cardiomyopathies have led to the discovery of several convergent phenotypes caused by different myofilament Ca2+-sensitizing mutations. For example, some myofilament mutations can affect the length dependence activation of the sarcomere (45–47) and/or blunt the myofilament response to β-AR stimulation (45, 48–51). However, the existence of several hypertrophic and restrictive cardiomyopathy mutations that lead to an increase in myofilament Ca2+ sensitivity suggests that unresolved mechanisms presumably dictate the final clinical outcome. Despite these unknown variables, we have extended our previous investigation (26) and report here the distinct molecular, cellular, and in vivo phenotypes associated with the cTnI-R21C mutation that ultimately converge to define the physiological basis of the diastolic dysfunction in older R21C KI mice.
HCM is often described as a compensatory disease in which several mechanisms are activated in response to a deficiency in CO. In the context of hypertrophic, dilated, and restrictive cardiomyopathies, all have been shown to increase circulating levels of catecholamines and/or down-regulate β-AR expression in humans (52, 53). Consequently, HR is usually elevated in order to sustain CO. Similar to HCM in humans, the HRs of older anesthetized and conscious R21C mice were significantly higher than the controls (Tables 1 and 2, respectively). Moreover, the administration of ISO in conscious older R21C mice could not further elevate the HR (Table 2). Our initial impression from these results was that the older R21C mice have compensated by constitutively turning on the β-adrenergic pathway. However, there is no change in total R21C β1-AR density in either the young or the old mice (Fig. 6A). Further evidence suggesting that β1-AR density may be normal stems from the ability of WT mice injected with ISO to decrease HRV and alter sympatho-vagal tone parameters to values that are no longer statistically different from those of the older R21C group exposed to ISO (Table 2). Taken together, these findings suggest that a reduction in parasympathetic activity exists that could leave the sympathetic nervous system considerably unopposed. These results coincide with those of Jimenez and Tardiff (54), who have shown that surface β1-AR density is normal in transgenic mice expressing the R92Q and Δ160 mutations in cTnI associated with HCM, despite the mutant mice having HRV values different from those of the non-transgenic controls. Based on their findings, Jimenez and Tardiff (54) concluded that β-adrenergic dysregulation occurs further downstream of the receptors. Furthermore, the cTnI-R21C mutation leads to an impaired baroreflex function, as indicated by a decrease in LF (Fig. 2D). Although reports have demonstrated that the baroreflex function is decreased in human patients with HCM (55), future studies are merited to determine how specific markers of baroreflex sensitivity and delay are affected in the R21C mice. Based on all of the above, β-AR density profiles between human patients and HCM rodent models may differ, but the physiological outcome seems the same, namely a blunted β-adrenergic response, dysautonomia, and a predisposition to arrhythmias.
As the number of mutations that affect intramyocyte signaling continues to grow, a developing paradigm emerges that associates an increase in the myofilament Ca2+ sensitivity with delays in the SL transient. For example, the R92Q (56), R145G (57), R145W (58), Δ160 (56), A172T (59), and R193H (9, 60) mutations in cTnI all increase the myofilament Ca2+ sensitivity and prolong SL relaxation times in isolated cardiomyocytes. Similarly, the older R21C mice have adapted by delaying the sarcomere relaxation time of myocytes, even in the absence of β-AR stimulation (Fig. 3B). These results indicate the existence of excitation-contraction uncoupling and directly coincide with the cTnI-R193H mutation, where sarcomere relaxation and Ca2+ decay were uncoupled in transgenic myocytes due to the increase in myofilament Ca2+ sensitivity (9).
Given that the incorporation of an HCM/restrictive cardiomyopathy mutant into the myofilament is expected to increase the Ca2+ sensitivity of contraction, investigation of the cTnI-R21C mutation poses a unique challenge because Ca2+ sensitization is transient, presumably occurring during periods of PKA activation that are triggered by physical activity and faster stimulation frequencies (61, 62). By rationale, it is no surprise that in the absence of β-AR stimulation or PKA, the cTnI-R21C mutation does not lead to big changes in the Ca2+ sensitivity of tension (26), relaxation rates in permeabilized cardiac fibers (Fig. 5), or changes in [Ca2+]i decay times in myocytes (Fig. 4B and Tables 3 and 4). Such transient beat-to-beat effects could account for the delayed onset of diastolic dysfunction and LV hypertrophy in the R21C mice. In support of this premise, the SL relaxation time was unaffected in myocytes isolated from the young R21C mice (Table 3) and corroborates the ECHO data from the young mice, which did not show any clear markers of diastolic dysfunction or hypertrophy. Although the mitral velocities (E and A; Table 1) of the young R21C hearts were elevated, the E/A ratio of the young R21C mice was comparable with that of the WT. This suggests that the young R21C hearts may be more compliant, capable of compensating for cardiac hemodynamic abnormalities. Nevertheless, these data cannot rule out the possibility that the elevated mitral velocities may represent an early manifestation of cardiac dysfunction in the young R21C mice.
Adaptations to intramyocyte signaling cannot fully account for all of the negative lusitropic effects associated with the cTnI-R21C mutation. When intracellular Ca2+ stores are washed out of cardiac fibers from R21C mice, the addition of PKA does not facilitate muscle relaxation (Fig. 5). Although myosin detachment is the rate-limiting step in mechanical relaxation (22, 63), the Ca2+ occupancy of the thin filament (dictated by the Ca2+ affinity of cTnC) allosterically regulates detachment kinetics (63, 65–67). Therefore, a transient increase in the Ca2+ sensitivity (in the absence of cTnI phosphorylation) will influence the myofilaments to buffer additional Ca2+ that will come off late in diastole (10). Consequently, this prolongs the decay of the Ca2+ transient in myocytes isolated from the older R21C mice when ISO is present (Fig. 4B) and is a contributing source of the relaxation impairments of older R21C hearts in vivo (Table 1).
The elevation of diastolic [Ca2+]i that manifests in the presence of β-AR stimulation (Fig. 4, A and D) in myocytes isolated from the older R21C mice is a distinct phenotype associated with the cTnI-R21C mutation. This elevation is not dependent on the pacing frequency; nor is it coupled to shorter diastolic SLs in myocytes (Table 4). This is in contrast to the frequency-dependent elevations in diastolic [Ca2+]i that have been reported in myocytes from human HCM patients (44), in rodent myocytes containing the troponin T-I79N (10), and cTnI-R193H (59) mutations (associated with cardiomyopathy) as well as the WT myocytes studied herein (Fig. 4D). Our data indicate that the down-regulation of CSQ (Fig. 6A) leads to the elevated diastolic [Ca2+]i in the older R21C myocytes by reducing the SR Ca2+ buffering capacity (Fig. 7). Caffeine-induced depletion of SR Ca2+ confirmed that the older R21C mice have a 2.9-fold reduction in stored Ca2+ compared with the WT when ISO is present (Fig. 7) and may explain why the older R21C mice have a ∼2.3-fold reduction in Ca2+ transient amplitudes (Fig. 4E). Altogether, these results indicate that long term intracellular adaptations have reduced the R21C myocyte ability to access sympathetic reserves (i.e. mobilization of SR Ca2+ stores). Notably, at 12–15 months of age, the R21C mice have not decompensated because parameters of systolic performance (e.g. systolic SL, SL amplitude, ejection fraction, and normalized aortic ejection time) are not statistically different from those in the WT mice.
The role of CSQ down-regulation has been investigated in heterozygous CSQ-null mice that only express 75% of the normal complement (68). Similar to the older R21C mice, the SR Ca2+ content from the CSQ-null mice was significantly reduced when exposed to ISO, which directly correlated with an increase in spontaneous SR Ca2+ leaks (68). These studies demonstrated that regardless of whether SERCA2a is up- or down-regulated, CSQ content dictates the SR Ca2+ buffering capacity and directly contributes to the reduction of the [Ca2+]i to normal diastolic levels (68).
Interestingly, myocytes isolated from the young R21C mice have opposite effects. The diastolic Ca2+ levels are significantly lower than the WT (Table 3), and the level of CSQ expression is comparable with that of the young WT hearts (Fig. 6A). This may explain why the mobilization of SR Ca2+ stores in the young R21C myocytes is unaffected in the presence and absence of ISO (data not shown). These results suggest that the inability to phosphorylate cTnI in the young R21C mice (having normal CSQ expression levels) increases the myofilament Ca2+ buffering capacity, which significantly reduces the diastolic [Ca2+]i (Table 3).
In normal conditions, frequency-dependent elevations in diastolic [Ca2+]i occur as a result of bursts of calcium dissociating from cardiac myofilaments in response to cellular and molecular mechanisms that hasten cross-bridge detachment (66). Upon β-AR stimulation or increasing the pacing frequency of cardiac myocytes, the phosphorylation of cTnI and cardiac MyBP-C accelerates relaxation (24, 62, 69). Changes in the relaxation rate associated with stepwise increases in frequency must be accompanied by proportional decreases in the myofilament Ca2+ sensitivity in order to prevent diastolic dysfunction (62, 69). It follows that frequency-dependent accelerated relaxation is associated with proportional increases in cTnI phosphorylation (61, 62). Based on the above, we would expect that when shorter SL relaxation times are required to keep up with faster pacing frequencies, the inability to transiently phosphorylate cTnI in R21C myocytes would ensure that no burst of calcium will dissociate from the myofilaments in response to PKA activation. Thus, myofilament Ca2+ sensitivity remains relatively static during the entire cardiac cycle, rendering the diastolic [Ca2+]i frequency-dissociated in the young and old R21C mice (Table 3 and Fig. 4D, respectively).
As the R21C mice progress with age, alterations to intramyocyte signaling and intracellular Ca2+ handling become more apparent. CSQ expression has decreased and leads to elevated diastolic Ca2+ levels (+ISO) and the prolongation of SL relaxation times (±ISO). Normally, the SR Ca2+ reuptake system is the predominant Ca2+ extrusion mechanism (70, 71), but the reduced SR Ca2+ buffering capacity in older R21C mice would impose higher demands on other Ca2+ extrusion mechanisms, such as NCX1. Several groups have reported that overexpression of NCX1 bestows a positive lusitropic effect by facilitating cytosolic Ca2+ extrusion (10, 44, 70, 72). It appears to be a trend associated with heart failure and ventricular hypertrophy in humans and animal models (73). Therefore, the up-regulation of NCX1 in this case seems to be a protective mechanism in place to enhance lusitropy by shifting the equilibrium in favoring Ca2+ dissociation from the myofilaments and hastening cross-bridge detachment.
Although cTnI is incapable of becoming phosphorylated at Ser-23 and Ser-24, several PKA signaling targets seem to be functional because we do not observe any changes in SERCA2a, PLB, or total β1-AR expression or any alterations in the phosphorylation of PLBSer-16 and MyBP-CSer-282 (Fig. 6B). This may explain why ISO is capable of endowing a positive lusitropic effect on Ca2+ decay and SL relaxation in the older R21C myocytes (Table 4). However, this cannot explain why CSQ is down-regulated and whether this would contribute to the inability of ISO to induce a positive chronotropic effect on the HRs of conscious mice (Fig. 2A). Altogether, our results indicate that dysregulation of the β-adrenergic pathway does occur downstream of the receptors (i.e. a reduction in cTnI phosphorylation, SR Ca2+ stores, and CSQ expression). However, these data alone cannot rule out the possibility that β1-AR desensitization also exists in the older R21C mice because an overactive sympathetic drive (e.g. due to decreased parasympathetic activity and/or increased catecholamine levels) can eventually blunt the β-adrenergic response as well. Therefore, additional cellular mechanisms may be involved in the modulation of the β-AR response in R21C mice. Although several mechanisms seem plausible, alterations to the Ca2+-calmodulin-dependent protein kinase II (CaMKII) signaling pathway may be involved because we observe a 50% reduction in phosphorylated PLBThr-17 in older R21C mice (Fig. 6B). This is in contrast to reports that have demonstrated increases in CaMKII activity and phosphorylated PLBThr-17 in heart failure samples (44, 71). We speculate that this inconsistency arises because there is no concomitant increase in PLBSer-16 phosphorylation (Fig. 6B), which is a criterion for CaMKII-mediated phosphorylation of PLBThr-17 (74). Alternatively, the up-regulation of phosphatases that occurs in heart failure and cardiomyopathy could presumably decrease the amount of PLBThr-17 as well (45, 75, 76). In any event, it would seem counterintuitive to have an increase in phosphorylated PLBThr-17 concomitantly with a down-regulation of CSQ in the R21C mice. Specifically, the myocyte would waste too many resources accelerating Ca2+ reuptake into the SR, which is incapable of buffering it properly. Further studies will have to address the implications of an altered CaMKII signaling pathway in the R21C mice as well as determine whether the levels of PLBThr-17 phosphorylation are indirectly coupled to the regulation of CSQ expression (and vice versa).
The role of the cTnI N-terminal extension and its phosphorylation has long been studied in mouse models. Constitutive expression of slow skeletal TnI in the mouse heart resulted in very similar phenotypes observed in the older R21C mice, namely diastolic dysfunction, which was accompanied by slower cardiomyocyte SL relaxation and [Ca2+]i decay kinetics in the presence of ISO (77). Skinned cardiac fibers from slow skeletal TnI mice were also incapable of desensitizing the myofilament to Ca2+ upon PKA incubation (77). Together with the R21C data, these findings reinforce the notion that the inability to enhance myofilament relaxation kinetics through cTnI phosphorylation predisposes the heart to abnormal diastolic function, especially during bouts of activity and faster frequencies. In contrast, transgenic mice engineered to express a phosphomimetic cTnI, where Ser-23 and Ser-24 were replaced with aspartic acids (cTnI-2D), had opposite effects (17, 19, 20). Unlike the slow skeletal TnI and R21C mice, the cTnI-2D transgenic mice had enhanced diastolic function with positive lusitropic effects on SL relaxation and [Ca2+]i decay times in isolated cardiomyocytes (19, 61). Although the cTnI-2D transgenic myocytes had lower systolic [Ca2+]i and Ca2+ amplitudes, twitch force was still comparable with that of the non-transgenic fibers (20). These results suggest that the phosphorylation of Ser-23 and Ser-24 is a critical component in modulating the kinetics of relaxation in the heart per se and not the peak force of contraction or systolic performance. In support of this argument, peak SL shortening and systolic function in vivo are comparable with those of the WT controls, although cTnI-R21C is incapable of phosphorylation by PKA and enhancing relaxation.
It is controversial whether changes in the level of phosphorylated cTnI in humans and rodent models are comparable (78, 79). Our group and others have not been able to identify phosphorylated sites in mouse cTnI other than at Ser-23 and Ser-24 (26, 64). In contrast, the existence of other phosphorylated sites in human cTnI samples have been reported as well as a mismatch in the phosphorylation levels among healthy and heart failure samples (50). In addition to the phosphorylation of Ser-23 and Ser-24, three novel phosphorylation sites in the N-terminal extension of human cTnI (i.e. Ser-4, Ser-5, and Tyr-25) were identified. Notably, all of these sites were significantly less phosphorylated in heart failure and cardiomyopathy samples (50). One explanation for the inconsistencies between human and mouse phosphorylation sites may lie within the differences in their primary cTnI sequences. Fig. 8 shows that the human and mouse cTnI sequences are 92% homologous; however, there are a few amino acid substitutions neighboring these new sites that may render them less phosphorylated in mice. Specifically, the replacement of glycine at position 3 in human cTnI with glutamic acid in mice and the introduction of an extra amino acid (i.e. alanine) after position 23 in the mouse cTnI sequence may affect the phosphorylation of Ser-4, Ser-5, and Tyr-25 in mice. Despite the controversy, these studies have shown that reduced levels of phosphorylated cTnI may represent a common biological phenomenon occurring in cardiomyopathy, thus reinforcing the notion that the phosphorylation of the cTnI N-terminal extension is critical in maintaining normal diastolic function. To this end, our data indicate that a reduction in the levels of phosphorylated cTnI predisposes the heart to diastolic dysfunction and may be a unifying mechanism that contributes to the deleterious effects of cardiomyopathy-linked mutations.
FIGURE 8.
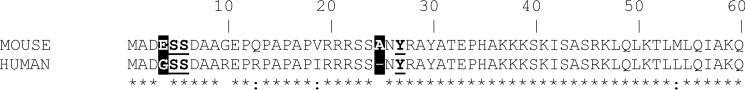
ClustalW alignment of the first 60 amino acids of mouse and human cTnI. There is a Gly to Glu amino acid replacement at position 3 in the mouse cTnI sequence. This substitution is one amino acid before the two novel Ser-4 and Ser-5 phosphorylation sites found in humans. Mouse cTnI has an extra alanine inserted after position 23 (excluding the initial methionine), which shifts the newly identified phosphorylation site to Tyr-26 in mice rather than Tyr-25 in humans (47). Asterisk, identical amino acid; colon, strongly similar amino acid; space, different amino acid.
Acknowledgments
We thank Ana Rojas and Yingcai Wang for assisting with the animal husbandry.
This work was supported, in whole or in part, by National Institutes of Health, NHLBI, Grants HL103840 (to J. R. P.) and 5R01 HL094849 (to J. M. H.).
- HCM
- hypertrophic cardiomyopathy
- β-AR
- β-adrenergic receptor
- CamKII
- Ca2+-calmodulin-dependent protein kinase II
- CM
- cardiomyocyte
- CO
- cardiac output
- cTn
- cardiac troponin
- cTnC
- cardiac troponin C
- cTnI
- cardiac troponin I
- cTnI-2D
- cTnI engineered with aspartic acid at positions 23 and 24
- CSQ
- calsequestrin
- ECG
- electrocardiogram
- ECHO
- echocardiography
- HR
- heart rate
- HRV
- heart rate variability
- ISO
- isoproterenol
- KI
- knock-in
- HF and LF
- high and low frequency components of the frequency domains, respectively
- LV
- left ventricle
- MyBP-C
- myosin-binding protein C
- NCX
- Na+-Ca2+ exchanger
- pCa
- −log[Ca2+]free
- PLB
- phospholamban
- SR
- sarcoplasmic reticulum
- SERCA
- SR Ca2+ transport ATPase
- TTP
- time from baseline to peak value
- SL
- sarcomere length
- TES
- 2-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}ethanesulfonic acid
- E
- early
- A
- atrial.
REFERENCES
- 1. Force T., Bonow R. O., Houser S. R., Solaro R. J., Hershberger R. E., Adhikari B., Anderson M. E., Boineau R., Byrne B. J., Cappola T. P., Kalluri R., LeWinter M. M., Maron M. S., Molkentin J. D., Ommen S. R., Regnier M., Tang W. H., Tian R., Konstam M. A., Maron B. J., Seidman C. E. (2010) Research priorities in hypertrophic cardiomyopathy: report of a Working Group of the National Heart, Lung, and Blood Institute. Circulation 122, 1130–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maron B. J., Maron M. S. (2013) Hypertrophic cardiomyopathy. Lancet 381, 242–255 [DOI] [PubMed] [Google Scholar]
- 3. Konno T., Chang S., Seidman J. G., Seidman C. E. (2010) Genetics of hypertrophic cardiomyopathy. Curr. Opin. Cardiol. 25, 205–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marian A. J. (2008) Genetic determinants of cardiac hypertrophy. Curr. Opin. Cardiol. 23, 199–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Robinson P., Griffiths P. J., Watkins H., Redwood C. S. (2007) Dilated and hypertrophic cardiomyopathy mutations in troponin and α-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ. Res. 101, 1266–1273 [DOI] [PubMed] [Google Scholar]
- 6. Dweck D., Hus N., Potter J. D. (2008) Challenging current paradigms related to cardiomyopathies. Are changes in the Ca2+ sensitivity of myofilaments containing cardiac troponin C mutations (G159D and L29Q) good predictors of the phenotypic outcomes? J. Biol. Chem. 283, 33119–33128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Willott R. H., Gomes A. V., Chang A. N., Parvatiyar M. S., Pinto J. R., Potter J. D. (2010) Mutations in troponin that cause HCM, DCM and RCM: what can we learn about thin filament function? J. Mol. Cell Cardiol. 48, 882–892 [DOI] [PubMed] [Google Scholar]
- 8. Frey N., Luedde M., Katus H. A. (2012) Mechanisms of disease: hypertrophic cardiomyopathy. Nat. Rev. Cardiol. 9, 91–100 [DOI] [PubMed] [Google Scholar]
- 9. Davis J., Yasuda S., Palpant N. J., Martindale J., Stevenson T., Converso K., Metzger J. M. (2012) Diastolic dysfunction and thin filament dysregulation resulting from excitation-contraction uncoupling in a mouse model of restrictive cardiomyopathy. J. Mol. Cell. Cardiol. 53, 446–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Knollmann B. C., Kirchhof P., Sirenko S. G., Degen H., Greene A. E., Schober T., Mackow J. C., Fabritz L., Potter J. D., Morad M. (2003) Familial hypertrophic cardiomyopathy-linked mutant troponin T causes stress-induced ventricular tachycardia and Ca2+-dependent action potential remodeling. Circ. Res. 92, 428–436 [DOI] [PubMed] [Google Scholar]
- 11. Solaro R. J. (2008) Multiplex kinase signaling modifies cardiac function at the level of sarcomeric proteins. J. Biol. Chem. 283, 26829–26833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gordon A. M., Homsher E., Regnier M. (2000) Regulation of contraction in striated muscle. Physiol. Rev. 80, 853–924 [DOI] [PubMed] [Google Scholar]
- 13. Kranias E. G., Solaro R. J. (1982) Phosphorylation of troponin I and phospholamban during catecholamine stimulation of rabbit heart. Nature 298, 182–184 [DOI] [PubMed] [Google Scholar]
- 14. MacLennan D. H., Kranias E. G. (2003) Phospholamban: a crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 4, 566–577 [DOI] [PubMed] [Google Scholar]
- 15. Sadayappan S., Gulick J., Osinska H., Barefield D., Cuello F., Avkiran M., Lasko V. M., Lorenz J. N., Maillet M., Martin J. L., Brown J. H., Bers D. M., Molkentin J. D., James J., Robbins J. (2011) A critical function for Ser-282 in cardiac myosin binding protein-C phosphorylation and cardiac function. Circ. Res. 109, 141–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pohlmann L., Kröger I., Vignier N., Schlossarek S., Krämer E., Coirault C., Sultan K. R., El-Armouche A., Winegrad S., Eschenhagen T., Carrier L. (2007) Cardiac myosin-binding protein C is required for complete relaxation in intact myocytes. Circ. Res. 101, 928–938 [DOI] [PubMed] [Google Scholar]
- 17. Yasuda S., Coutu P., Sadayappan S., Robbins J., Metzger J. M. (2007) Cardiac transgenic and gene transfer strategies converge to support an important role for troponin I in regulating relaxation in cardiac myocytes. Circ. Res. 101, 377–386 [DOI] [PubMed] [Google Scholar]
- 18. Solaro R. J., Moir A. J., Perry S. V. (1976) Phosphorylation of troponin I and the inotropic effect of adrenaline in the perfused rabbit heart. Nature 262, 615–617 [DOI] [PubMed] [Google Scholar]
- 19. Sakthivel S., Finley N. L., Rosevear P. R., Lorenz J. N., Gulick J., Kim S., VanBuren P., Martin L. A., Robbins J. (2005) In vivo and in vitro analysis of cardiac troponin I phosphorylation. J. Biol. Chem. 280, 703–714 [DOI] [PubMed] [Google Scholar]
- 20. Ramirez-Correa G. A., Cortassa S., Stanley B., Gao W. D., Murphy A. M. (2010) Calcium sensitivity, force frequency relationship and cardiac troponin I: critical role of PKA and PKC phosphorylation sites. J. Mol. Cell Cardiol. 48, 943–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Robertson S. P., Johnson J. D., Holroyde M. J., Kranias E. G., Potter J. D., Solaro R. J. (1982) The effect of troponin I phosphorylation on the Ca2+-binding properties of the Ca2+-regulatory site of bovine cardiac troponin. J. Biol. Chem. 257, 260–263 [PubMed] [Google Scholar]
- 22. Zhang R., Zhao J., Mandveno A., Potter J. D. (1995) Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation. Circ. Res. 76, 1028–1035 [DOI] [PubMed] [Google Scholar]
- 23. Kentish J. C., McCloskey D. T., Layland J., Palmer S., Leiden J. M., Martin A. F., Solaro R. J. (2001) Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ. Res. 88, 1059–1065 [DOI] [PubMed] [Google Scholar]
- 24. Colson B. A., Patel J. R., Chen P. P., Bekyarova T., Abdalla M. I., Tong C. W., Fitzsimons D. P., Irving T. C., Moss R. L. (2012) Myosin binding protein-C phosphorylation is the principal mediator of protein kinase A effects on thick filament structure in myocardium. J. Mol. Cell Cardiol. 53, 609–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Arad M., Penas-Lado M., Monserrat L., Maron B. J., Sherrid M., Ho C. Y., Barr S., Karim A., Olson T. M., Kamisago M., Seidman J. G., Seidman C. E. (2005) Gene mutations in apical hypertrophic cardiomyopathy. Circulation 112, 2805–2811 [DOI] [PubMed] [Google Scholar]
- 26. Wang Y., Pinto J. R., Solis R. S., Dweck D., Liang J., Diaz-Perez Z., Ge Y., Walker J. W., Potter J. D. (2012) Generation and functional characterization of knock-in mice harboring the cardiac troponin I-R21C mutation associated with hypertrophic cardiomyopathy. J. Biol. Chem. 287, 2156–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gomes A. V., Harada K., Potter J. D. (2005) A mutation in the N-terminus of troponin I that is associated with hypertrophic cardiomyopathy affects the Ca2+-sensitivity, phosphorylation kinetics and proteolytic susceptibility of troponin. J. Mol. Cell Cardiol. 39, 754–765 [DOI] [PubMed] [Google Scholar]
- 28. Oh J. K., Hatle L., Tajik A. J., Little W. C. (2006) Diastolic heart failure can be diagnosed by comprehensive two-dimensional and Doppler echocardiography. J. Am. Coll. Cardiol. 47, 500–506 [DOI] [PubMed] [Google Scholar]
- 29. Desai K. H., Sato R., Schauble E., Barsh G. S., Kobilka B. K., Bernstein D. (1997) Cardiovascular indexes in the mouse at rest and with exercise: new tools to study models of cardiac disease. Am. J. Physiol. 272, H1053–H1061 [DOI] [PubMed] [Google Scholar]
- 30. Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology (1996) Heart rate variability: standards of measurement, physiological interpretation and clinical use. Circulation 93, 1043–1065 [PubMed] [Google Scholar]
- 31. Thireau J., Zhang B. L., Poisson D., Babuty D. (2008) Heart rate variability in mice: a theoretical and practical guide. Exp. Physiol. 93, 83–94 [DOI] [PubMed] [Google Scholar]
- 32. Rahman F., Pechnik S., Gross D., Sewell L., Goldstein D. S. (2011) Low frequency power of heart rate variability reflects baroreflex function, not cardiac sympathetic innervation. Clin. Auton. Res. 21, 133–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Khan S. A., Skaf M. W., Harrison R. W., Lee K., Minhas K. M., Kumar A., Fradley M., Shoukas A. A., Berkowitz D. E., Hare J. M. (2003) Nitric oxide regulation of myocardial contractility and calcium cycling: independent impact of neuronal and endothelial nitric oxide synthases. Circ. Res. 92, 1322–1329 [DOI] [PubMed] [Google Scholar]
- 34. Dulce R. A., Yiginer O., Gonzalez D. R., Goss G., Feng N., Zheng M., Hare J. M. (2013) Hydralazine and organic nitrates restore impaired excitation-contraction coupling by reducing calcium leak associated with nitroso-redox imbalance. J. Biol. Chem. 288, 6522–6533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bassani R. A., Bassani J. W., Bers D. M. (1992) Mitochondrial and sarcolemmal Ca2+ transport reduce [Ca2+]i during caffeine contractures in rabbit cardiac myocytes. J. Physiol. 453, 591–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grynkiewicz G., Poenie M., Tsien R. Y. (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450 [PubMed] [Google Scholar]
- 37. Shannon T. R., Ginsburg K. S., Bers D. M. (2002) Quantitative assessment of the SR Ca2+ leak-load relationship. Circ. Res. 91, 594–600 [DOI] [PubMed] [Google Scholar]
- 38. Güth K., Potter J. D. (1987) Effect of rigor and cycling cross-bridges on the structure of troponin C and on the Ca2+ affinity of the Ca2+-specific regulatory sites in skinned rabbit psoas fibers. J. Biol. Chem. 262, 13627–13635 [PubMed] [Google Scholar]
- 39. Simnett S. J., Johns E. C., Lipscomb S., Mulligan I. P., Ashley C. C. (1998) Effect of pH, phosphate, and ADP on relaxation of myocardium after photolysis of diazo 2. Am. J. Physiol. 275, H951–H960 [DOI] [PubMed] [Google Scholar]
- 40. Dweck D., Reyes-Alfonso A., Jr., Potter J. D. (2005) Expanding the range of free calcium regulation in biological solutions. Anal. Biochem. 347, 303–315 [DOI] [PubMed] [Google Scholar]
- 41. Zhou Y. Q., Foster F. S., Parkes R., Adamson S. L. (2003) Developmental changes in left and right ventricular diastolic filling patterns in mice. Am. J. Physiol. Heart Circ. Physiol. 285, H1563–H1575 [DOI] [PubMed] [Google Scholar]
- 42. Syed F., Diwan A., Hahn H. S. (2005) Murine echocardiography: a practical approach for phenotyping genetically manipulated and surgically modeled mice. J. Am. Soc. Echocardiogr. 18, 982–990 [DOI] [PubMed] [Google Scholar]
- 43. Alves M. L., Gaffin R. D., Wolska B. M. (2010) Rescue of familial cardiomyopathies by modifications at the level of sarcomere and Ca2+ fluxes. J. Mol. Cell. Cardiol. 48, 834–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Coppini R., Ferrantini C., Yao L., Fan P., Del Lungo M., Stillitano F., Sartiani L., Tosi B., Suffredini S., Tesi C., Yacoub M., Olivotto I., Belardinelli L., Poggesi C., Cerbai E., Mugelli A. (2013) Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation 127, 575–584 [DOI] [PubMed] [Google Scholar]
- 45. Sequeira V., Wijnker P. J., Nijenkamp L. L., Kuster D. W., Najafi A., Witjas-Paalberends E. R., Regan J. A., Boontje N., Ten Cate F. J., Germans T., Carrier L., Sadayappan S., van Slegtenhorst M. A., Zaremba R., Foster D. B., Murphy A. M., Poggesi C., Dos Remedios C., Stienen G. J., Ho C. Y., Michels M., van der Velden J. (2013) Perturbed length-dependent activation in human hypertrophic cardiomyopathy with missense sarcomeric gene mutations. Circ. Res. 112, 1491–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liang B., Chung F., Qu Y., Pavlov D., Gillis T. E., Tikunova S. B., Davis J. P., Tibbits G. F. (2008) Familial hypertrophic cardiomyopathy-related cardiac troponin C mutation L29Q affects Ca2+ binding and myofilament contractility. Physiol. Genomics 33, 257–266 [DOI] [PubMed] [Google Scholar]
- 47. van Dijk S. J., Paalberends E. R., Najafi A., Michels M., Sadayappan S., Carrier L., Boontje N. M., Kuster D. W., van Slegtenhorst M., Dooijes D., dos Remedios C., ten Cate F. J., Stienen G. J., van der Velden J. (2012) Contractile dysfunction irrespective of the mutant protein in human hypertrophic cardiomyopathy with normal systolic function. Circ. Heart Fail. 5, 36–46 [DOI] [PubMed] [Google Scholar]
- 48. Bayliss C. R., Jacques A. M., Leung M. C., Ward D. G., Redwood C. S., Gallon C. E., Copeland O., McKenna W. J., Dos Remedios C., Marston S. B., Messer A. E. (2013) Myofibrillar Ca2+ sensitivity is uncoupled from troponin I phosphorylation in hypertrophic obstructive cardiomyopathy due to abnormal troponin T. Cardiovasc. Res. 97, 500–508 [DOI] [PubMed] [Google Scholar]
- 49. Messer A. E., Jacques A. M., Marston S. B. (2007) Troponin phosphorylation and regulatory function in human heart muscle: dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. J. Mol. Cell. Cardiol. 42, 247–259 [DOI] [PubMed] [Google Scholar]
- 50. Zhang P., Kirk J. A., Ji W., dos Remedios C. G., Kass D. A., Van Eyk J. E., Murphy A. M. (2012) Multiple reaction monitoring to identify site-specific troponin I phosphorylated residues in the failing human heart. Circulation 126, 1828–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang J., Guy M. J., Norman H. S., Chen Y. C., Xu Q., Dong X., Guner H., Wang S., Kohmoto T., Young K. H., Moss R. L., Ge Y. (2011) Top-down quantitative proteomics identified phosphorylation of cardiac troponin I as a candidate biomarker for chronic heart failure. J. Proteome Res. 10, 4054–4065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sucharov C. C., Mariner P. D., Nunley K. R., Long C., Leinwand L., Bristow M. R. (2006) A β1-adrenergic receptor CaM kinase II-dependent pathway mediates cardiac myocyte fetal gene induction. Am. J. Physiol. Heart Circ. Physiol. 291, H1299–H1308 [DOI] [PubMed] [Google Scholar]
- 53. Schotten U., Filzmaier K., Borghardt B., Kulka S., Schoendube F., Schumacher C., Hanrath P. (2000) Changes of β-adrenergic signaling in compensated human cardiac hypertrophy depend on the underlying disease. Am. J. Physiol. Heart Circ. Physiol. 278, H2076–H2083 [DOI] [PubMed] [Google Scholar]
- 54. Jimenez J., Tardiff J. C. (2011) Abnormal heart rate regulation in murine hearts with familial hypertrophic cardiomyopathy-related cardiac troponin T mutations. Am. J. Physiol. Heart Circ. Physiol. 300, H627–H635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Katarzynska-Szymanska A., Ochotny R., Oko-Sarnowska Z., Wachowiak-Baszynska H., Krauze T., Piskorski J., Gwizdala A., Mitkowski P., Guzik P. (2013) Shortening baroreflex delay in hypertrophic cardiomyopathy patients: an unknown effect of β-blockers. Br. J. Clin. Pharmacol. 75, 1516–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Haim T. E., Dowell C., Diamanti T., Scheuer J., Tardiff J. C. (2007) Independent FHC-related cardiac troponin T mutations exhibit specific alterations in myocellular contractility and calcium kinetics. J. Mol. Cell Cardiol. 42, 1098–1110 [DOI] [PubMed] [Google Scholar]
- 57. Reis S., Littwitz C., Preilowski S., Mügge A., Stienen G. J., Pott L., Jaquet K. (2008) Expression of cTnI-R145G affects shortening properties of adult rat cardiomyocytes. Pflugers Arch. 457, 17–24 [DOI] [PubMed] [Google Scholar]
- 58. Wen Y., Xu Y., Wang Y., Pinto J. R., Potter J. D., Kerrick W. G. (2009) Functional effects of a restrictive-cardiomyopathy-linked cardiac troponin I mutation (R145W) in transgenic mice. J. Mol. Biol. 392, 1158–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Davis J., Wen H., Edwards T., Metzger J. M. (2008) Allele and species dependent contractile defects by restrictive and hypertrophic cardiomyopathy-linked troponin I mutants. J. Mol. Cell Cardiol. 44, 891–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li Y., Charles P. Y., Nan C., Pinto J. R., Wang Y., Liang J., Wu G., Tian J., Feng H. Z., Potter J. D., Jin J. P., Huang X. (2010) Correcting diastolic dysfunction by Ca2+ desensitizing troponin in a transgenic mouse model of restrictive cardiomyopathy. J. Mol. Cell Cardiol. 49, 402–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Takimoto E., Soergel D. G., Janssen P. M., Stull L. B., Kass D. A., Murphy A. M. (2004) Frequency- and afterload-dependent cardiac modulation in vivo by troponin I with constitutively active protein kinase A phosphorylation sites. Circ. Res. 94, 496–504 [DOI] [PubMed] [Google Scholar]
- 62. Varian K. D., Janssen P. M. (2007) Frequency-dependent acceleration of relaxation involves decreased myofilament calcium sensitivity. Am. J. Physiol. Heart Circ. Physiol. 292, H2212–H2219 [DOI] [PubMed] [Google Scholar]
- 63. Stein R. B., Bobet J., Oğuztöreli M. N., Fryer M. (1988) The kinetics relating calcium and force in skeletal muscle. Biophys. J. 54, 705–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ayaz-Guner S., Zhang J., Li L., Walker J. W., Ge Y. (2009) In vivo phosphorylation site mapping in mouse cardiac troponin I by high resolution top-down electron capture dissociation mass spectrometry: Ser22/23 are the only sites basally phosphorylated. Biochemistry 48, 8161–8170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zot A. S., Potter J. D. (1989) Reciprocal coupling between troponin C and myosin crossbridge attachment. Biochemistry 28, 6751–6756 [DOI] [PubMed] [Google Scholar]
- 66. Baker A. J., Weiner M. W. (1997) Force decline during muscle relaxation promotes calcium release to the cytosol. Am. J. Physiol. 273, C85–C91 [DOI] [PubMed] [Google Scholar]
- 67. Wang Y., Xu Y., Guth K., Kerrick W. G. (1999) Troponin C regulates the rate constant for the dissociation of force-generating myosin cross-bridges in cardiac muscle. J. Muscle Res. Cell Motil. 20, 645–653 [DOI] [PubMed] [Google Scholar]
- 68. Chopra N., Kannankeril P. J., Yang T., Hlaing T., Holinstat I., Ettensohn K., Pfeifer K., Akin B., Jones L. R., Franzini-Armstrong C., Knollmann B. C. (2007) Modest reductions of cardiac calsequestrin increase sarcoplasmic reticulum Ca2+ leak independent of luminal Ca2+ and trigger ventricular arrhythmias in mice. Circ. Res. 101, 617–626 [DOI] [PubMed] [Google Scholar]
- 69. Varian K. D., Kijtawornrat A., Gupta S. C., Torres C. A., Monasky M. M., Hiranandani N., Delfin D. A., Rafael-Fortney J. A., Periasamy M., Hamlin R. L., Janssen P. M. (2009) Impairment of diastolic function by lack of frequency-dependent myofilament desensitization rabbit right ventricular hypertrophy. Circ. Heart Fail. 2, 472–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bers D. M., Pogwizd S. M., Schlotthauer K. (2002) Upregulated Na/Ca exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic Res. Cardiol. 97, I36–I42 [DOI] [PubMed] [Google Scholar]
- 71. Mattiazzi A., Mundiña-Weilenmann C., Guoxiang C., Vittone L., Kranias E. (2005) Role of phospholamban phosphorylation on Thr17 in cardiac physiological and pathological conditions. Cardiovasc. Res. 68, 366–375 [DOI] [PubMed] [Google Scholar]
- 72. Yao A., Su Z., Nonaka A., Zubair I., Lu L., Philipson K. D., Bridge J. H., Barry W. H. (1998) Effects of overexpression of the Na+-Ca2+ exchanger on [Ca2+]i transients in murine ventricular myocytes. Circ. Res. 82, 657–665 [DOI] [PubMed] [Google Scholar]
- 73. de Tombe P. P. (1998) Altered contractile function in heart failure. Cardiovasc. Res. 37, 367–380 [DOI] [PubMed] [Google Scholar]
- 74. Luo W., Chu G., Sato Y., Zhou Z., Kadambi V. J., Kranias E. G. (1998) Transgenic approaches to define the functional role of dual site phospholamban phosphorylation. J. Biol. Chem. 273, 4734–4739 [DOI] [PubMed] [Google Scholar]
- 75. Huke S., Bers D. M. (2007) Temporal dissociation of frequency-dependent acceleration of relaxation and protein phosphorylation by CaMKII. J. Mol. Cell Cardiol. 42, 590–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Neumann J. (2002) Altered phosphatase activity in heart failure, influence on Ca2+ movement. Basic Res. Cardiol. 97, I91–I95 [DOI] [PubMed] [Google Scholar]
- 77. Fentzke R. C., Buck S. H., Patel J. R., Lin H., Wolska B. M., Stojanovic M. O., Martin A. F., Solaro R. J., Moss R. L., Leiden J. M. (1999) Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. J. Physiol. 517, 143–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Solaro R. J., van der Velden J. (2010) Why does troponin I have so many phosphorylation sites? Fact and fancy. J. Mol. Cell Cardiol. 48, 810–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Marston S. B., de Tombe P. P. (2008) Troponin phosphorylation and myofilament Ca2+-sensitivity in heart failure: increased or decreased? J. Mol. Cell Cardiol. 45, 603–607 [DOI] [PMC free article] [PubMed] [Google Scholar]