

Background: Interleukin-1 receptor-associated kinases (IRAKs) play a critical role in TLR signaling and thus innate immunity.
Results: A coding IRAK2 variant, rs35060588, affects TLR signaling and colorectal cancer survival.
Conclusion: IRAK2 rs35060588 is a functional, disease-relevant variant.
Significance: IRAK2 and its variant rs35060588 may serve as a point of therapeutic intervention and predictive biomarker, respectively.
Keywords: Colorectal Cancer, Innate Immunity, Interleukin Receptor-associated Kinase (IRAK), Signal Transduction, Toll-like Receptor (TLR), Genetic Variant, Single Nucleotide Polymorphism
Abstract
Within innate immune signaling pathways, interleukin-1 receptor-associated kinases (IRAKs) fulfill key roles downstream of multiple Toll-like receptors and the interleukin-1 receptor. Although human IRAK4 deficiency was shown to lead to severe immunodeficiency in response to pyogenic bacterial infection during childhood, little is known about the role of human IRAK2. We here identified a non-synonymous IRAK2 variant, rs35060588 (coding R214G), as hypofunctional in terms of NF-κB signaling and Toll-like receptor-mediated cytokine induction. This was due to reduced ubiquitination of TRAF6, a key step in signal transduction. IRAK2 rs35060588 occurs in 3–9% of individuals in different ethnic groups, and our studies suggested a genetic association of rs35060588 with colorectal cancer survival. This for the first time implicates human IRAK2 in a human disease and highlights the R214G IRAK2 variant as a potential novel and broadly applicable biomarker for disease or as a therapeutic intervention point.
Introduction
The mammalian innate immune system relies on Toll-like receptors (TLRs)5 for the detection of invading microbes based on microbe-associated molecular patterns. For example, TLR2 detects bacterial lipoproteins, TLR4 lipopolysaccharide (LPS), and TLR7/8 bacterial and viral RNA (1). TLR activation by microbe-associated molecular patterns triggers cytoplasmic signaling culminating in the production of proinflammatory cytokines and interferons (IFNs), which, by transcriptional regulation, initiate and shape adaptive immune responses (1). The adaptor molecule MyD88 (myeloid differentiation primary response gene 88) plays a key role in integrating and diversifying signals elicited by all TLRs except TLR3 and by the interleukin (IL)-1 receptor (IL-1R) (2), which mediates the biological effects of the important inflammatory cytokine IL-1β (3). MyD88 cooperates with kinases of the IL-1R-associated kinase (IRAK) family (4). The pivotal importance of MyD88-IRAK signaling is evidenced by the fact that human carriers of rare loss-of-function mutations for MYD88 or IRAK4 suffer from severe susceptibility to pyogenic bacterial infection during childhood (5). Moreover, in various B cell malignancies, MyD88-IRAK signaling has been implicated in oncogenesis (6). The IRAK family in humans and mice consists of four members each, IRAK1, IRAK2, IRAK3 (also termed IRAK-M), and IRAK4 (4). All IRAKs share an N-terminal death domain (DD), enabling interactions with the DD-containing MyD88, and a central kinase domain (KD) (4). IRAK1 to -3 also feature a C-terminal extension that contains motifs required for subsequent TRAF6 (tumor necrosis factor receptor-associated factor 6) recruitment (4). MyD88-IRAK interactions take place in the context of the so-called “Myddosome” postreceptor complex, a hierarchical DD assembly of MyD88, IRAK4, and IRAK2 (7). Myddosome formation is initiated by MyD88 DD oligomerization into a ringlike structure that enables the recruitment of a second DD ring consisting of IRAK4 DD (8), a process blocked by naturally occurring MYD88 mutations (9). The four IRAK4 DDs in turn provide a charge-complementary docking site for a homo-oligomeric ring of four IRAK2 DDs (8). The precise molecular events of signal transduction still remain elusive, but DD-mediated Myddosome formation is supposed to enable proximity-induced activation of IRAK4 and IRAK1 or IRAK2, which, by an unknown mechanism, induces TRAF6 ubiquitination, a step required for subsequent downstream signaling leading to the activation of nuclear factor κB (NF-κB), mitogen-activated protein kinases, and protein kinase B (1). Although murine IRAKs have been well characterized in knock-out models, the respective roles and relative contributions of human IRAKs, apart from IRAK4, within the human immune system remain surprisingly enigmatic. In the only study published on the role of IRAK2 in primary human immune cells, IRAK2 was shown as essential for the induction of TNF upon stimulation of TLR4 and TLR8, suggesting that IRAK2 may play an important role in TLR signaling in humans (10). It is unclear if IRAK2 contributes to signaling through kinase activity because several critical residues in the putative catalytic cleft differ from the consensus motif, and IRAK2 does not autophosphorylate like IRAK1 or -4 (11). Other data imply that mouse IRAK2 kinase activity is required for signaling in reconstitution experiments in knock-out Irak2 cells (12). Because no additional functional analyses in human cells have been conducted and no associations of IRAK2 with diseases in humans have been reported, we sought to clarify the role of human IRAK2 and studied reported IRAK2 genetic variants (non-synonymous single-nucleotide polymorphisms (SNPs)) functionally and epidemiologically with regard to autoimmune and malignant diseases. We discovered IRAK2 rs35060588 (R214G) as a hypofunctional IRAK2 variant showing a diminished ability to induce TRAF6 ubiquitination and subsequent cytokine induction. Additionally, epidemiological association studies showed that rs35060588 may be a genetic factor impacting colorectal cancer (CRC) survival.
EXPERIMENTAL PROCEDURES
Reagents and Cells
Reagents were from Sigma unless otherwise stated. The following TLR ligands were used: Pam2CSK4 (Axxora or Invivogen), poly(I:C) (Sigma), R848 (Invivogen), LPS (Invivogen), flagellin (Imgenex), and CpG-ODN (MWG Biotech). Antibodies were as follows: rabbit anti-HA (Cell Signaling, catalog no. 3724), mouse anti-HA (Sigma, H3663), rabbit anti-TRAF6 (Santa Cruz Biotechnology, Inc., sc-7221), mouse anti-TRAF6 (Santa Cruz Biotechnology, sc-8409), rabbit anti-FLAG (Sigma, F7425), mouse anti-ubiquitin (Santa Cruz Biotechnology, sc-8017) and mouse anti-tubulin (Sigma, T4026), and anti-mouse (Promega) or anti-rabbit HRP conjugates (Vector PI). Human embryonic kidney 293T (HEK293T) cells (A. Dalpke, University of Heidelberg, Germany) were cultured as described (7). Immortalized Irak2-deficient macrophage-like cells were generated by Katherine Fitzgerald (University of Massachusetts Medical School, Worcester, MA) from primary macrophages isolated from Irak2-deficient C57BL/6 mice generated by James Watson (University of Texas Southwestern, Houston, TX) and cultured as described (13).
Polymorphism Information
A list of reported SNPs in human IRAK2 (Gene ID: 4615), was obtained from NCBI, National Institutes of Health (Table 1). HapMap data were from the International HapMap Project (14).
TABLE 1.
Overview of reported IRAK2 non-synonymous SNPs
For an overview of IRAK2 functional domains, see Ref. 4. Frequency is according to NCBI across all studies carried out so far. Boldface type highlights rs35060588 investigated here in detail.
| Amino acid exchange | rs number | Average frequency | Location |
|---|---|---|---|
| R43Q | 34945585 | 0.0005 | Death domain |
| S47Y | 11465864 | 0.01 | Death domain |
| L78M | 11709928 | Unknown | Death domain |
| I99V | 55898544 | 0.01 | Linker/ProST region |
| R214G | 35060588 | 0.04 | Kinase domain |
| L358M | 77590560 | 0.001 | Kinase domain |
| L392V | 3844283 | 0.30 | Kinase domain |
| D431E | 708035 | 0.21 | Kinase domain |
| L439V | 11465927 | 0.015 | Kinase domain |
| R498G | 75132222 | 0.08 | C terminus |
| E501G | 12486661 | 0.001 | C terminus |
| L503I | 9854688 | 0.03 | C terminus |
Plasmids, Cloning, and Site-directed Mutagenesis
Mutations in IRAK2 were introduced into human IRAK2 (AAC50954; Imagenes IRCMP5012D0935D) using a QuikChange XL Kit (Agilent). Gateway-compatible entry clones for IRAK2 or IRAK4 full-length or DD-only or TRAF6 (gift from S. Wiemann, German Cancer Research Center) were transferred via Gateway LR-reaction (Invitrogen) to a pT-Rex-Dest30-based plasmid containing N-terminal Renilla or Protein A tags (9). Alternatively, the IRAK2 full-length and DD constructs were transferred to a pcDNA5/FRT/TO-based plasmid to add an N- or C-terminal Strep-HA tag (T. Bürckstümmer (CeMM, Vienna) and M. Gstaiger (ETH Zurich)). To generate FLAG-TRAF6, an entry clone was transferred to a FLAG-containing Gateway plasmid provided by Stefan Pusch (Heidelberg University). PCR and mutagenesis primer sequences are available upon request.
Retroviral Transduction and Functional Analysis of Immortalized Irak2-deficient Macrophages
pMXs-IP-puro (Moloney murine leukemia virus LTR) (Kevin-Michael Dennehy, University of Tübingen), carrying either the empty multiple cloning site (mock), HA-tagged WT, or R214G, was used to transduce Irak2-deficient cells. Following puromycin selection (10 μg/ml), HA-IRAK2 expression was verified by anti-HA immunoblot. For TNF ELISA (Biolegend) experiments, macrophages were seeded in 96-well plates (2.5 × 104 cells/well) and stimulated with 1 μg/ml Pam2CSK4, 10 μg/ml poly(I:C), 0.05 μg/ml LPS, 50 ng/ml flagellin, 1 μg/ml R848, or 0.5 μm CpG for 16 h. For quantitative real-time PCR, 1.5 × 105 reconstituted macrophage cells were stimulated with 1 μg/ml Pam2CSK4, 0.05 μg/ml LPS, and 1 μg/ml R848 for 3 h; total RNA was extracted (RNeasy kit, Qiagen); DNA was digested by DNase treatment (Ambion); cDNA was synthesized (Applied Biosystems high capacity RNA-to-cDNA kit); and quantitative PCR was performed in duplicates using the TaqMan Universal Master Mix (Invitrogen). Tbp (TATA-binding protein) was used as a reference gene. Gene-specific TaqMan assays (Invitrogen) were used: TNFα Mm00443260_g1, IL-6 Mm00446190_m1, CCL5 Mm01302427_m1, Tbp Mm00446971_m1.
HEK293T Signaling Assays and ELISA
HEK293T were transfected with an NF-κB firefly luciferase reporter (100 ng; Stratagene), pRL-Tk Renilla luciferase control reporter (10 ng; Promega), pC1-EGFP (100 ng; Clontech), and IRAK2 plasmids (as indicated) and assayed as described (9). Luciferase measurements were performed in triplicate ± S.D. Using HEK293T cells transfected with pC1-EGFP (100 ng; Clontech) and IRAK2 plasmids (as indicated), IL-8 in supernatants was determined 48 h later by ELISA (Biolegend).
Co-immunoprecipitation and Expression Analysis
Immunoprecipitation experiments were done as described (9). TRAF6 ubiquitination analysis was performed as described (15).
Luminescence-based Mammalian Interactome Mapping (LUMIER)
As described in a recent Myddosome study (9), the avidity of a protein-protein interaction is measured by co-expressing two interacting proteins with Protein A (bait) and Renilla luciferase (prey) tags, respectively (see the legend to Fig. 2), and subsequently determining the level of specific binding of the Renilla partner upon Protein A purification. 104 HEK293T cells/well in 96-well plates were thus transfected in triplicates with 20 ng each of Protein A and Renilla fusion plasmids (Lipofectamine 2000, Invitrogen). 48 h post-transfection, cells were lysed, raw Renilla activity was measured, and Protein A-tagged proteins and bound Renilla-tagged interactors were purified using magnetic IgG beads. Bound Renilla luciferase activity was measured, and an interaction signal was processed by dividing the activity of bound Renilla luciferase by the amount of raw Renilla activity for each transfection and then normalized to the specific signal for a Protein A-only (no bait) condition.
FIGURE 2.
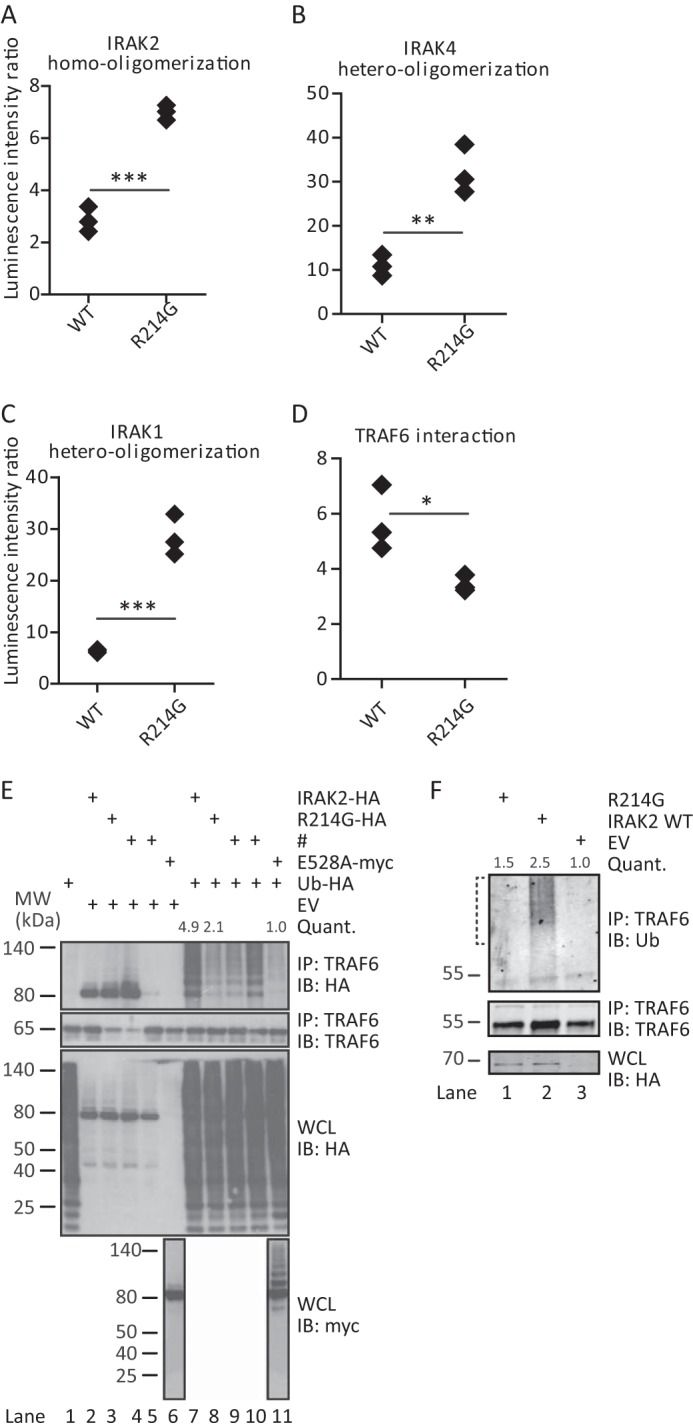
R214G is trapped in the Myddosome and fails to induce TRAF6 ubiquitination. Homo-oligomerization (A) and hetero-oligomerization with IRAK4 (B) and IRAK1 (C) are increased for R214G compared with WT. Conversely, interactions with TRAF6 (D) are reduced. Shown is LUMIER luciferase analysis from HEK293T cells transfected with Protein A-tagged IRAK2 WT or R214G and Renilla luciferase-tagged IRAK2 WT, IRAK4, IRAK1, or TRAF6. In each case, one representative experiment of at least three independent experiments done in triplicates is shown. E and F, R214G fails to induce ubiquitination of TRAF6. E, upon expression of empty vector (EV), Strep-HA-tagged IRAK2 WT or variant constructs, Myc-IRAK2 E528A, and HA-ubiquitin in HEK293T cells, protein complexes were immunoprecipitated (IP) using anti-TRAF6 antibody. Precipitates and whole cell lysates (WCL) were analyzed by immunoblot (IB) as indicated. One representative experiment of five independent experiments is shown. F, analysis of endogenous ubiquitination of TRAF6. Upon expression of Strep-HA-tagged IRAK2 WT or variant constructs in HEK293T cells, protein complexes were immunoprecipitated using anti-TRAF6 antibody. Precipitates and whole cell lysates were analyzed by immunoblot as indicated. High molecular weight smears indicate TRAF6 ubiquitination. One representative experiment of four independent experiments is shown. The brackets indicate the areas of ubiquitination quantification. # (E), transfection with constructs irrelevant to this study.
Sequence Alignments, Structural Analysis, and Homology Modeling
Protein sequences from NCBI were aligned with ClustalX. The homology model for the IRAK2 KD was generated using the MODELLER package (16) as described (9) based on the structure of the IRAK4 KD (Protein Data Bank code 2NRU) (17).
Autoimmune Disease Case-Control Study and Genotyping
The study involved 1,123 patients with systemic lupus erythematosus (SLE) fulfilling at least four of the American College of Rheumatology 1982 criteria for the classification of SLE and 1,184 ethnicity-matched healthy control subjects from a European multicenter collaboration (18). The SNP was genotyped using a TaqMan HT7500 FAST (Invitrogen) instrument.
Colorectal Cancer Cohort and Genotyping
The 613 CRC patients belonged to the population-based PopGen project in Schleswig-Holstein, Germany (19) and did not include hereditary non-polyposis colorectal cancer patients. Genotyping was performed using KASPar assays on demand (KBiosciences, Hoddesdon, UK).
Statistical Analysis
For functional experiments, p values were determined using an unpaired Student's t test in Figs. 1 (B, C, and F), 2 (A–D), and 3D. p values are designated as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001. Statistical analyses for epidemiological associations were performed with SPSS version 20.0 (SPSS) or with SAS version 9.2 (SAS Institute). The significance of differences was assessed in contingency tables using Pearson's χ2 test and Fischer's exact test. All tests were two-sided, and p values less than 0.05 were considered to be statistically significant. CRC analysis of prognostic significance was done by the univariate Cox proportional hazard model. Follow-up time was calculated from the date of CRC diagnosis to CRC-specific death or to the end of follow-up. Survival curves were derived using the Kaplan-Meier method and compared using log-rank testing.
FIGURE 1.
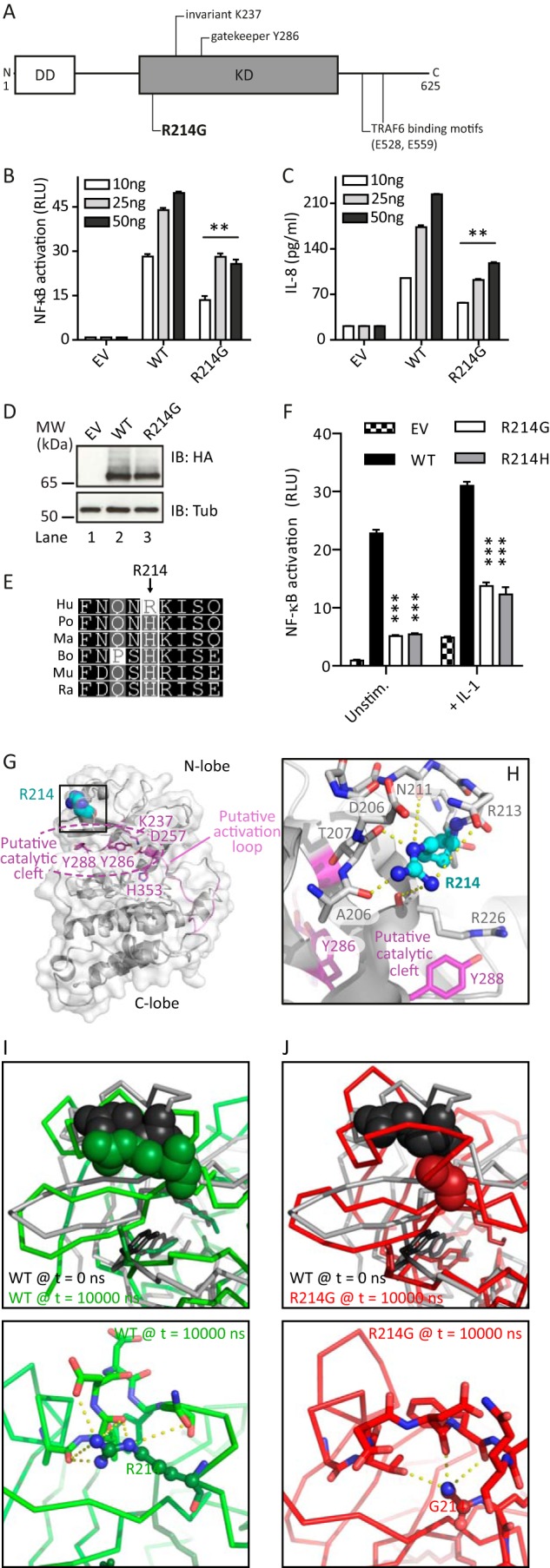
IRAK2 R214G overexpression shows reduced NF-κB activation. A, schematic overview of human IRAK2 domains with the non-synonymous SNP R214G highlighted. B, IRAK2 R214G shows reduced NF-κB induction (quantified by dual luciferase assay in triplicates ± S.D.) compared with WT in HEK293T cells transiently transfected with the indicated amounts of Strep-HA-tagged IRAK2 constructs or empty vector (EV). C, IL-8 secretion is reduced for R214G compared with WT. HEK293T cells were transiently transfected with IRAK2 constructs, and secreted IL-8 levels were quantified by ELISA (triplicates ± S.D.). D, expression of IRAK2 R214G is comparable with that of WT. HEK293T cells were transfected with Strep-HA-tagged expression constructs and analyzed by anti-HA immunoblot. E, sequence conservation of IRAK2 Arg-214. Shown are multiple-sequence alignments of IRAK2 homologues from different species (see “Experimental Procedures”). Hu, human; Po, Pongo pongo; Ma, Macaca mulatta; Bo, Bos taurus; Mu, Mus musculus; Ra, Rattus norvegicus. F, HEK293T cells were transfected as before with 10 ng of Strep-HA-tagged IRAK2 constructs or empty vector (EV), and NF-κB activation was quantified by a dual-luciferase assay in triplicates ± S.D. in the absence of stimulation or the presence of recombinant IL-1β (50 ng/ml) for 16 h. In B–D and F, one representative experimental of three independent experiments is shown. G and H, three-dimensional homology model of the IRAK2 kinase domain based on IRAK4. Putative catalytic residues are in magenta, the putative activation loop is in pink, and Arg-214 is in cyan. Arg-214 is surface-exposed above the potential catalytic cleft. Predicted hydrogen bonds are shown as yellow dashes. I and J, three-dimensional models for WT IRAK2 KD (I) and the R214G variant (J) were subjected to 10-ns molecular dynamics simulations. Top panels, the final frames for WT (green, I) and R214G (red, J) were compared with the starting three-dimensional structure of WT IRAK2 KD (gray). Position 214 is shown as spheres, the protein backbone is shown as a ribbon, and residues potentially required for catalytic activity are highlighted in stick representations. Bottom panels, possible hydrogen bonds emanating from Arg-214 (I) or Gly-214 (J) were analyzed.
FIGURE 3.
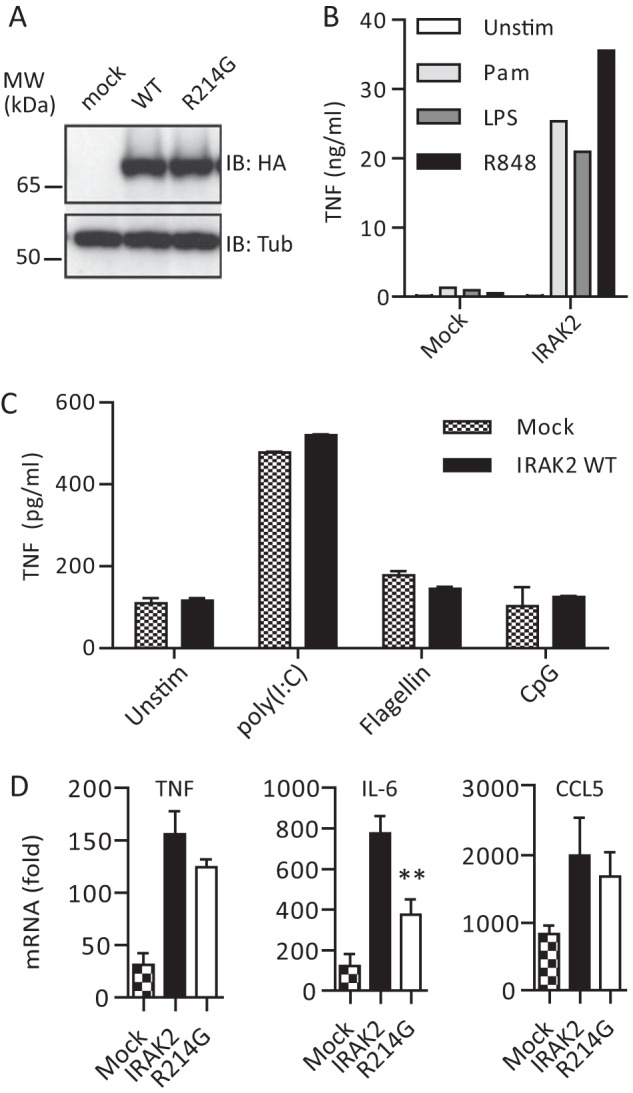
Irak2-deficient macrophages reconstituted with human IRAK2 R214G show altered cytokine responses upon stimulation with TLR2 ligands. A, anti-HA immunoblot analysis of whole cell lysates of mock, HA-IRAK2 WT, and HA-R214G reconstituted macrophages (see “Experimental Procedures”). B and C, reconstitution of Irak2-deficient murine macrophages with human IRAK2 is functional as assessed by TNF secretion analyzed by ELISA (triplicates ± S.D. (error bars)) for a 16-h stimulation with 1 μg/ml Pam2CSK4 (light gray), 0.05 μg/ml LPS (dark gray), and 1 μg/ml R848 (black) (B) but not with 10 μg/ml poly(I:C) (TLR3 ligand), 50 ng/ml flagellin (TLR5 ligand), and 0.5 μm CpG 1668 (TLR9 ligand) (C) or 50 ng/ml recombinant murine IL-1β (not shown). One of four (B) or two (C) representative experiments is shown. D, TNF, IL-6, and CCL5 mRNA induction in mock (checkered), WT human HA-IRAK2 (black), and R214G (white) reconstituted macrophages upon stimulation with Pam2CSK4 for 6 h, relative to unstimulated (triplicates ± S.D. of one representative experiment of two independent experiments shown).
RESULTS
The IRAK2 Variant R214G Is Associated with Reduced NF-κB Activation
When searching public databases for reported IRAK2 variants, we identified 12 non-synonymous variants with an estimated frequency of >0.1% in Caucasian populations (see Fig. 1A and Table 1). Due to its moderately high reported frequency (9% in Caucasians), we focused here on the rs35060588 (coding arginine to glycine at position 214, R214G). To investigate its ability for downstream signaling, we measured NF-κB activation in the HEK293T cell line upon overexpression of Strep-HA-tagged IRAK2 WT and R214G. It was shown earlier that IRAK2 overexpression in HEK293T cells, which do not express TLRs, drives NF-κB activation and allows assessment of signaling pathways downstream of IRAK2 (20). Compared with WT IRAK2, R214G showed NF-κB activation decreased (hypofunctional) by ∼45% (Fig. 1B), which also translated to 45% reduced IL-8 secretion from HEK293T cells (Fig. 1C), despite comparable expression levels (Fig. 1D). That reduced NF-κB activation is not due to lower expression was confirmed by measuring NF-κB activation upon expression of Renilla-tagged IRAK2 WT and R214G constructs; when NF-κB-dependent firefly activity values were normalized to Renilla firefly activity (corresponding to the respective expression levels), this ratio was consistently lower for R214G (data not shown). Interestingly, Arg-214, which mapped to the IRAK2 KD coding region (Fig. 1A), is only found in human IRAK2 (other species show a histidine at this position; see Fig. 1E), and the change from Arg to Gly or His is expected to be tolerated in the structure according to Polyphen-2 and SIFT server predictions (not shown). Interestingly, the R214H mutant was similarly reduced in NF-κB activation as R214G (Fig. 1F). Reduced signaling was also observed for both mutants in the presence of recombinant IL-1β, which also acts via IRAK2 (20) (Fig. 1F). To gain a molecular insight into the potential impact of amino acid exchange, we generated a three-dimensional model of the IRAK2 KD (Fig. 1G) based on IRAK4 (17). In this model, Arg-214 is surface-exposed and coordinates, via multiple hydrogen bonds, a loop region above the cleft harboring residues that could be involved in the putative activity of IRAK2 as a bona fide kinase (Fig. 1, G and H). Because the issue of whether IRAK2 is an active kinase is still under debate (4) and putative substrates are unknown, we were unable to assess an effect on kinase activity experimentally but instead conducted molecular dynamics simulations. These suggested that hydrogen bonds present in WT IRAK2 (Fig. 1I) would be lost upon amino acid exchange to glycine (Fig. 1J), leading to a destabilization of this protein region, potentially impacting kinase activity and/or protein-protein interactions of IRAK2.
R214G Reduces TRAF6 Ubiquitination
To test the latter possibility, we analyzed the impact of the R214G amino acid change in terms of known protein-protein interactions of IRAK2 within the so-called Myddosome, which is considered a key postreceptor complex in innate immunity (4, 15, 21). Here IRAK2 docks to the ring of IRAK4 DD already assembled upon upstream activation of MyD88 via TLRs. The unique role of IRAK2 is to then recruit TRAF6 to the Myddosome via C-terminal binding motifs (Fig. 1A), thus promoting the induction of TRAF6 ubiquitination (4, 15). IRAK2-induced TRAF6 ubiquitination and dissociation of an IRAK-TRAF6 complex are considered critical steps for activating the IKK complex for NF-κB activation (1, 4). The former steps are blocked by the negative regulator IRAK-M (22). To analyze whether these molecular interactions were affected by the R214G exchange, we employed LUMIER, a technique recently used to quantify Myddosome interactions (9). In brief, the avidity of a protein-protein interaction is measured by co-expressing two interacting proteins with Protein A (bait) and Renilla luciferase (prey) tags, respectively (see the legend to Fig. 2) and subsequently determining the level of specific binding of the Renilla partner upon Protein A purification. This analysis showed that R214G displays a significantly increased ability to homo-oligomerize (Fig. 2A) and bind to IRAK4 (Fig. 2B) and IRAK1 (Fig. 2C), suggesting that it is bound more tightly to the Myddosome. On the other hand, IRAK2 R214G showed slightly reduced TRAF6 binding in LUMIER analysis (Fig. 2D). In immunoprecipitations, which are less sensitive and accurate in terms of quantification, the TRAF6-IRAK2 interaction was not reduced for R214G compared with WT (quantified from Fig. 2E, lane 2 versus lane 3) (data not shown). However, ligation of TRAF6 with exogenously expressed HA-ubiquitin (Fig. 2E) and endogenous ubiquitin (Fig. 2F) was severely compromised. This reduction is consistent with a previously described mutant of IRAK2, E528A, that completely fails to induce TRAF6 ubiquitination (15) (Fig. 2E, lane 11). Collectively, our data suggest that R214G IRAK2 is more strongly recruited into the Myddosome, preventing the dissociation of the IRAK2-TRAF6 complex for full TRAF6 ubiquitination, a key step required for optimal signal transduction.
R214G Shows Reduced Cytokine Transcription upon TLR Stimulation
In order to gain a first insight into the influence of R214G upon endogenous TLR signal transduction in terms of cytokine induction, we reconstituted murine immortalized macrophages from Irak2-deficient mice retrovirally with human HA-IRAK2, as done recently for the analysis of MyD88 (13) and Mal (23) genetic variants. Upon transduction of Irak2-deficient macrophages with a mock or HA-IRAK2 WT- or R214G-internal ribosome entry site-puromycin cassette-containing retrovirus, puromycin-selected macrophages showed similar HA-IRAK2 expression levels (Fig. 3A) and localization (not shown). We validated reconstitution functionally by assessing TNF secretion by ELISA upon stimulation of the different puromycin-selected lines with agonist for TLR2 (Pam2CSK4), TLR3 (poly(I:C)), TLR4 (LPS), TLR5 (flagellin), TLR7 (R848), and TLR9 (CpG 1668). Conversely to mock-reconstituted macrophages, IRAK2-reconstituted macrophages were highly responsive to TLR2, TLR4, and TLR7 stimulation in terms of TNF secretion (Fig. 3, B and C), consistent with siRNA-mediated IRAK2 knock-down data from human peripheral blood mononuclear cells, which affected TLR4 and TLR7/8 signaling (10) (Fig. 3B). In contrast, TLR3, TLR5, and TLR9 signaling was unaffected by IRAK2 WT reconstitution (Fig. 3C) and was therefore not analyzed further. None of the generated macrophage lines responded to recombinant IL-1β (not shown). From these results it can be concluded that Irak2−/− macrophage reconstitution with human IRAK2 provided an adequate means to assess the impact of R214G on human IRAK2-dependent TLR2, TLR4, and TLR7 signaling. Because TLR2 signaling is entirely MyD88-IRAK-dependent in macrophages, we focused on Pam2CSK4-stimulated TNF, CCL5 (RANTES), and IL-6 cytokine induction elicited in mock and IRAK2 WT- and R214G-reconstituted macrophages by quantitative PCR after 3 or 6 h of stimulation. For TNF and CCL5, a non-significant trend toward lower cytokine induction was visible at 3 h (not shown) and 6 h (Fig. 3D). However, IL-6 induction was significantly (p = 0.0037), reduced to about 50% at the 6 h time point in keeping with the reported role of Irak2 in Il6 promoter stimulation and the late (>3 h) requirement for Irak2 in cytokine induction (12, 24). Collectively, these data indicate that some but not all IRAK2-dependent cytokines may be negatively affected by the R214G variant, in line with the hypofunctional phenotype observed in the HEK293T system. Given that in humans and wild-derived mice IRAK2 has a more pronounced positive role in signaling compared with laboratory mice (25), in primary cells from human carriers, R214G homozygosity might lead to modulated TLR-induced cytokine responses, and/or the susceptibility of variant carriers to diseases with TLR involvement might be affected.
R214G Modulates Colorectal Cancer Survival
Despite its interesting functional phenotype, there is little known about the hypofunctional R214G IRAK2 variant (which occurs in 3% of African Americans and 9% of Europeans), let alone genetic associations of IRAK2 with disease in general. To test whether the functional phenotype might entail an association with certain diseases, we investigated allele frequencies in well documented collectives (see “Experimental Procedures” for details) for the autoimmune disease SLE and for CRC, in both of which TLR pathways have been implicated (26, 27). First, IRAK2 association with SLE was analyzed in a case-control study of 1,123 patients and 1,184 controls. Interestingly, the rs35060588 GG genotype was less frequent in cases (0.0026%) than controls (0.0068%), but due the low frequency of homozygosity, this difference was not statistically significant (data not shown). Second, CRC survival analysis using the Cox proportional hazard models in a German cohort (n = 613) indicated a 60% increased risk of death by CRC for the heterozygous carriers of the rs35060588 G allele (HR 1.60 (1.01–2.52), p = 0.04; Table 2), supported by Kaplan-Meier analysis (CG versus CC log-rank p = 0.03; Fig. 4). Because only four persons were homozygous for the G allele, no firm conclusions could be drawn regarding homozygous carriage of the G allele, despite the fact that none of them died during the study period. No significant deviations from Hardy-Weinberg equilibrium were observed. Collectively, our data thus indicate that IRAK2 rs35060588, due to a functional phenotype, may affect disease risk or outcome.
TABLE 2.
Frequencies of IRAK2 35060588 and CRC-specific survival
HR, hazard ratio; CI, confidence interval. The genotype call rate was 97.4%. Boldface type indicates statistical significance at the 5% level.
| rs number | Genotype | CRC-specific survival |
|||
|---|---|---|---|---|---|
| No. at risk | No. died (%) | HR (95% CI) | p value | ||
| rs35060588 | CC | 524 | 124 (23.66) | 1.00 | |
| CG | 69 | 22 (31.88) | 1.60 (1.01–2.52) | 0.04 | |
| GG | 4 | 0 (0.00) | |||
| CG + GG | 73 | 22 (30.14) | 1.52 (0.96–2.40) | 0.07 | |
| Overall | 0.13 | ||||
FIGURE 4.

R214G is associated with altered CRC survival. Shown is a Kaplan-Meier graph for CRC survival for rs35060588 (R214G) alleles depicting WT (CC) (blue line), heterozygote (GC) (red dashed line), and homozygous minor allele (GG) (green dashed line), respectively. The log-rank p values were 0.03 for CG versus CC and 0.08 for the three-genotype model.
DISCUSSION
In the present work, we present the first study of an IRAK2 variant combining both functional analyses and genetic epidemiology. Functionally speaking, R214G was associated in vitro with modulated cytokine induction, namely IL-6, 6 h poststimulation (cf. Fig. 3D). The latter finding correlates with a recent report showing that murine Irak2 function is required for NF-κB recruitment to the Il6 promoter upon TLR2 stimulation (12) and that Irak2 is required for late cytokine transcription upon TLR activation (24). Our functional analysis points to reduced TRAF6 binding and ubiquitination as the mechanistic reason for the reduced NF-κB signaling and cytokine transcription. Thus, the ubiquitination of TRAF6 appears to be the defining event in downstream TLR signaling and a step that may have implications for disease in humans, as suggested by the disease association of rs35060588. At first glance, the increased strengths of interaction of IRAK2 R214G with IRAK4, IRAK2, and IRAK1 in the context of the Myddosome (cf. Fig. 2, C and D) seem surprising. However, it has been shown that the mechanism of IRAK-M to block TLR signaling is to prevent the IRAK-TRAF6 complex from dissociating from MyD88 (22). Thus inter-Myddosome interaction affinities must be low enough to accommodate this step. These insights are especially valuable because LUMIER allows assessment of protein-protein interactions in a quantitative way and within minutes of cell lysis, unlike many other techniques. Based on the current literature, our data suggest that increased IRAK interactions prevent IRAK2-TRAF6 dissociation from the membrane-proximal Myddosome to a subcellular location where TRAF6 ubiquitination occurs or is sensed (e.g. by the ubiquitin binding regulatory TAB2/TAB3 subunits of the TAK1 complex, which initiates IKK activation) (28, 29). Trapping of IRAK-TRAF6 would thus prevent optimal signal transduction. Using the R214G IRAK2 variant as a probe, our data suggest that the evidenced intricate hierarchical assembly of the Myddosome as a multiprotein complex (21) may thus be the result of fine-tuning in terms of association and dissociation equilibriums. That, according to our study, at least three steps (Myddosome association, dissociation, and TRAF6 ubiquitination, each involving multiple partners) appear to contribute to downstream signaling will be important to consider in approaches targeting the different IRAKs pharmacologically (4). Future investigations into the question of IRAK2 kinase status may be able to dissect to what extent TRAF6 interaction requires IRAK2 kinase activity. R214G may serve as a useful functional probe in kinase assays and biochemical reconstitution experiments combined with biophysical techniques that will be required to unravel the precise determinants of each step in IRAK2-dependent signal transduction.
Recently identified strongly hypofunctional MYD88 and IRAK4 mutations, which predispose children to severe pyogenic bacterial infection (5, 30), strikingly illustrated how genetic variants affecting the TLR/IL-1R signaling system may severely predispose specific individuals to disease. Because these variants are extremely rare (<0.001%) in the population at large, they contribute little to general disease susceptibility. Rather, functionally less severely attenuated but frequent variants, like the newly identified IRAK2 rs35060588, may contribute more broadly to susceptibility to different diseases, especially when acting in concert with other frequent functional variants (31). Many innate immune genes have been analyzed for such associations. To our knowledge, we present here the first study investigating an epidemiological association of an IRAK2 SNP with human disease. Although the IRAK2 rs35060588 GG genotype showed a reduced frequency in cases versus controls for SLE, this difference was not statistically significant. A reduced SLE risk for homozygous R214G carriers would be plausible because these individuals would be expected to have reduced TLR7/9 signaling, a disease contributing factor (27). Future genotyping studies and/or functional analyses in plasmacytoid dendritic cells or B cells from homozygous rs35060588 carriers will be required to unequivocally investigate whether IRAK2 is a disease-relevant factor in SLE. Of note, heterozygous rs35060588 carriage was found to correlate with worse survival in CRC patients, leading to a 60% higher risk of death (HR 1.60). The observed HR is within the range expected for a relatively frequent SNP. These findings are noteworthy because, despite numerous published studies in murine models, this study is one of the few studies addressing the role of TLR pathways in human CRC. Our study is also among the largest studies evaluating associations between genetic variants and CRC survival. Nevertheless, we had a restricted power to detect an association with CRC survival among rare homozygotes (cf. Fig. 4). This will require replication in additional studies. Opposite differences in outcome for heterozygous versus homozygous carriage are not unprecedented. For example, heterozygous carriage of a dysfunctional Mal/TIRAP variant, rs8177374 (coding S180L), was protective for several infectious diseases, whereas homozygous carriage was detrimental (32). In macrophage reconstitution experiments, which would more closely resemble a homozygous scenario, R214G showed reduced levels of IL-6. In CRC, high IL-6 levels in serum and tumor tissue were found to link with an unfavorable prognosis due to a tumor-promoting role of IL-6 via STAT3 (33). Thus, it will be interesting to determine how IL-6 induction is affected in homo- versus heterozygotes in future studies in CRC patients and healthy subjects. Future studies should also assess IL-1β induction in R214G carriers, not least because the cellular effects of IL-1β, like TLR signals, rely on IRAK2 (20). Unfortunately, reconstituted macrophages did not respond to IL-1β (not shown), but in HEK293T cells, the presence of R214G showed reduced NF-κB activation upon IL-1β stimulation compared with WT (cf. Fig. 1F). It will require further investigations in a more readily IL-1β-responsive system to verify in the natural setting to what extent the effects caused by a functional IRAK2 SNP may be potentiated at the level of IL-1β signaling. At this stage, it would not be wise to overextrapolate from the impact on signaling and initial cytokine induction upon TLR stimulation observed here to long term effects in a slowly progressing disease like CRC. Nevertheless, our data provide a strong rationale for the future clinical and epidemiological study of IRAK2 in additional disease cohorts and ethnic groups, for which rs35060588 could be a useful probe.
In conclusion, in the present study, we identified a functional human IRAK2 variant that may genetically affect a human disease, CRC, with considerable mortality and morbidity (34). Functionally, the variant also showed an impairment of downstream IRAK signaling at the level of TRAF6 ubiquitination, resulting in subsequent modulation of cytokine transcription. Our study highlights the still relatively enigmatic human IRAK2 as an important mediator in TLR postreceptor signaling and thus calls for further mechanistic and immunological studies. Furthermore, the data presented here also warrant the inclusion of IRAK2 rs35060588 in future genetic association studies and the exploration of its functional phenotype in human carriers. IRAK inhibitors are already under development for the treatment of inflammation and B cell malignancies (6, 35). In a dawning age of individualized medicine and straightforward sequence acquisition, broader studies into the genetic predisposition of infectious, autoimmune, and malignant diseases will help to characterize the potential value of rs35060588 and potentially other IRAK2 variants as biomarkers of disease or points of therapeutic intervention (e.g. in CRC). Given the moderately high prevalence of variants like rs35060588 in Caucasians and other ethnic groups, such approaches may be widely applicable.
Acknowledgments
We thank Birgit Kaiser, Beate Pömmerl, Celine Coppard, Manfred Kögl, Lisa Waggoner, Kevin Dennehy, Stefan Wiemann, Stefan Pusch, Matthias Gstaiger, Tilmann Bürckstümmer, and Hans-Peter Duerr for technical support, helpful discussions, reagents, and statistics advice. Kate Fitzgerald and James Watson kindly provided Irak2-deficient cells.
This work was supported by grants from the German Cancer Research Center DKFZ (to H. W., J. G., A. V. K., and A. N. R. W.), Deutsche Forschungsgemeinschaft Grants We-4195/1-1 (to H. W., J. G., A. V. K., and A. N. R. W.) and INST 37/888-1 CRC 685 “Immunotherapy” (to A. N. R. W.), the Chinese Scholarship Council CSC (to H. W.), the University of Tübingen (to H. W., S. D., T. P., and A. N. R. W.), Science Foundation Ireland Grants 07/IN1/B934 and 11/PI/1056 (to S. M. F. and A. G. B.).
- TLR
- Toll-like receptor
- CCL
- chemokine (C-C motif) ligand 5
- CRC
- colorectal cancer
- DD
- death domain
- HEK
- human embryonic kidney
- IL-1R
- IL-1 receptor
- IRAK
- IL-1R-associated kinase
- KD
- kinase domain
- LUMIER
- luminescence-based mammalian interactome mapping
- NF-κB
- nuclear factor κB
- RANTES
- regulated and normal T cell expressed and secreted
- SLE
- systemic lupus erythematosus
- HR
- hazard ratio.
REFERENCES
- 1. Kawai T., Akira S. (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650 [DOI] [PubMed] [Google Scholar]
- 2. Iwasaki A., Medzhitov R. (2004) Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5, 987–995 [DOI] [PubMed] [Google Scholar]
- 3. O'Neill L. A. (2008) The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol. Rev. 226, 10–18 [DOI] [PubMed] [Google Scholar]
- 4. Flannery S., Bowie A. G. (2010) The interleukin-1 receptor-associated kinases: critical regulators of innate immune signalling. Biochem. Pharmacol. 80, 1981–1991 [DOI] [PubMed] [Google Scholar]
- 5. Picard C., von Bernuth H., Ghandil P., Chrabieh M., Levy O., Arkwright P. D., McDonald D., Geha R. S., Takada H., Krause J. C., Creech C. B., Ku C. L., Ehl S., Maródi L., Al-Muhsen S., Al-Hajjar S., Al-Ghonaium A., Day-Good N. K., Holland S. M., Gallin J. I., Chapel H., Speert D. P., Rodriguez-Gallego C., Colino E., Garty B. Z., Roifman C., Hara T., Yoshikawa H., Nonoyama S., Domachowske J., Issekutz A. C., Tang M., Smart J., Zitnik S. E., Hoarau C., Kumararatne D. S., Thrasher A. J., Davies E. G., Bethune C., Sirvent N., de Ricaud D., Camcioglu Y., Vasconcelos J., Guedes M., Vitor A. B., Rodrigo C., Almazán F., Méndez M., Aróstegui J. I., Alsina L., Fortuny C., Reichenbach J., Verbsky J. W., Bossuyt X., Doffinger R., Abel L., Puel A., Casanova J. L. (2010) Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine 89, 403–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ngo V. N., Young R. M., Schmitz R., Jhavar S., Xiao W., Lim K. H., Kohlhammer H., Xu W., Yang Y., Zhao H., Shaffer A. L., Romesser P., Wright G., Powell J., Rosenwald A., Muller-Hermelink H. K., Ott G., Gascoyne R. D., Connors J. M., Rimsza L. M., Campo E., Jaffe E. S., Delabie J., Smeland E. B., Fisher R. I., Braziel R. M., Tubbs R. R., Cook J. R., Weisenburger D. D., Chan W. C., Staudt L. M. (2011) Oncogenically active MYD88 mutations in human lymphoma. Nature 470, 115–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Motshwene P. G., Moncrieffe M. C., Grossmann J. G., Kao C., Ayaluru M., Sandercock A. M., Robinson C. V., Latz E., Gay N. J. (2009) An oligomeric signalling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK4. J. Biol. Chem. 284, 25404–25411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lin S. C., Lo Y. C., Wu H. (2010) Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 465, 885–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. George J., Motshwene P. G., Wang H., Kubarenko A. V., Rautanen A., Mills T. C., Hill A. V., Gay N. J., Weber A. N. (2011) Two human MYD88 variants, S34Y and R98C, interfere with MyD88-IRAK4-Myddosome assembly. J. Biol. Chem. 286, 1341–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Flannery S. M., Keating S. E., Szymak J., Bowie A. G. (2011) Human interleukin-1 receptor-associated kinase-2 is essential for Toll-like receptor-mediated transcriptional and post-transcriptional regulation of tumor necrosis factor α. J. Biol. Chem. 286, 23688–23697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wesche H., Gao X., Li X., Kirschning C. J., Stark G. R., Cao Z. (1999) IRAK-M is a novel member of the Pelle/interleukin-1 receptor-associated kinase (IRAK) family. J. Biol. Chem. 274, 19403–19410 [DOI] [PubMed] [Google Scholar]
- 12. Kawagoe T., Sato S., Matsushita K., Kato H., Matsui K., Kumagai Y., Saitoh T., Kawai T., Takeuchi O., Akira S. (2008) Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat. Immunol. 9, 684–691 [DOI] [PubMed] [Google Scholar]
- 13. Nagpal K., Plantinga T. S., Sirois C. M., Monks B. G., Latz E., Netea M. G., Golenbock D. T. (2011) Natural loss-of-function mutation of myeloid differentiation protein 88 disrupts its ability to form Myddosomes. J. Biol. Chem. 286, 11875–11882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. International HapMap Consortium (2003) The International HapMap Project. Nature 426, 789–796 [DOI] [PubMed] [Google Scholar]
- 15. Keating S. E., Maloney G. M., Moran E. M., Bowie A. G. (2007) IRAK-2 participates in multiple Toll-like receptor signaling pathways to NFκB via activation of TRAF6 ubiquitination. J. Biol. Chem. 282, 33435–33443 [DOI] [PubMed] [Google Scholar]
- 16. Sali A., Overington J. P. (1994) Derivation of rules for comparative protein modeling from a database of protein structure alignments. Protein Sci. 3, 1582–1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang Z., Liu J., Sudom A., Ayres M., Li S., Wesche H., Powers J. P., Walker N. P. (2006) Crystal structures of IRAK-4 kinase in complex with inhibitors: a serine/threonine kinase with tyrosine as a gatekeeper. Structure 14, 1835–1844 [DOI] [PubMed] [Google Scholar]
- 18. Löfgren S. E., Delgado-Vega A. M., Gallant C. J., Sánchez E., Frostegård J., Truedsson L., de Ramón Garrido E., Sabio J. M., González-Escribano M. F., Pons-Estel B. A., D'Alfonso S., Witte T., Lauwerys B. R., Endreffy E., Kovács L., Vasconcelos C., Martins da Silva B., Martín J., Alarcón-Riquelme M. E., Kozyrev S. V. (2010) A 3′-untranslated region variant is associated with impaired expression of CD226 in T and natural killer T cells and is associated with susceptibility to systemic lupus erythematosus. Arthritis Rheum. 62, 3404–3414 [DOI] [PubMed] [Google Scholar]
- 19. Castro F. A., Försti A., Buch S., Kalthoff H., Krauss C., Bauer M., Egberts J., Schniewind B., Broering D. C., Schreiber S., Schmitt M., Hampe J., Hemminki K., Schafmayer C. (2011) TLR-3 polymorphism is an independent prognostic marker for stage II colorectal cancer. Eur. J. Cancer 47, 1203–1210 [DOI] [PubMed] [Google Scholar]
- 20. Muzio M., Ni J., Feng P., Dixit V. M. (1997) IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science 278, 1612–1615 [DOI] [PubMed] [Google Scholar]
- 21. Gay N. J., Gangloff M., O'Neill L. A. (2011) What the Myddosome structure tells us about the initiation of innate immunity. Trends Immunol. 32, 104–109 [DOI] [PubMed] [Google Scholar]
- 22. Kobayashi K., Hernandez L. D., Galán J. E., Janeway C. A., Jr., Medzhitov R., Flavell R. A. (2002) IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 110, 191–202 [DOI] [PubMed] [Google Scholar]
- 23. Nagpal K., Plantinga T. S., Wong J., Monks B. G., Gay N. J., Netea M. G., Fitzgerald K. A., Golenbock D. T. (2009) A TIR domain variant of MyD88 adapter-like (Mal)/TIRAP results in loss of MyD88 binding and reduced TLR2/TLR4 signaling. J. Biol. Chem. 284, 25742–25748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pauls E., Nanda S. K., Smith H., Toth R., Arthur J. S., Cohen P. (2013) Two phases of inflammatory mediator production defined by the study of IRAK2 and IRAK1 knock-in mice. J. Immunol. 191, 2717–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Conner J. R., Smirnova I. I., Poltorak A. (2009) A mutation in Irak2c identifies IRAK-2 as a central component of the TLR regulatory network of wild-derived mice. J. Exp. Med. 206, 1615–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. El-Omar E. M., Ng M. T., Hold G. L. (2008) Polymorphisms in Toll-like receptor genes and risk of cancer. Oncogene 27, 244–252 [DOI] [PubMed] [Google Scholar]
- 27. Barrat F. J., Coffman R. L. (2008) Development of TLR inhibitors for the treatment of autoimmune diseases. Immunol. Rev. 223, 271–283 [DOI] [PubMed] [Google Scholar]
- 28. Qian Y., Commane M., Ninomiya-Tsuji J., Matsumoto K., Li X. (2001) IRAK-mediated translocation of TRAF6 and TAB2 in the interleukin-1-induced activation of NFκB. J. Biol. Chem. 276, 41661–41667 [DOI] [PubMed] [Google Scholar]
- 29. Windheim M., Stafford M., Peggie M., Cohen P. (2008) Interleukin-1 (IL-1) induces the Lys63-linked polyubiquitination of IL-1 receptor-associated kinase 1 to facilitate NEMO binding and the activation of IkappaBalpha kinase. Mol. Cell. Biol. 28, 1783–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. von Bernuth H., Picard C., Jin Z., Pankla R., Xiao H., Ku C. L., Chrabieh M., Mustapha I. B., Ghandil P., Camcioglu Y., Vasconcelos J., Sirvent N., Guedes M., Vitor A. B., Herrero-Mata M. J., Aróstegui J. I., Rodrigo C., Alsina L., Ruiz-Ortiz E., Juan M., Fortuny C., Yagüe J., Antón J., Pascal M., Chang H. H., Janniere L., Rose Y., Garty B. Z., Chapel H., Issekutz A., Maródi L., Rodriguez-Gallego C., Banchereau J., Abel L., Li X., Chaussabel D., Puel A., Casanova J. L. (2008) Pyogenic bacterial infections in humans with MyD88 deficiency. Science 321, 691–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schork N. J., Murray S. S., Frazer K. A., Topol E. J. (2009) Common vs. rare allele hypotheses for complex diseases. Curr. Opin. Genet. Dev. 19, 212–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khor C. C., Chapman S. J., Vannberg F. O., Dunne A., Murphy C., Ling E. Y., Frodsham A. J., Walley A. J., Kyrieleis O., Khan A., Aucan C., Segal S., Moore C. E., Knox K., Campbell S. J., Lienhardt C., Scott A., Aaby P., Sow O. Y., Grignani R. T., Sillah J., Sirugo G., Peshu N., Williams T. N., Maitland K., Davies R. J., Kwiatkowski D. P., Day N. P., Yala D., Crook D. W., Marsh K., Berkley J. A., O'Neill L. A., Hill A. V. (2007) A Mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nat. Genet. 39, 523–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Waldner M. J., Foersch S., Neurath M. F. (2012) Interleukin-6: a key regulator of colorectal cancer development. Int. J. Biol. Sci. 8, 1248–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jemal A., Bray F., Center M. M., Ferlay J., Ward E., Forman D. (2011) Global cancer statistics. CA Cancer J. Clin. 61, 69–90 [DOI] [PubMed] [Google Scholar]
- 35. Wang Z., Wesche H., Stevens T., Walker N., Yeh W. C. (2009) IRAK-4 inhibitors for inflammation. Curr. Top. Med. Chem. 9, 724–737 [DOI] [PMC free article] [PubMed] [Google Scholar]