

Background: Contribution of MAPK members to integrin adhesion is not established.
Results: Inhibition of B-Raf function or expression selectively regulates integrin α4β1 in T cells.
Conclusion: B-Raf is a signaling component for integrin α4β1 cytoskeletal association, cell spreading, and adhesion.
Significance: This novel association of B-Raf and integrin α4β1 suggests new therapeutic targets in T cells and indicates potential off-target effects of sorafenib.
Keywords: Anticancer Drug, Cell Adhesion, Cytoskeleton, Extracellular Signal-regulated Kinase (ERK), Fibronectin, Integrins, Raf Kinase, Shear Stress, T-cell
Abstract
The regulation of integrin-mediated adhesion is of vital importance to adaptive and innate immunity. Integrins are versatile proteins and mediate T cell migration and trafficking by binding to extracellular matrix or other cells as well as initiating intracellular signaling cascades promoting survival or activation. The MAPK pathway is known to be downstream from integrins and to regulate survival, differentiation, and motility. However, secondary roles for canonical MAPK pathway members are being discovered. We show that chemical inhibition of RAF by sorafenib or shRNA-mediated knockdown of B-Raf reduces T cell resistance to shear stress to α4β1 integrin ligands vascular cell adhesion molecule 1 (VCAM-1) and fibronectin, whereas inhibition of MEK/ERK by U0126 had no effect. Microscopy showed that RAF inhibition leads to significant inhibition of T cell spreading on VCAM-1. The association of α4β1 integrin with the actin cytoskeleton was shown to be dependent on B-Raf activity or expression, whereas α4β1 integrin affinity for soluble VCAM-1 was not. These effects were shown to be specific for α4β1 integrin and not other integrins, such as α5β1 or LFA-1, or a variety of membrane proteins. We demonstrate a novel role for B-Raf in the selective regulation of α4β1 integrin-mediated adhesion.
Introduction
The dynamic regulation of T cell adhesion is a critical component of the adaptive immune response (1). Events such as T cell trafficking into lymphoid organs or sites of inflammation are generally mediated by β1 integrins (α4β1 integrin/VLA-4) binding to vascular cell adhesion molecule 1 (VCAM-1)2 and fibronectin (FN), or β2 integrins (αLβ2 integrin/LFA-1) binding to intercellular adhesion molecule 1 (ICAM-1) (2). Integrin-mediated adhesion is regulated by a process known as inside-out signaling and occurs either by a conformational change of the integrin determining the affinity of integrin for ligand or by avidity modulation due to receptor clustering (3). Integrin-mediated adhesion is also regulated by events following receptor occupancy that stabilize adhesion and coordinate cell spreading and migration, such as linkage of the integrin to the actin cytoskeleton and initiation of intracellular signaling (outside-in signaling) (4–6). This dynamic process is incompletely understood, more so when considering that each specific α/β integrin heterodimer or distinctly differentiated subset of activated lymphocytes may use a unique combination of interacting proteins to regulate each of these adhesive events (7).
The mitogen-activated protein kinase (MAPK) pathway is part of the outside-in signaling cascade initiated by integrin-mediated adhesion (8). The highly conserved Ras/Raf/MEK/ERK signaling module relays signals from the extracellular membrane to cellular effectors that regulate cell fate by influencing cellular proliferation, differentiation, apoptosis, and motility (9). Receptor stimulation activates Ras family small GTPases, subsequently recruiting RAF family kinases to the plasma membrane for activation. Activated RAF then binds and activates MEK to bind and activate ERK, which then translocates to the nucleus and activates transcription factor complexes. The RAF family of serine/threonine protein kinases consists of A-Raf, B-Raf, and C-Raf (Raf-1), which share a common modular structure and some binding partners but are unique in many aspects (10, 11).
There is accumulating evidence that members of the MAPK pathway have secondary roles outside their described signaling module regulating transcription (11, 12). For example, Raf-1 can regulate mitosis and apoptosis independent from MEK, and MEK can regulate autophagy independent from RAF and ERK (13–15). In adherent cell lines, MAPK pathway members were shown to regulate motility and cytoskeletal dynamics by interacting with paxillin, myosin light chain kinase, and the Rho family of small GTPases (16–19). However, our understanding of the role of MAPK in the regulation of lymphocyte adhesion or the direct regulation of integrin activity is still limited.
We set out to study secondary roles of the MAPK pathway members as regulators of integrin function in T cells. Inhibition of RAF but not MEK/ERK reduces the adhesion of Jurkat T cells to VCAM-1 under shear stress, demonstrating the independence of this function from downstream signaling of the MAPK pathway. shRNA-mediated knockdown of B-Raf reproduced the effect of chemical RAF inhibition, confirming a role for B-Raf in lymphocyte adhesion.
EXPERIMENTAL PROCEDURES
Reagents and Cells
The human T cell line, Jurkat, was cultured in complete medium (RPMI 1640 supplemented with 10% fetal bovine serum, 100 μg/ml penicillin, and 100 μg/ml streptomycin). Human fibronectin was purified from plasma (Gulf Coast Blood Center, Houston, TX) as described (20). Normal human T cells were negatively selected from healthy donors (Gulf Coast Blood Center, Houston, TX) using the RosetteSep Human T-cell enrichment mixture (Stemcell Technologies Inc., Vancouver, Canada) and then maintained in complete medium supplemented with human recombinant IL-2 (20 units/ml), anti-CD3 (1 μg/ml), and anti-CD28 (1 μg/ml). Human VCAM-1 was purified from the supernatant of CHO cells engineered to express soluble VCAM-1-Fc (21). AlexaFluor- and HRP-conjugated secondary antibodies, nonspecific rabbit and mouse IgGs, and mouse anti-GM1 were purchased (Invitrogen). The rabbit anti-p44/42 MAPK (Erk1/2), anti-phospho-p44/42 MAPK, anti-phospho-B-Raf (pSER445), and anti-β-actin were purchased (Cell Signaling, Danvers, MA). Rabbit anti-B-Raf (clone EP152Y) (Epitomics, Burlingame, CA), mouse anti-CD59, and anti-α5 integrin (clone P1D6) (Santa Cruz Biotechnology, Inc.) were purchased. The mouse anti-α4 integrin (clone L25), anti-LFA-1 (clone MHM24), anti-CD3 (clone OKT3), anti-CD4 (clone OKT4), anti-β1 integrin (clone 33B6), anti-α4β1 integrin (clone 19H8), anti-αL integrin (clone 32E6), anti-CD43 (clone IB7), and anti-CD28 (clone 95F12) were purified from mouse ascites (22–25). Doxycycline, puromycin, saponin, U0126 (Sigma-Aldrich), recombinant human ICAM-1/Fc (R&D Systems, Minneapolis, MN), and sorafenib (LC Laboratories, Woburn, MA) were purchased.
Generation of Inducible B-Raf Knockdown Cells
Knockdown cells were generated using shRNA-pTRIPZ clones (Open Biosystems, Huntsville, AL). Lentivirus was generated by Lipofectamine 2000 transfection of the packaging cell line, 293T-METR, with packaging plasmids containing pΔR8.91, CMV-pVSVG, and either scrambled non-silencing controls or a BRAF targeting sequence. Viral supernatants were collected at 48 h and concentrated by ultracentrifugation. Jurkat cells (1 × 106) were transduced and then selected in medium containing puromycin (1 μg/ml). After 1 week, cells were divided into a no treatment group and a doxycycline (1 μg/ml)-treated group. After 72 h, doxycycline-induced fluorescent red (TurboRFP) cells were purified with a FACS Aria IIu high-speed cell sorter (BD Biosciences) and cultured in complete medium with doxycycline.
Parallel Plate Flow Detachment Assay
The detachment assay was performed as described (26). In brief, human FN (5 μg/ml), VCAM-1 (10 μg/ml), or mAbs (1 μg/ml) were immobilized to plastic slides, washed with PBS, blocked with 2% BSA in PBS, and assembled to a parallel plate flow chamber. Cells (4 × 106) in running buffer (10 mm Tris, 103 mm NaCl, 24 mm NaHCO3, 5.5 mm glucose, 5.4 mm KCl, and 0.2% BSA, pH 7.4) were injected into the flow chamber and allowed to settle on the slide for 10 min. A computer-controlled syringe pump (Harvard Apparatus) was used to apply an increasing linear gradient of fluid flow to the cells for 300 s and recorded by digital microscopy. Shear stress calculations were determined every 50 s, and the shear stress in dynes/cm2 was defined as (6μQ)/(wh2), where μ is the viscosity of the medium (0.007), Q is the flow rate in cm3/s, w is the width of the chamber (0.3175 cm), and h is the height of the chamber (0.0254 cm).
Bright Field Microscopy
Human VCAM-1 was immobilized to 6-well non-tissue culture-treated plates (Falcon), washed with PBS, and blocked with 2% BSA in PBS. Cells (1 × 106) in complete medium were added, incubated at 37 °C for 10 min, and then fixed with 2% paraformaldehyde in PBS for 20 min at room temperature. Images were captured at ×20 magnification using a Nikon Diaphot-TMD microscope, equipped with a VI-470 CCD video camera (Optronics Engineering). Images were analyzed using Slidebook software (version 5.0) to distinguish spread cells from non-spread cells by creating a mask of spread cells and counting all cells that were larger or smaller than the threshold.
Super-resolution Immunofluorescence
Human VCAM-1 (10 μg/ml) was immobilized to glass coverslips, washed with PBS, and blocked with 2% BSA in PBS. Cells (5 × 105) in complete medium were added and incubated at 37 °C for 10 min and then fixed with 2% paraformaldehyde in PBS for 20 min at room temperature. Cells were permeabilized by adding saponin to a concentration of 0.1% for 30 min at room temperature. Cells were washed three times with PBS, 2% BSA, 0.1% saponin, stained for total B-Raf (AlexaFluor 647) and β1 integrin (AlexaFluor 488), and mounted to slides using Prolong Gold anti-fade reagent (Invitrogen). Images were acquired at room temperature using the OMX Blaze V4 structured illumination microscope (Applied Precision) with a ×100 numerical aperture 1.40 objective lens, two EM-CCD Photometrics Evolve 512 cameras, and DeltaVision OMX acquisition software. The images were reconstructed and rotated in three dimensions by 90°, and the height of cells was measured using the softWoRx software (version 6.0 beta 19). The image stacks were then transferred to either Slidebook software (version 5.0) to measure the area of contact of the cell with the glass coverslip or Imaris Bitplane software (version 7.6.1) to measure the colocalization of β1 integrin and B-Raf. The colocalization was quantified from the reconstructed three-dimensional image using the spot detection function for absolute fluorescence of both β1 integrin and B-Raf channels. Spots were generated with a 200-nm maximum xy diameter and a 500-nm maximum z diameter, identifying between 2000 and 15,000 spots for each channel per reconstructed image. Then the spots-to-spots colocalization function was used to identify all spots within 300 nm of spots from the other channel.
Soluble VCAM-1 Binding Assay
The soluble VCAM-1 binding assay was modified from a previous procedure (27). In brief, cells (1 × 106) in 100 μl of serum-free medium were incubated with human VCAM-1-Fc (10 μg/ml) at 37 °C for 10 min. The cells were then diluted and fixed by adding 2 ml of RPMI 1640 with 2% paraformaldehyde for 20 min at room temperature. The cells were washed twice with 2% BSA in PBS and incubated with AlexaFluor 488-conjugated rabbit anti-mouse for 20 min at room temperature. The cells were then washed and analyzed by flow cytometry using a FACSCalibur flow cytometer (BD Biosciences).
Cytoskeletal Stabilization Assay
The quantification of integrin-cytoskeleton attachment was modified from a previous procedure (26–28). Cells (2 × 106) in 100 μl of complete medium were incubated with mAb (1 μg/ml) at 4 °C for 30 min, and then either they were left untreated or AlexaFluor-conjugated rabbit anti-mouse was added at 4 °C for 30 min. The cells were incubated at either 4 or 37 °C for 10 min. The cells were then washed and resuspended in cytoskeletal stabilizing buffer (50 mm NaCl, 2 mm MgCl, 0.22 mm EGTA, 13 mm Tris, 1 mm PMSF, 10 mm iodacetamide, and 2% FBS, pH 8.0) with or without 0.1% Nonidet P-40. After 5 min at room temperature, 1 ml of cytoskeletal stabilizing buffer was added, and cells were immediately pelleted and fixed with 2% paraformaldehyde in PBS for 20 min at room temperature. The cells were then washed three times in PBS, and the amount of remaining bound mAb was determined by flow cytometry using a FACSCalibur (BD Biosciences).
RESULTS
RAF Inhibition, but Not ERK Inhibition, Leads to Decreased Adhesion of T Cells to Fibronectin
The parallel plate flow assay was used to measure adhesion and investigate the role of ERK signaling in integrin-mediated adhesion to human FN under conditions of shear stress. Two inhibitors of the ERK pathway were used, U0126, a MEK inhibitor, and sorafenib, a RAF inhibitor. Jurkat cell adhesion to FN was unchanged by 1 h of 1 μm U0126 treatment (Fig. 1A). The U0126- and vehicle (DMSO)-treated cells show very similar rates of detachment, with ∼49% of the initial cells remaining at 45 dynes/cm2 (maximum shear) (Fig. 1B). The phosphorylation of ERK was inhibited in these cells (Fig. 1C). In contrast to MEK/ERK inhibition, adhesion to FN was significantly reduced by 50 nm sorafenib when compared with vehicle control (MeOH) (Fig. 1D). The cells pretreated with vehicle show 47.5% of cells remaining at maximum shear, whereas the cells pretreated with sorafenib show an increased rate of detachment with only 11.9% of cells remaining, a 75% inhibition (Fig. 1E). The phosphorylation of B-Raf was inhibited by 50 nm sorafenib (Fig. 1F). These results suggest that RAF, and not MEK/ERK, contributes to T cell resistance to shear stress after adhesion to FN.
FIGURE 1.

Sorafenib reduces adhesion to fibronectin. A and D, parallel plate flow detachment assay to slides coated with human FN (5 μg/ml) of Jurkat cells incubated for 1 h with U0126 (1 μm) and DMSO vehicle control (A) or sorafenib (50 nm) and methanol vehicle control (D). B and E, percentage of cells remaining at 45 dynes/cm2 (maximum shear) from the parallel plate flow assay of cells incubated with U0126 (1 μm) and DMSO vehicle control (B) or sorafenib (50 nm) and methanol vehicle control (E). C and F, Western blots using SDS-PAGE (10% gel) of whole cell lysates (1 × 107 cells) after 10-min adhesion to FN and pretreated for 1 h with U0126 (1 μm) and DMSO vehicle control and probed for phosphorylated (p-) and total ERK (C) or sorafenib (50 nm) and methanol vehicle control and probed for phosphorylated and total B-Raf (F). Error bars, S.E. *, p < 0.01 using Student's t test.
RAF Inhibition Decreases α4β1 Integrin-mediated Adhesion to VCAM-1
Both α5β1 and α4β1 integrin bind FN, so to address the specificity of the adhesion, we tested adhesion to VCAM-1, which is a ligand for α4β1 and not α5β1. Adhesion to VCAM-1 was unchanged by 1 h of 1 μm U0126 treatment (Fig. 2A). The U0126- and vehicle (DMSO)-treated cells show very similar rates of detachment to VCAM-1, with ∼40% of cells remaining at maximum shear (Fig. 2B). The phosphorylation of ERK was inhibited in these cells (Fig. 2C). However, adhesion to VCAM-1 was reduced by 50 nm sorafenib (Fig. 2D). The cells pretreated with either vehicle or 10 nm sorafenib show very similar rates of detachment to VCAM-1, with ∼65% of the initial cells remaining at maximum shear, whereas the cells pretreated with 50 nm sorafenib show an increased rate of detachment with only 29.1% of the initial cells remaining, a 57% inhibition (Fig. 1E). The phosphorylation of B-Raf was inhibited at the 50 nm concentration of sorafenib (Fig. 2F). Sorafenib is most specific for Raf-1 (6 nm IC50) and B-Raf (22 nm IC50) (29), and combined, these data suggest that B-Raf phosphorylation contributes to α4β1 integrin-mediated adhesion to VCAM-1.
FIGURE 2.

Sorafenib reduces adhesion to VCAM-1. A and D, parallel plate flow detachment assay to slides coated with human VCAM-1 (10 μg/ml) of Jurkat cells incubated for 1 h with U0126 (1 μm) and DMSO vehicle control (A) or sorafenib (10 or 50 nm) and methanol vehicle control (D). B and E, percentage of cells remaining at maximum shear from the parallel plate flow assay of cells incubated with U0126 (1 μm) and DMSO vehicle control (B) or sorafenib (10 or 50 nm) and methanol vehicle control (E). C and F, Western blot using SDS-PAGE (10% gel) of whole cell lysates (1 × 107 cells) after 10-min adhesion to VCAM-1 and pretreated for 1 h with U0126 (1 μm) and DMSO vehicle control and probed for phosphorylated (p-) and total ERK (C) or sorafenib (10 or 50 nm) and methanol vehicle control and probed for phosphorylated and total B-Raf (F). Error bars, S.E. *, p < 0.01 using Student's t test.
RAF Inhibition Leads to Decreased Adhesion of Normal T Cells to VCAM-1 and Fibronectin
Given the reported differences between Jurkat and non-transformed T cells, we investigated the response of normal human T cells to sorafenib under conditions of shear stress after adhesion to α4β1 integrin ligands. Adhesion to both VCAM-1 (Fig. 3A) and FN (Fig. 3B) was significantly reduced by 50 nm sorafenib, compared with MeOH vehicle control. Three healthy donors were tested, and these results demonstrate that sorafenib reduces adhesion of normal human T cells to α4β1 integrin ligands.
FIGURE 3.
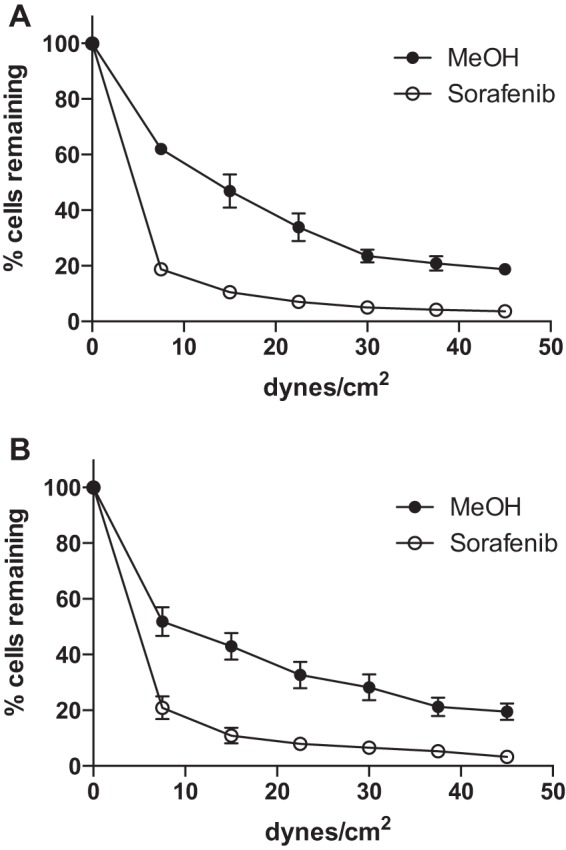
Sorafenib reduces adhesion of activated normal human T cells to VCAM-1 and fibronectin. A and B, parallel plate flow detachment assay of activated normal human T cells pretreated for 1 h with methanol vehicle control or sorafenib (50 nm) to slides coated with human VCAM-1 (10 μg/ml) (A) or human FN (5 μg/ml) (B). Data are shown for an experiment performed in triplicate from one healthy donor. Error bars, S.E.
BRAF Knockdown Decreases Adhesion to VCAM-1 and Fibronectin
To confirm the role of B-Raf in α4β1 integrin-mediated adhesion, Jurkat cells were stably transduced with doxycycline-inducible shRNA specific for BRAF or a scrambled shRNA vector control sequence. B-Raf protein expression was reduced in the B-Raf knockdown (KD) cells cultured with doxycycline (Fig. 4A). B-Raf knockdown cells (KD+D) compared with control cells (KD-NT, VC-NT, and VC+D) show decreased adhesion to both VCAM-1 (Fig. 4B) and FN (Fig. 4C). The control cells show very similar rates of detachment to VCAM-1, with ∼55% of the initial cells remaining at maximum shear, whereas the KD+D cells show an increased rate of detachment with only 4.9% of the initial cells remaining (Fig. 4D). Similarly, with FN, the control cells show equal rates of detachment, with ∼36% of the initial cells remaining at maximum shear, whereas the KD+D cells show an increased rate of detachment, with only 7.4% of the initial cells remaining (Fig. 4E). These results demonstrate that knockdown of B-Raf leads to decreased adhesion to α4β1 integrin ligands.
FIGURE 4.

B-Raf knockdown reduces adhesion to VCAM-1 and fibronectin. A, Western blot using SDS-PAGE (10% gel) of whole cell lysates (1 × 107 cells) of Jurkat, vector control (VC), and B-Raf KD cells cultured with and without doxycycline (±D), and probed for total B-Raf or β-actin. B and C, parallel plate flow detachment assay of vector control and KD cells cultured with doxycycline (+D) or without doxycycline (NT) to slides coated with human VCAM-1 (10 μg/ml) (B) or human FN (5 μg/ml) (C). D and E, percentage of cells remaining at maximum shear from the parallel plate flow assay of vector control and KD cells to slides coated with VCAM-1 (D) or FN (E). Error bars, S.E. *, p < 0.01 using Student's t test.
B-Raf Is Unessential for T Cell Proliferation or α4 and β1 Integrin Expression
A reduction in viability or α4β1 integrin expression of B-Raf knockdown cells could account for decreased adhesion. To confirm that B-Raf knockdown did not produce a defect in cell viability, the proliferation of the transduced or control cells cultured with or without doxycycline was measured for 10 days and found to remain unchanged (Fig. 5A). To confirm that B-Raf knockdown does not lead to reduced surface expression of integrin subunits, α4 or β1 integrin subunits were measured by flow cytometry and were unchanged (Fig. 5B). Thus, the decreased adhesion observed under conditions of B-Raf knockdown is not due to reduced viability or α4β1 integrin expression.
FIGURE 5.

Cell proliferation, integrin expression, and affinity for VCAM-1 are not affected by B-Raf knockdown or sorafenib. A, proliferation measured by trypan blue exclusion of Jurkat, shRNA vector control (VC), and B-Raf KD cells cultured with and without doxycycline (±D) for 10 days. B, flow cytometry using a FACSCalibur (BD Bioscience) of isotype control (ISO) (nonspecific mouse IgG) and α4 (mAb L25) or β1 (mAb 33B6) integrin subunits of vector control (VC) and B-Raf KD cells cultured with (+D) and without (NT) doxycycline stained with Alexa Fluor 488 goat anti-mouse secondary antibody. C and D, soluble VCAM-1 (10 μg/ml) binding assay of Jurkat cells incubated for 1 h with sorafenib (50 nm) or methanol vehicle control (C) and vector control (VC) and B-Raf KD cells cultured with doxycycline (+D) or without doxycycline (NT) (D). For C and D, cells were incubated with secondary (2′) AlexaFluor 488 rabbit anti-mouse IgG and no soluble VCAM-1 to show background fluorescence. Error bars, S.E. MFI, mean fluorescence intensity.
α4β1 Integrin Affinity Is Not Affected by B-Raf Knockdown or Sorafenib
Another mechanism of regulating the adhesion strength of integrins is the modulation of binding affinity. To test the role of B-Raf in the regulation of α4β1 integrin affinity, the binding affinity for soluble VCAM-1 was measured and found to be unchanged by pretreatment with 50 nm sorafenib (Fig. 5C) or by B-Raf knockdown (Fig. 5D). Cells were incubated with Mn2+ as a positive control for maximal integrin affinity, and the binding affinity for VCAM-1 of cells incubated with 1 mm Mn2+ was increased over the cells that did not receive Mn2+ but unchanged by B-Raf knockdown or sorafenib, indicating that the ability of the integrin to achieve a maximal affinity conformation is unaffected. These results indicate that B-Raf is unessential to α4β1 integrin affinity for soluble VCAM-1, and the reduced adhesion is not due to affinity regulation of α4β1.
Sorafenib or B-Raf Knockdown Specifically Reduces Adhesion Mediated by α4β1 Integrin
The fact that α4β1 affinity was shown to be unaffected suggested that we could bypass physiologic ligand coupling by using adhesion to immobilized mAbs to address the specificity of B-Raf inhibition on resistance to shear stress. Jurkat cell adhesion to an anti-α4β1 integrin mAb (19H8) was significantly reduced by 50 nm sorafenib when compared with vehicle control, whereas adhesion to an anti-CD28 mAb was unchanged by sorafenib (Fig. 6A). Adhesion was measured to mAbs to α4β1 integrin (19H8), β1 integrin (33B6), α5 integrin, LFA-1 (32E6 and MHM24), CD28, GM1, CD43, CD59, CD3 (OKT3), and CD4 (OKT4) (Fig. 6B). Adhesion was only reduced by sorafenib to α4β1 or β1 integrin mAbs. Adhesion was also measured to the LFA-1 natural ligand ICAM-1 and found to be unaffected by sorafenib (Fig. 6B). The shRNA vector control cells (VC+D) and B-Raf knockdown cells (KD+D) show equal detachment when bound to immobilized anti-LFA-1 (32E6), anti-CD3 (OKT3), and ICAM-1 (Fig. 6C). However, the adhesion of B-Raf knockdown cells to the anti-α4β1 integrin mAb (19H8) was significantly reduced when compared with shRNA vector control cells (Fig. 6C). These results indicate that B-Raf specifically regulates α4β1 integrin and not other integrins or a variety of membrane components.
FIGURE 6.
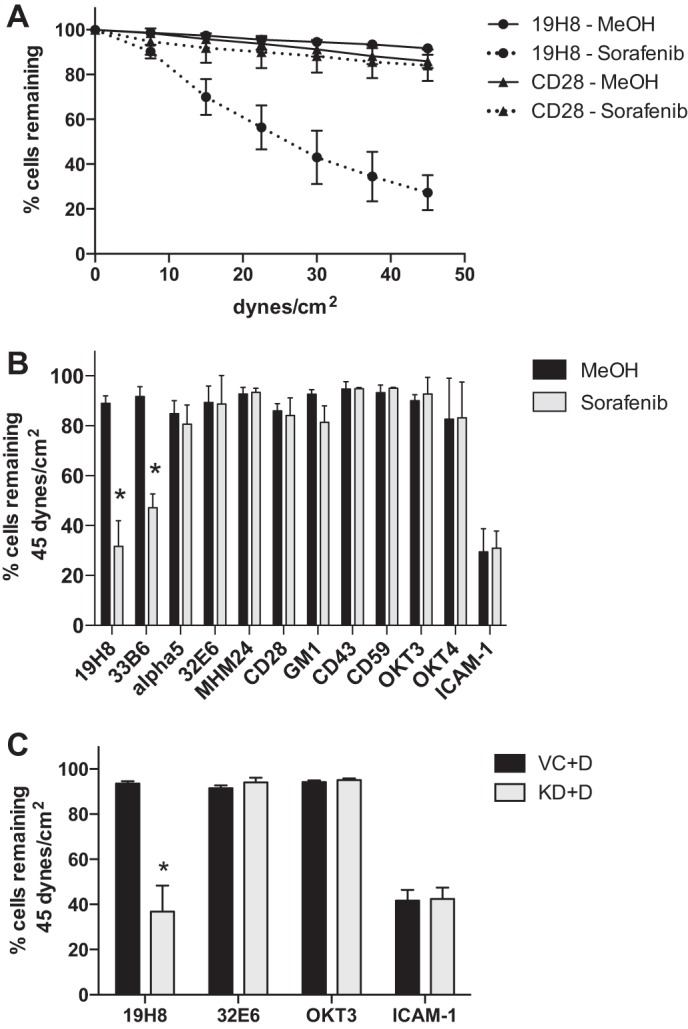
Sorafenib or B-Raf knockdown specifically reduces adhesion mediated by α4β1 integrin. A, parallel plate flow detachment assay of Jurkat cells incubated for 1 h with sorafenib (50 nm) and methanol vehicle control to slides coated with anti-α4β1 integrin mAb (19H8) or anti-CD28 mAb (1 μg/ml). B, percentage of cells incubated for 1 h with sorafenib (50 nm) and methanol vehicle control remaining at maximum shear to slides coated with anti-α4β1 integrin (19H8) (n = 8), anti-β1 integrin (33B6) (n = 6), anti-α5 integrin (n = 4), anti-LFA-1 (32E6) (n = 4), anti-LFA-1 (MHM24) (n = 3), anti-CD3 (OKT3) (n = 3), anti-CD4 (OKT4) (n = 3), anti-CD28 (n = 6), anti-CD43 (n = 3), anti-CD59 (n = 3), anti-GM1 (n = 3), or human ICAM-1 (5 μg/ml) (n = 6). C, percentage of B-Raf KD or vector control (VC) cells cultured with doxycycline (+D) remaining at maximum shear to slides coated with anti-α4β1 integrin (19H8) (n = 5), anti-LFA-1 (32E6) (n = 4), anti-CD3 (OKT3) (n = 4), or human ICAM-1 (5 μg/ml) (n = 6). Error bars, S.E. *, p < 0.01 using Student's t test.
Sorafenib or B-Raf Knockdown Inhibits Cell Spreading after Adhesion to VCAM-1
Bright field microscopy was used to investigate the effects of RAF inhibition on cell morphology after adhesion to VCAM-1. Images were captured and quantified after 10 min of adhesion to VCAM-1 (Fig. 7A). An average of 84.4% of MeOH vehicle-treated cells were spread, compared with 9% of cells treated with 50 nm sorafenib (Fig. 7B). Similarly, an average of 77.3% of shRNA vector control cells (VC+D) were spread, compared with 7.9% of B-Raf knockdown cells (KD+D) (Fig. 7C). These results demonstrate that B-Raf activity or expression is required for efficient α4β1 integrin-driven cell spreading.
FIGURE 7.

Sorafenib or B-Raf knockdown inhibits cell spreading on VCAM-1. A, bright field microscopy after 10-min adhesion to VCAM-1 (10 μg/ml) of Jurkat cells pretreated for 1 h with methanol vehicle control or sorafenib (50 nm) (left panels) and shRNA vector control (VC) or B-Raf knockdown (KD) cells cultured with doxycycline (+D) (right panels). Images were quantified, and results are shown as the mean percentage of cell spreading from two independent experiments performed in triplicate for vehicle- and sorafenib-treated cells (B) and control and B-Raf KD cells (C). Error bars, S.E. *, p < 0.001 using Student's t test.
Sorafenib Inhibits Cell Spreading and β1 Integrin Colocalization with B-Raf after Adhesion to VCAM-1
Super-resolution microscopy was used to investigate RAF inhibition on cell morphology and localization of β1 integrin and B-Raf after 10-min adhesion to VCAM-1. Cell spreading after pretreatment with 50 nm sorafenib was measured using the height of cells (Fig. 8A) and the area of cellular contact with the glass coverslip (Fig. 8B) as indicators of spreading. The average height of cells pretreated with vehicle control (MeOH) was 5.5 μm, whereas the average height of cells pretreated with sorafenib was 10.2 μm (Fig. 8C). The average area of cellular contact with VCAM-1 of control cells was 512 μm2, whereas the average with sorafenib was 110 μm2, a 78.4% reduction (Fig. 8D). The colocalization of β1 integrin and B-Raf was quantified from reconstructed three-dimensional images using the spots generated from absolute fluorescence (Fig. 8E). The control cells had 31.6% of β1 integrin colocalize with B-Raf, and the cells pretreated with sorafenib had only 13.3% of β1 integrin colocalize with B-Raf, a 58% reduction (Fig. 8F). A trend was observed in the decreased amount of B-Raf to colocalize with β1 integrin in sorafenib-treated cells, but it failed to reach significance (Fig. 8G). These results demonstrate that sorafenib inhibits cell spreading on VCAM-1 and reduces β1 integrin colocalization with B-Raf after adhesion to VCAM-1.
FIGURE 8.

Sorafenib inhibits cell spreading and β1 integrin colocalization with B-Raf after adhesion to VCAM-1. A and B, super-resolution immunofluorescence of β1 integrin (green) and B-Raf (red) of Jurkat cells incubated for 1 h with methanol vehicle control or sorafenib (50 nm) and then fixed after 10 min of adhesion to glass coverslips coated with VCAM-1 (10 μg/ml). A, representative maximum intensity projection of three-dimensional reconstructed images rotated by 90° to illustrate the height of cells; B, representative maximum intensity projection of the five z-sections closest to the interface with the coverslip to illustrate the area of contact with VCAM-1. C, images were analyzed for the height of the cells when rotated by 90° in three dimensions; D, the area of contact at the interface of the glass coverslip. E, colocalization of β1 integrin and B-Raf using the spot detection function of absolute fluorescence for total β1 integrin (green) and β1 integrin colocalized with B-Raf (yellow) (left) and total B-Raf (red) and B-Raf colocalized with β1 integrin (yellow) (right). The percentage of colocalization was determined by dividing the number of colocalized spots by the total number of spots for β1 integrin (F) and B-Raf (G). Data are shown from one experiment containing individual cells pretreated with methanol (n = 10) or sorafenib (n = 14), and the experiment was repeated with methanol (n = 10) and sorafenib (n = 12). Scale bars, 4 μm; error bars, S.E. *, p < 0.01 using Student's t test.
Sorafenib Prevents β1 Integrin Association with the Cytoskeleton
Integrin association with the actin cytoskeleton increases post-ligand binding and is necessary for focal adhesion formation and cellular resistance to shear stress. It has been shown that antibody cross-linking of α4 integrin on Jurkat cells leads to a significant increase in the amount of anti-α4 mAb able to resist solubilization by a non-ionic detergent, and this is interpreted to result from an increase in α4 integrin association with the cytoskeleton (30). The association of β1 integrin with the cytoskeleton after activation by cross-linking of an anti-β1 mAb was measured. Cells pretreated for 1 h with 50 nm sorafenib show significantly reduced β1 integrin association with the cytoskeleton after activation at 37 °C (5.2%) compared with control (23.6%), a 78% reduction (Fig. 9A). These results show that RAF inhibition reduces β1 integrin association with the cytoskeleton and indicate a role for B-Raf phosphorylation in this pathway.
FIGURE 9.
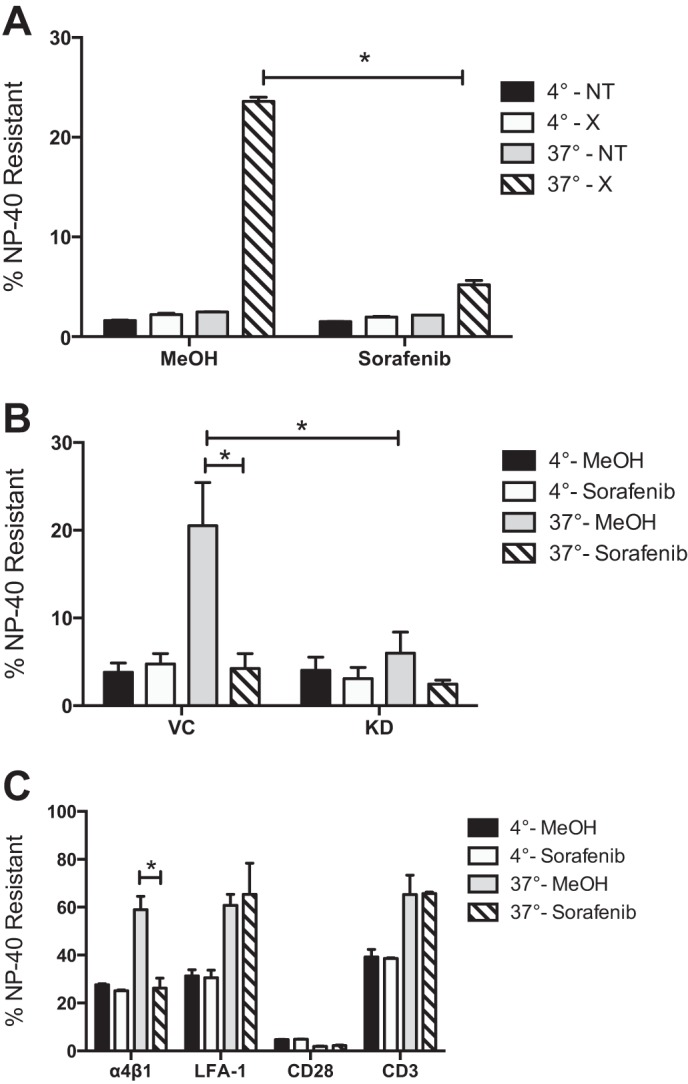
Sorafenib or B-Raf knockdown specifically reduces β1 and α4β1 integrin association with the cytoskeleton. A, cytoskeletal stabilization assay of Jurkat cells incubated for 1 h with sorafenib (50 nm) and methanol vehicle control and then incubated with AF-488-conjugated anti-β1 integrin mAb and either cross-linked with an anti-mouse secondary mAb (×) or left untreated (NT) at 4 or 37 °C. B, cytoskeletal stabilization assay of vector control (VC) or B-Raf knockdown (KD) cells incubated for 1 h with sorafenib (50 nm) and methanol vehicle control and then incubated with AF-488-conjugated anti-β1 integrin mAb and anti-mouse secondary mAb at 4 or 37 °C. C, Cytoskeletal stabilization assay of Jurkat cells incubated for 1 h with sorafenib (50 nm) and methanol vehicle control and then incubated with AF-647-conjugated anti-mouse secondary mAb and either anti-α4β1 (19H8), anti-LFA-1 (32E6), anti-CD3, or anti-CD28 mAb at 4 or 37 °C. The percentage of Nonidet P-40-resistant mAb was determined by dividing the mean fluorescence intensity of Nonidet P-40-treated cells by the MFI of cells that received no Nonidet P-40. Error bars, S.E. *, p < 0.001 using Student's t test.
B-Raf Is Essential for β1 Integrin Association with the Cytoskeleton
The association of β1 integrin with the cytoskeleton was measured in B-Raf knockdown or shRNA vector control cells pretreated with 50 nm sorafenib or vehicle control. Consistent with results in Fig. 9A, shRNA vector control cells pretreated for 1 h with sorafenib show significantly reduced β1 integrin association with the cytoskeleton after activation at 37 °C (4.2%) compared with vehicle control (20.5%), a 79.6% reduction (Fig. 9B). In comparison with vector control cells (20.5%), B-Raf knockdown cells show significantly reduced β1 integrin association with the cytoskeleton (6%) after activation at 37 °C, a 71.8% reduction. These results indicate that B-Raf is essential for β1 integrin association with the cytoskeleton.
Sorafenib Specifically Prevents α4β1 Integrin Association with the Cytoskeleton
The resistance to Nonidet P-40 solubilization was measured for mAbs to α4β1 integrin, LFA-1, CD28, and CD3. Compared with control cells at 4 °C, cross-linking at 37 °C significantly increased the amount of detergent-resistant α4β1 (from 27.6 to 59%), LFA-1 (from 31.3 to 60.8%), and CD3 (from 39.2 to 65.3%), but there was very little CD28 association with the cytoskeleton (∼4%) (Fig. 9C). Treatment with 50 nm sorafenib had little effect on the amount of detergent-resistant LFA-1 (65.3%), CD28 (∼2%), or CD3 (65.7%), whereas the amount of detergent-resistant α4β1 mAb was significantly reduced (from 59 to 26.2%). These results show that sorafenib inhibits α4β1 integrin association with the cytoskeleton and indicate that phosphorylation of B-Raf is specifically mediating the α4β1 integrin association with the cytoskeleton while having no effect on LFA-1, CD28, or CD3 association with the cytoskeleton.
DISCUSSION
This work has demonstrated a novel role for B-Raf in the direct regulation of the α4β1 integrin in lymphocytes. Both the chemical inhibition and knockdown of B-Raf lead to decreased resistance to shear stress of T cells after adhesion to α4β1 ligands and decreased α4β1 association with the cytoskeleton after mAb cross-linking. These effects were both specific for α4β1 integrin and independent from affinity regulation or downstream MEK/ERK activity. We have also shown that sorafenib inhibits α4β1 integrin-driven cell spreading and β1 integrin colocalization with B-Raf. The increased height and reduced area of cellular contact with VCAM-1 of cells treated with sorafenib suggest that these cells are experiencing greater shear stress distributed over a smaller area of anchorage, probably contributing to decreased adhesion in our laminar flow system. Cell adhesion has been shown to be dependent on the dynamics of the actin cytoskeleton and cell spreading, promoting adhesion strengthening and migration, in part by providing a more streamlined shape for the cell to reduce the shear stress imposed by laminar flow (6, 31, 32).
At present, it is not clear on the molecular level how B-Raf regulates α4β1 integrin function. Chemokine binding to G protein-coupled receptors induces the activation of phospholipase C and calcium signaling, leading to the rapid up-regulation of α4β1 and LFA-1 integrin affinity (33, 34). Downstream of calcium signaling, Rap1 from the Ras family of small GTPases plays an important role in β2 integrin affinity regulation and adhesion, but this has not been established for α4β1 integrin (4, 35). It has been shown that Rap1 mediates phorbol-ester (phorbol 12-myristate 13-acetate)-stimulated adhesion to FN, but Rap1 does not mediate SDF-1α-stimulated affinity up-regulation of α4β1 integrin for VCAM-1 or adhesion to VCAM-1 after SDF-1α or phorbol 12-myristate 13-acetate stimulation (36, 37). However, Rap1 specifically activates B-Raf, not Raf-1, and precedence for RAF family members in adhesion regulation was provided by studies demonstrating that H-Ras activation of Raf-1 suppressed integrin activation in CHO cells, but there were conflicting results concerning whether the suppression was independent of ERK (38–42). Therefore, a secondary role for B-Raf in the direct regulation of integrin-mediated adhesion was feasible.
Identification of the specific B-Raf-containing complex and binding partners of this mechanism will be important to the understanding of lymphocyte adhesion. Our results indicate that B-Raf is specifically regulating the α4β1 integrin and not LFA-1 or a variety of other membrane proteins. Integrin cytoplasmic tails binding to integrin-associated proteins mediate the events following receptor occupancy, such as adhesion strengthening and outside-in signaling. It has been shown that talin regulates cytoskeletal association, resistance to shear stress, and affinity of both α4β1 and LFA-1 (43, 44). However, it is established that different integrin heterodimers expressed by the same cells can utilize distinct signaling components and downstream effectors. For example, paxillin binding to α4 integrin cytoplasmic domains is known to regulate cytoskeletal association and resistance to shear stress but not affinity of α4β1, whereas paxillin does not bind to αL of LFA-1 (5, 30, 45).
In addition to possible Rap1 interaction, novel binding partners of B-Raf in T cells have been found (46). Of interest, B-Raf was shown to interact with both Dock2 (dedicator of cytokinesis protein 2), a guanine nucleotide exchange factor known to specifically activate Rac, and IQGAP1 (Ras GTPase-activating-like protein 1), a scaffolding protein known to interact with actin, Rac1, calmodulin, and Src (46–48). Alternatively, B-Raf regulates cytoskeletal dynamics in melanoma cells by mediating cross-talk with the Rho/ROCK pathway through Rnd3 and in fibroblasts through the ROCKII/LIMK/cofilin pathway (19, 49). Also, whereas an ERK-paxillin complex has since been identified in human adenocarcinoma cells, it was previously shown that Ras-induced serine phosphorylation of paxillin was mediated by induced expression of an activated B-Raf construct in a variety of cell types (17, 50). However, we found that sorafenib or B-Raf knockdown did not affect the induced phosphorylation of paxillin (Tyr-118) after adhesion to VCAM-1 (data not shown), a residue essential for actin cytoskeleton-dependent cell spreading and motility in lymphocytes (51–53), whereas other studies using adherent cell lines have identified Raf-1 association with vimentin, myosin phosphatase, and the Rho-effector Rok-α, raising the possibility of similar interactions for B-Raf (54–56). Altogether, and given the inherent differences between how adherent cell types and lymphocytes regulate adhesion, more studies are required to explore the molecular details of how B-Raf and α4β1 integrin interact in T cells.
The activities of all Raf isoforms are subject to complex regulation but have been shown to be dependent on Ras activity for the initiation of Raf activation. The activation of both A-Raf and Raf-1 requires phosphorylation on the B-Raf corresponding residue Ser-455, but this residue has been shown to be constitutively phosphorylated on B-Raf in many cell types (57, 58). Therefore, B-Raf activation is solely dependent on Ras activity and phosphorylation of Thr-598/Ser-601 (11). Constitutive activation of MAPK signaling by mutant forms of B-Raf (i.e. V600E) is observed broadly in solid cancers of multiple primary sites (59, 60). Leukemias of lymphoid origin express B-Raf, and other than the recent discovery of hairy cell leukemia, mutations of the BRAF gene are very rare, suggesting a fundamental and conserved role in T cell leukemia and possibly normal T cell function (61–67). Although there are few studies that focus on the importance of B-Raf to T cell physiology, it has been shown that MAPK signaling during T cell development progression beyond the CD4-CD8 double positive stage requires B-Raf, and rescue experiments with B-Raf can restore proliferation through MAPK signaling in anergic T cells (40, 67).
Specific inhibitors of mutant B-Raf were approved for treatment of melanoma in 2011, whereas general RAF inhibitors have been clinically used since 2007 (68–70). Chemical inhibition of MEK or RAF results in the inhibition of ERK and is sufficient to stop proliferation of many cancer cells (71). Sorafenib is a multikinase inhibitor once higher concentrations have been reached, but it is most specific for C-Raf (6 nm IC50) and B-Raf (22 nm IC50) and is still used in the clinic for the treatment of advanced renal cell carcinoma or hepatocellular carcinoma (29). The findings presented in this work should cause a reevaluation of clinical use of sorafenib having potential off-target effects on T cells (72–74). The effects of sorafenib on α4β1 integrin could impact T cell migration and homing to sites of inflammation or potentially effector functions (75–77). This is significant because the selectivity of this interaction may provide a therapeutic target for α4 integrin-related diseases, such as asthma, multiple sclerosis, rheumatoid arthritis, inflammatory bowl disease, and certain leukemias and lymphomas (78). Given that we have focused on sorafenib inhibition of B-Raf Ser-445 and the rarity of B-Raf mutations in T cells, this work also suggests that specific V600E B-Raf inhibitors may have fewer off-target effects on T cells (79).
We propose a novel secondary role for B-Raf in the up-regulation of cytoskeletal association of α4β1 and cell spreading mediated by the α4β1 integrin to regulate resistance to shear stress. Interestingly, this effect is independent from downstream MEK/ERK signaling, and within the scope of our studies, this association is unique to α4β1 integrin and not to other integrins or unrelated surface proteins. Both α4β1 integrin and B-Raf play important roles in human diseases, and understanding the mechanisms of their functions will provide important insights into the adaptive immune response and design of therapeutic strategies.
Acknowledgments
We thank Tomasz Zal, Ph.D., for super-resolution microscopy advice and Drs. Qing Ma, Peter Vanderslice, and Darren Woodside for providing reagents.
This work was supported, in whole or in part, by National Institutes of Health Grant 2-T32-CA-09598 (to W. S. B.), National Institutes of Health High-End Instrumentation Grant 1S10RR029552-01 (to the Immunology Imaging Core facility), and NCI, National Institutes of Health, Cancer Center Support Grant P30CA16672 (to the Flow Cytometry Core Facility at the University of Texas MD Anderson Cancer Center).
- VCAM-1
- vascular cell adhesion molecule 1
- FN
- fibronectin
- ICAM-1
- intercellular adhesion molecule 1
- KD
- knockdown
- GM1
- monosialotetrahexosylganglioside.
REFERENCES
- 1. Pribila J. T., Quale A. C., Mueller K. L., Shimizu Y. (2004) Integrins and T cell-mediated immunity. Annu. Rev. Immunol. 22, 157–180 [DOI] [PubMed] [Google Scholar]
- 2. Luster A. D., Alon R., von Andrian U. H. (2005) Immune cell migration in inflammation: present and future therapeutic targets. Nat. Immunol. 12, 1182–1190 [DOI] [PubMed] [Google Scholar]
- 3. Kim C., Ye F., Ginsberg M. H. (2011) Regulation of integrin activation. Annu. Rev. Cell Dev. Biol. 27, 321–345 [DOI] [PubMed] [Google Scholar]
- 4. Abram C. L., Lowell C. A. (2009) The ins and outs of leukocyte integrin signaling. Annu. Rev. Immunol. 27, 339–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hyduk S. J., Rullo J., Cano A. P., Xiao H., Chen M., Moser M., Cybulsky M. I. (2011) Talin-1 and Kindlin-3 regulate α4β1 integrin-mediated adhesion stabilization, but not G protein-coupled receptor-induced affinity upregulation. J. Immunol. 187, 4360–4368 [DOI] [PubMed] [Google Scholar]
- 6. Rullo J., Becker H., Hyduk S. J., Wong J. C., Digby G., Arora P. D., Cano A. P., Hartwig J., McCulloch C. A., Cybulsky M. I. (2012) Actin polymerization stabilizes α4β1 integrin anchors that mediate monocyte adhesion. J. Cell Biol. 197, 115–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zaidel-Bar R., Geiger B. (2010) The switchable integrin adhesome. J. Cell Sci. 123, 1385–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ramos J. W. (2008) The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int. J. Biochem. Cell Biol. 40, 2707–2719 [DOI] [PubMed] [Google Scholar]
- 9. Chang L., Karin M. (2001) Mammalian MAP kinase signaling cascades. Nature 410, 37–40 [DOI] [PubMed] [Google Scholar]
- 10. Wellbrock C., Karasarides M., Marais R. (2004) The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 5, 875–885 [DOI] [PubMed] [Google Scholar]
- 11. Matallanas D., Birtwistle M., Romano D., Zebisch A., Rauch J., von Kriegsheim A., Kolch W. (2011) Raf family kinases: old dogs have learned new tricks. Genes Cancer 2, 232–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yoon S., Seger R. (2006) The extracellular signal-related kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24, 21–44 [DOI] [PubMed] [Google Scholar]
- 13. Mielgo A., Seguin L., Huang M., Camargo M. F., Anand S., Franovic A., Weis S. M., Advani S. J., Murphy E. A., Cheresh D. A. (2011) A MEK-independent role for CRAF in mitosis and tumor progression. Nat. Med. 17, 1641–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. O'Neill E., Rushworth L., Baccarini M., Kolch W. (2004) Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science 306, 2267–2270 [DOI] [PubMed] [Google Scholar]
- 15. Wang J., Whiteman M. W., Lian H., Wang G., Singh A., Huang D., Denmark T. (2009) A non-conical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J. Biol. Chem. 284, 21412–21424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Viala E., Pouysségur J. (2004) Regulation of tumor cell motility by ERK mitogen-activated protein kinases. Ann. N.Y. Acad. Sci. 1030, 208–218 [DOI] [PubMed] [Google Scholar]
- 17. Pribic J., Brazill D. (2012) Paxillin phosphorylation and complexing with Erk and FAK are regulated by PLD activity in MDA-MD-231 cells. Cell. Signal. 24, 1531–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Klemke R. L., Cai S., Giannini A. L., Gallagher P. J., de Lanerolle P., Cheresh D. A. (1997) Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 137, 481–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Klein R. M., Spofford L. S., Abel E. V., Ortiz A., Aplin A. E. (2008) B-RAF regulation of Rnd3 participates in actin cytoskeletal and focal adhesion organization. Mol. Biol. Cell. 19, 498–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wooten D. K., Teague T. K., McIntyre B. W. (1999) Separation of integrin-dependent adhesion from morphological changes based on differential PLC specificities. J. Leukoc. Biol. 65, 127–136 [DOI] [PubMed] [Google Scholar]
- 21. Woodside D. G., Kram R. M., Mitchell J. S., Belsom T., Billard M. J., McIntyre B. W., Vanderslice P. (2006) Contrasting roles for domain 4 of VCAM-1 in the regulation of cell adhesion and soluble VCAM-1 binding to integrin α4β1. J. Immunol. 176, 5041–5049 [DOI] [PubMed] [Google Scholar]
- 22. Bednarczyk J. L., Szabo M. C., Wygant J. N., Lazarovits A. I., McIntyre B. W. (1994) Identification of a combinatorial epitope expressed by the integrin α4β1 heterodimer involved in the regulation of cell adhesion. J. Biol. Chem. 269, 8348–8354 [PubMed] [Google Scholar]
- 23. Mitchell J. S., Kanca O., McIntyre B. W. (2002) Lipid microdomain clustering induces a redistribution of antigen recognition and adhesion molecules on human T lymphocytes. J. Immunol. 168, 2737–2744 [DOI] [PubMed] [Google Scholar]
- 24. Champe M., McIntyre B. W., Berman P. W. (1995) Monoclonal antibodies that block the activity of leukocyte function-associated antigen 1 recognize three discreet epitopes in the inserted domain of CD11a. J. Biol. Chem. 270, 1388–1394 [DOI] [PubMed] [Google Scholar]
- 25. Miyamoto Y. J., Wann E. R., Fowler T., Duffield E., Höök M., McIntyre B. W. (2001) Fibronectin binding protein A of Staphylococcus aureus can mediate human T lymphocyte adhesion and coactivation. J. Immunol. 166, 5129–5138 [DOI] [PubMed] [Google Scholar]
- 26. Mitchell J. S., Brown W. S., Woodside D. G., Vanderslice P., McIntyre B. W. (2009) Clustering T-cell GM1 lipid rafts increases cellular resistance to shear on fibronectin through changes in integrin affinity and cytoskeletal dynamics. Immunol. Cell Biol. 87, 324–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rose D. M., Grabovsky V., Alon R., Ginsberg M. H. (2001) The affinity of integrin α4β1 governs lymphocyte migration. J. Immunol. 167, 2824–2830 [DOI] [PubMed] [Google Scholar]
- 28. Geppert T. D., Lipsky P. E. (1991) Association of various T cell-surface molecules with the cytoskeleton: effect of cross-linking and activation. J. Immunol. 146, 3298–3305 [PubMed] [Google Scholar]
- 29. Wilhelm S. M., Carter C., Tang L., Wilkie D., McNabola A., Rong H., Chen C., Zhang X., Vincent P., McHugh M., Cao Y., Shujath J., Gawlak S., Eveleigh D., Rowley B., Liu L., Adnane L., Lynch M., Auclair D., Taylor I., Gedrich R., Voznesensky A., Riedl B., Post L. E., Bollag G., Trail P. A. (2004) BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 64, 7099–7109 [DOI] [PubMed] [Google Scholar]
- 30. Alon R., Feigelson S. W., Manevich E., Rose D. M., Schmitz J., Overby D. R., Winter E., Grabovsky V., Shinder V., Matthews B. D., Sokolovsky-Eisenberg M., Ingber D. E., Benoit M., Ginsberg M. H. (2005) α4β1-dependent adhesion strengthening under mechanical strain is regulated by paxillin association with the α4-cytoplasmic domain. J. Cell Biol. 171, 1073–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. D'Souza-Schorey C., Boettner B., Van Aelst L. (1998) Rac regulates integrin-mediated spreading and increased adhesion of T lymphocytes. Mol. Cell. Biol. 18, 3936–3946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maqueda A., Moyano J. V., Gutiérrez-López M. D., Ovalle S., Rodríguez-Frade J. M., Cabañas C., Garcia-Pardo A. (2006) Activation pathways of α4β1 integrin leading to distinct T-cell cytoskeleton reorganization, Rac1 regulation and Pyk2 phosphorylation. J. Cell. Physiol. 207, 746–756 [DOI] [PubMed] [Google Scholar]
- 33. Hyduk S. J., Chan J. R., Duffy S. T., Chen M., Peterson M. D., Waddell T. K., Digby G. C., Szaszi K., Kapus A., Cybulsky M. I. (2007) Phospholipase C, calcium, and calmodulin are critical for α4β1 integrin affinity up-regulation and monocyte arrest triggered by chemoattractants. Blood 109, 176–184 [DOI] [PubMed] [Google Scholar]
- 34. Constantin G., Majeed M., Giagulli C., Piccio L., Kim J. Y., Butcher E. C., Laudanna C. (2000) Chemokines trigger immediate β2 integrin affinity and mobility changes: differential regulation and roles in lymphocyte arrest under flow. Immunity 13, 759–769 [DOI] [PubMed] [Google Scholar]
- 35. de Bruyn K. M., Rangarajan S., Reedquist K. A., Figdor C. G., Bos J. L. (2002) The small GTPase Rap1 is required for Mn2+- and antibody-induced LFA-1- and VLA-4-mediated cell adhesion. J. Biol. Chem. 277, 29468–29476 [DOI] [PubMed] [Google Scholar]
- 36. Liu L., Schwartz B. R., Tupper J., Lin N., Winn R. K., Harlan J. M. (2002) The GTPase Rap1 regulates phorbol 12-myristate 13-acetate-stiumlated but not ligand-induced β1 integrin-dependent leukocyte adhesion. J. Biol. Chem. 277, 40893–40900 [DOI] [PubMed] [Google Scholar]
- 37. Ghandour H., Cullere X., Alvarez A., Luscinskas F. W., Mayadas T. N. (2007) Essential role for Rap1 GTPase and its guanine exchange factor CalDAG-GEFI in LFA-1 but not VLA-4 integrin mediated human T-cell adhesion. Blood 110, 3682–3690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carey K. D., Watson R. T., Pessin J. E., Stork P. J. (2003) The requirement of specific membrane domains for Raf-1 phosphorylation and activation. J. Biol. Chem. 278, 3185–3196 [DOI] [PubMed] [Google Scholar]
- 39. Jin A., Kurosu T., Tsuji K., Mizuchi D., Arai A., Fujita H., Hattori M., Minato N., Miura O. (2006) BCR/ABL and IL-3 activate Rap1 to stimulate the B-Raf/MEK/ERK and Akt signaling pathways and to regulate proliferation, apoptosis, and adhesion. Oncogene 25, 4332–4340 [DOI] [PubMed] [Google Scholar]
- 40. Dillon T. J., Karpitski V., Wetzel S. A., Parker D. C., Shaw A. S., Stork P. J. (2003) Ectopic B-Raf expression enhances extracellular signal-regulated kinase (ERK) signaling in T cells and prevents antigen-presenting cell-induced anergy. J. Biol. Chem. 278, 35940–35949 [DOI] [PubMed] [Google Scholar]
- 41. Hughes P. E., Renshaw M. W., Pfaff M., Forsyth J., Keivens V. M., Schwartz M. A., Ginsberg M. H. (1997) Suppression of integrin activation: a novel function of a Ras/Raf-initiated MAP kinase pathway. Cell 88, 521–530 [DOI] [PubMed] [Google Scholar]
- 42. Hughes P. E., Oertli B., Hansen M., Chou F. L., Willumsen B. M., Ginsberg M. H. (2002) Suppression of integrin activation by activated Ras or Raf does not correlate with bulk activation of ERK MAP kinase. Mol. Biol. Cell. 13, 2256–2265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Simonson W. T., Franco S. J., Huttenlocher A. (2006) Talin1 regulates TCR-mediated LFA-1 activation. J. Immunol. 177, 7707–7714 [DOI] [PubMed] [Google Scholar]
- 44. Calderwood D. A., Campbell I. D., Critchley D. R. (2013) Talins and kindlins: partners in integrin-mediated adhesion. Nat. Rev. Mol. Cell Biol. 14, 503–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Han J., Rose D. M., Woodside D. G., Goldfinger L. E., Ginsberg M. H. (2003) Integrin α4β1-dependent T cell migration requires both phosphorylation and dephosphorylation of the α4 cytoplasmic domain to regulate reversible binding of paxillin. J. Biol. Chem. 278, 348–353 [DOI] [PubMed] [Google Scholar]
- 46. Deswal S., Meyer A., Fiala G. J., Eisenhardt A. E., Schmitt L. C., Salek M., Brummer T., Acuto O., Schamel W. W. (2013) Kidins220/ARMS associates with B-Raf and the TCR, promoting sustained ERK signaling in T cells. J. Immunol. 190, 1927–1935 [DOI] [PubMed] [Google Scholar]
- 47. White C. D., Erdemir H. H., Sacks D. B. (2012) IQGAP1 and its binding proteins control diverse cellular functions. Cell. Signal. 24, 826–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ren J. G., Li Z., Sacks D. B. (2008) IQGAP1 integrates Ca2+/calmodulin and B-Raf signaling. J. Biol. Chem. 283, 22972–22982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pritchard C. A., Hayes L., Wojnowski L., Zimmer A., Marais R. M., Norman J. C. (2004) B-Raf acts via the ROCKII/LIMK/cofilin pathway to maintain actin stress fibers in fibroblasts. Mol. Cell. Biol. 24, 5937–5952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Woodrow M. A., Woods D., Cherwinski H. M., Stokoe D., McMahon M. (2003) Ras-induced serine phosphorylation of the focal adhesion protein paxillin is mediated by the RAF → MEK → ERK pathway. Exp. Cell Res. 287, 325–338 [DOI] [PubMed] [Google Scholar]
- 51. Nakamura K., Yano H., Uchida H., Hashimoto S., Schaefer E., Sabe H. (2000) Tyrosine phosphorylation of paxillin α is involved in temporospatial regulation of paxillin-containing focal adhesion formation and F-actin organization in motile cells. J. Biol. Chem. 275, 27155–27164 [DOI] [PubMed] [Google Scholar]
- 52. Tsubouchi A., Sakakura J., Yagi R., Mazaki Y., Schaefer E., Yano H., Sabe H. (2002) Localized suppression of RhoA activity by Tyr31/118-phosphorylated paxillin in cell adhesion and migration. J. Cell Biol. 159, 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Romanova L. Y., Hashimoto S., Chay K. O., Blagosklonny M. V., Sabe H., Mushinski J. F. (2004) Phosphorylation of paxillin tyrosines 31 and 118 controls polarization and motility of lymphoid cells and is PMA-sensitive. J. Cell Sci. 117, 3759–3768 [DOI] [PubMed] [Google Scholar]
- 54. Janosch P., Kieser A., Eulitz M., Lovric J., Sauer G., Reichert M., Gounari F., Büscher D., Baccarini M., Mischak H., Kolch W. (2000) The Raf-1 kinase associates with vimentin kinases and regulates the structure of vimentin kinases. FASEB J. 14, 2008–2021 [DOI] [PubMed] [Google Scholar]
- 55. Broustas C. G., Grammatikakis N., Eto M., Dent P., Brautigan D. L., Kasid U. (2002) Phosphorylation of the myosin-binding subunit of myosin phosphatase by Raf-1 and inhibition of phosphatase activity. J. Biol. Chem. 277, 3053–3059 [DOI] [PubMed] [Google Scholar]
- 56. Ehrenreiter K., Piazzolla D., Velamoor V., Sobczak I., Small J. V., Takeda J., Leung T., Baccarini M. (2005) Raf-1 regulates Rho signaling and cell migration. J. Cell Biol. 168, 955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tran N. H., Wu X., Frost J. A. (2005) B-Raf and Raf-1 are regulated by distinct autoregulatory mechanisms. J. Biol. Chem. 280, 16244–16253 [DOI] [PubMed] [Google Scholar]
- 58. Mason C. S., Springer C. J., Cooper R. G., Superti-Furga G., Marshall C. J., Marais R. (1999) Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J. 18, 2137–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Davies H., Bignell G. R., Cox C., Stephens P., Edkins S., Clegg S., Teague J., Woffendin H., Garnett M. J., Bottomley W., Davis N., Dicks E., Ewing R., Floyd Y., Gray K., Hall S., Hawes R., Hughes J., Kosmidou V., Menzies A., Mould C., Parker A., Stevens C., Watt S., Hooper S., Wilson R., Jayatilake H., Gusterson B. A., Cooper C., Shipley J., Hargrave D., Pritchard-Jones K., Maitland N., Chenevix-Trench G., Riggins G. J., Bigner D. D., Palmieri G., Cossu A., Flanagan A., Nicholson A., Ho J. W., Leung S. Y., Yuen S. T., Weber B. L., Seigler H. F., Darrow T. L., Paterson H., Marais R., Marshall C. J., Wooster R., Stratton M. R., Futreal P. A. (2002) Mutations of the BRAF gene in human cancer. Nature 417, 949–954 [DOI] [PubMed] [Google Scholar]
- 60. Dhillon A. S., Hagan S., Rath O., Kolch W. (2007) MAP kinase signaling pathways in cancer. Oncogene 26, 3279–3290 [DOI] [PubMed] [Google Scholar]
- 61. Tiacci E., Trifonov V., Schiavoni G., Holmes A., Kern W., Martelli M. P., Pucciarini A., Bigerna B., Pacini R., Wells V. A., Sportoletti P., Pettirossi V., Mannucci R., Elliott O., Liso A., Ambrosetti A., Pulsoni A., Forconi F., Trentin L., Semenzato G., Inghirami G., Capponi M., Di Raimondo F., Patti C., Arcaini L., Musto P., Pileri S., Haferlach C., Schnittger S., Pizzolo G., Foà R., Farinelli L., Haferlach T., Pasqualucci L., Rabadan R., Falini B. (2011) BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 364, 2305–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Davidsson J., Lilljebjörn H., Panagopoulos I., Fioretos T., Johansson B. (2008) BRAF mutations are very rare in B- and T-cell pediatric acute lymphoblastic leukemias. Leukemia 22, 1619–1621 [DOI] [PubMed] [Google Scholar]
- 63. Eychène A., Dusanter-Fourt I., Barnier J. V., Papin C., Charon M., Gisselbrecht S., Calothy G. (1995) Expression and activation of B-Raf kinase isoforms in human and murine leukemia cell lines. Oncogene 10, 1159–1165 [PubMed] [Google Scholar]
- 64. Tsukamoto H., Irie A., Nishimura Y. (2004) B-Raf contributes to sustained extracellular signal-regulated kinase activation associated with interleukin-2 production stimulated through the T cell receptor. J. Biol. Chem. 279, 48457–48465 [DOI] [PubMed] [Google Scholar]
- 65. Tsukamoto H., Irie A., Chen Y. Z., Takeshita K., Kim J. R., Nishimura Y. (2006) TCR ligand avidity determines the mode of B-Raf/Raf-1/ERK activation leading to the activation of human CD4+ T cell clone. Eur. J. Immunol. 36, 1926–1937 [DOI] [PubMed] [Google Scholar]
- 66. Hindley A., Kolch W. (2007) Raf-1 and B-Raf promote protein kinase C θ interaction with BAD. Cell. Signal. 19, 547–555 [DOI] [PubMed] [Google Scholar]
- 67. Tsukamoto H., Irie A., Senju S., Hatzopoulos A. K., Wojnowski L., Nishimura Y. (2008) B-Raf-mediated signaling pathway regulates T cell development. Eur. J. Immunol. 38, 518–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lito P., Rosen N., Solit D. B. (2013) Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 19, 1401–1409 [DOI] [PubMed] [Google Scholar]
- 69. Johnson D. B., Sosman J. A. (2013) Update on the targeted therapy of melanoma. Curr. Treat. Options Oncol. 14, 280–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ibrahim N., Yu Y., Walsh W. R., Yang J. L. (2012) Molecular targeted therapies for cancer: sorafenib mono-therapy and its combination with other therapies (review). Oncol. Rep. 27, 1303–1311 [DOI] [PubMed] [Google Scholar]
- 71. Montagut C., Settleman J. (2009) Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 283, 125–134 [DOI] [PubMed] [Google Scholar]
- 72. Boni A., Cogdill A. P., Dang P., Udayakumar D., Njauw C. N., Sloss C. M., Ferrone C. R., Flaherty K. T., Lawrence D. P., Fisher D. E., Tsao H., Wargo J. A. (2010) Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 70, 5213–5219 [DOI] [PubMed] [Google Scholar]
- 73. Zhao W., Gu Y. H., Song R., Qu B. Q., Xu Q. (2008) Sorafenib inhibits activation of human peripheral blood T cells by targeting LCK phosphorylation. Leukemia 22, 1226–1233 [DOI] [PubMed] [Google Scholar]
- 74. Cabrera R., Ararat M., Xu Y., Brusko T., Wasserfall C., Atkinson M. A., Chang L. J., Liu C., Nelson D. R. (2013) Immune modulation of effector CD4+ and regulatory T cell function by sorafenib in patients with hepatocellular carcinoma. Cancer Immunol. Immunother. 62, 737–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Houben R., Voigt H., Noelke C., Hofmeister V., Becker J. C., Schrama D. (2009) MAPK-independent impairment of T-cell responses by the multikinase inhibitor sorafenib. Mol. Cancer Ther. 8, 433–440 [DOI] [PubMed] [Google Scholar]
- 76. Cao M., Xu Y., Youn J. I., Cabrera R., Zhang X., Gabrilovich D., Nelson D. R., Liu C. (2011) Kinase inhibitor sorafenib modulates immunosuppressive cell populations in a murine liver cancer model. Lab. Invest. 91, 598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Desar I. M., Jacobs J. H., Hulsbergen-vandeKaa C. A., Oyen W. J., Mulders P. F., van der Graaf W. T., Adema G. J., van Herpen C. M., de Vries I. J. (2011) Sorafenib reduces the percentage of tumor infiltrating regulatory T cells in renal cell carcinoma patients. Int J Cancer. 129, 507–512 [DOI] [PubMed] [Google Scholar]
- 78. Rose D. M., Han J., Ginsberg M. H. (2002) α4 integrins and the immune response. Immunol. Rev. 186, 118–124 [DOI] [PubMed] [Google Scholar]
- 79. Halaban R., Zhang W., Bacchiocchi A., Cheng E., Parisi F., Ariyan S., Krauthammer M., McCusker J. P., Kluger Y., Sznol M. (2010) PLX4032, a selective BRAF(V600E) inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAF melanoma cells. Pigment Cell Melanoma Res. 23, 190–200 [DOI] [PMC free article] [PubMed] [Google Scholar]