

Background: The pro-angiogenic endothelial fatty acid-binding protein 4 (FABP4) is regulated by VEGFA.
Results: VEGFA-induced FABP4 was dependent on Delta-like ligand4 (DLL4)-NOTCH signaling and FOXO1.
Conclusion: DLL4-NOTCH together with Foxo1 are key regulators of FABP4.
Significance: The results link NOTCH and angiogenic signaling directly to FABP4 induction and potentially to FABP4-mediated fatty acid metabolism and may provide a bypass mechanism for anti-VEGFA therapy.
Keywords: Angiogenesis, Endothelial Cell, Fatty Acid-binding Protein, FOXO, Metabolism, Notch Pathway, Vascular Endothelial Growth Factor (VEGF), DLL4
Abstract
Fatty acid-binding protein 4 (FABP4) is an adipogenic protein and is implicated in atherosclerosis, insulin resistance, and cancer. In endothelial cells, FABP4 is induced by VEGFA, and inhibition of FABP4 blocks most of the VEGFA effects. We investigated the DLL4-NOTCH-dependent regulation of FABP4 in human umbilical vein endothelial cells by gene/protein expression and interaction analyses following inhibitor treatment and RNA interference. We found that FABP4 is directly induced by NOTCH. Stimulation of NOTCH signaling with human recombinant DLL4 led to FABP4 induction, independently of VEGFA. FABP4 induction by VEGFA was reduced by blockade of DLL4 binding to NOTCH or inhibition of NOTCH signal transduction. Chromatin immunoprecipitation of the NOTCH intracellular domain showed increased binding to two specific regions in the FABP4 promoter. The induction of FABP4 gene expression was dependent on the transcription factor FOXO1, which was essential for basal expression of FABP4, and FABP4 up-regulation following stimulation of the VEGFA and/or the NOTCH pathway. Thus, we show that the DLL4-NOTCH pathway mediates endothelial FABP4 expression. This indicates that induction of the angiogenesis-restricting DLL4-NOTCH can have pro-angiogenic effects via this pathway. It also provides a link between DLL4-NOTCH and FOXO1-mediated regulation of endothelial gene transcription, and it shows that DLL4-NOTCH is a nodal point in the integration of pro-angiogenic and metabolic signaling in endothelial cells. This may be crucial for angiogenesis in the tumor environment.
Introduction
Fatty acid-binding protein 4 (FABP4) facilitates the compartmental distribution of fatty acids inside the cell. Processes regulated by FABP4 are various and not restricted to one cell type. They include nuclear translocation of fatty acids and regulation of transcription in adipocytes and macrophages, protein-protein interaction and regulation, as well as transcellular transport of fatty acids and feeding of adjacent tissues such as tumor tissue (1–3).
Increased plasma levels of FABP4 are positively associated with atherosclerosis, diabetes, and endothelial dysfunction (1, 4, 5). For instance, exogenous FABP4 interferes with insulin-stimulated production of nitric oxide in endothelial cells, thereby interfering with vasodilatory functions and demonstrating a causative role in the development of insulin resistance (6).
Endothelial FABP4 production is up-regulated in response to the angiogenic growth factor VEGFA (7). Gene expression studies suggest that FABP4 in response to VEGFA3 promotes integral signaling pathways such as p38, endothelial nitric-oxide synthase, and c-kit (8). Down-regulation of FABP4 reduces endothelial cell proliferation, migration, and sprouting in response to VEGFA. These are processes required for blood vessel formation (7, 8).
The Delta-like ligand (DLL)4-NOTCH signaling pathway limits the endothelial response to VEGFA and counteracts excessive blood vessel formation, for example in tumor tissue. VEGFA binding to the VEGF receptor 2 on endothelial cells leads to increased DLL4 on the cell surface. DLL4 is a ligand for the NOTCH receptor on adjacent cells. DLL4-NOTCH binding induces intracellular proteolytic cleavages of the NOTCH receptor by ADAM10 and ADAM17 and by presenilin/γ-secretase. NOTCH intracellular domain (NICD) is released and translocates to the nucleus to regulate gene transcription in a complex with other transcription factors such as the DNA binding and NOTCH-responsive recombination signal binding protein for immunoglobulin κJ region (RBPJκ).
Gene transcription effects of the NICD transcription factor complex include induction of Hairy/Enhancer of split (HES) and HES-related genes (HEY, CHF, HRT, and HESR), DLL4, and VEGF receptor 1 and suppression of VEGF receptor 2, which ultimately limits the response to VEGFA and amplifies NOTCH signaling across the endothelial cell layer. Therefore, the balance of expression of the VEGFA and/or NOTCH receptors and of the respective downstream signaling is a fine-tuning of the angiogenic response toward outside cues.
Increased DLL4-NOTCH signaling generates resistance to anti-VEGFA therapy in tumor xenografts (9) but is also indicated in the development of atherosclerosis, insulin resistance, and fat accumulation (10).
In this study, we show that DLL4-NOTCH directly regulates FABP4 gene expression, by binding of NICD to specific regions of the FABP4 promoter. The FABP4 response to VEGFA is dependent on the NOTCH pathway, as inhibition of DLL4 binding to NOTCH and inhibition of NOTCH cleavage leads to FABP4 reduction in response to VEGFA. Furthermore, DLL4-NOTCH-induced FABP4 is dependent on the insulin-responsive FOXO1 transcription factor, providing a nodal point for the integration of angiogenic and metabolic signaling.
EXPERIMENTAL PROCEDURES
Drugs
Drugs used were the γ-secretase inhibitor dibenzazepine (Sigma), the ADAM17 inhibitor INCB004298 (Incyte), the ADAM17/10 inhibitor INCB003619 (Incyte), and the AKT inhibitor X (AKTiX, Sigma). The DLL4 blocking antibody was from Genentech.
Cell Culture
Human umbilical vein endothelial cells (HUVECs) were cultured in endothelial basal medium 2 with supplements (EGM2) (Lonza) in incubators at 37 °C and atmosphere at 5% CO2. To activate DLL4-NOTCH signaling by recombinant human DLL4 (rhDLL4, R & D Systems), tissue cultureware was coated with 1 μg/ml rhDLL4 or 1 μg/ml BSA (Sigma) in 0.2% gelatin in PBS for 16 h at 4 °C. HUVECs were seeded on pre-warmed coated tissue cultureware and grown for 16–48 h. To stimulate HUVECs with VEGFA, HUVECS were cultured in endothelial basal medium with 2% FCS, without supplements, for 16 h prior to addition of VEGFA. Recombinant human VEGFA (Invitrogen) or BSA was added at 50 ng/ml, and HUVECs were grown for 24–48 h.
Western Blot Analysis
Western blot analysis was performed using the Novex® NuPAGE® SDS-PAGE gel system and semi-dry blotter (Invitrogen), according to the manufacturer's protocols. Band densitometry analysis was performed using the ImageQuant TL software (GE Healthcare).
RNA Isolation and cDNA Synthesis
RNA isolation was performed using the TRIzol method, according to the manufacturer's protocols. cDNA synthesis was performed using the High Capacity cDNA RT kit (Applied Biosystems), according to the manufacturer's protocols.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed using the EZ-ChIPTM-chromatin immunoprecipitation kit (Millipore) according to the manufacturer's protocol. Briefly, 2.5 × 106 cells were plated on BSA- or rhDLL4-coated 150-mm dishes, using two dishes per condition to obtain sufficient cell numbers, while remaining at 50–70% cell confluence to reduce cell-cell contact-induced Notch signaling. After 16 h in media with or without DBZ, cells were trypsinized and counted. For each condition, 5 × 106 cells were cross-linked in 1% paraformaldehyde for 10 min. Glycine was added to quench unreacted paraformaldehyde. Cells were washed in ice-cold PBS containing protease inhibitors and lysed in the supplied SDS-lysis buffer at 1 × 107 cells per ml. To shear cellular DNA into 200–1000 bp, samples were sonicated in 100-μl aliquots containing 1 × 106 cells, for 12 cycles of 30-s pulses at high power, using the Bioruptor Plus (Diagenode). An aliquot of each of the samples was taken and used as input for normalization purposes. Immunoprecipitation was performed overnight at 4 °C, using a NOTCH1 (D1E11) antibody (Cell Signaling Technology) dilution of 1:100 or rabbit IgG (Cell Signaling technology) as a negative control. Antibody-antigen-DNA complexes were collected with protein G-agarose and eluted using appropriate buffers as provided. After cross-link reversal and DNA purification, the samples were analyzed by quantitative real time PCR using the primers indicated below. Raw Ct values were analyzed using the percent input method. Ct values obtained from the ChIP samples were divided by Ct values obtained from the appropriate input samples (1% of the starting chromatin), using the formula 100 × 2^ ((Ct input − log2(100)) − Ct IP) to obtain percent input values (% input). The obtained percent input values were then divided by the percent input values of the control condition (BSAc with DMSO as vehicle control) of the respective gene promoter region to obtain fold changes.
Antibodies for Western Blot Analysis
Anti-DLL4 (rabbit), anti-phosphorylated AKT (serine 473, rabbit), anti-total AKT (rabbit), and anti-FOXO1 (rabbit) were purchased from Cell Signaling Technology. Anti-FABP4 (rabbit) was purchased from Sigma (Prestige). Anti-ADAM17 (rabbit) and anti-ADAM10 (rabbit) were purchased from ProSci Inc. and Abcam, respectively. Monoclonal anti-β-actin-peroxidase (Sigma) was used to control for protein loading. Anti-rabbit HRP (DAKO) was used as a secondary antibody.
Quantitative Real Time PCR and Primer Sequences
Quantitative real time PCR was performed using the SensiFASTTM SYBR No-ROX kit (Bioline) according to the manufacturer's protocols. Raw Ct values were analyzed using the 2(−ΔΔ Ct) method and normalized to the housekeeping gene ACTB. The normalized expression values of each gene of interest were then divided by the normalized gene of interest expression value of the respective control condition to obtain fold changes. Primer sequences used for analysis of mRNA expression were synthesized by Invitrogen and as follows: FABP4 (forward 5′-GCGAACTTCAGTCCAGGTCAAC-3′ and reverse 5′-ACGAGAGGATGATAAACTGGTGG-3′); DLL4 (forward 5′-CCCTGGCAATGTACTTGTGAT-3′ and reverse 5′-TGGTGGGTGCAGTAGTTGAG-3′); HEY1 (forward 5′-CGAGCTGGACGAGCCCAT-3′ and reverse 5′-GGAACCTAGAGCCGAACTCA-3′); PPARγ (forward 5′-GACAGGAAAGACAACAGACAAATC-3′ and reverse 5′-GGGGTGATGTGTTTGAACTTG-3′); FOXO1 (forward 5′-TGGACATGCTCAGCAGACATC-3′ and reverse 5′-CTTGGGTCAGGCGGTTCA-3′); and ACTB (forward 5′-GAGGAGGCACCGGTAAATG-3′ and reverse 5′-GTCACTCACTGGGACATAGGC-3′). Primer sequences used for detection of immunoprecipitated DNA sequences were synthesized by Invitrogen and as follows: DLL4 promoter (forward 5′-GACGCTTAGCTTGGCCTGGAGCTG-3′ and reverse 5′-TGTGTAAAATACAGGAAGGGGCCCGTCAGT-3′) (11); HEY1 promoter (forward 5′-AATTCAGCGGCGCGAGA-3′ and reverse 5′-CTCACGCTTTGCCTCTGGTTA-3′, from Ohnuki et al. (12)); FABP4 R-F1 (forward 5′-TTTTGCAGCAACATAGGTGAA-3′ and reverse 5′-ACCTTCCCACCTTTTGGAGT-3′); FABP4 R-2 (forward 5′-GAGACTCCAAAAGGTGGGAAG-3′ and reverse 5′-CTCCCAAAGTGCTGGGATTA-3′); and UBC (forward 5′-TTGCTGGCAAATATCAGACG-3′ and reverse 5′-GCAAGACCATCACCCTTGAG-3′).
RNA Interference
Transfection of siRNA duplexes was performed using Lipofectamine RNAiMax (Invitrogen) according to the manufacturer's protocols. ON-TARGETplus SMARTpool targeting FOXO1 (Dharmacon) was used, and the ON-TARGETplus nontargeting pool (Dharmacon) was used as a control. siRNAs were used at a final concentration of 20 nm.
Statistical Analysis
Results are expressed as the mean ± S.D. of at least three independent experiments. Statistical analysis was performed by analysis of variance with the Bonferroni post hoc test unless stated otherwise. p < 0.05 was considered statistically significant.
RESULTS
DLL4-NOTCH Signaling Is a Regulator of FABP4 mRNA and Protein
Preliminary analysis of mRNA expression arrays of immortalized human endothelial microvascular endothelial cells overexpressing DLL4 showed a 1.6-fold up-regulation of FABP4. We validated DLL4-NOTCH-dependent FABP4 expression in primary HUVECs. To induce NOTCH signaling, HUVECs were grown on plates coated with rhDLL4 protein (DLL4c) or BSA (BSAc) for 16–48 h and analyzed for mRNA and protein expression of FABP4. FABP4 mRNA expression was significantly induced by DLL4c stimulation after 16 h (Fig. 1A). FABP4 protein expression was significantly induced after 48 h (Fig. 1B, band densitometry analysis in Fig. 1C).
FIGURE 1.

DLL4-NOTCH signaling is a regulator of FABP4 mRNA and protein expression. HUVECs were grown on BSA- or DLL4-coated (BSAc or DLL4c) dishes for the indicated time points. Cells were harvested and analyzed for mRNA expression of FABP4 (A) or protein expression of DLL4 and FABP4 (B), and band densitometry analysis of FABP4 expression relative to the loading control in three independent experiments was performed (C). Values are expressed as mean ± S.D., *, p < 0.05; **, p < 0.01; ***, p < 0.001 (A). β-Actin was used as a loading control (B).
FABP4 Expression in Response to DLL4-NOTCH Is Down-regulated by γ-Secretase Inhibition and Oxygen Deprivation
Because FABP4 has been reported to be regulated by VEGFA (7), and oxygen deprivation strongly induces VEGFA signaling, we investigated FABP4 expression in HUVECs at 21, 1, or 0.1% oxygen, in response to DLL4-NOTCH stimulation. Additionally, to assess NOTCH-dependent regulation, HUVECs were treated with the γ-secretase inhibitor DBZ. Protein expression of DLL4 and FABP4 are shown in Fig. 2A. Band densitometry analysis of FABP4 relative to the loading control is shown in Fig. 2B. DLL4-NOTCH signaling in HUVECS stimulated with DLL4c up-regulated FABP4 at 21% and 1% oxygen. FABP4 in response to DLL4-NOTCH signaling was reduced at 1% oxygen and further reduced at 0.1% oxygen, compared with 21% oxygen (Fig. 2, A and B). DBZ reduced FABP4 induction in response to DLL4-NOTCH at 21 and 1%, indicating a DLL4-NOTCH-dependent regulation of FABP4 in normoxic and hypoxic conditions (Fig. 2, A and B).
FIGURE 2.
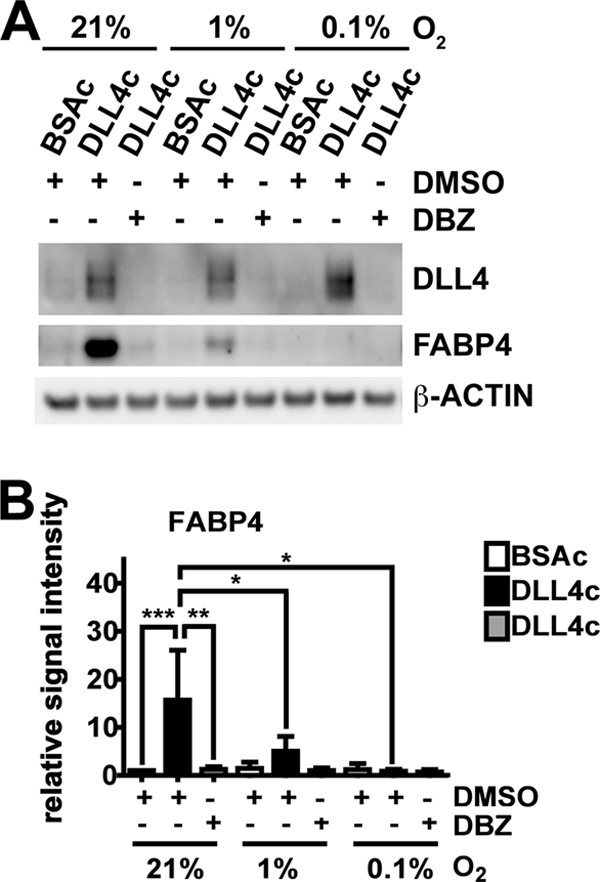
DLL4-NOTCH-induced FABP4 is down-regulated by NOTCH signaling inhibition and by oxygen deprivation. HUVECs were grown on BSA- or DLL4-coated (BSAc or DLL4c) dishes in 21% oxygen, 1% oxygen, or 0.1% oxygen. Cells were treated with DBZ to inhibit NOTCH cleavage or vehicle control (DMSO). Cells were harvested and analyzed for protein expression of DLL4 and FABP4 (A), and band densitometry analysis of FABP4 protein expression relative to the loading control of three independent experiments was performed (B). β-Actin was used as a loading control. Values are expressed as mean ± S.D., *, p < 0.05; **, p < 0.01; ***, p < 0.001 (B).
Inhibition of DLL4-NOTCH Binding by a DLL4 Blocking Antibody Reduces FABP4 Induction by VEGFA
Next, we investigated whether DLL4-NOTCH was required for FABP4 induction by VEGFA. To investigate the requirement of DLL4 binding to NOTCH for induction of FABP4, HUVECs were treated with a DLL4-blocking antibody (anti-DLL4) at 1 μg/ml to prevent DLL4 from binding to the NOTCH receptor in the presence of VEGFA or BSA. DLL4 and FABP4 mRNA induction in response to VEGFA was significantly down-regulated by treatment with anti-DLL4 (Fig. 3). These results indicate that induction of NOTCH signaling in response to VEGFA is dependent on DLL4-NOTCH binding and signal transduction and that FABP4 induction in response to VEGFA is dependent on DLL4-NOTCH signaling.
FIGURE 3.

Inhibition of DLL4-NOTCH binding by a DLL4 blocking antibody reduces FABP4 induction by VEGFA. HUVECs were growth factor-starved, stimulated with BSA or VEGFA (50 ng/ml), and treated with DLL4 blocking antibody (anti-DLL4) or IgG control. Cells were harvested and analyzed for mRNA expression of DLL4 and FABP4. Values are expressed as mean ± S.D., *, p < 0.05; **, p < 0.01; ***, p < 0.001.
VEGFA-induced FABP4 Is Down-regulated by γ-Secretase Inhibition
Based on the previous results indicating that FABP4 was dependent on DLL4-NOTCH, VEGFA-stimulated HUVECs were treated with DBZ to inhibit NOTCH cleavage, and FABP4 expression was monitored. FABP4 protein and mRNA were up-regulated by VEGFA. This up-regulation was significantly reduced by DBZ treatment, indicating that VEGFA-mediated FABP4 induction was through activation of DLL4-NOTCH signaling (Fig. 4, A and B). DLL4 protein expression in response to VEGFA was down-regulated by DBZ (Fig. 4B), and VEGFA-mediated up-regulation of DLL4 and HEY1 mRNA was sensitive to DBZ treatment (Fig. 4C). The previously reported DLL4-NOTCH target PPARγ, an inducer of FABP4, was not significantly up-regulated by VEGFA (Fig, 4C). Thus, VEGFA leads to DBZ-sensitive induction of canonical NOTCH targets and FABP4 mRNA and protein.
FIGURE 4.
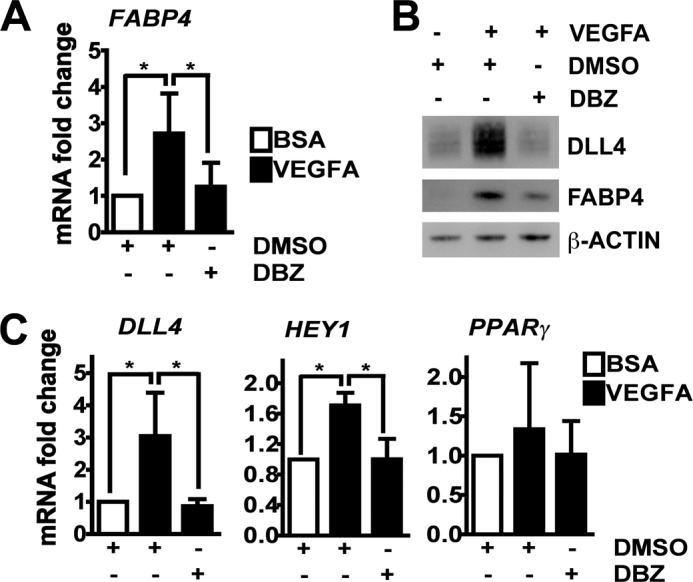
VEGFA-induced FABP4 is down-regulated by γ-secretase inhibition. HUVECs were growth factor-starved, stimulated with BSA or VEGFA (50 ng/ml), and treated with DBZ (10 nm) or vehicle control (DMSO). Cells were harvested and analyzed for mRNA expression of FABP4 (A), DLL4, HEY1, and PPARγ (C), or protein expression of DLL4 and FABP4 (B). β-Actin was used as a loading control (B). Values are expressed as mean ± S.D., *, p < 0.05 (A and C).
Inhibition of ADAM10 and ADAM17 Reduces FABP4 Induction by VEGFA
VEGFA has been reported to induce activation of ADAM10 and ADAM17 and thereby regulate the shedding of VEGF receptor 2, neuropilin1, and several other receptors (13). ADAM10 and ADAM17, required for NICD release following DLL4-Notch binding, are expressed as inactive enzyme precursors (premature form), and removal of the prodomains results in maturation or activation of the enzymes (14). To investigate whether the mechanism of VEGFA-mediated NOTCH signaling induction is related to increased activation of ADAM10 and/or ADAM17 and thus increased NOTCH cleavage, the premature versus mature forms of ADAM10 or ADAM17 were analyzed in HUVECs treated with VEGFA. VEGFA treatment did not lead to increased maturation of ADAM10 or ADAM17 (Fig. 5A), and band densitometry analysis did not show a significant change in the ratio of the premature (pADAM) and mature form (mADAM) of ADAM10 or ADAM17 (Fig. 5B). This indicated that increased activity of ADAM10 or ADAM17 is not responsible for increased NOTCH signaling in response to VEGFA.
FIGURE 5.

Inhibition of ADAM10 and ADAM17 reduces FABP4 induction by VEGFA. HUVECs were growth factor-starved, stimulated with BSA or VEGFA (50 ng/ml), and treated with DBZ (A and B), INCB4298 (10 μm, targets ADAM17) (C), INCB3619 (5 μm, targets ADAM17/10) (C), or vehicle control (DMSO) as indicated. Cells were harvested and analyzed for protein expression of ADAM10 and ADAM17 (A, premature form = pADAM and mature form = mADAM), band intensity (three independent experiments (B)), or analyzed for mRNA expression of DLL4 and FABP4 (C). β-Actin was used as a loading control (A). Values are expressed as mean ± S.D., *, p < 0.05; **, p < 0.01 (C).
Because basal activities of ADAM17 and ADAM10 are required for NICD release and transduction of NOTCH signaling, we tested whether VEGFA-induced FABP4 was dependent on ADAM17 and ADAM10 activity. For this, VEGFA-stimulated HUVECs were treated with the ADAM17 inhibitor INCB4298 or the ADAM17/10 inhibitor INCB3619. DLL4 and FABP4 mRNA expressions in response to VEGFA were significantly down-regulated by ADAM17 or ADAM17/10 inhibitor treatment (Fig. 5B). This shows that basal activities of ADAM10 and especially ADAM17 are required for NOTCH signaling and FABP4 induction in response to VEGFA.
AKT Inhibition Increases FABP4 Expression
Elmasri et al. (7) reported that FABP4 expression in response to VEGFA was dependent on the mammalian target of the rapamycin complex 1 (MTORC1) pathway, inhibition of which can inhibit a negative feedback loop to AKT kinase (15). Because FABP4 gene expression was repressed in gene expression microarrays of HUVECs overexpressing AKT (16), we hypothesized that the FABP4 expression was sensitive to changes of AKT activity. To investigate this hypothesis, HUVECs stimulated with DLL4c or BSAc were treated with AKT kinase inhibitor X (AKTiX) and analyzed for protein expression of FABP4 and the effect on AKT phosphorylation (Fig. 6A). Furthermore, band densitometry analysis was performed on FABP4, compared with the loading control, and on phosphorylated AKT, compared with total levels of AKT (Fig. 6B). Statistical analysis showed an overall significant change of AKT phosphorylation, and FABP4 expression, in response to the treatments (Friedman test p < 0.05 for phospho-AKT, p < 0.01 for FABP4, and Dunn's post hoc test not significant). AKTiX treatment leads to a consistent decrease of AKT phosphorylation in BSAc-treated and DLL4c-treated HUVEC. We observed that DLL4c treatment itself leads to a slight decrease of AKT phosphorylation. AKTiX treatment leads to a consistent increase of FABP4 expression compared with DMSO in BSAc- or in DLL4c-stimulated cells. These results indicate that the regulation of FABP4 is dependent on the phosphorylation status of AKT and that increased AKT phosphorylation and activity are possible mechanisms of FABP4 repression.
FIGURE 6.

AKT inhibition increases FABP4 expression. HUVECs were grown on BSA- or DLL4-coated (BSAc or DLL4c) dishes and treated with AKT inhibitor X (AKTiX) or vehicle control (DMSO). Cells were harvested and analyzed for protein expression of DLL4, AKT, and FABP4 and for the serine 473 phosphorylation status of AKT (pAKT). β-Actin was used as a loading control (A), and band densitometry analysis of three independent experiments was performed (B). Statistical analysis was performed by Friedman test for nonparametric data (pAKT p < 0.05; FABP4 p < 0.01).
Regulation of FABP4 Downstream of VEGFA or Direct DLL4-NOTCH Activation Is Dependent on FOXO1
Activation of AKT signaling leads to phosphorylation of the forkhead transcription factor FOXO1 and renders FOXO1 inactive by trapping it in the cytoplasm (17). Gene expression microarray studies in HUVECs indicated that AKT and FOXO1 regulate FABP4 in a reciprocal manner (16), and FABP4 was shown to be regulated by FOXO1 in macrophages (18). Therefore, we hypothesized that FOXO1 may be required for FABP4 transcription in HUVECs. We investigated FABP4 expression in BSA- or VEGFA-stimulated HUVEC (A) or in BSAc- or DLL4c-stimulated HUVEC (B and C), treated with control or FOXO1 targeting RNAi (Fig. 7). RNAi of FOXO1 led to >50% reduction of FOXO1 mRNA in BSA-, VEGFA-, BSAc-, or DLL4c-stimulated cells (Fig. 7, A and B). FABP4 mRNA levels were significantly reduced by FOXO1 suppression to less than 20% in cells stimulated with BSA or VEGFA and BSAc or DLL4c (Fig. 7, A and B). FABP4 protein levels in response to DLL4c were repressed by FOXO1 RNAi (Fig. 7C).
FIGURE 7.

Regulation of FABP4 is dependent on FOXO1. HUVECs were growth factor-starved and stimulated with BSA or VEGFA (50 ng/ml) (A) or grown on BSA- or DLL4-coated (BSAc or DLL4c) (B and C) and were transfected with siRNA targeting FOXO1 (siFOXO1) or scrambled siRNA (control = ctr). Cells were harvested after 48 h and analyzed for mRNA expression of FOXO1 and FABP4 (A and B) and protein expression of DLL4, FOXO1 and FABP4 (C). β-Actin was used as a loading control (C). Values are expressed as mean ± S.D., *, p < 0.05; ***, p < 0.001 (A and B).
NICD Binds to Specific FABP4 Promoter Regions Adjacent to the FOXO1-binding Motif
In hepatic cells, there is evidence for a NOTCH-FOXO1 co-dependent mechanism of gene transcription (19, 20). Our results showed that FOXO1 is required for FABP4 expression in response to DLL4-NOTCH in HUVECs. Therefore, we investigated the following: 1) whether the FABP4 gene promoter contains binding sites for the RBPJκ-NICD complex in proximity to FOXO1-binding sites, and 2) whether NICD associates with RBPJκ-binding sites to promote FABP4 gene transcription.
In the NICD transcription factor complex, NICD indirectly binds to DNA by associating with the co-factor RBPJκ. Therefore, we searched the promoter region of FABP4 for RBPJκ- and FOXO1-binding sites. The motifs were derived from Castel et al. and Fan et al. (21, 22), respectively. The FABP4 gene promoter sequence (gi|42516792) was searched for RBPJκ- and FOXO1-binding motifs and their variations. As highlighted in Fig. 8B, an RBPJκ-binding motif was present at −2097 bp, directly adjacent to a FOXO1-binding motif (in red). We referred to this region as RBPJκ (R)-FOXO1 (F)-1 (R-F-1). Furthermore, there were RBPJκ-binding motifs at −1935 bp (three adjacent repeats, referred to as R-2) and −1284 bp (three repeats, separated by several base pairs, referred to as R-3A, R-3B, and R-3C) (Fig. 8A).
FIGURE 8.

NICD binds to specific FABP4 promoter regions adjacent to the FOXO1-binding motif. Location of RBPJκ-binding motifs (underlined, R-F-1, R-2, R-3A, R-3B, and R-3C) or adjacent RBPJκ- and FOXO1-binding motifs (R-F-1, FOXO1-binding motif in red) in the FABP4 promoter sequence are shown (A). HUVECs were grown on BSA- or DLL4-coated (BSAc or DLL4c) dishes for 16 h and were treated with DBA or vehicle control (DMSO). Cells were fixed and analyzed for NICD binding to the predicted binding sites R-F-1 and R2, known binding sites in DLL4 and HEY1 promoters, and a region in UBC promoter by chromatin immunoprecipitation (ChIP) (B). Values are expressed relative to percent input of the control condition (BSAc and DMSO) and as mean ± S.D., *, p < 0.05; **, p < 0.01 (B). Statistical analysis was performed by Friedman test for nonparametric data, and Dunn's post hoc test.
We then investigated the regions R-F-1 and R-2 in more detail, because of their proximity to the FOXO1-binding motif. To control for the induction of NOTCH target genes in our assay, we also analyzed known RBPJκ binding regions in the promoters of the NOTCH target genes DLL4 and HEY1 as positive controls and a region in the promoter of the UBC gene, which is not regulated by NOTCH, as a negative control.
To analyze the association of NICD to the predicted RBPJκ-binding sites, ChIP was performed in HUVECs stimulated with DLL4c or BSAc for 16 h and treated with or without DBZ or DMSO to inhibit NICD release and induction of NOTCH signaling. An antibody targeting NOTCH1 was used to detect NICD. According to the manufacturer's instructions, the antibody used here recognizes intracellular epitopes between 2400 and 2500 amino acids of human NOTCH1, and it does not detect the extracellular domain of NOTCH1 once it is cleaved by ADAM-like proteases. Rabbit IgG was used as a negative control.
NICD IP and rabbit IgG IP results are shown in one graph for each promoter region of interest (Fig. 8B). The ChIP results of the regions of interests R-F-1 and R-2 are shown in the two upper panels. The ChIP results of the Notch target genes DLL4 and HEY1 are shown in Fig. 8B, middle two panels, and the results of the UBC gene are shown in Fig. 8B, lower panel.
ChIP of NICD showed increased binding to R-F-1 and R-2 in response to DLL4c stimulation, compared with BSAc, which was significantly reduced by DBZ treatment (Fig. 8B, upper panels). ChIP of NICD also showed increased binding to DLL4 and HEY1 promoter regions in DLL4c-stimulated HUVECs compared with BSAc, which was reduced by DBZ treatment (Fig. 8B, middle panels). No increased binding of NICD to the UBC promoter in response to DLL4c stimulation was observed (Fig. 8B, lower panel). The IP with rabbit IgG showed no significant binding to any of the analyzed regions in response to DLL4c stimulation.
These findings show that NICD associates with RBPJκ-binding motifs in the FABP4 gene promoter, which are directly adjacent to a FOXO1-binding motif. These findings further indicate that FABP4 transcription is induced by NICD release and its association with the FABP4 promoter via RBPJκ and that FABP4 is a target gene of the DLL4-NOTCH signaling pathway.
DISCUSSION
Previous studies by Elmasri et al. (7) have shown that FABP4 is induced by VEGFA and suggested an MTORC1-dependent regulatory axis. However, we have found that there are additional regulatory mechanisms of FABP4 induction. VEGFA acts indirectly by inducing DLL4, which then activates NOTCH signaling and initiates FABP4 gene transcription. FABP4 expression is sensitive to AKT activity. We suggest that an increase of AKT activity renders the transcription factor FOXO1 in the cytoplasm, preventing it from promoting FABP4 gene transcription. A proposed model of FABP4 regulation in endothelial cells is shown in Fig. 9.
FIGURE 9.

Regulation and mechanism of FABP4 induction in endothelial cells is dependent on DLL4-NOTCH signaling and FOXO1. VEGFA induces DLL4 expression in endothelial cells, thereby promoting the paracrine activation of NOTCH signaling in adjacent cells and inducing indirect binding of NICD via a transcription factor complex to promoter regions of the FABP4 gene, to induce FABP4 transcription. Under basal levels of AKT activity, FOXO1 is present in the nucleus and promotes DLL4-NOTCH-dependent FABP4 transcription. AKT activation induces FOXO1 phosphorylation and cytoplasmic translocation and diminishes FABP4 transcription. AKT inhibition leads to FOXO1 being trapped in the nucleus and further promoting FABP4 transcription.
Inhibition of each step required for NOTCH signaling activation led to reduced induction of FABP4 expression in response to VEGFA. Additionally, inhibition of intracellular cleavage also led to reduction of NICD binding to two specific regions in the FABP4 promoter, consequently suggesting that DLL4-NOTCH signaling is a direct promoter of FABP4 gene transcription. Basal levels and VEGFA or DLL4-NOTCH-stimulated FABP4 were suppressed by FOXO1 knockdown. Thus, we identify that FOXO1 is crucial for maintaining FABP4 expression. This suggests a NOTCH and FOXO1 co-dependent regulation of FABP4.
FABP4 indirectly affects gene transcription by delivering free fatty acids to the nucleus, which serve as ligands for peroxisome proliferator-activated receptors PPARs and regulate gene transcription (1). FABP4 also delivers fatty acids to other compartments of the cell such as the mitochondria for further utilization. Nieman et al. (3) have shown a role of FABP4 in omental ovarian cancer metastasis. FABP4 was required for increased fatty acid oxidation in ovarian cancer cells adjacent to the omentum and up-regulated in the adjacent adipocytes. FABP4−/− mice injected with ovarian cancer cells had a smaller tumor burden with decreased microvessel density (3). Additionally, high FABP4 expression has been described in several cancers (23, 24), indicating therapeutic relevance.
Elmasri et al. (7) suggested an MTORC1-p70S6 kinase-dependent positive regulation of FABP4. However, MTORC1-p70S6 kinase inhibition triggers a negative feedback mechanism, resulting in the activation of AKT signaling via IGF-1R/PI3K signaling (25), and also the activation of ERK (15). Up-regulation of FOXO1-dependent gene transcription is a candidate mechanism for resistance to therapy targeting AKT and ERK in cancer (26). In our study, AKT inhibition led to up-regulation of FABP4, and FABP4 was suppressed by FOXO1 knockdown. Thus, FABP4 expression induced by DLL4-NOTCH can be modulated by AKT, MTORC1, and ERK signaling pathways, which is important to consider in combination therapies.
It has been shown that FABP4 mediates VEGFA-dependent pro-angiogenic effects. FABP4 suppression inhibited VEGFA-induced proliferation, migration, and inhibited sprouting in the aortic ring model. Furthermore, it affected endothelial gene transcription, for example of stem cell factor/c-kit and endothelial nitric-oxide synthase (7, 8). Our findings show that, paradoxically, a VEGFA target with pro-angiogenic features is downstream of angiogenesis-restricting NOTCH signaling. Therefore, up-regulation of FABP4 by increased DLL4-NOTCH and the resulting FABP4 pro-angiogenic effects may overcome effects of VEGFA inhibition in tumor tissue, and therefore result in therapy resistance. Endothelial FABP4 down-regulation in hypoxia may indicate that endothelial FABP4 function becomes significant in (re-)oxygenated areas, when growing tumor blood vessels become perfused and need maintenance. This stresses why a combination of DLL4 blockade and VEGFA inhibition may have additional therapeutic effects in tumor angiogenesis by blocking endothelial cell proliferation and differentiation.
FOXO1 and NOTCH have been shown to regulate hepatic gene transcription of gluconeogenic genes in a co-dependent manner, and both FOXO1- and RBPJκ-binding sites were required for the induction of target genes. This study also linked NOTCH directly to insulin resistance (19). Furthermore, atherosclerosis has been linked to aberrant NOTCH signaling and is responsive to therapeutic DLL4 blockade in vivo (10). With regard to direct endothelial cell function, DLL4-NOTCH signaling restricts angiogenesis, and its up-regulation is a possible mechanism of resistance to anti-VEGFA therapy in tumors. The mechanism involves decreased cell proliferation, increased cell differentiation, and increased blood vessel size and perfusion, which can be reversed by treatment with a DLL4-blocking antibody (9). FOXO1 limits migratory and sprouting angiogenesis (27).
Our study suggests that DLL4-NOTCH and FOXO1 co-regulate gene transcription of FABP4 in endothelial cells. FABP4 regulation by NOTCH is a candidate mechanism for NOTCH-dependent effects in metabolic disease such as insulin resistance, atherosclerosis, as well as tumor angiogenesis. It will be exciting to investigate the consequence of NOTCH-dependent mechanisms of FABP4 induction for fatty acid metabolism in tumor endothelial cells. As recent findings have established that endothelial glucose metabolism is critical for sprouting angiogenesis (28), it is of relevance to investigate whether NOTCH and FOXO1 are co-regulators of endothelial gene transcription and fatty acid metabolism and whether that is a main pathway for controlling sprouting angiogenesis in development and disease.
In summary we show that FABP4 induction by VEGFA is through a DLL4-NOTCH-dependent mechanism and may be modulated by AKT pathways via FOXO1. It is possible that there are other genes in the VEGFA transcriptome dependent on this DLL4 feed-forward loop. Furthermore, FOXO1 and NOTCH co-dependent gene regulation, in response to DLL4-NOTCH signal transduction, may be a general mechanism of gene transcription in endothelial cells, which embeds environmental and metabolic cues into the angiogenic process. DLL4-NOTCH-mediated regulation of FABP4 may be integral for its implications in cancer, angiogenesis, and metabolic disease. Furthermore, FABP4 up-regulation should be considered in mechanisms of resistance to anti-angiogenic tumor therapy, because of its pro-angiogenic features in VEGFA abundant environments.
This work was supported by Cancer Research United Kingdom, Breast Cancer Research Foundation, and National Health Service Biomedical Research Centre.
- VEGFA
- vascular endothelial growth factor A
- HUVEC
- human umbilical vein endothelial cell
- NICD
- NOTCH intracellular domain
- DBZ
- dibenzazepine
- rhDLL4
- recombinant human DLL4
- PPAR
- peroxisome proliferator-activated receptor
- IP
- immunoprecipitation.
REFERENCES
- 1. Kralisch S., Fasshauer M. (2013) Adipocyte fatty acid-binding protein: a novel adipokine involved in the pathogenesis of metabolic and vascular disease? Diabetologia 56, 10–21 [DOI] [PubMed] [Google Scholar]
- 2. Jenkins-Kruchten A. E., Bennaars-Eiden A., Ross J. R., Shen W. J., Kraemer F. B., Bernlohr D. A. (2003) Fatty acid-binding protein-hormone-sensitive lipase interaction. Fatty acid dependence on binding. J. Biol. Chem. 278, 47636–47643 [DOI] [PubMed] [Google Scholar]
- 3. Nieman K. M., Kenny H. A., Penicka C. V., Ladanyi A., Buell-Gutbrod R., Zillhardt M. R., Romero I. L., Carey M. S., Mills G. B., Hotamisligil G. S., Yamada S. D., Peter M. E., Gwin K., Lengyel E. (2011) Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 17, 1498–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Djoussé L., Gaziano J. M. (2012) Plasma levels of FABP4, but not FABP3, are associated with increased risk of diabetes. Lipids 47, 757–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Holm S., Ueland T., Dahl T. B., Michelsen A. E., Skjelland M., Russell D., Nymo S. H., Krohg-Sørensen K., Clausen O. P., Atar D., Januzzi J. L., Aukrust P., Jensen J. K., Halvorsen B. (2011) Fatty acid-binding protein 4 is associated with carotid atherosclerosis and outcome in patients with acute ischemic stroke. PLoS One 6, e28785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aragonès G., Saavedra P., Heras M., Cabré A., Girona J., Masana L. (2012) Fatty acid-binding protein 4 impairs the insulin-dependent nitric oxide pathway in vascular endothelial cells. Cardiovasc. Diabetol. 11, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Elmasri H., Karaaslan C., Teper Y., Ghelfi E., Weng M., Ince T. A., Kozakewich H., Bischoff J., Cataltepe S. (2009) Fatty acid-binding protein 4 is a target of VEGF and a regulator of cell proliferation in endothelial cells. FASEB J. 23, 3865–3873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Elmasri H., Ghelfi E., Yu C. W., Traphagen S., Cernadas M., Cao H., Shi G. P., Plutzky J., Sahin M., Hotamisligil G., Cataltepe S. (2012) Endothelial cell-fatty acid-binding protein 4 promotes angiogenesis: role of stem cell factor/c-kit pathway. Angiogenesis 15, 457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li J. L., Sainson R. C., Oon C. E., Turley H., Leek R., Sheldon H., Bridges E., Shi W., Snell C., Bowden E. T., Wu H., Chowdhury P. S., Russell A. J., Montgomery C. P., Poulsom R., Harris A. L. (2011) DLL4-Notch signaling mediates tumor resistance to anti-VEGF therapy in vivo. Cancer Res. 71, 6073–6083 [DOI] [PubMed] [Google Scholar]
- 10. Fukuda D., Aikawa E., Swirski F. K., Novobrantseva T. I., Kotelianski V., Gorgun C. Z., Chudnovskiy A., Yamazaki H., Croce K., Weissleder R., Aster J. C., Hotamisligil G. S., Yagita H., Aikawa M. (2012) Notch ligand δ-like 4 blockade attenuates atherosclerosis and metabolic disorders. Proc. Natl. Acad. Sci. U.S.A. 109, E1868–E1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hayashi H., Kume T. (2008) Foxc transcription factors directly regulate Dll4 and Hey2 expression by interacting with the VEGF-Notch signaling pathways in endothelial cells. PLoS One 3, e2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ohnuki H., Inoue H., Takemori N., Nakayama H., Sakaue T., Fukuda S., Miwa D., Nishiwaki E., Hatano M., Tokuhisa T., Endo Y., Nose M., Higashiyama S. (2012) BAZF, a novel component of cullin3-based E3 ligase complex, mediates VEGFR and Notch cross-signaling in angiogenesis. Blood 119, 2688–2698 [DOI] [PubMed] [Google Scholar]
- 13. Swendeman S., Mendelson K., Weskamp G., Horiuchi K., Deutsch U., Scherle P., Hooper A., Rafii S., Blobel C. P. (2008) VEGF-A stimulates ADAM17-dependent shedding of VEGFR2 and crosstalk between VEGFR2 and ERK signaling. Circ. Res. 103, 916–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Endres K., Anders A., Kojro E., Gilbert S., Fahrenholz F., Postina R. (2003) Tumor necrosis factor-α converting enzyme is processed by proprotein-convertases to its mature form which is degraded upon phorbol ester stimulation. Eur. J. Biochem. 270, 2386–2393 [DOI] [PubMed] [Google Scholar]
- 15. Chen X. G., Liu F., Song X. F., Wang Z. H., Dong Z. Q., Hu Z. Q., Lan R. Z., Guan W., Zhou T. G., Xu X. M., Lei H., Ye Z. Q., Peng E. J., Du L. H., Zhuang Q. Y. (2010) Rapamycin regulates Akt and ERK phosphorylation through mTORC1 and mTORC2 signaling pathways. Mol. Carcinog. 49, 603–610 [DOI] [PubMed] [Google Scholar]
- 16. Daly C., Wong V., Burova E., Wei Y., Zabski S., Griffiths J., Lai K. M., Lin H. C., Ioffe E., Yancopoulos G. D., Rudge J. S. (2004) Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1). Genes Dev. 18, 1060–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brunet A., Bonni A., Zigmond M. J., Lin M. Z., Juo P., Hu L. S., Anderson M. J., Arden K. C., Blenis J., Greenberg M. E. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868 [DOI] [PubMed] [Google Scholar]
- 18. Nakae J., Cao Y., Oki M., Orba Y., Sawa H., Kiyonari H., Iskandar K., Suga K., Lombes M., Hayashi Y. (2008) Forkhead transcription factor FoxO1 in adipose tissue regulates energy storage and expenditure. Diabetes 57, 563–576 [DOI] [PubMed] [Google Scholar]
- 19. Pajvani U. B., Shawber C. J., Samuel V. T., Birkenfeld A. L., Shulman G. I., Kitajewski J., Accili D. (2011) Inhibition of Notch signaling ameliorates insulin resistance in a FoxO1-dependent manner. Nat. Med. 17, 961–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kitamura T., Kitamura Y. I., Funahashi Y., Shawber C. J., Castrillon D. H., Kollipara R., DePinho R. A., Kitajewski J., Accili D. (2007) A Foxo/Notch pathway controls myogenic differentiation and fiber type specification. J. Clin. Invest. 117, 2477–2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Castel D., Mourikis P., Bartels S. J., Brinkman A. B., Tajbakhsh S., Stunnenberg H. G. (2013) Dynamic binding of RBPJ is determined by Notch signaling status. Genes Dev. 27, 1059–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fan W., Morinaga H., Kim J. J., Bae E., Spann N. J., Heinz S., Glass C. K., Olefsky J. M. (2010) FoxO1 regulates Tlr4 inflammatory pathway signalling in macrophages. EMBO J. 29, 4223–4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cataltepe O., Arikan M. C., Ghelfi E., Karaaslan C., Ozsurekci Y., Dresser K., Li Y., Smith T. W., Cataltepe S. (2012) Fatty acid-binding protein 4 is expressed in distinct endothelial and non-endothelial cell populations in glioblastoma. Neuropathol. Appl. Neurobiol. 38, 400–410 [DOI] [PubMed] [Google Scholar]
- 24. Boiteux G., Lascombe I., Roche E., Plissonnier M. L., Clairotte A., Bittard H., Fauconnet S. (2009) A-FABP, a candidate progression marker of human transitional cell carcinoma of the bladder, is differentially regulated by PPAR in urothelial cancer cells. Int. J. Cancer 124, 1820–1828 [DOI] [PubMed] [Google Scholar]
- 25. Wan X., Harkavy B., Shen N., Grohar P., Helman L. J. (2007) Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 26, 1932–1940 [DOI] [PubMed] [Google Scholar]
- 26. Roy S. K., Srivastava R. K., Shankar S. (2010) Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of FOXO transcription factor, leading to cell cycle arrest and apoptosis in pancreatic cancer. J. Mol. Signal. 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Potente M., Urbich C., Sasaki K., Hofmann W. K., Heeschen C., Aicher A., Kollipara R., DePinho R. A., Zeiher A. M., Dimmeler S. (2005) Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J. Clin. Invest. 115, 2382–2392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De Bock K., Georgiadou M., Schoors S., Kuchnio A., Wong B. W., Cantelmo A. R., Quaegebeur A., Ghesquière B., Cauwenberghs S., Eelen G., Phng L. K., Betz I., Tembuyser B., Brepoels K., Welti J., Geudens I., Segura I., Cruys B., Bifari F., Decimo I., Blanco R., Wyns S., Vangindertael J., Rocha S., Collins R. T., Munck S., Daelemans D., Imamura H., Devlieger R., Rider M., Van Veldhoven P. P., Schuit F., Bartrons R., Hofkens J., Fraisl P., Telang S., Deberardinis R. J., Schoonjans L., Vinckier S., Chesney J., Gerhardt H., Dewerchin M., Carmeliet P. (2013) Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154, 651–663 [DOI] [PubMed] [Google Scholar]