

Background: Adenoviruses use the short non-coding transcript VA RNAI to inhibit host antiviral kinase PKR.
Results: VA RNAI contains a pH- and Mg2+-sensitive tertiary structure that, unexpectedly, is not required for PKR inhibition.
Conclusion: Structural requirements for an RNA inhibitor of PKR are simpler than appreciated previously.
Significance: These findings explain how non-coding RNAs of varied sequence and structure can efficiently inhibit PKR.
Keywords: Adenovirus, Isothermal Titration Calorimetry (ITC), Mutagenesis, Protein Kinase RNA-activated (PKR), RNA Structure, RNA-Protein Interaction, Translation Control, SHAPE Structure Probing, UV Melting
Abstract
Virus-associated RNA I (VA RNAI) is a short (∼160-nucleotide) non-coding RNA transcript employed by adenoviruses to subvert the innate immune system protein double-stranded RNA-activated protein kinase (PKR). The central domain of VA RNAI is proposed to contain a complex tertiary structure that contributes to its optimal inhibitory activity against PKR. Here we use a combination of VA RNAI mutagenesis, structural analyses, as well as PKR activity and binding assays to dissect this tertiary structure and assess its functional role. Our results support the existence of a pH- and Mg2+-dependent tertiary structure involving pseudoknot formation within the central domain. Unexpectedly, this structure appears to play no direct role in PKR inhibition. Deletion of central domain sequences within a minimal but fully active construct lacking the tertiary structure reveals a crucial role in PKR binding and inhibition for nucleotides in the 5′ half of the central domain. Deletion of the central domain 3′ half also significantly impacts activity but appears to arise indirectly by reducing its capacity to assist in optimally presenting the 5′ half sequence. Collectively, our results identify regions of VA RNAI critical for PKR inhibition and reveal that the requirements for an effective RNA inhibitor of PKR are simpler than appreciated previously.
Introduction
The double-stranded RNA-activated protein kinase (PKR)3 is a central component of innate cellular immunity. Upon binding double-stranded RNA (dsRNA) produced as a consequence of viral replication, PKR initiates the shutdown of general translation, blocking synthesis of viral proteins. As a countermeasure to this activity, adenoviruses produce copious quantities (∼108/cell) of an ∼160-nucleotide, non-coding transcript VA RNAI that binds to PKR and prevents it from exerting its antiviral effect (1, 2).
In addition to its well established role as a proviral, non-coding RNA, VA RNAI is a substrate of Dicer and, therefore, an effective inhibitor of the RNAi system as its high cellular concentration results in saturation of the RNA-induced silencing complex (3, 4). Specific regulation of host genes by VA RNAI-derived microRNAs has been proposed (5, 6), but a precise functional role in adenoviral replication has not yet been established (7). The remaining portion of the Dicer-processed VA RNAI is a truncated form of the transcript, lacking the viral microRNA sequences at both the 5′ and 3′ ends, that retains full activity against PKR (8). Paradoxically, full-length VA RNAI activates a second protein of the innate immune response, 2′-5′-oligoadenylate synthetase 1 (OAS1), leading to downstream activation of RNaseL and degradation of viral and cellular RNAs (9). Recently, however, Dicer processing has been shown to largely reverse this stimulatory activity because the truncated form of VA RNAI acts as a pseudoinhibitor of OAS1 (10). Therefore, a single VA RNAI transcript possesses multiple activities. It can inhibit two antiviral proteins (PKR and OAS1), potentially regulate gene expression via the cellular RNAi system, and saturate the RNA-processing enzyme Dicer.
VA RNAI molecules from different adenovirus serotypes vary considerably at the sequence level (11). Although differences in the details of their secondary structures also likely exist, all possess three major structural domains: apical stem, central domain, and terminal stem (11, 12). The apical stem serves as the primary binding site for the two N-terminal dsRNA binding domains of PKR, whereas the central domain is believed to be responsible for inhibition of the kinase (13–20). However, identification of specific central domain sequence(s) or structure(s) that form the direct contacts with PKR required to impart this activity has remained elusive. Despite this, the notion persists that the central domain folds into a complex tertiary structure and that its placement adjacent to the primary PKR binding site in the apical stem confers upon VA RNAI its inhibitory activity against PKR.
On the basis of a wealth of prior mutagenesis studies, RNA structure probing, and functional analyses, a model for the central domain was proposed for adenovirus type 2 VA RNAI in which the two loops within the central domain interact to form a pseudoknot structure (Fig. 1A, loops connected by a dashed line) (21). At the center of this pseudoknot structure in the helical junction of the central domain are two highly conserved and base-paired tetranucleotide sequences that appear critical in both their sequence and orientation for optimum VA RNAI activity (Fig. 1A, nucleotides in outline font) (11, 12, 21). We have shown that the stability of the central domain structure is also highly sensitive to solution pH, suggesting the involvement of a protonated base-base tertiary interaction (8). Finally, Mg2+ and PKR have been shown to induce similar structural rearrangements in the central domain (14), and Mg2+ appears to fine-tune the mode and affinity of the PKR-VA RNAI interaction (22). Together, these features lend strong support to the presence of a tertiary structure within the central domain.
FIGURE 1.

The VA RNAI central domain contains putative tertiary interactions that are stabilized by low pH, Mg2+, and PKR. A, TSΔ21, the truncated form of adenovirus type 2 VA RNAI, used as the starting point in this study. This RNA construct lacks all terminal stem nucleotides (TSΔ21) and has a six-nucleotide deletion in the apical stem (A2dl2). The proposed pseudoknot interaction between loop II and loop III (dashed lines) and conserved complementary tetranucleotide sequences (outline font) are highlighted. B, UV melting analysis of VA RNAI with combined TSΔ21 and A2dl2 deletions. Melting profiles depict the first derivative of the UV melting curve (inset) at pH 5.5 (red) and pH 7.5 (black). Apparent transitions (“peaks”) in the melting profile correspond to unfolding of structures within the VA RNAI apical stem (partly visible at >90 °C) and central domain (three at pH 7.5 and two at pH 5.5 over the range of 20–80 °C) are indicated. Of the central domain transitions, the first is assigned to the putative RNA tertiary structure and is stabilized dramatically at low pH (rightward shift of profile in red dashed line). Arrowheads denote temperatures used for SHAPE probing of this RNA. The first apparent transition is also stabilized by both Mg2+ ion (C) and PKR (D). The concentration of divalent ion or molar equivalents (eq.) of PKR included in the experiment is indicated.
Our initial objectives in this study were to address two important remaining questions regarding the VA RNAI central domain. What is the nature of its tertiary structure, and what role does it play in PKR inhibition? The analyses presented here reveal the defining features of the central domain tertiary structure but demonstrate that it is not required for activity against PKR. Instead, inhibition requires only a subset of the secondary structural elements of the central domain. Therefore, the requirements for optimal inhibitory activity against PKR are far simpler than appreciated previously, offering a satisfying explanation for the consistently observed inhibition of PKR by VA RNAI molecules from different adenovirus serotypes despite their significant differences at the sequence and secondary structural levels.
EXPERIMENTAL PROCEDURES
RNA in Vitro Transcription
The RNA construct used as the starting point for these studies (Fig. 1A) possesses both a deleted terminal stem, previously termed TSΔ21 RNA (8), and a deletion in the apical stem (A2dl2) that removes conformational heterogeneity in this region (23). This RNA and its derivatives generated in this study are expected to form a 1:1 complex with PKR (8). All new RNAs prepared here contain the A2dl2 mutation, but for brevity they are named only for their terminal deletion or point mutation(s) made within the TSΔ21-A2dl2 context.
New templates were created by direct ligation of 5′ end phosphorylated double-stranded DNA into a plasmid created previously for the transcription of target RNAs with a 3′ end-fused hepatitis delta virus (3′-HDV) ribozyme (24, 25). RNAs were produced by runoff in vitro transcription using T7 RNA polymerase under optimal conditions for VA RNAI (26), purified by preparative denaturing PAGE, and recovered as described previously (8, 27). TSΔ21 RNA was also transcribed without the fused 3′-HDV ribozyme by insertion of an additional guanosine immediately after the VA RNAI coding sequence, creating a new NaeI restriction enzyme site for plasmid linearization at the authentic 3′ end of the TSΔ21 encoding sequence (GCC ↓ GGC, the inserted G is underlined). No difference in gel mobility, melting profile, or PKR-inhibitory activity was observed for RNA produced in this manner (data not shown). The A123U construct was made exclusively with the NaeI runoff site. RNAs for SHAPE and Tb3+ probing were generated by the same direct ligation approach but using a modified plasmid preloaded with 5′ and 3′ flanking sequences encoding a “structure cassette” (25, 28).
PKR Expression and Purification
PKR was expressed from plasmid pET-PKR/PPase (29) as described previously (8). Protein was expressed in Escherichia coli Rosetta 2 (DE3) and purified sequentially using heparin affinity, poly(I)·poly(C) RNA affinity, and gel filtration chromatographies on an ÄKTA Purifier10 system (GE Healthcare). Protein from the gel filtration column was obtained in 20 mm HEPES buffer (pH 7.5) containing 150 mm NaCl, 0.1 mm EDTA, 10 mm β-mercaptoethanol, and 10% (v/v) glycerol.
RNA UV Melting Analysis
UV melting curves at 260 and 280 nm were collected on a Cary 400 UV-visible spectrophotometer (Varian) with a six-cell multichanger and an in-cell temperature probe placed in a buffer-only sample. Samples contained 20–25 μg of RNA in a solution of 50 mm KCl and 10 mm MOPS buffer (pH 7.5), or 10 mm MES buffer (pH 5.5). Experiments with Mg2+ were performed in the pH 7.5 buffer solution with addition of 0.01–5.0 mm MgCl2. Experiments with PKR were performed using RNA in the pH 7.5 buffer solution mixed with an equal volume of PKR in gel filtration buffer (to give 0.5, 1, and 2 molar equivalents of protein). To facilitate comparison between different RNAs or solution conditions, the UV melting data are presented as “melting profiles” corresponding to the first derivative of the UV absorbance curve.
Selective 2′-Hydroxyl Acylation Analyzed by Primer Extension (SHAPE) Probing
RNA was annealed in 0.5× Tris-EDTA (TE) buffer at 65 °C for 10 min, cooled to room temperature over 10 min, and then placed on ice. Samples were examined by 50% (w/v) urea denaturing and native PAGE to evaluate their purity and conformational homogeneity, respectively. SHAPE RNA probing with N-methylisatoic anhydride (NMIA) was carried out as described previously (28) but with the following modifications. Reactions were initiated by addition of 1 μl of 10× NMIA (100 mm in dimethyl sulfoxide) or 1 μl of dimethyl sulfoxide for controls without NMIA and allowed to proceed for five NMIA hydrolysis half-lives, i.e. 0.5, 10, 30, and 240 min at 85, 50, 40, and 20 °C, respectively (28). Reverse transcription was performed with Superscript III reverse transcriptase (Invitrogen) using a 5′ end γ32P-labeled DNA primer (0.12 μm final concentration) corresponding to the sequence of the 3′ flanking region of the structure cassette (25) in a total volume of 10 μl in 100 mm HEPES buffer (pH 8.0) containing 100 mm NaCl. Reactions were stopped by addition of 35 μl of a 4:25 (v/v) mixture of 1 m unbuffered Tris-HCl and gel loading solution (85% formamide, 0.5× TBE (pH 8.0) and 50 mm EDTA with trace bromphenol blue and xylene cyanol dyes) and heating to 95 °C for 5 min. A 3-μl aliquot of each RT reaction was resolved on an 8% acrylamide, 50% (w/v) urea, 1× TBE PAGE sequencing gel. Gels were imaged using a Typhoon Trio PhosphorImager (GE Healthcare) and analyzed using ImageQuant software. Background subtraction, band normalization, and generation of cutoff values for no (<5.5%), low (5.5–11%), medium (11–22%), and high (>22%) reactivity were done as described previously (30).
SHAPE probing reactions with PKR were performed in 30 μl of total volume and contained 1 μl of protein in gel filtration column buffer (giving a final PKR:RNA molar ratio of 2:1). Equivalent reactions without PKR were also performed in the same way but using 1 μl of gel filtration column buffer with no protein. After initiation by addition of 3 μl of 10× NMIA (or dimethyl sulfoxide for controls) and incubation at 20 °C, reactions were diluted by addition of 200 μl of 0.5× TE and extracted twice with two volumes of premixed phenol:chloroform:isoamyl alcohol (25:24:1) and once with two volumes of chloroform. The RNA was recovered by ethanol precipitation in the presence of glycogen (20 μg/ml) at −80 °C for 30 min, resuspended in 10 μl of 0.5× TE, and subjected to RT analysis as described above.
Tb3+ RNA Probing
Tb3+ probing reactions were performed in a total volume of 10 μl in solution containing 100 mm HEPES buffer (pH 8.0) and 100 mm NaCl. TSΔ21 RNA (100 nm) was incubated with TbCl3 (0–0.25 mm) at 37 °C for 15 min. RNA in each probing reaction was ethanol-precipitated, recovered, and analyzed by RT as described for SHAPE probing.
PKR Activity Assays
Assays of PKR activation contained PKR (0.1 μg), alone or with eIF2α (0.25 μg), in 50 mm Tris buffer (pH 7.8) containing poly(I)·poly(C) dsRNA (0–100 μg/ml), 20 μm ATP, 10 μCi [γ32P]ATP (10 mCi/ml, 6000 Ci/mmol), 50 mm KCl, 2 mm MgCl2, 2.5 mm DTT, and 10% glycerol in a total reaction volume of 10 μl. Reactions were incubated at 25 °C for 10 min and then stopped by addition of gel loading dye (for SDS-PAGE analysis) or 400 μl of ice-cold phosphate-buffered saline containing 200 μm ATP (for slot blot analysis). In the latter case, quenched samples were applied promptly to a Bio-Dot SF (Bio-Rad) microfiltration system, and proteins were bound to a nitrocellulose membrane under vacuum. The membrane was washed to remove unreacted [γ32P]ATP and then air-dried for 15 min. For both membranes and dried SDS-PAGE gels, the extent of PKR and eIF2α phosphorylation was determined by exposure to a phosphor storage screen and analysis using a Typhoon FLA 7000 PhosphorImager and ImageQuant software (GE Healthcare).
PKR inhibition assays were performed in a similar manner but included a 10-min preincubation step at 25 °C with the VA RNAI variant (0–10 μm) and PKR (0.1 μg), either alone or with eIF2α (0.25 μg), in 50 mm Tris buffer (pH 7.8) containing 50 mm KCl, 2.5 mm DTT, and 10% glycerol. Reactions (10-μl final volume) were initiated by addition of 0.04 μg/ml poly(I)·poly(C) dsRNA, 20 μm ATP, 10 μCi [γ32P]ATP (10 mCi/ml, 6000 Ci/mmol), and 2 mm MgCl2. After a 10-min incubation at 25 °C, the reactions were quenched, and the products were quantified as described above for the activation assays.
Isothermal Titration Calorimetry (ITC)
Binding affinities for mutant VA RNAI-PKR interaction were measured using an Auto-iTC200 microcalorimeter (GE Healthcare). Protein and RNA samples were individually dialyzed against 50 mm Tris buffer (pH 7.5) containing 100 mm NaCl. PKR (60 μm, syringe) was titrated into RNA (6 μm, sample cell) at 25 °C in 16 × 2.5 μl injections with 150-s spacing. Titration curves were fit by a non-linear least-squares method in MicroCal Origin software using the single site binding model.
RESULTS AND DISCUSSION
A Putative pH-dependent VA RNAI Central Domain Tertiary Structure Is Stabilized by Mg2+ and PKR
A new VA RNAI variant was created (Fig. 1A) that combined two previously characterized mutations: a 21 base pair truncation of the terminal stem (TSΔ21) that mirrors that produced by Dicer processing in vivo (8) and a six-nucleotide deletion in the apical stem (A2dl2) (31). These independent mutations are within functionally separate domains, and each retains the full inhibitory activity of wild-type VA RNAI (8, 18). The global folding of this new RNA was assessed by UV melting analysis (Fig. 1B), and the melting profiles obtained were found to be entirely consistent with previous studies of the individual deletions (8, 23). First, the region of the melting profile corresponding to the central domain is essentially identical to that observed for VA RNAI with only the TSΔ21 deletion (8). Second, the A2dl2 mutation stabilizes the apical stem, significantly increasing its apparent Tm (to >90 °C) relative to the wild-type sequence (apparent Tm ∼86 °C). Third, the absence of any additional effects on other regions of the profile resulting from the A2dl2 mutation corroborates our previous finding that the apical stem and central domain structures and stabilities are completely independent (27). This new construct, henceforth termed TSΔ21 for brevity (see “Experimental Procedures”), is ideal for detailed analysis of the central domain structure because the stabilizing effect of the A2dl2 mutation fully resolves the apparent transitions corresponding to the central domain and apical stem (Fig. 1B).
Our previous analysis (8) showed that the TSΔ21 central domain unfolds in at least two overlapping apparent transitions between 30 °C and 80 °C. Here a low temperature starting point of ∼15 °C allowed us to identify an additional broad apparent unfolding transition centered on ∼30 °C that is present only at pH 7.5 (Fig. 1B). The dramatic stabilization of the central domain structure at low pH (8) shifts this apparent transition to a significantly higher Tm at pH 5.5. We hypothesized that this newly identified apparent transition corresponds to the unfolding of the putative central domain tertiary structure. The dramatic rightward shift and coupling of this and the second apparent transition (centered on ∼45 °C at pH 7.5) into a single apparent transition (centered on ∼57 °C at pH 5.5) is consistent with a low-stability (apparent Tm ∼30 °C) tertiary structure at pH 7.5 that is stabilized via a protonated tertiary contact. Through the largely hierarchical nature of RNA folding, this stabilized tertiary structure additionally stabilizes the central domain secondary structure components from which it is formed.
Previous VA RNAI structure probing studies in the presence of Mg2+ or PKR have shown that both induce similar structural changes in the central domain (14). More recently, the interaction between PKR and VA RNAI has been shown to be sensitive to the presence of Mg2+ (22). Having identified a new apparent transition that potentially corresponds to the RNA tertiary structure, we next examined the effect of Mg2+ and PKR on the RNA melting profile. As expected, addition of Mg2+ stabilizes all elements of the VA RNAI structure, corresponding to a rightward shift in all apparent transitions (Fig. 1C). The first apparent transition is, however, especially sensitive to addition of Mg2+, with a significant shift in apparent Tm at only 10–25 μm and complete coupling of the first and second apparent transitions at 0.1–0.2 mm and higher. Melting experiments with PKR are complicated by precipitation of the protein at ∼50 °C but, again, indicate that the putative tertiary structure, corresponding to the broad first apparent transition, is stabilized by PKR (Fig. 1D).
The behavior of this newly identified, first low-temperature apparent transition in the melting profile of TSΔ21 RNA at low pH and in the presence of Mg2+ is consistent with it representing the unfolding of an RNA tertiary structure. Therefore, from these various lines of evidence, we conclude that the central domain does, as proposed previously, contain a set of tertiary interactions, including a protonated base, and that its stability is influenced by specific binding of one or more Mg2+ or PKR. This conclusion is further corroborated by the results of the SHAPE and mutagenesis experiments described below.
SHAPE Analysis of the VA RNAI Central Domain Tertiary Structure
To identify the regions of the central domain involved in forming its tertiary structure, we used SHAPE to probe nucleotide flexibility in TSΔ21 RNA at two temperatures that define the lower (20 °C) and upper (40 °C) boundaries of the tertiary structure unfolding transition identified in the melting experiments (Fig. 1B, black arrowheads). These conditions should, therefore, reflect nucleotide flexibility in the presence and absence of the folded tertiary structure.
At 20 °C, most nucleotides predicted to be base-paired display low SHAPE reactivity, as expected (Fig. 2, A and B). The only exceptions are nucleotides bordering loops I and III and those at the 3′ end (nts 129–131). Most notably, a much lower reactivity than expected was observed for the majority of nucleotides in loops II and III (nts 104–107 and 123–127), indicating lower nucleotide flexibility and, therefore, involvement in interactions beyond those depicted in the secondary structure model. In contrast, nts 115–117 of the 3′ side of loop I display the high reactivity expected for non-base-paired nucleotides. At 40 °C, reactivity of these loop II and III nucleotides increases substantially, along with a general increase in reactivity of paired and loop I nucleotides in the 3′ half of the central domain (nts 111–131, Fig. 2B, inset). The low reactivity of the loop II and III nucleotides and their concomitant increase in reactivity at a temperature above the identified tertiary structure unfolding event support the conclusion that these partially complementary nucleotides base-pair directly to form a pseudoknot structure within the central domain.
FIGURE 2.

Identification of tertiary interactions and Mg2+ binding sites within the VA RNAI central domain. A, representative gel showing SHAPE probing of TSΔ21 RNA below (20 °C) and above (40 °C) the apparent transition corresponding to the central domain tertiary structure unfolding (marked with arrowheads in Fig. 1B). Lanes with (+) and without (−) SHAPE reagent (NMIA) and RT reactions with dideoxynucleotides (U, A, C, G) for RNA sequencing are indicated. VA RNAI nucleotide numbering is shown on the left, and bands corresponding to central domain loops I-III, the apical stem (AS) loop, and full-length product (FL) are indicated on the right. B, quantification of SHAPE reactivity at 20 (top panel) and 40 °C (bottom panel) and changes between these temperatures (center panel). Normalized band intensities were averaged from three sets of reactions and categorized according to the reactivity keys shown. C, Tb3+ probing of the VA RNAI central domain with sites of RNA cleavage enhanced by increasing concentrations of Tb3+ (0–0.25 mm) indicated (right), and shown on the secondary structure map in B (center panel). Sequencing lanes (Seq) contain the complementary dideoxynucleotide. FL*, full-length product with contrast reduced 5×.
Role of Mg2+ and Protonation in Central Domain Tertiary Structure Formation
To further explore the role of Mg2+ in the formation of the central domain tertiary structure, we used Tb3+ probing, a method applied previously to both the mapping of ion binding sites and monitoring RNA folding (32). TSΔ21 RNA was incubated with a range of TbCl3 concentrations, and the induced strand cleavages were examined using reverse transcription. Two major sites of strand scission were identified, A123 and G97 (Fig. 2, B and C). Each site was present at the lowest Tb3+ concentration used and increased consistently in intensity with increasing Tb3+ concentration. These results point to the presence of at least one Mg2+ binding site in the proximity of loop III and, unexpectedly, a base-paired nucleotide in the middle of the central domain stem-loop structure. A third, weaker strand scission is also noted at A103 within loop II that may indicate the presence of an additional Mg2+ site or that this nucleotide is in proximity to the ion located at G97/A123.
Next, we exploited the strong influence of solution pH on central domain stability to further dissect the features critical for formation of its tertiary structure. We hypothesized that pH dependence arises through specific protonation of at least one central domain nucleotide involved in a direct tertiary contact. Further, mutation of this nucleotide should result in loss of pH dependence and, therefore, identical RNA melting profiles at pH 5.5 and pH 7.5. The pKa of cytosine N3 is ∼4.5 in the free nucleotide, making this site on one or more of the single-stranded cytosine residues of the central domain the most likely candidate for the origin of its pH sensitivity. Each of these residues, C104, C105, C107, and C116, was mutated individually to U, and the effect on pH dependence in the RNA melting profile was assessed (Fig. 3, A and B). Notably, none of the individual mutations eliminated the strong stabilization of the tertiary structure in the pH 5.5 melting profile, indicating that none of these residues harbors the putative site of protonation. However, the role of loop II nucleotides in forming the central domain tertiary structure is further corroborated by these mutations. First, the broad low temperature apparent transition corresponding to the tertiary structure at pH 7.5 is absent in C104U, C105U, and C107U RNAs (Fig. 3B, compare black and blue solid lines between 20–40 °C). Second, although folding of the central domain in each of these RNAs is driven by protonation at pH 5.5 (rightward shift of the first apparent transition), the melting profiles show some change in apparent Tm, transition shape, and/or hypochromicity, indicative of additional subtle structural alterations. The C116U mutant is distinct in that it appears to retain the tertiary fold, but a component of the secondary structure is destabilized at pH 7.5. In contrast, at lower pH, where central domain folding is driven by the base protonation, the melting profile of this mutant is identical to that of the parent TSΔ21 construct.
FIGURE 3.

A123 is the protonated nucleotide within the VA RNAI central domain. A, central domain secondary structure highlighting the four individual single-stranded C-to-U mutants (blue circles), mutation of all three loop II C nucleotides to U (3U RNA, green), and the A123-to-U point mutant (red circle). B and C, UV melting profiles of C104U, C105U, C107U, C116U (blue), A123U (red), and 3U RNA (green). The melting profile for each RNA is shown at pH 7.5 (solid line) and pH 5.5 (dashed line) and compared with those of TSΔ21 RNA (black). The region corresponding to unfolding of the TSΔ21 RNA pH-dependent tertiary structure at pH 7.5 is marked with a horizontal bar. D, the cis Watson-Crick/Hoogsteen A+·G base pair proposed for A123+·G97, highlighting the available base edges for potential additional interactions as discussed in the text.
Nucleotide A123 was implicated by our Tb3+ probing experiments in Mg2+ binding and, like the adjacent nucleotides involved in the central domain pseudoknot structure, displays lower than expected SHAPE reactivity. We therefore mutated A123 to U (A123U RNA) to determine whether it harbors the protonation site (adenine N1, pKa ∼3.8). The melting profile for A123U RNA at pH 7.5 lacks the broad, low-temperature apparent transition corresponding to the tertiary structure (Fig. 3C) but was otherwise unchanged, aside from a small increase in hypochromicity in the highest Tm (∼65 °C) apparent transition. At pH 5.5, the profile was nearly identical to that observed at pH 7.5, with only small changes in hypochromicity of each apparent transition and a small decrease in apparent Tm for the highest Tm (∼65 °C) apparent transition (Fig. 3C). Critically, neither pH-dependent stabilization nor the reappearance of an apparent transition corresponding to the tertiary unfolding was observed. We conclude that A123 is the protonated base within the central domain critical for the formation of its tertiary structure.
Though none of the individual mutations in loop II (C104U, C105U, and C107U) resulted in melting profiles indicative of the loss of tertiary structure, a result similar to that observed for A123U was obtained when all three cytosines of loop II were mutated to uridines (3U RNA, Fig. 3C). We interpret this as being a more significant disruption of the loop II-loop III pseudoknot tertiary interaction such that A123 protonation-driven tertiary folding cannot compensate as it does for an individual C-to-U mutation at each location.
Protonated adenine bases have been observed in non-Watson-Crick base pairs with either guanine or cytidine partners in RNA crystal structures. For the VA RNAI central domain, the latter possibility is largely eliminated because we demonstrated the existence of a pseudoknot structure in which C104 and C105 are base-paired with G124 and G125, and the mutation of each single-stranded C residue does not affect the structural pH dependence. Formation of the pseudoknot structure between loops II and III potentially creates a G·A pair between G106 and A123. However, although the G106A mutation resulted in an expected loss of the broad tertiary structure unfolding at pH 7.5, it did not eliminate the central domain folding dependence upon pH (data not shown). This result does not exclude an interaction between G106 and A123 as part of the pseudoknot but shows that it is not dependent upon pH. Although G97 is base-paired with C111 in the secondary model, our Tb3+ probing data implicate this guanine residue, together with A123, in coordinating a Mg2+ within the central domain. Of the known base pairings in RNA, we speculate that a parallel strand orientation, cis Watson-Crick/Hoogsteen A123+·G97 base pair (family 3 (33), Fig. 3D) would meet the requirement for a protonated A123 interaction within the central domain structure without the need for strand reversal. This pairing would also leave open the Watson-Crick face of G97 for base pairing with C111 and the Hoogsteen face of A123 for (non-protonated) interaction with G106 as part of the loop II-loop III pseudoknot. Although a high-resolution structure will be required to fully resolve the interactions of A123 in VA RNAI, such A+·G base pairs are found within several RNA structures in the PDB (Protein Data Bank) (34), including both eukaryotic and prokaryotic ribosomal subunits (35, 36), where they are engaged in complex tertiary structures stabilized by Mg2+.
Minimum Sequence Requirements for Central Domain Tertiary Structure Formation
Having defined several critical features of the VA RNAI central domain structure, including the formation of a pseudoknot interaction, we sought to determine the minimal sequence required to form the tertiary structure using additional deletion mutants. The first, TSΔ25 RNA, lacks nucleotides at both the 5′ and 3′ ends (nts 32–36 and 123–131, respectively) compared with the original TSΔ21 construct, corresponding to loop III and the terminal base-paired nucleotides (Fig. 4). As might be expected, this substantial deletion destabilizes central domain folding, resulting in a profile for TSΔ25 RNA with significantly lowered hypochromicity and broad, flat features in which only the highest Tm apparent transition (∼65 °C) of the central domain appears largely unaffected (Fig. 4, top panel). Critically, the TSΔ25 RNA melting profiles at pH 7.5 and 5.5 are identical, demonstrating that the central domain folding in this RNA is pH-independent and, therefore, that the tertiary structure interactions are absent. This was expected because the TSΔ25 mutation removes the protonated base A123 and all loop III nucleotides involved in forming the pseudoknot structure.
FIGURE 4.
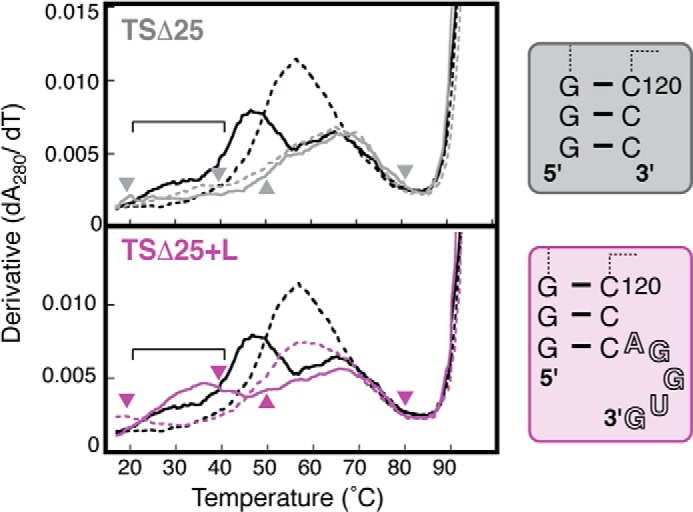
Loop III nucleotides 123–127 are sufficient to form the central domain pseudoknot structure. Melting profiles of TSΔ25+L (purple) and TSΔ25 (gray) RNAs shown at pH 7.5 (solid line) and pH 5.5 (dashed line) and compared with those of TSΔ21 RNA (black). The region corresponding to unfolding of the pH-dependent tertiary structure present in TSΔ25+L RNA, but absent in TSΔ25 RNA, is marked with a horizontal bar. Arrowheads on each profile denote temperatures used for SHAPE probing of the corresponding RNA. Loop III nucleotides are shown in outline font in the structure of TSΔ25+L RNA (right panel).
A second mutant, TSΔ25+L RNA, was created that also lacked the terminal base-paired nucleotides deleted in TSΔ25 RNA but retained the five residues of loop III (nts 123–127) as a 3′ end single-stranded extension (Fig. 4). The melting profile of TSΔ25+L RNA is similarly altered with broad, flat features. However, in sharp contrast to TSΔ25, it retains a clear additional broad apparent transition centered on ∼35 °C at pH 7.5. Further, TSΔ25+L RNA also retains a strong dependence upon pH, with a profile at pH 5.5 identical to that of TSΔ21 RNA but with a lowered hypochromicity that correlates with its reduction in length and secondary structural content.
To verify that this apparent restoration of tertiary structure and its associated pH dependence is due to specific interaction between nucleotides of loops II and III, we created a final mutant, TSΔ25xL RNA. This RNA possesses an identical secondary structure to that of TSΔ25+L but with mutation of the 3′ single-stranded nucleotides corresponding to loop III to the sequence UUGCG. The melting profile of this RNA was identical to that observed for TSΔ25 (data not shown), indicating that no interaction occurs between the loop II and the mutated loop III nucleotides. These results show that the terminal base-paired nucleotides absent in TSΔ25 RNA play no role in tertiary structure formation and that the specific sequence of loop III is necessary and sufficient to form the central domain pseudoknot tertiary interaction.
Conformational Heterogeneity within the Mutant VA RNAI Central Domain
The substantial changes in melting profile between TSΔ21 and the TSΔ25/TSΔ25+L constructs prompted us to explore the central domain secondary structures of these new RNAs using SHAPE. As before, each RNA structure was probed at pH 7.5 at temperatures corresponding to boundaries of apparent transitions for central domain unfolding (Fig. 4, arrowheads). Reactions were performed at 20 °C and 50 °C to identify changes corresponding to the broad lower temperature apparent transition and at 85 °C to identify additional changes that occur upon complete central domain unfolding (Fig. 5). Each RNA was also probed at 40 °C to allow direct comparison to the equivalent experiments with TSΔ21 RNA (probed at 20 and 40 °C, Fig. 2).
FIGURE 5.

SHAPE analysis of TSΔ25+L and TSΔ25 RNAs. A, representative gel showing SHAPE probing of TSΔ25+L RNA at temperatures below (20 °C), between (40 and 50 °C), and above (85 °C) the apparent transitions corresponding to the unfolding of the central domain structure. Lanes with (+) and without (−) SHAPE reagent (NMIA) and RT reactions with dideoxynucleotides (U, A, C, and G) for RNA sequencing are indicated. VA RNAI nucleotide numbering is shown on the left, and bands corresponding to central domain loops I-III, the apical stem (AS) loop, and full-length product (FL) are indicated on the right. B, quantification of SHAPE reactivity from low to high temperature (top to bottom) with relative changes shown between each pair of temperatures (dashed boxes). Normalized band intensities were averaged from three sets of reactions and categorized according to the reactivity key shown. C, nucleotide flexibility at 20 °C mapped on the nucleotide sequence of TSΔ25+L RNA in the currently established central domain secondary structure (top panel) and redrawn on the basis of observed SHAPE reactivities. D, E, and F, as A–C for TSΔ25 RNA.
The temperature-dependent SHAPE reactivities of nucleotides in loop II (nts 105–107) and the 3′ side of loop III (nts 124–128) in TSΔ25+L RNA are very similar to those observed for TSΔ21 RNA. These two regions show a lower than expected reactivity with NMIA at 20 °C and a concomitant increase in reactivity at 50 °C, where the tertiary, but not secondary structures, are unfolded (Fig. 5, A and B). This suggests that these nucleotides maintain tertiary pairing interactions that, in conjunction with a protonated A123, participate in the formation of the central domain tertiary structure of TSΔ25+L RNA. In further support of this, in TSΔ25 RNA, where the 3′ end nucleotides of loop III are deleted, the reactivity of loop II nucleotides is high, even at 20 °C (Fig. 5, D and E).
Although the loop II-loop III pseudoknot interaction appears to be maintained in TSΔ25+L RNA, other substantial changes in nucleotide flexibility are observed. Both RNAs lack the exceptionally strong reactivity of loop I (nts 115–117) in TSΔ21 RNA. Additionally, a much higher than expected SHAPE reactivity is observed for the 3′ half of the central domain stem loop (nts 108–114), even at the lowest temperature. This implies that, in both TSΔ25 and TSΔ25+L RNAs, these nucleotides are not base-paired with nts 94–99, suggesting that the changes in secondary structure of these RNAs are more complex than a simple erasure of nucleotides from the model of TSΔ21 (Fig. 5, C and F, top panels). Therefore, we revised the secondary structure models of these RNAs to reflect our experimentally determined nucleotide flexibilities (Fig. 5, C and F, bottom panels).
Because TSΔ25+L RNA appears to retain only some elements of the central domain structure, we next asked whether PKR could directly influence its structure. We hypothesized that PKR binding to TSΔ25+L RNA might result in reorganization of the central domain to resemble that of TSΔ21 RNA so that both RNAs ultimately use the same structure to inhibit PKR. This, however, was not the case because we found the structure of TSΔ25+L RNA determined by SHAPE to be identical in the absence or presence of PKR (data not shown). We conclude that TSΔ25+L and TSΔ25 RNAs adopt a central domain secondary structure, distinct from TSΔ21 RNA, that is maintained when bound by PKR and that TSΔ25+L RNA contains tertiary interactions that are built on this altered scaffold.
The VA RNAI Central Domain Tertiary Structure Is Not Required for PKR Inhibition
TSΔ21 RNA is an effective inhibitor of PKR despite lacking the entire terminal stem of wild-type VA RNAI (8). Additionally, recent evidence demonstrates that the central domain acts in concert with the apical stem to initiate Mg2+-dependent, high-affinity PKR binding (37) in addition to its established role in inhibition. Having established the minimal context in which the VA RNAI central domain adopts a pH-dependent tertiary structure (TSΔ25+L RNA) and a small deletion that eliminates it (TSΔ25 RNA), we next used these RNAs to directly test the role of the central domain tertiary structure in PKR inhibition.
To assess the activity of each RNA, we developed a PKR inhibition assay using a slot blot apparatus analogous to the established SDS-PAGE analysis, allowing for higher throughput (see “Experimental Procedures”). This new assay was first validated by comparative analysis of PKR activation (autophosphorylation) by poly(I)·poly(C) dsRNA and inhibition by TSΔ21 RNA between SDS-PAGE and the slot blot apparatus (Fig. 6, A and B). For both activation and inhibition, results obtained by each method were essentially identical (Fig. 6, C and D). Additionally, we confirmed that inhibition of eIF2α substrate phosphorylation by TSΔ21 RNA correlates well with PKR inhibition (Fig. 6, A–D), solidifying our ability to link in vitro PKR activation to eIF2α modification and shutdown of general translation. As expected, with its intact pH-dependent tertiary structure, TSΔ25+L RNA inhibited PKR almost as well as TSΔ21 (Fig. 6E). Surprisingly, however, TSΔ25 RNA, which lacks the pH-dependent tertiary structure, inhibited PKR equally well. To test the possibility that the altered secondary structure of these two RNAs might be responsible for this unexpected activity, we tested the A123U mutant, which lacks the pH-dependent central domain tertiary structure within the TSΔ21 context. We found that A123U RNA also retains full inhibitory activity against PKR, comparable with TSΔ21 (Fig. 6E). This result excludes the central domain structural rearrangements as the origin of the activity of these RNAs in the absence of tertiary structure. Correlating with the comparable activity of these RNAs against PKR, we found that PKR binding affinity was not altered significantly by the presence or absence of the tertiary structure (Kd 28–41 nm, Fig. 6F).
FIGURE 6.

The intact VA RNAI central domain tertiary structure is not required for PKR inhibition or binding. A, comparison of representative assays of phosphorylation activation by poly(I)·poly(C) dsRNA in the presence of [γ32P]ATP with PKR/eIF2α (top panel) and PKR alone (center panel) using SDS-PAGE and with PKR alone (bottom panel) using the newly established slot blot method (SB). B, as A but for inhibition of phosphorylation by TSΔ21 RNA. C and D, quantification of replicate experiments shown in A and B, respectively, demonstrating the reproducibility of the approach and correlation between PKR autophosphorylation (blue, SDS-PAGE; black, slot blot) and phosphorylation of the substrate eIF2α (green). The arrowhead in C indicates the concentration of PKR used in all subsequent kinase inhibition assays. E, quantification of [γ32P]ATP slot blot PKR autophosphorylation inhibition assays using the VA RNAI variants: TSΔ21, TSΔ25, TSΔ25+L, and A123U RNAs at a fixed concentration of poly(I)·poly(C) dsRNA activator. F, representative ITC experiments for titration of PKR into (left to right): TSΔ21, TSΔ25+L, and TSΔ25 RNAs.
From these data, we conclude that the central domain tertiary structure does not contribute to PKR binding and is not required for full inhibition of PKR. Taken together, SHAPE probing and these functional analyses suggest that substantially different secondary structures can be appended to the VA RNAI apical stem to produce potent PKR inhibitors. This observation is significant because it explains how activity against PKR is maintained despite extensive variation in the VA RNAI central domain sequence and secondary structure among different adenovirus serotypes (11).
PKR Inhibition Requires Only the VA RNAI Apical Stem and Three-helix Junction
Collectively, the results presented above demonstrate that, although the VA RNAI central domain possesses a complex tertiary structure, it is not required for PKR inhibition. Given the well established role of the central domain in PKR inhibition, the critical question remains: what are the features of the VA RNAI central domain that truly define it as an inhibitor and, therefore, an effective mechanism for circumventing innate cellular immunity? We made six further mutations in the TSΔ25 RNA context to dissect the contributions made to PKR inhibition by the pair of conserved tetranucleotide sequences within the central domain (21) as well as the structures within its 5′ and 3′ halves (Fig. 7A). 5′-mut and 5′/3′-bp RNAs contain mutations in the 5′-conserved tetranucleotide sequence, without and with compensatory changes on the 3′ side, respectively. Deletion mutants Δ5′(minor), Δ5′(major), Δ3′(minor), and Δ3′(major) RNAs lack part or all of one half of the central domain.
FIGURE 7.

Role of the central domain 5′-strand nucleotides and three-helix junction in optimal PKR inhibition. A, VA RNAI central domain mutants made in the TSΔ25 context: two terminus sequence variants, 5′-mut and 5′/3′-bp RNAs, and four deletion mutants, Δ5′(minor), Δ5′(major), Δ3′(minor), and Δ3′(major) RNAs. B, quantification of slot blot PKR autophosphorylation inhibition assays for the 5′-mut (dashed purple line) and 5′/3′-bp (solid purple line) sequence variants in the presence of [γ32P]ATP and poly(I)·poly(C) dsRNA activator. TSΔ21 (black) and TSΔ25 (gray) are shown for comparison. C, as B but for the deletion mutants Δ5′(minor) RNA (cyan), Δ5′(major) RNA (green), Δ3′(minor) RNA (orange), and Δ3′(major) RNA (red). D, UV melting profiles of each deletion mutant (colored as in A and C). The melting profile for each RNA is shown at pH 7.5 (solid line) and pH 5.5 (dashed line) and compared with those of TSΔ21 RNA (black). The region corresponding to unfolding of the TSΔ21 RNA pH-dependent tertiary structure at pH 7.5 is marked with a horizontal bar. E, representative SDS-PAGE analysis (top panel) and quantification of replicate (bottom panel) PKR (solid line) and eIF2α (dashed line) phosphorylation inhibition experiments with Δ5′(major) RNA (green) in the presence of poly(I)·poly(C) dsRNA activator and [γ32P]ATP. Equivalent TSΔ21 RNA data shown for comparison are the same as in Fig. 6D. F, representative ITC experiments for titration of PKR into Δ5′(major) (left) and Δ3′(major) (right) RNAs.
We first assessed the ability of each RNA to inhibit PKR autophosphorylation using the slot blot apparatus as described above. Alteration of the 5′ half of the conserved tetranucleotide sequence with or without compensatory changes in the 3′ half, in 5′/3′-bp and 5′-mut RNAs respectively, had no effect on inhibitory activity compared with TSΔ25 RNA (Fig. 7B). This result was unexpected given the previous demonstration that, in the context of the full-length RNA, mutation of these nucleotides caused loss of activity in a cell-based translational rescue assay that could be partially restored by compensatory mutations (21). Additionally, in that study, the same mutations were found to cause major changes in the nuclease sensitivity of loops II and III, consistent with their role in tertiary structure formation. Despite this discrepancy, the activity of our mutated RNAs in the TSΔ25 RNA context is consistent with our finding that the tertiary structure plays no role in PKR inhibition, and, collectively, these observations point to additional roles for the central domain tertiary structure in a cellular context.
In contrast, each of the central domain deletion mutants impacted PKR inhibitory capacity, ranging from a very modest effect with the smaller 5′ end deletion, Δ5′(minor), to the greatest decrease in activity with the larger 5′ end deletion in Δ5′(major) (Fig. 7C). Both 3′ end deletions had an essentially identical intermediate impact on PKR inhibition. This latter observation suggests that the 3′ half of the central domain may contribute to inhibitory activity through its interaction with the critical region located at the base of the apical stem on its 5′ side.
Because short hairpin RNAs with single-stranded 5′ and 3′ extensions (ss-ds-RNAs) are dependent upon a 5′ end triphosphate for potent PKR activation (38), we considered the possibility that our Δ5′ and Δ3′ variants might resemble such RNAs and activate poorly because their terminal extensions are suboptimal for activation (absent, too short, or base-paired). We reasoned that this might result in pseudoinhibition because the poorer VA RNAI mutant activator outcompetes the more potent poly(I)·poly(C) dsRNA at higher concentrations. However, other than a modest change in the activity of Δ3′(minor) RNA, no differences were observed between equivalent RNAs with a 5′-triphosphate or 5′-hydroxyl (generated by calf intestinal alkaline phosphatase treatment, data not shown).
Each of the four deletions represents a substantial truncation of the central domain structure. To assess the global folding of each RNA for any unexpected effects on its structure, we examined the UV melting profile of each RNA (Fig. 7D). However, as expected, all four RNAs lack the first (pH-dependent) and second apparent transitions (as for TSΔ25 RNA, Fig. 4) with further loss of hypochromicity in each of the larger deletions, corresponding to the greater reduction in structural content. With its almost complete deletion of the central domain, other than the short single-stranded 5′ end sequence, Δ3′(major) RNA has essentially no hypochromicity in the regions of the profile corresponding to central domain unfolding (Fig. 7D, fourth panel).
Finally, we assessed the functional impacts of the largest central domain deletion mutants. As expected, the ability of Δ5′(major) to inhibit downstream phosphorylation of eIF2α by PKR was decreased markedly, correlating directly with its substantially reduced inhibition of PKR autophosphorylation (Fig. 7E). We also measured the binding affinity of Δ5′(major) and Δ3′(major) RNAs for PKR using ITC. Despite its substantial impact on PKR inhibition, deletion of the entire 3′ half of the central domain resulted in only a ∼2-fold reduction in affinity, whereas deletion of the 5′ end resulted in a more dramatic decrease in Kd (Fig. 7F).
These results, together with the kinase assays, indicate that the 5′ half of the central domain is most critical for mediating high-affinity PKR binding and conferring full inhibitory activity. Although the conserved 5′ side tetranucleotide sequence (nts 37–40) does not appear as critical as we had anticipated, the larger deletion in Δ5′(major) RNA partially overlaps with an unusually thermodynamically stable region at the base of the apical stem that is resistant to denaturation and results in aberrant migration of VA RNAI in denaturing polyacrylamide gels (27). Whether this structure contributes to binding and/or inhibition will require additional detailed structural characterization of the PKR-VA RNAI complex. The observation that disconnecting the 5′ and 3′ termini in Δ3′(minor) RNA results in as great a reduction in activity as deletion of the entire 3′ half of the central domain suggests that the three-helix junction is required for full inhibitory activity. Whether this is due to direct interactions at the junction or indirectly via optimal positioning of the 5′ strand of the central domain also remains to be determined.
CONCLUSIONS
The results presented here provide strong evidence for the formation of a pH- and Mg2+-dependent tertiary structure within the adenovirus type 2 VA RNAI central domain. In sharp contrast to our expectation, however, this structure is entirely dispensable for inhibition of PKR. Instead, the overall organization of the central domain as a three-helix junction, in concert with the 5′ strand at the base of the apical stem, is critical for activity. During preparation of this manuscript, two reports were published describing PKR interactions with VA RNAI using analytical ultracentrifugation and small angle x-ray scattering with molecular modeling (37, 39). The observation that the central domain contributes to high-affinity PKR binding (37), potentially through placement of dsRBM2 at the helical junction (39), is entirely compatible with our finding of the direct and critical role for the 5′ half of this domain.
Collectively, our results reveal that requirements for RNA-mediated inhibition of PKR are substantially simpler than appreciated previously and highlight the need for an expanded view of RNA features required for PKR inhibition. Our findings offer a satisfying explanation for the ability of VA RNAI from different serotypes with varying sequences and structures to effectively inhibit PKR and, thereby, ensure successful viral replication (11). Relaxed criteria in this sense reconcile the inclusion of seemingly disparate non-coding RNAs, such as the Epstein-Barr virus RNA EBER-1 (40) and the cellular RNA nc886 (41), with VA RNAI in a collection of non-coding RNAs that exert an inhibitory effect against PKR. In light of our finding that the highly conserved central domain structure reported previously as necessary for optimal VA RNAI-mediated translational rescue (21) has a minimal impact on PKR inhibition, speculation on alternative roles for the VA RNAI central domain raises several intriguing possibilities. For example, the central domain tertiary structure might protect the critical region of the helical junction from degradation, could influence Dicer processing of the terminal stem (3, 4), or may mask structures or sequences that might cause unwanted regulation of other dsRNA binding proteins.
Proteins of the innate immune system must maintain features that render them sufficiently promiscuous for detection of a range of invading viruses but not so much so that they elicit an autoimmune response. That an even wider range of structures than anticipated can fulfill an inhibitory role is in agreement with multiple lines of evidence pointing to the functional flexibility of PKR in its interactions with activating and inhibitory RNAs.
Acknowledgments
We thank Dr. Christine M. Dunham for critical reading of the manuscript and discussions and members of the Conn and Dunham laboratories for discussions.
This work was supported, in whole or in part, by National Institutes of Health grant R21-AI097803. This work was also supported by Emory University Research Council Grant URC 2010050. The Auto-iTC200 instrument was purchased with support from National Science Foundation MRI Program Grant 104177, the Winship Cancer Institute shared resource program, and the Biochemistry Department of Emory University.
- PKR
- double-stranded RNA-activated protein kinase
- dsRNA
- double-stranded RNA
- VA RNAI
- virus-associated RNA I
- SHAPE
- selective 2′-hydroxyl acylation analyzed by primer extension
- NMIA
- N-methylisatoic anhydride
- TBE
- Tris borate-EDTA
- ITC
- isothermal titration calorimetry
- nt
- nucleotide
- TE
- Tris-EDTA.
REFERENCES
- 1. Mathews M. B., Shenk T. (1991) Adenovirus virus-associated RNA and translation control. J. Virol. 65, 5657–5662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. García M. A., Gil J., Ventoso I., Guerra S., Domingo E., Rivas C., Esteban M. (2006) Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 70, 1032–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andersson M. G., Haasnoot P. C., Xu N., Berenjian S., Berkhout B., Akusjärvi G. (2005) Suppression of RNA interference by adenovirus virus-associated RNA. J. Virol. 79, 9556–9565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lu S., Cullen B. R. (2004) Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and microRNA biogenesis. J. Virol. 78, 12868–12876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aparicio O., Carnero E., Abad X., Razquin N., Guruceaga E., Segura V., Fortes P. (2010) Adenovirus VA RNA-derived miRNAs target cellular genes involved in cell growth, gene expression and DNA repair. Nucleic Acids Res. 38, 750–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aparicio O., Razquin N., Zaratiegui M., Narvaiza I., Fortes P. (2006) Adenovirus virus-associated RNA is processed to functional interfering RNAs involved in virus production. J. Virol. 80, 1376–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kamel W., Segerman B., Öberg D., Punga T., Akusjärvi G. (2013) The adenovirus VA RNA-derived miRNAs are not essential for lytic virus growth in tissue culture cells. Nucleic Acids Res. 41, 4802–4812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wahid A. M., Coventry V. K., Conn G. L. (2008) Systematic deletion of the adenovirus-associated RNAI terminal stem reveals a surprisingly active RNA inhibitor of double-stranded RNA-activated protein kinase. J. Biol. Chem. 283, 17485–17493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Desai S. Y., Patel R. C., Sen G. C., Malhotra P., Ghadge G. D., Thimmapaya B. (1995) Activation of interferon-inducible 2′-5′ oligoadenylate synthetase by adenoviral VAI RNA. J. Biol. Chem. 270, 3454–3461 [DOI] [PubMed] [Google Scholar]
- 10. Meng H., Deo S., Xiong S., Dzananovic E., Donald L. J., van Dijk C. W., McKenna S. A. (2012) Regulation of the interferon-inducible 2′-5′-oligoadenylate synthetases by adenovirus VA(I) RNA. J. Mol. Biol. 422, 635–649 [DOI] [PubMed] [Google Scholar]
- 11. Ma Y., Mathews M. B. (1996) Structure, function, and evolution of adenovirus-associated RNA: a phylogenetic approach. J. Virol. 70, 5083–5099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ma Y., Mathews M. B. (1993) Comparative analysis of the structure and function of Adenovirus virus associated RNAs. J. Virol. 67, 6605–6617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Furtado M. R., Subramanian S., Bhat R. A., Fowlkes D. M., Safer B., Thimmappaya B. (1989) Functional dissection of Adenovirus VAI RNA. J. Virol. 63, 3423–3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clarke P. A., Mathews M. B. (1995) Interactions between the double stranded RNA binding motif and RNA: definition of the binding site for the interferon-induced protein kinase DAI (PKR) on Adenovirus VA RNA. RNA 1, 7–20 [PMC free article] [PubMed] [Google Scholar]
- 15. Clarke P. A., Pe'ery T., Ma Y., Mathews M. B. (1994) Structural features of adenovirus 2 virus-associated RNA required for binding to the protein kinase DAI. Nucleic Acids Res. 22, 4364–4374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ghadge G. D., Malhotra P., Furtado M. R., Dhar R., Thimmapaya B. (1994) In vitro analysis of virus associated RNA-I (VAI RNA): inhibition of the double stranded RNA activated protein kinase PKR by VAI RNA mutants correlates with the in vivo phenotype and the structural integrity of the central domain. J. Virol. 68, 4137–4151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ghadge G. D., Swaminathan S., Katze M. G., Thimmapaya B. (1991) Binding of the Adenovirus VAI RNA to the interferon induced 68 kDa protein kinase correlates with function. Proc. Natl. Acad. Sci. U.S.A. 88, 7140–7144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mellits K. H., Mathews M. B. (1988) Effects of mutations in stem and loop regions on the structure and function of Adenovirus VA RNA1. EMBO J. 7, 2849–2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Spanggord R. J., Beal P. A. (2001) Selective binding by the RNA binding domain of PKR revealed by affinity cleavage. Biochemistry 40, 4272–4280 [DOI] [PubMed] [Google Scholar]
- 20. Spanggord R. J., Vuyisich M., Beal P. A. (2002) Identification of binding sites for both dsRBMs of PKR on kinase-activating and kinase-inhibiting RNA ligands. Biochemistry 41, 4511–4520 [DOI] [PubMed] [Google Scholar]
- 21. Ma Y., Mathews M. B. (1996) Secondary and tertiary structure in the central domain of adenovirus type 2 VA RNA(I). RNA 2, 937–951 [PMC free article] [PubMed] [Google Scholar]
- 22. Launer-Felty K., Wong C. J., Wahid A. M., Conn G. L., Cole J. L. (2010) Magnesium-dependent interaction of PKR with Adenovirus VAI. J. Mol. Biol. 402, 638–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wahid A. M., Coventry V. K., Conn G. L. (2009) The PKR-binding domain of adenovirus VA RNAI exists as a mixture of two functionally non-equivalent structures. Nucleic Acids Res. 37, 5830–5837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Walker S. C., Avis J. M., Conn G. L. (2003) General plasmids for producing RNA in vitro transcripts with homogeneous ends. Nucleic Acids Res. 31, e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vachon V. K., Conn G. L. (2012) Plasmid template design and in vitro transcription of short RNAs within a “structure cassette” for structure probing experiments. Methods Mol. Biol. 941, 157–169 [DOI] [PubMed] [Google Scholar]
- 26. Pe'ery T., Mathews M. B. (1997) Synthesis and purification of single-stranded RNA for use in experiments with PKR and in cell-free translation systems. Methods 11, 371–381 [DOI] [PubMed] [Google Scholar]
- 27. Coventry V. K., Conn G. L. (2008) Analysis of adenovirus VA RNA(I) structure and stability using compensatory base pair modifications. Nucleic Acids Res. 36, 1645–1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilkinson K. A., Merino E. J., Weeks K. M. (2006) Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 1, 1610–1616 [DOI] [PubMed] [Google Scholar]
- 29. Lemaire P. A., Lary J., Cole J. L. (2005) Mechanism of PKR activation: dimerization and kinase activation in the absence of double-stranded RNA. J. Mol. Biol. 345, 81–90 [DOI] [PubMed] [Google Scholar]
- 30. Merino E. J., Wilkinson K. A., Coughlan J. L., Weeks K. M. (2005) RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE). J. Am. Chem. Soc. 127, 4223–4231 [DOI] [PubMed] [Google Scholar]
- 31. Fowlkes D. M., Shenk T. (1980) Transcriptional control regions of the adenovirus VAI RNA gene. Cell 22, 405–413 [DOI] [PubMed] [Google Scholar]
- 32. Hargittai M. R., Musier-Forsyth K. (2000) Use of terbium as a probe of tRNA tertiary structure and folding. RNA 6, 1672–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leontis N. B., Westhof E. (2001) Geometric nomenclature and classification of RNA base pairs. RNA 7, 499–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Petrov A. I., Zirbel C. L., Leontis N. B. (2011) WebFR3D: a server for finding, aligning and analyzing recurrent RNA 3D motifs. Nucleic Acids Res. 39, W50–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ben-Shem A., Garreau de Loubresse N., Melnikov S., Jenner L., Yusupova G., Yusupov M. (2011) The structure of the eukaryotic ribosome at 3.0 A resolution. Science 334, 1524–1529 [DOI] [PubMed] [Google Scholar]
- 36. Klein D. J., Moore P. B., Steitz T. A. (2004) The roles of ribosomal proteins in the structure assembly, and evolution of the large ribosomal subunit. J. Mol. Biol. 340, 141–177 [DOI] [PubMed] [Google Scholar]
- 37. Launer-Felty K., Cole J. L. (2014) Domain interactions in adenovirus VAI RNA mediate high-affinity PKR binding. J. Mol. Biol. 426, 1285–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nallagatla S. R., Hwang J., Toroney R., Zheng X., Cameron C. E., Bevilacqua P. C. (2007) 5′-triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science 318, 1455–1458 [DOI] [PubMed] [Google Scholar]
- 39. Dzananovic E., Patel T. R., Chojnowski G., Boniecki M. J., Deo S., McEleney K., Harding S. E., Bujnicki J. M., McKenna S. A. (2014) Solution conformation of adenovirus virus associated RNA-I and its interaction with PKR. J. Struct. Biol. 185, 48–57 [DOI] [PubMed] [Google Scholar]
- 40. Sharp T. V., Schwemmle M., Jeffrey I., Laing K., Mellor H., Proud C. G., Hilse K., Clemens M. J. (1993) Comparative analysis of the regulation of the interferon-inducible protein kinase PKR by Epstein Barr virus RNAs EBER-1 and EBER-2 and Adenovirus VA(I) RNA. Nucleic Acids Res. 21, 4483–4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jeon S. H., Lee K., Lee K. S., Kunkeaw N., Johnson B. H., Holthauzen L. M., Gong B., Leelayuwat C., Lee Y. S. (2012) Characterization of the direct physical interaction of nc886, a cellular non-coding RNA, and PKR. FEBS Lett. 586, 3477–3484 [DOI] [PubMed] [Google Scholar]