

Background: Metformin exhibits anti-inflammatory effects.
Results: In murine macrophages, metformin induces activating transcription factor-3 (ATF-3) in parallel with protective effects against LPS-induced inflammation.
Conclusion: Anti-inflammatory action of metformin is at least partly mediated via ATF-3 induction.
Significance: This finding provides a new perspective on metformin action and novel therapeutic means of treating inflammation-related diseases, i.e. ATF-3 modulation.
Keywords: Inflammation, Interleukin 6 (IL-6), Lipopolysaccharide (LPS), Macrophage, Tumor Necrosis Factor (TNF), ATF-3, Metformin
Abstract
Metformin, a well known antidiabetic agent that improves peripheral insulin sensitivity, also elicits anti-inflammatory actions, but its mechanism is unclear. Here, we investigated the mechanism responsible for the anti-inflammatory effect of metformin action in lipopolysaccharide (LPS)-stimulated murine macrophages. Metformin inhibited LPS-induced production of tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) in a concentration-dependent manner and in parallel induction of activating transcription factor-3 (ATF-3), a transcription factor and member of the cAMP-responsive element-binding protein family. ATF-3 knockdown abolished the inhibitory effects of metformin on LPS-induced proinflammatory cytokine production accompanied with reversal of metformin-induced suppression of mitogen-activated protein kinase (MAPK) phosphorylation. Conversely, AMP-activated protein kinase (AMPK) phosphorylation and NF-κB suppression by metformin were unaffected by ATF-3 knockdown. ChIP-PCR analysis revealed that LPS-induced NF-κB enrichments on the promoters of IL-6 and TNF-α were replaced by ATF-3 upon metformin treatment. AMPK knockdown blunted all the effects of metformin (ATF-3 induction, proinflammatory cytokine inhibition, and MAPK inactivation), suggesting that AMPK activation by metformin is required for and precedes ATF-3 induction. Oral administration of metformin to either mice with LPS-induced endotoxemia or ob/ob mice lowered the plasma and tissue levels of TNF-α and IL-6 and increased ATF-3 expression in spleen and lungs. These results suggest that metformin exhibits anti-inflammatory action in macrophages at least in part via pathways involving AMPK activation and ATF-3 induction.
Introduction
Metformin is a widely used antidiabetic drug that increases the peripheral uptake of glucose (1, 2) and decreases hepatic glucose production (3, 4). However, the mechanism responsible for the effect of metformin on glucose homeostasis remains unclear. LKB1-AMPK3 activation followed by transcriptional activation of gluconeogenic genes has been suggested as a molecular mechanism of metformin action (5–7). Conversely, other reports suggest that metformin inhibits complex 1 of the mitochondrial respiratory chain (8, 9), and its antidiabetic effects are independent of the LKB1-AMPK pathway via a decrease in hepatic energy state (10).
Chronic low grade inflammation is associated with the development of insulin resistance, and obesity-driven progressive infiltration of macrophages into adipose tissues and subsequent activation of macrophages lead to a considerable amplification of the inflammatory states via production of a variety of proinflammatory cytokines, impairing insulin signaling in insulin-sensitive tissues (11, 12). Notably, clinical outcomes suggest that metformin may alter inflammation independently of its effect on glycemic control (13, 14). Furthermore, reports have described the anti-inflammatory effects of metformin on various cell types including human vascular smooth muscle cells and endothelial cells (15–17). However, the mechanisms underlying the anti-inflammatory effects of metformin and the relative contribution of its anti-inflammatory action to ameliorating insulin resistance remain to be fully elucidated.
Activating transcription factor-3 (ATF-3) is a member of the ATF/cAMP-responsive element-binding protein family of basic leucine zipper-type transcription factors (18) and is markedly induced by various stresses including LPS in endothelial cells, smooth muscle cells, and macrophages (19). The biological role of ATF-3 depends on cell type and stimulus, and it can act as either a transcriptional activator or repressor (20, 21). Recent studies based on systems biologic approaches have shown that ATF-3 acts as a negative regulator of toll-like receptor 4 (TLR-4) by directly binding to the promoter region of IL-6 (22). Furthermore, Gilchrist et al. (22) found that circulating levels of IL-6 and IL-12b were significantly higher in ATF-3 knock-out mice treated with intraperitoneal LPS than in wild type mice, thus confirming the negative effect of ATF-3 on LPS response in vivo. Intriguingly, ATF-3 has also been shown to attenuate saturated free fatty acid and TLR-4 signaling and macrophage activation in obese adipose tissues in vivo (23).
Recently, it was shown that the tyrosine kinase c-Src regulates TLR signaling via ATF-3 induction (24), and c-Src activation may be involved in metformin action via the PI3K-LKB1-AMPK pathway (6). Thus, we hypothesized that the anti-inflammatory effects of metformin in macrophages are due to the induction of ATF-3. The anti-inflammatory mechanism of metformin was investigated using RAW264.7 murine macrophages and peritoneal macrophages, and inflammatory conditions were induced by treating cells with LPS, a bacterial product that induces metabolic inflammation in vivo (25). It was found that metformin protected against LPS-induced production of TNF-α and IL-6 in murine macrophages at least in part via ATF-3 induction in an AMPK-dependent manner. Notably, oral metformin induced both ATF-3 expression and AMPK phosphorylation in lungs and spleen and concomitantly reduced the plasma and tissue levels of TNF-α and IL-6. These results provide first evidence that the pharmacological modulation of ATF-3 may have an anti-inflammatory effect and ameliorate insulin resistance.
EXPERIMENTAL PROCEDURES
Animals
Male C57BL/6J mice (6 weeks old) were obtained from Orient Bio (Seongnam, Korea). Animals were housed under specific pathogen-free conditions in an air-conditioned room at 23 ± 2 °C. Food and water were supplied ad libitum. C57BL/6J-Lepob leptin-deficient mice (ob/ob mice, male, 6 weeks old) were inbred in the Korean Research Institute of Chemical Technology (Taejeon, Korea). Mice were randomly allocated to control and metformin groups. All animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (National Academy, 1996) and approved by the Animal Care and Use Committee at Gachon University.
Materials
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), penicillin, glutamine, streptomycin, Lipofectamine 2000, and Opti-MEM medium were obtained from Invitrogen. LPS (Escherichia coli 055:B5), metformin, 5-aminoimidazolecarboxamide riboside (AICAR), and all other chemicals were from Sigma. TNF-α, IL-1β, and IL-6 ELISA kits were from Enzo Life Sciences (New York, NY). Antibodies against phospho-AMPK-α (Thr172), AMPK, phospho-JNK (Thr183/Tyr185), phospho-acetyl-CoA carboxylase (Thr172), and phospho-p38 (Thr180/Tyr182) were from Cell Signaling Technology (Beverly, MA). Antibodies against IgG, β-actin, ATF-3, p65, phospho-ERK1/2 (Tyr204), and ERK1/2 were from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell Culture and Treatment
RAW264.7 mouse macrophages (Korea Cell Line Bank) were seeded in 12-well plates at a density of 5 × 105 cells/well in DMEM supplemented with 2 mm l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% (v/v) heat-inactivated fetal bovine serum in a humidified 5% CO2 atmosphere at 37 °C. Mouse peritoneal macrophages were isolated from C57BL/6J mice after stimulation with an intraperitoneal injection of 2% thioglycolate solution (3 ml/mouse).
Real Time Quantitative (qPCR) and Reverse Transcription (RT)-PCR
Total RNA was isolated from cells and tissues using easy-BLUE Total RNA extraction kits (iNtRON Inc., Korea). Reverse transcription of total RNA (1 μg) was performed using AccuPower RT PreMix (Bioneer Inc., Korea). The primers for RT-PCR and qPCR to amplify TNF-α, IL-6, ATF-3, and GAPDH (internal standard) were as follows (where F represents forward and R represents reverse): for RT-PCR: ATF-3 (F), TTG CTA ACC TGA CAC CCT TTG; ATF-3 (R), CGG TGC AGG TTG AGC ATG TA; GAPDH (F), TTC ACC ACC ATG GAG AAG GC; GAPDH (R), GGC ATG GAC TGT GGT CAT GA; for qPCR: TNF-α (F), CTG TAG CCC ACG TCG TAG C; TNF-α (R), TTG AGA TCC ATG CCG TTG; IL-6 (F), TCT AAT TCA TAT CTT CAA CCA AGA GC; IL-6 (R), TGG TCC TTA GCC ACT CCT TC; ATF-3 (F), GCT GGA GTC AGT TAC CGT CAA; ATF-3 (R), CGC CTC CTT TTC CTC TCA; GAPDH (F), CCT TGA GAT CAA CAC GTA CCA G; GAPDH (R), CGC CTG TAC ACT CCA CCA C. Reverse transcription-PCR was conducted using 35 cycles of denaturation at 94 °C for 1 min, annealing at 60 °C for 1 min, and extension at 72 °C for 1 min followed by extension at 72 °C for 10 min. After amplification, PCR mixtures were electrophoresed on 1.5% agarose gels and visualized by Gel Red (Elpis Biotech, Taejeon, Korea) staining under UV light. qPCR was done using LightCycler DNA Master SYBR Green I on a Roche LightCycler 2.0 (Roche Applied Science). Relative abundances of mRNA were calculated with respect to GAPDH.
Western Blotting
Cells were lysed in lysis buffer containing 50 mm HEPES (pH 7.0), 250 mm NaCl, 5 mm EDTA, 0.1% Nonidet P-40, 1 mm phenylmethylsulfonyl fluoride, 0.5 mm dithiothreitol, 5 mm sodium fluoride, 0.5 mm sodium orthovanadate, 5 μg/ml leupeptin, and 5 μg/ml aprotinin by incubating for 10 min at 4 °C. Following centrifugation at 12,000 rpm and denaturation, 50 μg of total proteins was loaded into an 8% sodium dodecyl sulfate-polyacrylamide gel and subsequently transferred to nitrocellulose membranes (Amersham Biosciences). Blocked membranes were then incubated with the indicated antibodies and then treated with secondary antibodies conjugated with horseradish peroxidase (HRP). Immunoreactive bands were visualized by enhanced chemiluminescence (Amersham Biosciences) and quantified using UN-SCAN-IT Gel 5.1 software (Silk Scientific Inc., Orem, UT). Protein concentrations were determined using Bio-Rad protein assay reagent according to the manufacturer's instructions.
ATF-3 and AMPK Knockdown
Transfection of small interfering RNA (siRNA) (100 nm each/well) against α1-AMPK, ATF-3, or negative control siRNA (Santa Cruz Biotechnology) into primary peritoneal macrophages (5 × 105 cells/well at 70–80% confluence) was accomplished using Lipofectamine 2000 reagent (6 μl/well). Cells were then incubated in Opti-MEM medium for 6 h, and media were replaced with DMEM containing 10% FBS. Eighteen hours later, cells were treated with LPS (10 ng/ml) in the presence or absence of metformin (2 mm, 24 h).
Chromatin Immunoprecipitation (ChIP)-PCR
ChIP assays were performed as described previously (26). Briefly, treated RAW264.7 cells (5 × 105 cells/well) were cross-linked with 1% formaldehyde for 10 min at 37 °C. After washing cells with ice-cold PBS, nuclei were collected and sonicated, and then chromatin solution (500 μg) was incubated with Dynabeads protein A (Invitrogen) and 5 μg of rabbit anti-ATF-3 or rabbit anti-p65 at 4 °C overnight. Antibody-bound complexes were obtained, and DNA fragments extricated from these complexes were purified using a QIAquick PCR Purification kit (Qiagen, Shanghai, China). The purified DNA samples were analyzed by conventional PCR. Primers for PCR are as follows: Cyclo (F), TGGAGAGCACCAAGACAGACA, Cyclo (R)-TGCCGGAGTCGACAATGAT; TNF-α (F), AGAAGAAACCGAGACAGAGGTGTAG; TNF-α (R), TTCAACCCTCGGAAAACTTCCT; IL-6 (F), GATAAGGGCAACTCTCACAGAGACT; IL-6 (R), TCAAGCAATCTTGGCTTCCA.
In Vivo Study
C57BL/6J mice were orally administered either vehicle (saline, 200 μl/20 g, n = 5) or metformin (250 or 500 mg/kg, bid, 200 μl/20 g, n = 5 each dose) for 3 days. On day 4, LPS (20 mg/kg, intraperitoneal) was administered 3 h after the last metformin administration. One hour later, mice were anesthetized with diethyl ether, and whole blood was collected by cardiac puncture. As another animal model, ob/ob mice were orally administered either vehicle (saline, 200 μl/40 g, n = 4) or metformin (250 mg/kg, bid, 200 μl/40 g, n = 5) for 3 weeks. Tail blood glucose concentrations were measured weekly (Accu-Chek, Roche Diagnostics GmbH). After 3 weeks, plasma was obtained from whole blood by centrifugation and stored at −20 °C until assayed. Plasma insulin concentration was determined by ELISA according to the manufacturer's instructions (Millipore, Billerica, MA). Lungs and spleens were removed and frozen immediately in liquid nitrogen until assayed. For immunohistochemistry, isolated tissues were embedded in paraffin, fixed, sectioned (4-μm thickness), and stained using a Dako LSAB-HRP kit (Dako, Carpinteria, CA). After deparaffinization in xylene and antigen retrieval in citrate buffer, tissue sections were treated with hydrogen peroxide (3%) and incubated with anti-ATF-3 polyclonal antibody (1:500) for 1 h followed by 30-min incubation with biotinylated linker as a secondary antibody. Antibody complexes were detected using 3,3′-diaminobenzidine tetrahydrochloride. The sections were then mounted for light microscopy after counterstaining with hematoxylin. For protein analysis, tissues were homogenized and lysed with a Prep-sol (iNtRON Inc.).
Statistical Analysis
Results are expressed as means ± S.E. Statistical significance was determined by one-way analysis of variance followed by Tukey's multiple comparison test. Statistical significance was accepted for p values <0.05.
RESULTS
Effects of Metformin on the LPS-induced TNF-α, IL-6, and ATF-3 Expression
LPS markedly increased the production of TNF-α and IL-6 in murine primary macrophages, and metformin (0.5–4 mm) concentration-dependently suppressed these LPS-induced increases (Fig. 1, A and B). The effects of metformin were also time-dependent (results not shown). Metformin alone had little effect on the production of TNF-α and IL-6 in the absence of LPS. Under this experimental condition, no metformin cytotoxicity was observed.
FIGURE 1.

Effects of metformin on LPS-induced proinflammatory cytokine production and ATF-3 induction. Primary macrophages (5 × 105 cells/well) were treated with metformin (Met) (0. 5–4 mm) for 20 h and then stimulated with 10 ng/ml LPS for 4 h. Levels of TNF-α (A) and IL-6 (B) in culture media were determined by ELISA. C, ATF-3 expression was determined by Western blotting, and densitometric analysis was carried out by using UN-SCAN-IT Gel 5.1 software. As a control, the cells were also treated with metformin alone in the absence of LPS. Results are expressed as means ± S.E. (error bars) of three independent experiments performed in triplicate. *, p < 0.05 versus LPS alone; #, p < 0.05 versus control. C, control.
As shown in Fig. 1C, LPS substantially increased ATF-3 protein as compared with control, and metformin additively increased ATF-3 expression in a concentration-dependent manner (left panel). Metformin itself increased ATF-3 expression at higher than 2 mm concentration (right panel). Based on these results, 2 mm metformin was chosen in further experiments.
Involvement of ATF-3 in the Action Mechanism of Metformin
To ascertain the involvement of ATF-3 in the protective effects of metformin against LPS-induced inflammation, we examined the effects of ATF-3 siRNA on metformin action. ATF-3 protein and mRNA were reduced by ∼70% after ATF-3 siRNA transfection as compared with control siRNA in primary macrophages (Fig. 2A). ATF-3 siRNA partially but significantly attenuated the inhibitory effects of metformin on the production of TNF-α (from 6.40 ± 3.49 to 68.8 ± 8.56% of control) and IL-6 (from 13.1 ± 0.01 to 88.2 ± 0.06% of control) (Fig. 2B).
FIGURE 2.
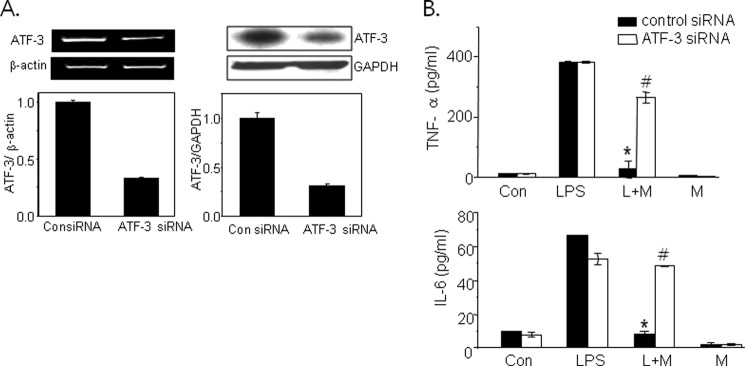
Effects of ATF-3 knockdown on the protective effects of metformin against LPS-induced inflammation. ATF-3 siRNA (100 nm) was transfected into primary macrophages, and the extent of ATF-3 knockdown was determined by RT-PCR (left panel) and Western blotting (right panel) (A). Then the cells were treated with 2 mm metformin (M) for 20 h followed by LPS (L) for 4 h. Levels of TNF-α and IL-6 in the culture media were determined by ELISA (B). Results are expressed as means ± S.E. (error bars) of three independent experiments performed in triplicate. *, p < 0.05 versus LPS alone; #, p < 0.05 versus control siRNA. Con, control.
Effects of ATF-3 Knockdown on Metformin-induced Suppression of TLR Signaling
We examined the effects of metformin on LPS-induced MAPK phosphorylation and the effects of ATF-3 siRNA on metformin action. Rapid increase of ERK, JNK, and p38 MAPK phosphorylation by LPS was suppressed by metformin in primary macrophages, and this effect was abolished by ATF-3 knockdown (Fig. 3A). However, ATF-3 siRNA had little effect on metformin-suppressed NF-κB activation by LPS (Fig. 3B). These results suggest that ATF-3 induction followed by MAPK inactivation is important for metformin action, but NF-κB suppression is unlikely to be the major mechanism of metformin action in murine macrophages.
FIGURE 3.

Effects of ATF-3 knockdown on the suppressive effects of metformin against LPS-induced MAPK phosphorylation. Control siRNA or ATF-3 siRNA was transfected into primary macrophages (A–C). Cells were then treated with metformin (M) (2 mm) for 20 h followed by LPS (L) for 30 min. Phosphorylated levels of p38 (Thr180/Tyr182) (p-p38), ERK (Tyr204) (p-ERK), JNK (Thr183/Tyr185) (p-JNK), IκBα (Ser32/36) (p-IκBα), and AMPK-α (Thr172) (p-AMPK) were determined by Western blotting. *, p < 0.05 versus control. D, ChIP-PCR was carried out after RAW264.7 cells were treated with LPS in the presence or absence of 2 mm metformin using either ATF-3 antibody or NF-κB antibody for immunoprecipitation. Relative mRNA levels of TNF-α and IL-6 were normalized versus IgG control. The experiment was repeated three times, and representative results are shown. Error bars represent S.E. *, p < 0.05 versus ATF-3 in LPS-treated cells; #, p < 0.05 versus NF-κB-p65 in LPS-treated cells. Con, control.
Next, we examined the effects of ATF-3 siRNA on the metformin-induced phosphorylation of AMPK. As shown in Fig. 3C, ATF-3 siRNA had little effect on AMPK-α-Thr172 phosphorylation, which is essential for AMPK activity, suggesting that AMPK activation by metformin precedes ATF-3 induction.
Because both LPS and metformin induce ATF-3 expression but produce opposite action with regard to proinflammatory cytokine production, we carried out ChIP-PCR analysis using RAW264.7 cells to define molecular mechanisms involved in this paradoxical action. As shown in Fig. 3D, ATF-3 binding to the promoters of TNF-α and IL-6 was enriched in the presence of metformin as compared with LPS alone, whereas LPS-induced NF-κB binding to each promoter was reduced by metformin, suggesting that metformin-induced ATF-3 appears to suppress proinflammatory cytokine production via competition with NF-κB for binding to TNF-α and IL-6 promoters.
Involvement of AMPK in Metformin Action against LPS-induced Inflammation
AMPK siRNA reduced the AMPK protein level by 68% in primary macrophages as compared with control siRNA (Fig. 4A). AMPK knockdown blunted metformin action including suppression of proinflammatory cytokine (Fig. 4B; TNFα, from 17.3 ± 1.30 to 92.9 ± 1.31% of control; IL-6, from 5.10 ± 2.50 to 73.8 ± 5.77% of control), ATF-3 induction (Fig. 4C), and MAPK inactivation (Fig. 4D).
FIGURE 4.

Effects of AMPK knockdown on metformin action against LPS-induced inflammation. AMPK siRNA (100 nm) or control siRNA was transfected into primary macrophages, and the extent of AMPK knockdown was determined by RT-PCR (left panel) and Western blotting (right panel) (A). Then the cells were treated with metformin (M) (2 mm) for 20 h and then stimulated with LPS (L) for 4 h. Levels of TNF-α and IL-6 in culture media were determined by ELISA (B). The effects of AMPK siRNA on ATF-3 expression (C) and MAPK phosphorylation (D) were determined by Western blotting. The effects of AICAR (E) and compound C (F) on the levels of ATF-3, phospho-AMPK-α (Thr172) (p-AMPK) (Western blotting) and cytokines (ELISA) were determined. The experiment was repeated three times in triplicate. Results are expressed as means ± S.E. (error bars). *, p < 0.05 versus LPS alone; **, p < 0.05 versus LPS and metformin (L+M); #, p < 0.05 versus control siRNA. Con, control; p-ERK, phospho-ERK; p-p38, phospho-p38; p-JNK, phospho-JNK; Met, metformin.
The involvement of AMPK in the ATF-3-mediated anti-inflammatory effects was further confirmed using AICAR (an AMPK activator) and compound C (an AMPK inhibitor) under similar experimental conditions. As shown in Fig. 4E, AICAR mimicked the effects of metformin vis-à-vis ATF-3 induction and suppression of the production of proinflammatory cytokines. In addition, compound C (5 μm) attenuated all the effects of metformin (i.e. ATF-3 and phospho-AMPK expression and suppression of TNF-α and IL-6 production) (Fig. 4F). These results suggest that metformin-induced AMPK activation is required for ATF-3 induction followed by suppression of LPS-induced MAPK phosphorylation and proinflammatory cytokine production.
Effects of Metformin in an LPS-induced Septic Shock Mouse Model
As shown in Fig. 5A, LPS increased the plasma levels of TNF-α and IL-6 (the effect was maximal at 1 h and then rapidly declined). The proinflammatory cytokine levels were significantly and dose-dependently reduced by metformin (TNF-α, 47.4 ± 3.62 and 71.9 ± 3.99% inhibition; IL-6, 24.9 ± 2.88 and 60.1 ± 3.91% inhibition at 250 and 500 mg/kg, bid, respectively). There was little change in body weights between treatments, and no apparent toxicity due to metformin administration was observed (Fig. 5B). AMPK and acetyl-CoA carboxylase phosphorylation was higher in the lungs and spleens (two major tissues in terms of macrophage production) of the metformin-treated group than in the LPS-treated group (Fig. 5, C and E). In parallel, the protein (Fig. 5, C and E) and mRNA levels (Fig. 5, D and F) of TNF-α and IL-6 were reduced upon metformin treatment in both tissues. As expected, ATF-3 protein levels were elevated in the lungs and spleens to 2.36- and 2.15-fold at 500 mg/kg metformin, respectively, as compared with the LPS-treated group, and this was further confirmed by immunohistochemistry (detected as brownish color; Fig. 5G). No difference in IgG control was detected.
FIGURE 5.

In vivo effects of metformin in an LPS-induced endotoxemia model. After 4 days of metformin treatment (250 (M250) and 500 (M500) mg/kg, n = 5 each dose, bid, oral), LPS (L) (20 mg/kg, intraperitoneal) was administered, 1 h later plasma was obtained by cardiac puncture, and levels of TNF-α and IL-6 were determined by ELISA (A). Changes in body weights are shown (B). The expressions of phospho-AMPK-α (Thr172) (p-AMPK), phospho-acetyl-CoA carboxylase (Thr172) (p-ACC), TNF-α, and IL-6 in lung and spleen tissues were determined by Western blotting (C, lung; E, spleen) and real time qPCR (D, lung; F, spleen). G, ATF-3 expression was detected by Western blotting (upper panel) and immunohistochemistry (lower panel). IgG was detected as a native control. The experiment was repeated twice, and results are expressed as means ± S.E. (error bars). *, p < 0.05; **, p < 0.01 versus LPS alone; #, p < 0.05 versus control. Con, control.
Effect of Metformin in the ob/ob Mouse Model
Ob/ob mice develop insulin resistance and inflammation in various tissues and represent a well established model of obesity and type 2 diabetes. To obtain further evidence that metformin modulates the inflammatory pathway via ATF-3 induction, we administered metformin orally to ob/ob mice at 250 mg/kg for 3 weeks and analyzed proinflammatory cytokines and ATF-3 in plasma and tissues. As shown in Fig. 6A, metformin significantly lowered proinflammatory cytokine levels in plasma (49.1 ± 2.51% inhibition of TNF-α and 46.3 ± 3.48% inhibition of IL-6) and decreased plasma glucose levels (Fig. 6B; 33.8 ± 6.58% inhibition) with decreased plasma insulin (from 3.05 ± 0.33 to 1.97 ± 0.17 ng/ml; Fig. 6C). Changes in body weight were insignificant (Fig. 6D). Metformin increased AMPK phosphorylation (2.41- and 5.26-fold) and ATF-3 expression (2.55- and 2.20-fold) in lung and spleen tissues, respectively, as compared with the vehicle-treated group with reduced TNF-α and IL-6 levels (TNF-α, 64.8 ± 3.54 and 61.0 ± 8.69% inhibition; IL-6, 61.3 ± 8.49 and 41.4 ± 10.9% inhibition in lungs and spleens, respectively) (Fig. 6E).
FIGURE 6.

In vivo effects of metformin in ob/ob mice. Metformin (250 mg/kg, bid, n = 5) or vehicle (saline, n = 4) was orally administered to ob/ob mice for 3 weeks. Plasma TNF-α and IL-6 levels (A), plasma glucose (B), and insulin (C) were determined by ELISA. Body weight changes are shown (D). The expression of ATF-3, phospho-AMPK-α (Thr172) (p-AMPK), TNF-α, and IL-6 in lung and spleen tissues was determined by Western blotting (E, left panel, lung; right panel, spleen). The experiment was repeated twice, and results are expressed as means ± S.E. (error bars). *, p < 0.05 versus vehicle.
DISCUSSION
The present study shows that metformin inhibits LPS-induced proinflammatory cytokine production in murine macrophages at least in part via an AMPK-ATF-3-dependent mechanism. In accordance with the previous finding that ATF-3 plays an important role as a negative regulator of TLR signaling (22), this study shows for the first time that metformin, an antidiabetic drug, suppresses TLR signaling by inducing ATF-3 expression.
Metformin is known to act as an insulin sensitizer in liver and skeletal muscle and has been reported to reduce cardiovascular complications in diabetic patients, which implies anti-inflammatory effects (13, 27, 28), but its mechanisms beyond glycemic control still remain poorly understood. Of note, metformin has been reported to ameliorate inflammation in various cells including endothelial cells (15, 16). AMPK activation appears to be involved in metformin action possibly via an indirect manner (8, 9) and has also been suggested to be an anti-inflammatory target. For example, AMPK activation is associated with endothelial NOS activation in endothelial cells (29) and with inducible NOS inhibition in myocytes, adipocytes, and mouse bone marrow-derived macrophages (30). Furthermore, metformin attenuates TNF-α-induced expression of proinflammatory and adhesion genes via AMPK activation in endothelial cells (15, 16). Conversely, the anti-inflammatory action of AICAR has been reported to be independent of AMPK (31, 32), which suggests that the action mechanisms of AICAR and metformin differ. In the present study, modulation of AMPK activation using AMPK siRNA or AICAR showed that AMPK activation may be responsible for the protective effects of metformin against LPS-induced inflammation in murine macrophages.
The functional significance of ATF-3 is unclear, and it has been reported to have both protective and detrimental effects. In a recent study, the transcriptional regulatory network activated by bacterial LPS in murine macrophages was analyzed, and ATF-3 was found to play novel roles during the inflammatory response (33). Upon LPS exposure in macrophages, NF-κB was initially activated via TLR-4 activation, and then ATF-3 expression was induced to curb inflammation (22). In accordance with this, we also observed that LPS induced ATF-3 expression and that the inflammatory response induced by LPS was further increased by ATF-3 knockdown although statistically insignificant (results not shown), suggesting that ATF-3 induction by LPS is likely an adaptive homeostatic mechanism, restoring cellular stress to the normal condition. Thus, it may be plausible that superinduction of ATF-3 produces protective effects against various stress conditions (34–36). In the present study, metformin was found to superinduce ATF-3 expression in a concentration- and time-dependent manner, and this superinduction was found to inhibit proinflammatory cytokine production. This finding suggests a novel anti-inflammatory action mechanism of metformin, i.e. induction of ATF-3 expression. In support of the present results, it was shown previously that IFN-γ suppressed the LPS-induced expression of matrix metalloproteinase in human monocytes via the superinduction of ATF-3 and the suppression of AP-1 in a STAT1-dependent manner (34).
Multiple signaling pathways are known to be activated in macrophages after LPS exposure, and of these pathways, ERK, JNK, and p38 MAPK pathways are involved in the production of proinflammatory cytokines (37). As was expected, in the present study, LPS-induced phosphorylation of all three MAPKs was inhibited by metformin, and this was prevented by either ATF-3 siRNA or AMPK siRNA and accompanied by the attenuation of metformin-suppressed proinflammatory cytokine production. When the effects of ATF-3 siRNA and AMPK siRNA on metformin action were compared, AMPK siRNA reversed all the metformin action, i.e. ATF-3 induction, cytokine suppression, and MAPK inactivation, whereas ATF-3 siRNA blocked metformin-induced suppression of proinflammatory cytokine production and MAPK inactivation but not AMPK phosphorylation. Thus, AMPK activation by metformin appears to occur prior to ATF-3 induction and to be a prerequisite for ATF-3 up-regulation, leading to MAPK inactivation. However, how AMPK activation by metformin leads to ATF-3 induction remains unclear. Interestingly, metformin improves hepatic insulin resistance through the AMPK-small heterodimer partner pathway (38). The role of small heterodimer partner and the possible interaction between small heterodimer partner and ATF-3 need further investigation. An alternative pathway involved in the LPS-induced inflammatory response is NF-κB activation, and previous reports have shown that metformin inhibits NF-κB-mediated inflammatory action in an AMPK-dependent manner in endothelial cells (15–17). Consistently, metformin inhibited LPS-induced NF-κB activation in the present study, but this effect was unaffected by ATF-3 knockdown. Thus, it seems that NF-κB suppression and ATF-3 induction are two independent pathways of metformin action and that the inhibitory action of metformin on LPS-induced NF-κB activation may not be the primary anti-inflammatory mechanism of metformin in murine macrophages.
In a recent study performed to elucidate the molecular mechanisms whereby ATF-3 induction leads to the inhibition of TNF-α and IL-6 production, it was found that ATF-3 and Rel (NF-κB subunit) jointly regulated LPS-induced IL-6 transcription in a temporal manner (22). At the early stage of LPS stimulus, Rel binds to the promoter region of IL-6 with acetylated histone, allowing IL-6 transcription, whereas delayed expression of ATF-3 was associated with histone deacetylase, resulting in the inhibition of IL-6 transcription, which suggests that ATF-3 and NF-κB compete for binding on the promoter of IL-6, producing opposite actions: ATF-3 down-regulates and NF-κB up-regulates IL-6 transcription. Our ChIP-PCR results support this hypothesis that LPS recruits NF-κB predominantly on the IL-6 promoter at 1 h and that metformin-superinduced ATF-3 occupies the IL-6 promoter region, excluding LPS-induced NF-κB from binding to the promoter. Interestingly, ATF-3 and NF-κB counter-regulation was demonstrated by Hua et al. (39), who showed that ATF-3 counteracts NF-κB-mediated antiapoptotic action via suppression of antiapoptotic gene transcription.
Two mouse models were used in the present study to confirm our in vitro findings. Intraperitoneally administered LPS boosted plasma levels of TNF-α and IL-6 within 1 h, and oral metformin administered for 3 consecutive days to LPS-induced endotoxemic mice reduced the plasma levels of both proinflammatory cytokines and increased AMPK phosphorylation and ATF-3 expression in lung and spleen tissues. Likewise, oral administration of metformin for 3 weeks to ob/ob mice reduced plasma proinflammatory cytokines and glucose levels with lower plasma insulin levels and concomitantly up-regulated ATF-3 expression and AMPK phosphorylation in tissues. Taken together, these in vivo results further support our in vitro findings that the anti-inflammatory action of metformin is mediated at least in part via ATF-3 induction in macrophages, possibly ameliorating insulin resistance. Interestingly, transgenic mice overexpressing human ATF-3 in macrophages had lower expression of TNF-α and IL-6 mRNAs in adipose tissues and in peritoneal macrophages compared with that in wild type mice (23), suggesting that ATF-3 overexpression attenuated macrophage activation in vivo, which is consistent with our results. However, at present, the possibility that other pathways are also involved in the action mechanism of metformin cannot be excluded because ATF-3 knockdown failed to fully restore the LPS response in vitro.
In summary, metformin reduces LPS-stimulated proinflammatory cytokine production in murine macrophages via up-regulation of ATF-3 expression. A proposed model of metformin action is presented in Fig. 7. This study provides the novel idea that pharmacological modulation of ATF-3 expression or activation may offer a potential therapeutic means for macrophage-driven inflammatory diseases and particularly metabolic disorders. Our findings suggest that studies to assess the contribution made by ATF-3 to the beneficial effects of metformin in myocytes and hepatocytes should be conducted. Finally, the clinical relevance of ATF-3 induction by metformin requires further investigation.
FIGURE 7.
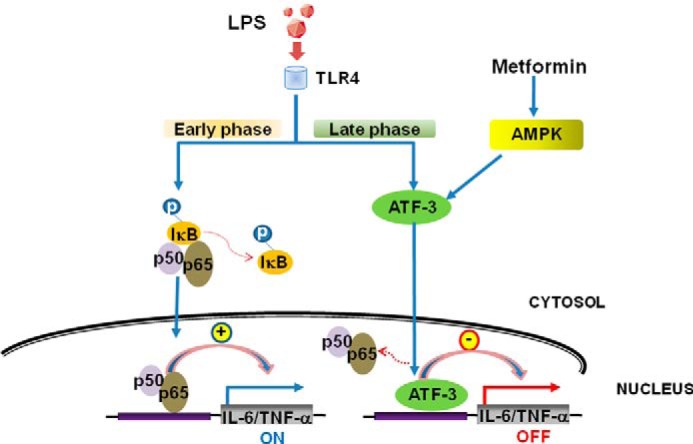
A proposed working model for the anti-inflammatory action of metformin in murine macrophages. Metformin induces ATF-3 expression via AMPK activation, resulting in protection against LPS-induced inflammatory responses.
This work was supported by the Basic Science Research Program of the Korean National Research Foundation (NRF) funded by the Korean Ministry of Education and Technology (Grant 2010-0007517).
- AMPK
- AMP-activated protein kinase
- ATF-3
- activating transcription factor-3
- TLR
- toll-like receptor
- AICAR
- 5-aminoimidazolecarboxamide riboside
- qPCR
- quantitative PCR
- bid
- twice a day.
REFERENCES
- 1. Hundal H. S., Ramlal T., Reyes R., Leiter L. A., Klip A. (1992) Cellular mechanism of metformin action involves glucose transporter translocation from an intracellular pool to the plasma membrane in L6 muscle cells. Endocrinology 131, 1165–1173 [DOI] [PubMed] [Google Scholar]
- 2. Galuska D., Nolte L. A., Zierath J. R., Wallberg-Henriksson H. (1994) Effects of metformin on insulin-stimulated glucose transport in isolated skeletal muscle obtained from patients with NIDDM. Diabetologia 37, 826–832 [DOI] [PubMed] [Google Scholar]
- 3. Stumvoll M., Nurjhan N., Perriello G., Dailey G., Gerich J. E. (1995) Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N. Engl. J. Med. 333, 550–554 [DOI] [PubMed] [Google Scholar]
- 4. Hundal R. S., Krssak M., Dufour S., Laurent D., Lebon V., Chandramouli V., Inzucchi S. E., Schumann W. C., Petersen K. F., Landau B. R., Shulman G. I. (2000) Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes 49, 2063–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou G., Myers R., Li Y., Chen Y., Shen X., Fenyk-Melody J., Wu M., Ventre J., Doebber T., Fujii N., Musi N., Hirshman M. F., Goodyear L. J., Moller D. E. (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 108, 1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zou M. H., Kirkpatrick S. S., Davis B. J., Nelson J. S., Wiles W. G., 4th, Schlattner U., Neumann D., Brownlee M., Freeman M. B., Goldman M. H. (2004) Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. J. Biol. Chem. 279, 43940–43951 [DOI] [PubMed] [Google Scholar]
- 7. Shaw R. J., Lamia K. A., Vasquez D., Koo S. H., Bardeesy N., Depinho R. A., Montminy M., Cantley L. C. (2005) The kinase LKB1 mediated glucose homeostasis in liver and therapeutic effects of metformin. Science 310, 1642–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. El-Mir M. Y., Nogueira V., Fontaine E., Avéret N., Rigoulet M., Leverve X. (2000) Diemthylbiguanide inhibits cell proliferation via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 275, 223–228 [DOI] [PubMed] [Google Scholar]
- 9. Owen M. R., Doran E., Halestrap A. P. (2000) Evidence that metformin exerts its antidiabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 348, 607–614 [PMC free article] [PubMed] [Google Scholar]
- 10. Foretz M., Hébrard S., Leclerc J., Zarrinpashneh E., Soty M., Mithieux G., Sakamoto K., Andreelli F., Viollet B. (2010) Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 120, 2355–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Greenberg A. S., McDaniel M. L. (2002) Identifying the links between obesity, insulin resistance and β cell function: potential role of adipocyte-derived cytokines in the pathogenesis of type 2 diabetes. Eur. J. Clin. Invest. 32, Suppl. 3, 24–34 [DOI] [PubMed] [Google Scholar]
- 12. Fujisaka S., Usui I., Bukhari A., Ikutani M., Oya T., Kanatani Y., Tsuneyama K., Nagai Y., Takatsu K., Urakaze M., Kobayashi M., Tobe K. (2009) Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice. Diabetes 58, 2574–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Caballero A. E., Delgado A., Aguilar-Salinas C. A., Herrera A. N., Castillo J. L., Cabrera T., Gomez-Perez F. J., Rull J. A. (2004) The differential effects of metformin on markers of endothelial activation and inflammation in subjects with impaired glucose tolerance: a placebo-controlled, randomized clinical trial. J. Clin. Endocrinol. Metab. 89, 3943–3948 [DOI] [PubMed] [Google Scholar]
- 14. Dandona P., Aljada A., Ghanim H., Mohanty P., Tripathy C., Hofmeyer D., Chaudhuri A. (2004) Increased plasma concentration of macrophage migration inhibitory factor (MIF) and MIF mRNA in mononuclear cells in the obese and the suppressive action of metformin. J. Clin. Endocrinol. Metab. 89, 5043–5047 [DOI] [PubMed] [Google Scholar]
- 15. Hattori Y., Suzuki K., Hattori S., Kasai K. (2006) Metformin inhibits cytokine-induced nuclear factor κB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension 47, 1183–1188 [DOI] [PubMed] [Google Scholar]
- 16. Huang N. L., Chiang S. H., Hsueh C. H., Liang Y. J., Chen Y. J., Lai L. P. (2009) Metformin inhibits TNF-α-induced IκB kinase phosphorylation, IκB-α degradation and IL-6 production in endothelial cells through PI3K-dependent AMPK phosphorylation. Int. J. Cardiol. 134, 169–175 [DOI] [PubMed] [Google Scholar]
- 17. Isoda K., Young J. L., Zirlik A., MacFarlane L. A., Tsuboi N., Gerdes N., Schönbeck U., Libby P. (2006) Metformin inhibits proinflammatory responses and nuclear factor-κB in human vascular wall cells. Arterioscler. Thromb. Vasc. Biol. 26, 611–617 [DOI] [PubMed] [Google Scholar]
- 18. Cai Y., Zhang C., Nawa T., Aso T., Tanaka M., Oshiro S., Ichijo H., Kitajima S. (2000) Homocysteine-responsive ATF3 gene expression in human vascular endothelial cells: activation of c-Jun NH2-terminal kinase and promoter response element. Blood 96, 2140–2148 [PubMed] [Google Scholar]
- 19. Hai T., Wolfgang C. D., Marsee D. K., Allen A. E., Sivaprasad U. (1999) ATF3 and stress responses. Gene Expr. 7, 321–335 [PMC free article] [PubMed] [Google Scholar]
- 20. Hashimoto Y., Zhang C., Kawauchi J., Imoto I., Adachi M. T., Inazawa J., Amagasa T., Hai T., Kitajima S. (2002) An alternatively spliced isoform of transcriptional repressor ATF3 and its induction by stress stimuli. Nucleic Acids Res. 30, 2398–2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mayr B., Montminy M. (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2, 599–609 [DOI] [PubMed] [Google Scholar]
- 22. Gilchrist M., Thorsson V., Li B., Rust A. G., Korb M., Roach J. C., Kennedy K., Hai T., Bolouri H., Aderem A. (2006) Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 441, 173–178 [DOI] [PubMed] [Google Scholar]
- 23. Suganami T., Yuan X., Shimoda Y., Uchio-Yamada K., Nakagawa N., Shirakawa I., Usami T., Tsukahara T., Nakayama K., Miyamoto Y., Yasuda K., Matsuda J., Kamei Y., Kitajima S., Ogawa Y. (2009) Activating transcription factor 3 constitutes a negative feedback mechanism that attenuates saturated fatty acid/toll-like receptor 4 signaling and macrophage activation in obese adipose tissue. Circ. Res. 105, 25–32 [DOI] [PubMed] [Google Scholar]
- 24. Nguyen T. T., Johnsen I. B., Knetter C. F., Drabløs F., Fitzgerald K. A., Lien E., Anthonsen M. W. (2010) Differential gene expression downstream of toll-like receptors (TLRs) Role of c-Src and activating transcription factor 3 (ATF3). J. Biol. Chem. 285, 17011–17019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burcelin R. (2012) Regulation of metabolism: a crosstalk between gut microbiota and its human host. Physiology 27, 300–307 [DOI] [PubMed] [Google Scholar]
- 26. Cha J. Y., Repa J. J. (2007) The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J. Biol. Chem. 282, 743–751 [DOI] [PubMed] [Google Scholar]
- 27. UK Prospective Diabetes Study (UKPDS) Group (1998) Effects of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 352, 854–865 [PubMed] [Google Scholar]
- 28. De Jager J., Kooy A., Lehert P., Bets D., Wulffelé M. G., Teerlink T., Scheffer P. G., Schalkwijk C. G., Donker A. J., Stehouwer C. D. (2005) Effects of short-term treatment with metformin on markers of endothelial function and inflammatory activity in type 2 diabetes mellitus: a randomized, placebo-controlled trial. J. Intern. Med. 257, 100–109 [DOI] [PubMed] [Google Scholar]
- 29. Zou M. H., Hou X. Y., Shi C. M., Nagata D., Walsh K., Cohen R. A. (2002) Modulation by peroxynitrite of Akt-and AMP-activated kinase-dependent Ser1179 phosphorylation of endothelial nitric oxide synthase. J. Biol. Chem. 277, 32552–32557 [DOI] [PubMed] [Google Scholar]
- 30. Pilon G., Dallaire P., Marette A. (2004) Inhibition of inducible nitric oxide synthase by activators of AMP-activated protein kinase: a new mechanism of action of insulin-sensitizing drugs. J. Biol. Chem. 279, 20767–20774 [DOI] [PubMed] [Google Scholar]
- 31. Jhun B. S., Jin Q., Oh Y. T., Kim S. S., Kong Y., Cho Y. H., Ha J., Baik H. H., Kang I. (2004) 5-Aminoimidazole-4-carboxamide riboside suppresses lipopolysaccharide-induced TNF-α production through inhibition of phosphatidylinositol 3-kinase/Akt activation in RAW264.7 murine macrophages. Biochem. Biophys. Res. Commun. 318, 372–380 [DOI] [PubMed] [Google Scholar]
- 32. Kuo C. L., Ho F. M., Chang M. Y., Prakash E., Lin W. W. (2008) Inhibition of lipopolysaccharide-induced inducible nitric oxide synthase and cyclooxygenase-2 gene expression by 5-aminoimidazole-4-carboxamide riboside is independent of AMP-activated protein kinase. J. Cell. Biochem. 103, 931–940 [DOI] [PubMed] [Google Scholar]
- 33. Nilsson R., Bajic V. B., Suzuki H., di Bernardo D., Björkegren J., Katayama S., Reid J. F., Sweet M. J., Gariboldi M., Carninci P., Hayashizaki Y., Hume D. A., Tegner J., Ravasi T. (2006) Transcriptional network dynamics in macrophage activation. Genomics 88, 133–142 [DOI] [PubMed] [Google Scholar]
- 34. Ho H. H., Antoniv T. T., Ji J. D., Ivashkiv L. B. (2008) Lipopolysaccharide-induced expression of matrix metalloproteinases in human monocytes is suppressed by IFN-γ via superinduction of ATF-3 and suppression of AP-1. J. Immunol. 181, 5089–5097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Whitmore M. M., Iparraguirre A., Kubelka L., Weninger W., Hai T., Williams B. R. (2007) Negative regulation of TLR-signaling pathways by activating transcription factor-3. J. Immunol. 179, 3622–3630 [DOI] [PubMed] [Google Scholar]
- 36. Gilchrist M., Henderson W. R., Jr., Clark A. E., Simmons R. M., Ye X., Smith K. D., Aderem A. (2008) Activating transcription factor 3 is a negative regulator of allergic pulmonary inflammation. J. Exp. Med. 205, 2349–2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Johnson G. L., Lapadat R. (2002) Mitogen-activated protein kinase pathways mediated by ERK, JNK and p38 protein kinases. Science 298, 1911–1912 [DOI] [PubMed] [Google Scholar]
- 38. Kim Y. D., Kim Y. H., Cho Y. M., Kim D. K., Ahn S. W., Lee J. M., Chanda D., Shong M., Lee C. H., Choi H. S. (2012) Metformin ameliorates IL-6-induced hepatic insulin resistance via induction of orphan nuclear receptor small heterodimer partner (SHP) in mouse models. Diabetologia 55, 1482–1494 [DOI] [PubMed] [Google Scholar]
- 39. Hua B., Tamamori-Adachi M., Luo Y., Tamura K., Morioka M., Fukuda M., Tanaka Y., Kitajima S. (2006) A splice variant of stress response gene ATF-3 counteracts NF-κB-dependent anti-apoptosis through inhibiting recruitment of CREB-binding protein/p300 coactivator. J. Biol. Chem. 281, 1620–1629 [DOI] [PubMed] [Google Scholar]