

Abstract
Invariant Natural Killer T-cells (iNKT) cells are emerging as key mediators of innate immune cellular and inflammatory responses to sepsis and peritonitis. iNKT-cells mediate survival following murine septic shock. Macrophages are pivotal to survival following sepsis. iNKT-cells have been shown to modulate various mediators of the innate immune system, including macrophages. Herein we demonstrate sepsis inducing iNKT-cell exodus from the liver appearing in the peritoneal cavity, the source of the sepsis. This migration was affected by Programmed Death Receptor-1(PD-1). PD-1 is an inhibitory immune receptor, reported as ubiquitously expressed at low levels on iNKT-cells. PD-1 has been associated with markers of human critical illness. PD-1 deficient iNKT-cells failed to demonstrate similar migration. To the extent that iNKT-cells affected peritoneal macrophage function we assessed peritoneal macrophages ability to phagocytose bacteria. iNKT−/− mice displayed dysfunctional macrophage phagocytosis and altered peritoneal bacterial load. This dysfunction was reversed when peritoneal macrophages from iNKT−/− mice were co-cultured with wild type iNKT-cells. Together, our results indicate that sepsis induces liver iNKT-cell exodus into the peritoneal cavity mediated by PD-1, and these peritoneal iNKT-cells appear critical to regulation of peritoneal macrophage phagocytic function. iNKT-cells offer therapeutic targets for modulating immune responses and detrimental effects of sepsis.
Keywords: Invariant Natural Killer T-cells (iNKT-cells), Programmed Death Receptor-1 (PD-1), Sepsis
INTRODUCTION
Sepsis remains a leading cause of death in Intensive Care Units (ICUs) worldwide. Despite aggressive ICU care, early resuscitation, and source control, mortality remains dismally high between 30 and 80%. Sepsis is a complex interplay between pro- and anti- inflammatory responses, coupled with activation and alterations in cellular components of both the innate and acquired arms of the immune system[1]. Furthermore, sepsis is associated with a dysfunction and loss of lymphocytes in both mice and humans[2]. However, as most studies have focused on the more overt T-lymphocyte sub-populations (as defined by CD3+) such as CD4+, CD8+ and/or more recently CD4+CD25+[2–4], comparatively little attention has been paid to the changes and/or contribution of some of the more minor innate-regulatory CD3+ T-cell sub-populations, such as the invariant Natural Killer T-cells (iNKT-cells) or gamma-delta TCR+ T-cell.
With respect to iNKT-cells, they are a pluri-potent subset of lymphocytes capable of bridging the innate and adaptive immune systems and producing either a Th-1 or a Th-2 response. Considerable evidence is emerging demonstrating not only a central role for iNKT-cells in many disease states, including cancer, infections[5], and auto-immune diseases, it is also becoming more evident that iNKT-cells are instrumental in the proper coordination of the immune response to sepsis. We have previously demonstrated a role for iNKT-cells in response to polymicrobial sepsis[6]. Response to and control of sepsis is dependent on an appropriate innate immune response, of which macrophages play an important role, especially in relation to their ability to phagocytose and clear bacteria[7].
Growing evidence suggests that in varying infectious and non-infectious disease states, iNKT-cells interact with and alter function of various phagocytic/myeloid cell members of the innate immune system[8–10], most notably macrophages, including during more indolent and chronic infections[6,11]. However the effect of iNKT-cells on macrophages during sepsis is not well understood. The exact role of iNKT-cells in infections is variable and appears to be highly dependent of whether the infection is intra- versus extra-cellular in origin.
We have previously demonstrated that polymicrobial sepsis is associated with an increased activation of both liver and circulating iNKT-cells[6]. This was coupled with the finding that the liver iNKT-cell population declined rapidly following the onset of sepsis. It has recently been shown that iNKT-cells are capable of rapid migration within the liver[10,12]. The iNKT-cell migration appeared to occur in a surveillance fashion. We hypothesized that the decline of iNKT-cells may indeed be a reflection of migration out of the liver and into the site of the infection, namely the peritoneal cavity in our model of cecal ligation and puncture (CLP)-induced peritonitis. Programmed death receptor-1 (PD-1) is a co-inhibitory surface protein, classically described as controlling T-cells in immune responses. PD-1 has been implicated in survival following sepsis[13–16], and has been shown to play a key role regulating iNKT-cell functioning[17]. Thus we set out to test the hypothesis that sepsis induces changes in iNKT-cell frequency and number reflective of a migration from the liver to the site of infection (peritoneal cavity), and this would be affected by PD-1. Further, we hypothesized that peritoneal macrophage functioning is dependent upon iNKT-cells.
MATERIALS AND METHODS
Animals
C57BL/6J mice (from The Jackson Laboratory) were used for wild type. C57BL/6 mice deficient in either iNKT cells (Jα18−/−) or in PD-1 (PD1−/−) (bred at Rhode Island Hospital) were used. Mice aged 10 to 14 weeks of age were used in all experimental procedures. 6–8 mice were used for each group. Research objectives and all animal protocols were approved by the Institutional Animal Care and Use Committee of Rhode Island Hospital and conducted in accordance with the Animal Welfare Act and National Institutes of Health guidelines for animal care and use.
Septic Model
The CLP protocol established by Baker et al.[18] and modified by us[19] was used to induce polymicrobial septic shock in mice. In brief, the cecum was exposed through a midline incision, ligated and subjected to two punctures with a 22-gauge needle. Sham surgery mice were subjected to anesthesia and midline laparotomy without ligation or puncture of the caecum. 12 hours after the procedures, mice were euthanized with an overdose of CO2. Blood was collected via cardiac puncture into heparinized syringes and peritoneal fluid was collected via peritoneal lavage.
Circulating and Peritoneal Lymphocyte levels
Whole blood and peritoneal fluid analysis was undertaken to assess the lymphocytic profile. Monoclonal antibodies directed against CD3, TCR pan- γδ and CD69 (a marker of early cell activation) were used, according to the manufacturer’s recommendations. To identify iNKT-cells we used α-GalCer pre-loaded CD1d tetramers conjugated to Allophycocyanin (APC) (specific for the Vα14Jα18-TCR). The control was unloaded tetramer, both of which have been obtained from the NIAID Tetramer Facility (Germantown, MD).
iNKT-cell isolation
Miltenyi magnetic bead separation techniques were applied according to manufacturer’s recommendations. Non-parenchymal cells (NPCs) were isolated from wild type livers over a Percoll gradient. The NPCs were washed and the pellet re-suspended. The T-cells were isolated using the Miltenyi MACS pan-T-cell isolation kit. These cells were collected and stained for CD1d tetramer APC. Anti-APC magnetic beads were conjugated against the CD1d allowing positive selection of iNKT-cells.
Peritoneal Macrophage Isolation and Functioning
Peritoneal aspirate from wild-type and iNKT−/− mice was collected following both sham and CLP procedures[13]. The aspirate was pelleted and resuspended at a concentration of 106 macrophages/ml. Macrophages were plated onto 24-well tissue culture plates at 1ml/well and incubated for 1 hour allowing for macrophage adherence. Following washing, 200 microliters of approximately 1×107 PE-labelled Escherichia coli in PBS was added and incubated. Plates were washed and adherent macrophages were detached. Flow cytometery was used to calculate the percentage of macrophages taking up E. coli.
Peritoneal Microbiology sampling
Peritoneal lavage fluid was aspirated and serially diluted in sterile saline. 100-microliter aliquots of 1:100 and 1:10,000 dilutions were then spread on tryptic soy agar (TSA) blood agar plates (Remel, Lenexa, KS) and analyzed 24 hours following incubation at 37 °C. Colonies were counted and expressed as colony forming units (CFU)/ml of peritoneal lavage samples.
Statistical Analysis
Data are expressed as mean and standard error of the mean. Logarithmic transformation of bacterial counts was undertaken for the purposes of analysis of bacterial load in the mouse peritoneal cavity. Categorical data was assessed using Chi-squared or Fisher’s exact test. Mann-Whitney U was used to assess continuous data across two groups. One way analysis of variance (ANOVA) with Holm-Sidak post-hoc analysis was used for continuous data across multiple groups. Alpha was set to 0.05.
RESULTS
We have previously documented that mice either deficient in iNKT-cells or who were administered CD1d-antibody blockade were noted to have decreased mortality in an experimental model of septic shock[6]. Furthermore sepsis was noted to induce a decline in liver iNKT-cells. We wished to therefore assess for a possible mechanism driving this finding. Inasmuch, it was initially important to determine to what extent iNKT-cell populations were changing in the blood and/or peritoneal cavity.
Following the onset of sepsis, circulating CD3+ lymphocytes were noted to be significantly decreased in both wild type and iNKT−/− mice. Unlike the blood, following CLP, the peritoneal cavity of wild type mice displayed no difference in the frequency of CD3+ lymphocytes. However, following CLP the iNKT−/− mice, unlike the wild-type controls, displayed an increase in peritoneal CD3+ lymphocytes (Figure 1).
Figure 1. Percentage of peripheral circulating and peritoneal CD3+-lymphocytes in wild type and iNKT−/− mice.

Following CLP, there was a decline in percentage of circulating CD3+-lymphocytes in both wild type (^ p<0.001) and iNKT−/− (# p=0.001) mice. With respect to the peritoneal cavity following CLP compared to Sham, there was no difference in wild type CD3+-lymphocytes; however iNKT−/− mice displayed an increase in percentage of peritoneal CD3+-lymphocytes (* p<0.001). Notably there was no difference in sham peritoneal CD3+-lymphocytes comparing wild type and iNKT−/− mice. However with respect to CLP, peritoneal CD3+ Lymphocytes were significantly greater in iNKT−/− mice compared to wild type (** p<0.001).
Intriguingly, following CLP there was an increase in the frequency of circulating CD1d-tetramer+ cells (iNKT-cells) as an absolute number and as a portion of CD3+ lymphocytes in septic mice (14.2 +/− 2.5% versus 4.8 +/− 0.9%; p=0.003). Furthermore, a significantly larger percent of circulating iNKT-cells were noted to express CD69, a marker of activation, in response to CLP (see Table 1a). We then assessed the iNKT-cell profile of the peritoneal fluid (as an index of the comparative response at site more proximal to the source of infection). Similar to the blood, there was an increase in iNKT-cells both in absolute number and as a percentage of CD3+ T-cells (7.9 +/− 1.6% versus 3.2 +/− 1.2%; p=0.03). Further, significantly more peritoneal iNKT-cells expressed the activation marker CD69 (73.2 +/−19% versus 6.9 +/−0.9%; p=0.004) (see Table 1b).
Table 1 a) and b). Levels of iNKT-cells in a) the peripheral circulation and b) peritoneal cavity of mice subjected to either sham laparotomy or CLP-induced sepsis.
An increase in the frequency of iNKT-cells in the circulation was noted following CLP, both as an absolute number and as a percentage of CD3+-lymphocytes. Significantly more circulating iNKT-cells were activated. Within the peritoneal cavity, there was also an increase in iNKT-cells following CLP, most of which were noted to be activated.
| 1a) – Peripheral Circulation | |||
|---|---|---|---|
| Peripheral Circulation | Sham | Sepsis | p value |
| Number of iNKT-cells | 8 +/− 0.6 ×103 | 19 +/− 1.5 ×103 | 0.02 |
| iNKT-cells as % of CD3+ cells | 4.8% (+/− 0.9) | 14.2% (+/− 2.5) | 0.003 |
| CD69+ iNKT-cells | 3.7% (+/− 1.2) | 22.1% (+/− 2.5) | <0.001 |
| 1b) – Peritoneal Cavity | |||
|---|---|---|---|
| Peritoneal Cavity | Sham | Sepsis | p value |
| Wild Type | |||
| Number of iNKT-cells | 6 +/− 0.7 ×103 | 31 +/− 2.1 ×103 | <0.01 |
| iNKT-cells as % of CD3+ cells | 3.2% (+/− 1.2) | 7.9% (+/− 1.6) | 0.03 |
| CD69+ iNKT-cells | 6.9% (+/− 0.9) | 73.2% (+/− 19) | 0.004 |
To assess the possible contribution of the inhibitory immune receptor PD-1 expression to this noted migration of iNKT-cells into the peritoneal cavity following CLP, we undertook an analysis of iNKT-cell populations following CLP in various anatomic locations in PD-1−/− mice. As previously observed[6], sepsis induced a loss of absolute number of hepatic iNKT-cells in wild type mice when compared with sham. In wild type mice, there was an increase in the percentage of iNKT-cells that were activated. However, in PD-1−/− mice, while sepsis was noted to induce an increase in absolute number of hepatic iNKT-cells when compared with sham, there was no subsequent change in the percentage of activated iNKT-cells (see Figure 2 and Table 2).
Figure 2. iNKT-cell profile within a) liver, b) blood, and c) peritoneum following CLP comparing wild type versus PD1−/− mice.
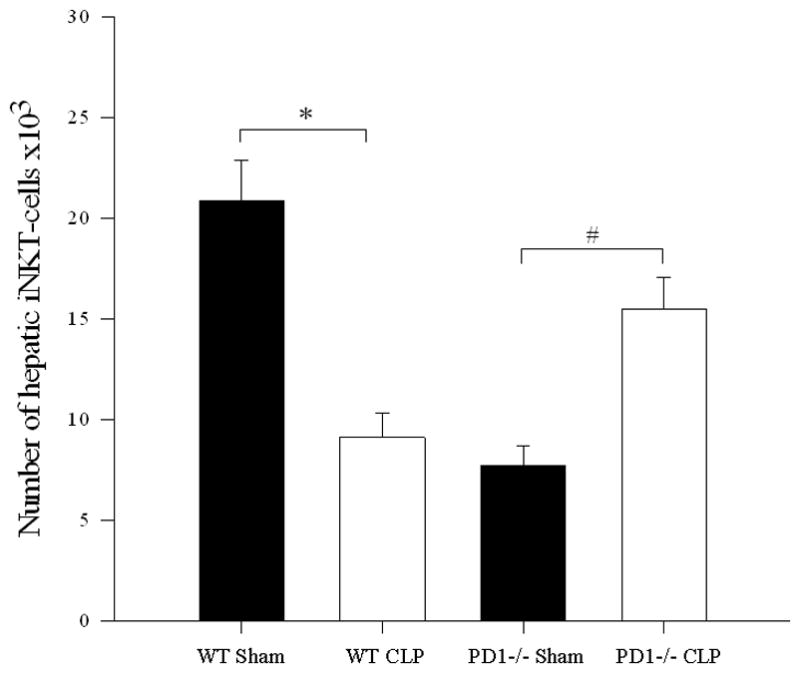
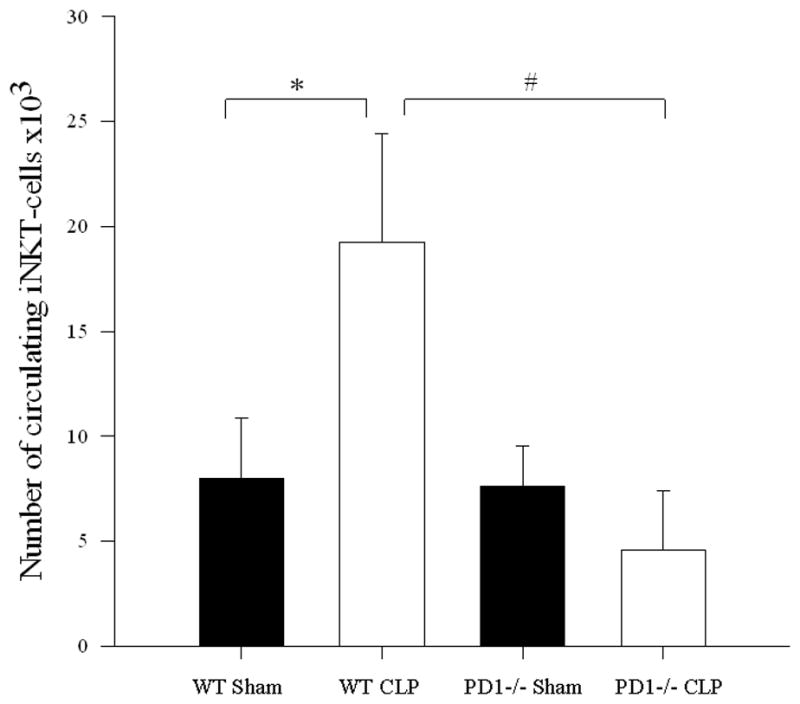
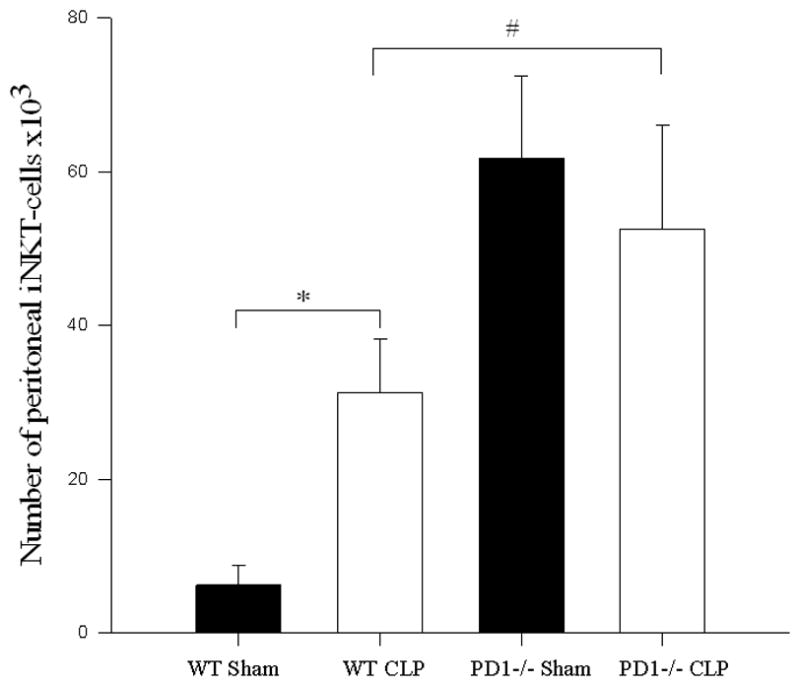
a) Following CLP, there was a decrease in hepatic iNKT-cells in wild type mice (* p<0.001), but an increase in hepatic iNKT-cells in PD1−/− mice (# p=0.004). b) In the peripheral circulation, although wild type mice displayed an increase in circulating iNKT-cells (* p<0.001), there was no difference in the number of circulating iNKT-cells between sham and CLP within PD1−/− mice. PD-1−/− mice had significantly fewer circulating iNKT-cells than wild type mice following CLP (# p<0.001). c) Within the peritoneal cavity, wild type mice displayed an increase in iNKT-cells following CLP (* p<0.001), whereas there was no change in the number of iNKT-cells in PD1−/− mice. Notably PD-1−/− mice had greater peritoneal iNKT-cells compared to wild type following CLP (# p<0.001).
Table 2. The effect of PD-1 deficiency upon iNKT-cell activation and migration.
PD-1 deficiency affected iNKT-cell migration and iNKT-cell activation. Although hepatic iNKT-cells were noted to have higher rates of activation following CLP, this was not evident in PD-1−/− mice. Conversely, in PD-1−/− mice, the rates of activation of circulating and peritoneal PD-1−/− iNKT-cells were significantly elevated following CLP.
| Sham | Sepsis | p-value | |
|---|---|---|---|
| Wild Type mice | |||
| iNKT-cells as a % of CD3 | 48 +/− 2.9% | 34 +/− 1.7% | 0.001 |
| % of hepatic CD69+ iNKT-cells | 28 +/− 2.3% | 42 +/− 4.4% | 0.014 |
| PD-1 −/− mice | |||
| Hepatic | |||
| iNKT-cells as a % of CD3 | 37 +/− 1.7% | 59 +/− 2.4% | 0.02 |
| % of CD69+ iNKT-cells | 19.7 +/− 2.6% | 18.9 +/− 4.2% | 0.87 |
| Circulating | |||
| iNKT-cells as a % of CD3 | 5.4 +/− 0.7% | 6.2 +/− 1.9% | 0.7 |
| % of CD69+ iNKT-cells | 1.5 +/− 0.4% | 19.0 +/− 5.4% | 0.006 |
| Peritoneal | |||
| iNKT-cells as a % of CD3 | 2.2 +/− 0.8% | 2.8 +/− 0.7% | 0.58 |
| % of CD69+ iNKT-cells | 24 +/− 3.5% | 41 +/− 5.8% | 0.02 |
Following CLP, circulating iNKT-cells were noted to be unchanged compared to sham animals either in absolute number or as a percentage of CD3+ lymphocytes from septic PD-1−/− mice. However, there was a significant increase in the frequency of activation in these circulating cells: in PD-1−/− mice, iNKT-cells from septic animals had significantly higher percent of CD69 expression compared to sham animals (see Figure 2 and Table 2). With respect to the peritoneal cavity, unlike wild type mice, there was no change in either the absolute number or percentage of iNKT-cells in the peritoneal cavity of PD-1−/− septic mice compared to sham. However, in PD-1−/− mice, significantly higher proportions of peritoneal iNKT-cells were CD69+ following sepsis compared with sham mice. Notably, PD-1−/− mice had greater numbers of iNKT-cells in the peritoneal cavity when comparing sham laparotomy to wild type sham as well as comparing CLP in PD-1−/− to CLP in wild type mice (see Figure 2 and Table 2).
To the extent that these changes in iNKT cell frequency and the degree of their activation (increased percentage of CD69+ expression) culminated in significant functional effect, we next attempted to determine if the ex vivo capacity of peritoneal macrophages to engulf E. coli was altered if they were derived from iNKT−/− or wild type background control mice 12 hours following sham laparotomy or CLP. Following induction of sepsis, in keeping with our prior report[13], we initially noted that wild type mouse-derived peritoneal macrophages were observed to have a decreased ability to uptake E. coli when compared to wild type sham mouse derived macrophages. However, it was noted that there conversely was an increase in the ability of iNKT−/− derived peritoneal macrophages to engulf E. coli (Figure 3).
Figure 3. Percentage of peritoneal macrophages engulfing E. coli following sham or sepsis.
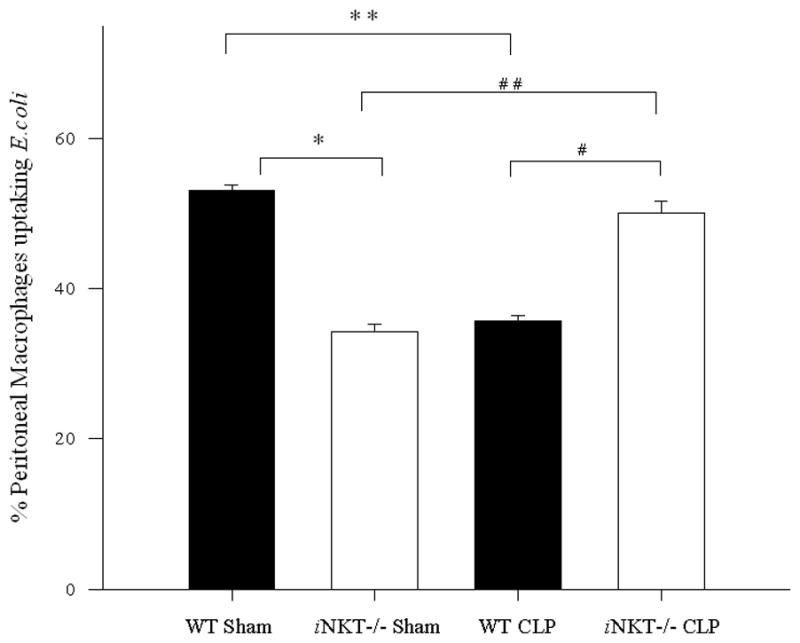
Wild type mice showed significantly decreased peritoneal macrophage uptake of E. coli comparing sham to CLP (** p<0.001), whereas iNKT−/− mice showed an increase in percentage of peritoneal macrophages up taking E. coli comparing sham to CLP (## p<0.001). Wild type mice, compared with iNKT−/− mice, showed significantly increased percentage of E. coli uptake by peritoneal macrophages following sham laparotomy (* p<0.001). However, wild type mice had significantly lower bacterial uptake following CLP compared to iNKT−/− mice. (# p<0.001).
To the extent that this change was a direct result of the absence of the iNKT-cells in the knockout animal from which they were derived, we found that when peritoneal macrophages from iNKT −/− mice were co-cultured with iNKT-cells isolated from septic wild type mice displayed a return to normal levels of engulfment compared with wild type (Figure 4).
Figure 4. E. coli engulfment by iNKT−/− peritoneal macrophages co-cultured with iNKT-cells.
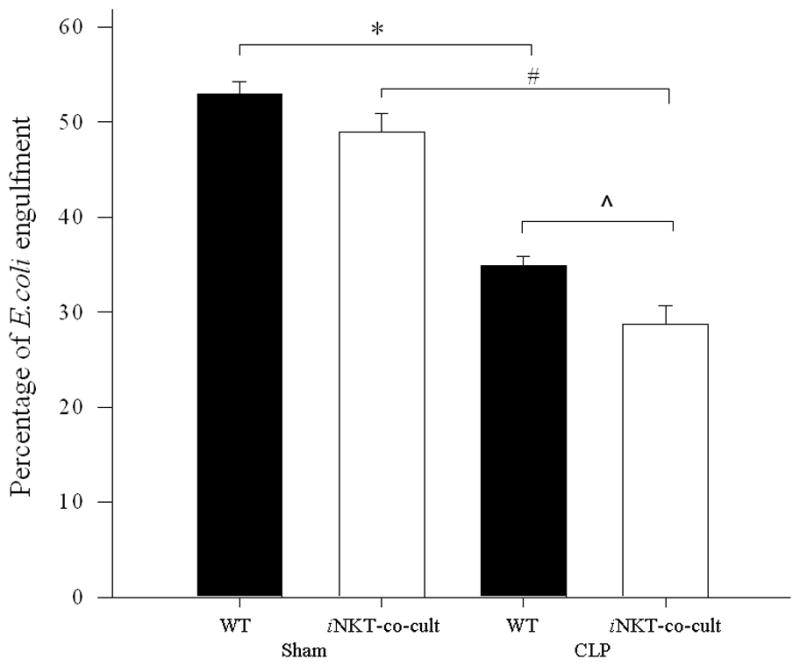
Peritoneal macrophages from iNKT−/− mice subjected to sham or sepsis were co-cultured with iNKT cells isolated from wild type mice. Macrophage uptake of E. coli uptake was assessed. There was no difference in percentage of E. coli uptake following sham laparotomy between wild type macrophages and iNKT−/− macrophages co-cultured with iNKT-cells. The previously noted decrease in E. coli uptake in wild type macrophages was now noted between sham and CLP for iNKT−/− macrophages co-cultured with iNKT-cells (# p<0.001). Notably the percentage of E. coli engulfment was lower for iNKT−/− macrophages co-cultured with iNKT-cells following CLP when compared with wild type sepsis peritoneal macrophages (^ p=0.023).
To the degree that these ex vivo effects on bacterial uptake reflect changes in bacterial clearance/killing we also assessed overall effect of the absence of iNKT-cells on the peritoneal bacterial burden in septic mice by culturing the peritoneal lavage fluid from these animals. Not surprisingly, we found there were no significant differences in the bacterial load in peritoneal fluid following sham protocol between iNKT knockout mice compared with wild type mice. Following CLP, iNKT −/− mice demonstrated slightly increased bacterial load compared to sham, but this was a significantly reduced bacterial load when compared to wild type mice (Figure 5).
Figure 5. Bacterial load in the peritoneal cavity following sham or sepsis (CLP) in wild type versus iNKT−/− mice.
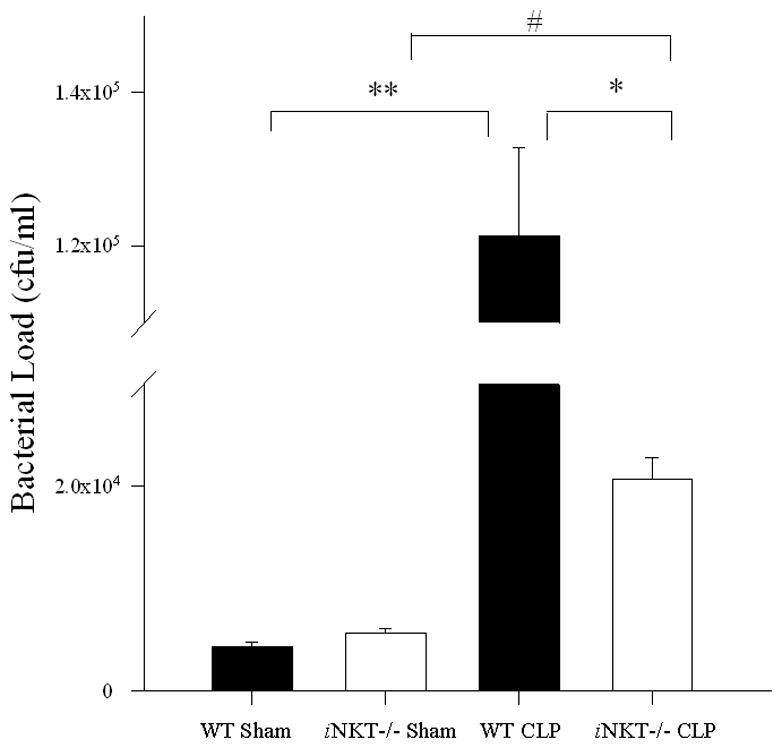
Compared to their respective sham groups, both wild type (** p<0.001) and iNKT−/− mice (* p<0.001) displayed increased bacterial load following CLP. However, iNKT−/− mice exhibited significantly lower peritoneal bacterial load when compared with wild type mice following CLP (# p<0.001).
DISCUSSION
Sepsis, a leading cause of death world-wide, induces dysfunction and loss of lymphocytes, which is thought to contribute to the overall morbidity of this condition[1,3,20]. Innate regulatory T-cells are emerging as key modulators of appropriate immune responses, specifically invariant Natural Killer T-cells (iNKT-cells). iNKT cells regulate immune response in many clinically significant infectious conditions[11, 21,22], and auto-immune disease models[23]. We have previously shown that iNKT-cells play a key role in modulating the immune response to sepsis. Following an otherwise fatal model of polymicrobial sepsis, mice either administered CD1d blocking antibody or genetically deficient in iNKT-cells (Jα18−/−) exhibited a survival rate of 35%, a survival advantage manifested as early as 48 hours[6].
iNKT-cells are noted to be relatively resistant to apoptosis, and that when activated there is an up-regulation of anti-apoptotic genes[24]. This led us to postulate that the decreased iNKT-cell liver population[6] may in fact be due to migration as opposed to loss via cell death. The results presented here expand on our findings by showing that, unlike the otherwise global loss of T-lymphocytes[25], iNKT-cells as a sub-population appear to be preserved in the peripheral circulation of mice, as well as in their peritoneal cavity, in response to an abdominal septic source. We believe that this data supports the hypothesis that acute sepsis induces an exodus of iNKT-cells from the liver, migrating through the blood stream and into the peritoneal cavity, the source of infection.
iNKT-cell / macrophage interactions have been noted in several chronic disease states and models. Macrophage influx and activity has been noted to be dependent upon iNKT-cells in diet induced obesity [26], progression of atherosclerotic lesions[27] and models of rheumatoid arthritis[28]. Further, iNKT-cells activate immune cells critical to successful responses to an infection[9,23,29,30] including natural killer cells, macrophages, dendritic cells, T-cells and B-cells. However this interaction appears to be bi-directional wherein macrophages have been noted to modulate iNKT-cell responses. The effector and target cells appear dependent upon both the model used, such as burns [31] versus abdominal sepsis, as well as the background mouse strain used.
This iNKT-cell / macrophage interaction appears to be mediated via Interferon (IFN)-γ and Tissue Necrosis Factor (TNF)-α, both found in abundance and mobilized from stimulated iNKT-cells [29], as well as through CD1-mediated antigen presentation in macrophages and dendritic cells. Emoto et al. demonstrated a significant role for iNKT-cells in peritoneal macrophage bactericidal activity, postulating that, following stimulation, iNKT-cells infiltrated the peritoneal cavity[9]. We expand upon their postulation by demonstrating an expansion of the peritoneal iNKT-cell population (7.9% versus 3.2%; p=0.03) in the setting of sepsis, most of which were noted to be activated (73.2% versus 6.9%; p=0.004).
Our data showing that iNKT-cells significantly affect peritoneal macrophage engulfment, a finding reversed by the addition of iNKT-cells, supports prior observations that iNKT-cells play key roles in mediating important cellular responses, specifically in relation to macrophage function[6,32] and cellular migration and/or recruitment to a site of infection[33]. We noted that in an acute polymicrobial infection, iNKT-cells affected peritoneal bacterial clearance with lower peritoneal bacterial burden following CLP in iNKT−/− mice. Despite a relatively minor change in the absolute number of iNKT-cells we still observed a significant biological effect in macrophage function. We believe that this further speaks to the regulatory nature of iNKT-cells wherein their relatively small numbers as a sub-population belies their ability to enact a large biological response.
The liver is the predominant source of iNKT-cells in mice[6]. Furthermore, it is noted that most facultative intracellular bacteria are trapped in the liver immediately after systemic infection. The high abundance of liver iNKT-cells, and their rapid and vigorous cytokine release in response to sepsis[6] is highly suggestive of a sentinel role for iNKT-cells in response to systemic infections. Prior studies have shown that not only do liver iNKT-cells interact with tissue macrophages to activate them, but that these liver macrophages contribute to the systemic pro-inflammatory cytokine response to sepsis[34].
iNKT-cells have recently been shown to be capable of both random and directional migration in a patrolling fashion within the liver in response to bacterial infection[10,12]. To explain a mechanism allowing the observed exodus from the liver and apparent migration into the peritoneal cavity, the source of the infection, we assessed the role of PD-1 in iNKT-cell migration. PD-1 is a co-inhibitory surface protein, classically described as controlling T-cells in immune responses. Monaghan et al., Guignant et al., and Zhang et al. demonstrated that surface expression of PD-1 on circulating cells in critically ill patients correlates with APACHE-II scores, an index of critical illness[16,35,36].
Interaction between PD-1 on T-cells and the ligands, PD-L1 and PD-L2, is classically thought to induce tolerance among T-cells. PD-1 can inhibit a robust immune response and has been noted to correlate with inadequately cleared and chronic viral and fungal infections[37]. It has been proposed that PD-1 on iNKT-cells may prevent excessive iNKT-cell associated inflammation. Activated iNKT-cells have increased cell surface expression of PD-1. Repeat α-GalCer stimulation of iNKT-cells has been noted to induce anergy, which is associated with very high levels of PD-1. Blockage of the interaction between PD-1 and its ligands (PD-L1 and PD-L2) at the time of α-GalCer administration prevented the induction of iNKT-cell anergy[17]. However, whether this is a direct or indirect effect of PD-1:PD-L ligation is not clear.
Active M. tuberculosis infection is associated with deficient NKT-cell proliferation and function, defects associated with elevated PD-1 expression. Blockade of PD-1 signaling enhances NKT-cell response to α-GalCer. However, Huang et al. demonstrated that mice lacking PD-1, or treated with PD-1 blocking antibody, had better survival following sepsis[13]. Indeed the authors noted PD-1 expression modulated bactericidal activity and bacterial clearance. Given the abundance of PD-1 on lymphocytes, we speculated that PD-1 may affect iNKT-cell migration, and that this in turn may offer an explanation for the reduced mortality noted in iNKT-cell knockout mice[6].
We demonstrated that PD-1−/− mice exhibited a significantly blunted migration of iNKT-cells following sepsis, with increased liver iNKT-cell populations, most of which were activated. This is coupled with the finding of no increase in circulating or peritoneal iNKT-cells following sepsis. However, PD-1 deficiency did not affect the sepsis-induced increase in the degree of iNKT-cell activation. Coupling our findings together offers a potential target for future therapy in septic patients. PD-1 blocking antibodies are already available in clinical trials in cancer[38]; thereby, in the future, blocking PD-1 may become a real possibility that might allow one to temporarily modulate the early iNKT-cell response to sepsis, allowing for greater bacterial clearance. In support of this speculation is the recent finding that PD-1 antibody blockade was protective against mortality from sepsis[15].
CONCLUSIONS
iNKT-cells play a key role in the immune response following sepsis. Their actions are mediated in part via their interaction with macrophages. iNKT-cells affect macrophage bacterial clearance. Finally, iNKT-cells migrate from the liver into the peritoneal cavity following CLP. Intriguingly, liver iNKT-cell transmigration (exodus) out of the liver is markedly affected by PD-1 gene expression.
Acknowledgments
Portions of this work were supported by grants from the Shock Society’s-Jr. Faculty Fellowship Award (D.S.H.), the Armand D. Versaci Research Scholar in Surgical Sciences Award (S.F.M. & R.K.T.) as well as from the NIH-NIGMS R01-GM046354 (A.A.).
We graciously acknowledge the provision by Drs. T Honjo [Kyoto University Graduate School of Medicine, Kyoto] and M. Sykes [Massachusetts General Hospital, Transplantation Biology Research Center, Boston] of PD-1 −/− mice, and the provision by Dr. H. Taniguchi, Kanagawa, Japan of iNKT−/− mice.
The manuscript does contain experiments involving animals and approval was obtained from the RIH IACUC (number – 0130-11). This manuscript does not contain human studies.
Abbreviations
- APC
Allophycocyanin
- CD
Cluster of differentiation
- CLP
Cecal ligation and puncture
- iNKT-cells
Invariant Natural Killer T-cells
- NPC
Non-parenchymal cells
- PD-1
Programmed Death Receptor-1
- PD-L1
Programmed Death Receptor Ligand-1
Footnotes
CONFLICTS OF INTEREST
This is to testify that none of the authors has any financial or academic conflict of interest to declare with respect to this manuscript.
References
- 1.Salomao R, Brunialti MK, Rapozo MM, Baggio-Zappia GL, Galanos C, Freudenberg M. Bacterial sensing, cell signaling, and modulation of the immune response during sepsis. Shock. 2012;38(3):227–242. doi: 10.1097/SHK.0b013e318262c4b0. [DOI] [PubMed] [Google Scholar]
- 2.Venet F, Davin F, Guignant C, Larue A, Cazalis MA, Darbon R, Allombert C, Mougin B, Malcus C, Poitevin-Later F, Lepape A, Monneret G. Early assessment of leukocyte alterations at diagnosis of septic shock. Shock. 2010;34(4):358–363. doi: 10.1097/SHK.0b013e3181dc0977. [DOI] [PubMed] [Google Scholar]
- 3.Thakkar R, Huang X, Lomas-Neira J, Heffernan D, Ayala A. Sepsis and the immune response. In: Eremin O, Sewell H, editors. Essential Immunology for Surgeons. Oxford: Oxford University Press; 2011. pp. 303–342. [Google Scholar]
- 4.Wisnoski N, Chung CS, Chen Y, Huang X, Ayala A. The contribution of CD4+ CD25+ T-regulatory-cells to immune suppression in sepsis. Shock. 2007;27(3):251–257. doi: 10.1097/01.shk.0000239780.33398.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olson CM, Jr, Bates TC, Izadi H, Radolf JD, Huber SA, Boyson JE, Anguita J. Local production of IFN-gamma by invariant NKT cells modulates acute Lyme carditis. J Immunol. 2009;182(6):3728–3734. doi: 10.4049/jimmunol.0804111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu CK, Venet F, Heffernan DS, Wang YL, Horner B, Huang X, Chung CS, Gregory SH, Ayala A. The role of hepatic invariant NKT cells in systemic/local inflammation and mortality during polymicrobial septic shock. J Immunol. 2009;182(4):2467–2475. doi: 10.4049/jimmunol.0801463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai TH, Chen SF, Huang TY, Tzeng CF, Chiang AS, Kou YR, Lee TS, Shyue SK. Impaired Cd14 and Cd36 expression, bacterial clearance, and Toll-like receptor 4-Myd88 signaling in caveolin-1-deleted macrophages and mice. Shock. 2011;35(1):92–99. doi: 10.1097/SHK.0b013e3181ea45ca. [DOI] [PubMed] [Google Scholar]
- 8.Wintermeyer P, Cheng CW, Gehring S, Hoffman BL, Holub M, Brossay L, Gregory SH. Invariant natural killer T cells suppress the neutrophil inflammatory response in a mouse model of cholestatic liver damage. Gastroenterology. 2009;136(3):1048–1059. doi: 10.1053/j.gastro.2008.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Emoto M, Yoshida T, Fukuda T, Kawamura I, Mitsuyama M, Kita E, Hurwitz R, Kaufmann SH, Emoto Y. Alpha-galactosylceramide promotes killing of Listeria monocytogenes within the macrophage phagosome through invariant NKT-cell activation. Infect Immun. 2010;78(6):2667–2676. doi: 10.1128/IAI.01441-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee WY, Moriarty TJ, Wong CH, Zhou H, Strieter RM, van Rooijen N, Chaconas G, Kubes P. An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells. Nat Immunol. 2010;11(4):295–302. doi: 10.1038/ni.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sada-Ovalle I, Chiba A, Gonzales A, Brenner MB, Behar SM. Innate invariant NKT cells recognize Mycobacterium tuberculosis-infected macrophages, produce interferon-gamma, and kill intracellular bacteria. PLoS Pathog. 2008;4(12):e1000239. doi: 10.1371/journal.ppat.1000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geissmann F, Cameron TO, Sidobre S, Manlongat N, Kronenberg M, Briskin MJ, Dustin ML, Littman DR. Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol. 2005;3(4):e113. doi: 10.1371/journal.pbio.0030113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, Swan R, Kherouf H, Monneret G, Chung CS, Ayala A. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci U S A. 2009;106(15):6303–6308. doi: 10.1073/pnas.0809422106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monaghan SF, Thakkar RK, Heffernan DS, Huang X, Chung CS, Lomas-Neira J, Cioffi WG, Ayala A. Mechanisms of indirect acute lung injury: a novel role for the coinhibitory receptor, programmed death-1. Ann Surg. 2012;255(1):158–164. doi: 10.1097/SLA.0b013e31823433ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brahmamdam P, Inoue S, Unsinger J, Chang KC, McDunn JE, Hotchkiss RS. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J Leukoc Biol. 2010;88(2):233–240. doi: 10.1189/jlb.0110037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monaghan SF, Thakkar RK, Tran ML, Huang X, Cioffi WG, Ayala A, Heffernan DS. Programmed death 1 expression as a marker for immune and physiological dysfunction in the critically ill surgical patient. Shock. 2012;38(2):117–122. doi: 10.1097/SHK.0b013e31825de6a3. [DOI] [PubMed] [Google Scholar]
- 17.Parekh VV, Lalani S, Kim S, Halder R, Azuma M, Yagita H, Kumar V, Wu L, Kaer LV. PD-1/PD-L blockade prevents anergy induction and enhances the anti-tumor activities of glycolipid-activated invariant NKT cells. J Immunol. 2009;182(5):2816–2826. doi: 10.4049/jimmunol.0803648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baker CC, Chaudry IH, Gaines HO, Baue AE. Evaluation of factors affecting mortality rate after sepsis in a murine cecal ligation and puncture model. Surgery. 1983;94(2):331–335. [PubMed] [Google Scholar]
- 19.Rhee RJ, Carlton S, Lomas JL, Lane C, Brossay L, Cioffi WG, Ayala A. Inhibition of CD1d activation suppresses septic mortality: a role for NK-T cells in septic immune dysfunction. J Surg Res. 2003;115(1):74–81. doi: 10.1016/s0022-4804(03)00220-8. [DOI] [PubMed] [Google Scholar]
- 20.Le Tulzo Y, Pangault C, Gacouin A, Guilloux V, Tribut O, Amiot L, Tattevin P, Thomas R, Fauchet R, Drénou B. Early circulating lymphocyte apoptosis in human septic shock is associated with poor outcome. Shock. 2002;18(6):487–494. doi: 10.1097/00024382-200212000-00001. [DOI] [PubMed] [Google Scholar]
- 21.Kawakami K, Yamamoto N, Kinjo Y, Miyagi K, Nakasone C, Uezu K, Kinjo T, Nakayama T, Taniguchi M, Saito A. Critical role of V alpha14+ natural killer T cells in the innate phase of host protection against Streptococcus pneumoniae infection. Eur J Immunol. 2003;33(12):3322–3330. doi: 10.1002/eji.200324254. [DOI] [PubMed] [Google Scholar]
- 22.Hazlett LD, Li Q, Liu J, McClellan S, Du W, Barrett RP. NKT cells are critical to initiate an inflammatory response after Pseudomonas aeruginosa ocular infection in susceptible mice. J Immunol. 2007;179(2):1138–1146. doi: 10.4049/jimmunol.179.2.1138. [DOI] [PubMed] [Google Scholar]
- 23.Taniguchi M, Seino K, Nakayama T. The NKT cell system: bridging innate and acquired immunity. Nat Immunol. 2003;4(12):1164–1165. doi: 10.1038/ni1203-1164. [DOI] [PubMed] [Google Scholar]
- 24.Seino K, Harada M, Taniguchi M. NKT cells are relatively resistant to apoptosis. Trends Immunol. 2004;25(5):219–221. doi: 10.1016/j.it.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 25.Kasten KR, Tschöp J, Adediran SG, Hildeman DA, Caldwell CC. T cells are potent early mediators of the host response to sepsis. Shock. 2010;34(4):327–336. doi: 10.1097/SHK.0b013e3181e14c2e. [DOI] [PubMed] [Google Scholar]
- 26.Ohmura K, Ishimori N, Ohmura Y, Tokuhara S, Nozawa A, Horii S, Andoh Y, Fujii S, Iwabuchi K, Onoé K, Tsutsui H. Natural killer T cells are involved in adipose tissues inflammation and glucose intolerance in diet-induced obese mice. Arterioscler Thromb Vasc Biol. 2010;30(2):193–199. doi: 10.1161/ATVBAHA.109.198614. [DOI] [PubMed] [Google Scholar]
- 27.Nakai Y, Iwabuchi K, Fujii S, Ishimori N, Dashtsoodol N, Watano K, Mishima T, Iwabuchi C, Tanaka S, Bezbradica JS, Nakayama T, Taniguchi M, Miyake S, Yamamura T, Kitabatake A, Joyce S, Van Kaer L, Onoé K. Natural killer T cells accelerate atherogenesis in mice. Blood. 2004;104(7):2051–2059. doi: 10.1182/blood-2003-10-3485. [DOI] [PubMed] [Google Scholar]
- 28.Miellot-Gafsou A, Biton J, Bourgeois E, Herbelin A, Boissier MC, Bessis N. Early activation of invariant natural killer T cells in a rheumatoid arthritis model and application to disease treatment. Immunology. 2010;130(2):296–306. doi: 10.1111/j.1365-2567.2009.03235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, Schreiber R, Mak TW, Bloom BR. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995;2(6):561–572. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 30.Szalay G, Ladel CH, Blum C, Brossay L, Kronenberg M, Kaufmann SH. Anti-CD1 monoclonal antibody treatment reverses the production patterns of TGF-beta 2 and Th1 cytokines and ameliorates listeriosis in mice. J Immunol. 1999;162(12):6955–6958. [PubMed] [Google Scholar]
- 31.Faunce DE, Gamelli RL, Choudhry MA, Kovacs EJ. A role for CD1d-restricted NKT cells in injury-associated T cell suppression. J Leukoc Biol. 2003;73(6):747–755. doi: 10.1189/jlb.1102540. [DOI] [PubMed] [Google Scholar]
- 32.Ji Y, Sun S, Xu A, Bhargava P, Yang L, Lam KS, Gao B, Lee CH, Kersten S, Qi L. Activation of natural killer T cells promotes M2 Macrophage polarization in adipose tissue and improves systemic glucose tolerance via interleukin-4 (IL-4)/STAT6 protein signaling axis in obesity. J Biol Chem. 2012;287(17):13561–13571. doi: 10.1074/jbc.M112.350066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kok WL, Denney L, Benam K, Cole S, Clelland C, McMichael AJ, Ho LP. Invariant NKT cells reduce accumulation of inflammatory monocytes in the lungs and decrease immune-pathology during severe influenza A virus infection. J Leukoc Biol. 2012;91(3):357–368. doi: 10.1189/jlb.0411184. [DOI] [PubMed] [Google Scholar]
- 34.O’Neill PJ, Ayala A, Wang P, Ba ZF, Morrison MH, Schultze AE, Reich SS, Chaudry IH. Role of Kupffer cells in interleukin-6 release following trauma-hemorrhage and resuscitation. Shock. 1994;1(1):43–47. doi: 10.1097/00024382-199401000-00008. [DOI] [PubMed] [Google Scholar]
- 35.Guignant C, Lepape A, Huang X, Kherouf H, Denis L, Poitevin F, Malcus C, Chéron A, Allaouchiche B, Gueyffier F, Ayala A, Monneret G, Venet F. Programmed death-1 levels correlate with increased mortality, nosocomial infection and immune dysfunctions in septic shock patients. Crit Care. 2011;15(2):R99. doi: 10.1186/cc10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Li J, Lou J, Zhou Y, Bo L, Zhu J, Zhu K, Wan X, Cai Z, Deng X. Upregulation of programmed death-1 on T cells and programmed death ligand-1 on monocytes in septic shock patients. Crit Care. 2011;15(1):R70. doi: 10.1186/cc10059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lázár-Molnár E, Gácser A, Freeman GJ, Almo SC, Nathenson SG, Nosanchuk JD. The PD-1/PD-L costimulatory pathway critically affects host resistance to the pathogenic fungus Histoplasma capsulatum. Proc Natl Acad Sci U S A. 2008;105(7):2658–2663. doi: 10.1073/pnas.0711918105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, Gilson MM, Wang C, Selby M, Taube JM, Anders R, Chen L, Korman AJ, Pardoll DM, Lowy I, Topalian SL. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]