

Abstract

There is growing interest in the topic of intrinsically disordered proteins (IDPs). Atomistic Metropolis Monte Carlo (MMC) simulations based on novel implicit solvation models have yielded useful insights regarding sequence-ensemble relationships for IDPs modeled as autonomous units. However, a majority of naturally occurring IDPs are tethered to ordered domains. Tethering introduces additional energy scales and this creates the challenge of broken ergodicity for standard MMC sampling or molecular dynamics that cannot be readily alleviated by using generalized tempering methods. We have designed, deployed, and tested our adaptation of the Nested Markov Chain Monte Carlo sampling algorithm. We refer to our adaptation as Hamiltonian Switch Metropolis Monte Carlo (HS-MMC) sampling. In this method, transitions out of energetic traps are enabled by the introduction of an auxiliary Markov chain that draws conformations for the disordered region from a Boltzmann distribution that is governed by an alternative potential function that only includes short-range steric repulsions and conformational restraints on the ordered domain. We show using multiple, independent runs that the HS-MMC method yields conformational distributions that have similar and reproducible statistical properties, which is in direct contrast to standard MMC for equivalent amounts of sampling. The method is efficient and can be deployed for simulations of a range of biologically relevant disordered regions that are tethered to ordered domains.
Introduction
A significant percentage of eukaryotic proteins are classified as being intrinsically disordered1. They are referred to as intrinsically disordered proteins or IDPs because, as autonomous units, they fail to fold into well-defined three-dimensional structures. Importantly, IDPs challenge the conventional structure–function paradigm given that they feature prominently in protein–protein and protein-nucleic acid interactions. Many IDPs can adopt ordered structures in specific bound complexes.2 The intrinsic heterogeneity in their unbound forms is reflected in their ability to adopt different folds in the context of different complexes.3 An additional area of interest is the ability of IDPs to mediate the formation of nonmembrane bound liquid-like or gel-like supramolecular assemblies via microscale phase transitions.4
Within the sequence-structure–function paradigm, functional annotation is enabled by the ability to assign a fold to an amino acid sequence.5 IDPs challenge this paradigm because they adopt heterogeneous ensembles of conformations in aqueous solutions for which no single structure or collection of structures provides an adequate description. This does not imply that the sequences of IDPs encode random conformational preferences. In fact, coarse grained6 and atomistic simulations7 in conjunction with single molecule,8 ensemble,9 and other10 experiments have provided synergistic descriptions of the conformational ensembles sampled by different categories of IDP sequences. These efforts have yielded a predictive “diagram of states”,7c which provides a framework for quantitative coarse grain polymeric descriptions of the type of conformational ensemble, such as rod, coil, globule, or chimera, that an IDP sequence will most likely access for given solution conditions.11 Additionally, the degree of heterogeneity in conformational ensembles of IDPs can also be quantified.12 Accordingly, descriptions of sequence-ensemble relationships and comparative assessments of conformational heterogeneity have enabled the use of de novo design approaches to modulate the conformational properties of IDPs. In reality, a majority of IDPs are in fact intrinsically disordered regions (IDRs) that are tethered to ordered domains (ODs).13 While the study of IDRs as autonomous units yields useful insights regarding their sequence-ensemble relationships, it is imperative that the biophysical properties of these regions be studied in their naturally occurring contexts, that is, tethered to ODs. Figure 1 shows schematics for three categories of IDRs. Based on the tethering mode IDRs are either bristles or linkers. If the number of bristles is greater than one, then the collection of tethered IDRs make up a polymer brush (see Figure 1).14
Figure 1.

Schematic representations of different modes that are expected for IDRs. These include the bristle (panel A), linker (panel B), and brush (panel C) modes. In each panel, the IDRs are sketched in black and the ordered domains are shown in green. Short Linear Motifs or SLiMs45 are shown as red triangles and those SLiMs that can be the target of post-translational modifications are depicted as blue triangles. In the bristle mode, a single IDR is tethered to an ordered domain. Panel B depicts the linker mode where the IDR connects two ordered domains. Panel C shows the polymer brush mode whereby multiple bristles are either tethered to a single ordered domain (left), a single filament (middle) as in intermediate filaments46/microtubules,47 or grafted onto the surface lining the interior of a pore (right) as is the case with nuclear pore complexes.48
Simulation Challenge for Tethered Systems
The amplitudes of conformational fluctuations of a disordered region are likely to be larger than the relatively rigid OD. Further, the energy landscapes of IDRs are expected to be “weakly funneled”,15 whereby conformational heterogeneity results from equivalent thermodynamic preferences of disordered regions for multiple, albeit conformationally distinct minima. In standard Metropolis Monte Carlo (S-MMC) simulations, we deploy a series of moves that are designed to be ergodic to enable the sampling of a broad range of conformations that are thermodynamically relevant for IDPs.16 Tethering an IDP to an OD introduces a new set of interactions viz., those between the IDR and the OD. Interactions within the IDR and between the IDR and OD can create deep energy traps. These traps can severely limit the range of thermodynamically relevant conformations that are sampled in a typical S-MMC simulation.
A predictive “diagram of states” for IDPs7c,7e that connects sequence compositional biases to conformational classes has emerged from high-throughput investigations whereby atomistic simulations were carried out for O(102) archetypal sequences drawn from databases such as DisProt.17 These ensembles of simulations, spanning multiple investigations, were enabled by the combination of a fast and accurate implicit solvation model,18 improved methods for Monte Carlo sampling that leverage the advantages of implicit solvation models,16 refinements to force field parameters,19 the deployment of thermal replica exchange (TREx)20 simulations to obtain conformational characterization for each of the archetypal sequences,7c−7e,21 and advanced weighted histogram analysis methods22 for analyzing the collection of simulation results. A similar high throughput approach is required for IDRs whereby all relevant combinations of archetypal IDRs and frequently occurring ODs are investigated in order to unmask the degree to which different modes of tethering affect the intrinsic conformational properties of tethered IDRs. These systems pose unique challenges because an IDR tethered to an OD is an archetype of a “hot” region tethered to a “cold” domain.
Naïve deployments of TREx20b,23 are not particularly useful because several conceptual and logistical issues confound the design of TREx simulations. These are as follows: (i) At higher simulation temperatures the ordered domains will unfold and the feature of a hot IDR and cold OD will be lost at higher temperatures. This is not ideal because our interest is in the range of conformations accessible to the IDR in the context of the OD rather than its unfolded counterpart. (ii) Of course, one could work around the difficulty of ODs unfolding at higher temperatures by adding suitable conformational restraints to maintain foldedness. However, this can only make sense for the temperatures where the OD should remain folded. Therefore, the design of TREx will have to incorporate replicas where the OD thermally unfolds, which creates a biphasic problem whereby some replicas involve the IDR tethered to a folded OD whereas higher temperatures involve replicas tethered to an unfolded OD. (iii) In order to ensure significant overlap of energy distributions between replicas corresponding to the two phases, we would need to incorporate a nonlinear increase in the number of replicas used for each TREx simulation. From a logistical standpoint, much of the sampling for a finite amount of computational resources will be invested in communication between replicas. Previous work has demonstrated the importance of performing multiple independent TREx or S-MMC simulations in order to minimize the statistical uncertainty on estimates of different moments of conformational distributions.7c−7e,16 This is difficult to obtain by dedicating large numbers of compute nodes to single TREx runs. (iv) Finally, the growth in system size further slows down the time taken per TREx simulation and together these constraints severely limit our ability to carry out high throughput assessments of sequence-ensemble relationships for a spectrum of tethered IDRs.
Here, we pursue a strategy to circumvent the logistical barriers posed by tempering methods such as TREx. Our design requirement is that we should be able to gather and analyze information from multiple, independent, “embarrassingly parallel” runs in order to obtain a robust description of the conformational ensembles for tethered systems. For such an approach to be reliable it is imperative that we obtain similar statistical distributions for a range of conformational properties from each independent simulation. Standard sampling methods cannot solve the problem of broken ergodicity and hence a naïve deployment of a large number of multiple, independent simulations will fail to meet the design specifications. We therefore pursue an alternative route that meets our design specifications while leveraging the intrinsic “hot” and “cold” nature of the IDR and OD, respectively.
New Sampling Method for Tethered IDRs
We have adapted a method based on the algorithm proposed by Gelb.24 Monte Carlo sampling on complex, multidimensional energy landscapes can be improved by the introduction of an auxiliary Markov chain that draws conformations from a Boltzmann distribution governed by an approximate potential energy function. Gelb’s proposal, referred to in the literature as Nested Markov Chain Monte Carlo or NMCMC,25 was motivated by the need to reduce computational costs associated with Markov Chain Monte Carlo sampling when using quantum mechanical and polarizable Hamiltonians that are expensive even for single point energy calculations. The formalism developed by Gelb satisfies detailed balance and can be adapted to any pair of Hamiltonians, where only one of the Hamiltonians is of interest. The other can, in theory, be any conceivable approximation, as long as it is useful and helps achieve efficient sampling. We refer to our adaptation of Gelb’s formalism as Hamiltonian Switch Metropolis Monte Carlo or HS-MMC. This terminology highlights the fact that we engineer a switch between distinct Hamiltonians (potential functions) and uses the Metropolis criterion for Markov Chain Monte Carlo simulations. In the following sections, we describe the details of this method and show three examples of realistic sampling problems that are overcome by its use.
Methods
Force Field, Degrees of Freedom, a Parameters
All simulations were performed using the ABSINTH implicit solvation model18 and force field paradigm as implemented in the CAMPARI software package (http://campari.sourceforge.net). The general functional form of the potential function is shown in eq 1. The simulations use atomistic descriptions of polypeptides, explicit representations of solution ions (Na+ and Cl–), and the ABSINTH implicit solvation model to capture solvent-mediated interactions. For any system that includes polypeptides and mobile ions, the backbone ϕ, ψ, ω, and side chain χ dihedral angles as well as the rigid-body coordinates for the polypeptide chains and solution ions constitute the degrees of freedom. For a specific configuration of polypeptide and solution ions, the energy function takes the form
| 1 |
Here, Wsolv is a multibody, direct mean field interaction term that captures the energetics of transferring a solute (polypeptide plus ions) in a specific configuration from the gas phase into the continuum solvent with dielectric constant of ε = 78; Wel denotes the mean field electrostatic term that accounts for dielectric inhomogeneities in the screening/descreening of interatomic electrostatic interactions; ULJ models van der Waals interactions between nonbonded pairs of atoms and Uother denotes the collection of terms used to model specific torsions, coupling between bond angles and torsions, the puckering of flexible rings, the coupling between ring puckering and backbone torsion angles, and any additional restraint terms that are included in the simulation.18,19
All of the force field parameters were taken from the abs_3.2_opls.prm file that is part of version 1 of the CAMPARI distribution. These parameters are based on the charges derived from the OPLS-AA/L force field.26 They include the default Lennard-Jones parameters from preceding versions and the modified parameters that were designed for simulations of sequences that include prolyl residues. In addition, the parameters used here replace the default Lennard-Jones parameters for alkali and halide ions with those developed by Mao and Pappu.19b
Choice of Ordered Domain
We selected the Src Homology domain (SH3) from the sequence of the multidomain human cytoplasmic noncatalytic region of the tyrosine kinase adaptor protein also known as NCK1 (Uniprot ID P16333, http://uniprot.org/). For simplicity, we refer to this domain as SH3. Panel A in Figure 2 shows a model for the structure of the SH3 domain that was derived from coordinates deposited in the protein data bank (http://rcsb.org), PDB code 2JS0.
Figure 2.
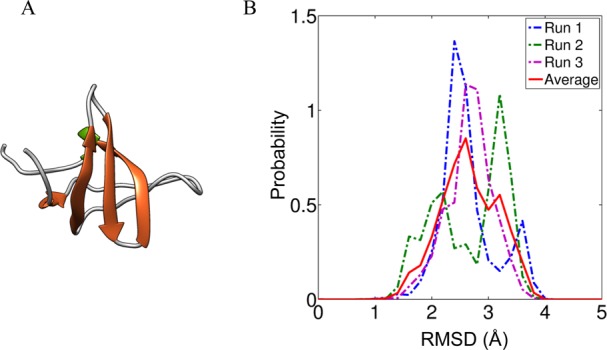
Structure (PDB code 2JS0) of the SH3(2) domain (panel A) from human cytoplasmic NCK1 (showing residues 107–165 from the sequence with UniProt ID P16333) drawn using the VMD package.49 The color-coding is based on secondary structures. Panel B quantifies the reliability of the force field and sampling paradigm for simulations of the folded protein. Here, we show histograms of root-mean-square deviations (RMSDs) from the folded structure that were observed for three different independent S-MMC simulations that start from the folded state of SH3, depicted as Runs 1–3. The curve in red is the histogram obtained by averaging across the three runs. Histograms were generated using bin widths of 0.2 Å.
Choice of IDRs
Sequence details of the IDRs used in this work are summarized in Figure 3. Two of the IDRs were extracted from the NCK1 protein. The third IDR, designated as IDR3 was derived from sequence shuffling of IDR2 based on the recent observation of Das and Pappu,7c who showed that the conformational properties of polyampholytic sequences can be modulated by altering the linear sequence distribution of oppositely charged residues. This linear sequence patterning is quantified using the parameter κ that is shown in Figure 3. Based on its amino acid composition, IDR1 is designated as a weak polyampholyte and is expected to prefer compact, globular conformations as an autonomous unit. Conversely, IDRs 2 and 3 are strong polyampholytes and their conformational properties are likely to correspond to semicompact hairpins and random-coils, respectively. These expectations are based on the combination of amino acid composition and κ values for IDR2 and IDR3.7c
Figure 3.

Sequence details of two naturally occurring IDRs excised from the sequence of human cytoplasmic NCK1. IDR1 and IDR2 span residues 166–192 and 62–106, respectively for the NCK1 sequence with UniProt ID P16333. The sequence depicted as IDR3 is a variant of IDR2 obtained by a redistribution of the residues along the linear sequence while keeping the amino acid composition fixed. The table summarizes various composition and sequence-specific parameters for each sequence including N, the chain length; f+ and f– the fraction of positive and negatively charged residues, respectively; NCPR = | f+ – f– |, the net charge per residue; FCR = (f+ + f–), the fraction of charged residues; and κ, the parameter that quantifies the segregation/mixing of oppositely charged residues within the linear sequence. Higher the value of κ, the more segregated the oppositely charged residues within the sequence—compare the sequence of IDR2 to IDR3. The bottom panel shows a snapshot of IDR2 tethered to the SH3 domain depicted using a surface electrostatic representation that was generated using the APBS tool50 that is built into the UCSF Chimera package.51 The calculated isopotential contour surfaces are plotted at ±2 kT/e where kT denotes thermal energy calculated using the Boltzmann constant k at temperature T, and e is electronic unit of charge. In the figure, red denotes a negative potential while blue denotes a positive potential. All the simulations described in this work were performed in bristle mode with each IDR tethered to the N-terminal end of the SH3 domain.
Simulation Setup
IDRs modeled as autonomous units or tethered to the N-terminus of the SH3 domain were placed inside large spherical droplets along with explicitly represented Na+ and Cl– ions to neutralize the net polypeptide charge and include excess ion pairs to mimic a 15 mM salt solution. All simulations were performed with a spherical droplet boundary condition. The droplet radius was 100 Å for tethered systems and 90 Å for IDRs modeled as autonomous units. The spatial cutoffs for the Lennard-Jones and electrostatic interactions between net-neutral charge groups were set at 10 and 14 Å, respectively. No cutoffs were employed for computing electrostatic interactions involving solution ions and side chain moieties that have a net charge. The N- and C-termini of all sequence constructs were N-acetylated and N′-methylamidated, respectively.
Standard Metropolis Monte Carlo (S-MMC) Protocol
The main sampling strategy is based on S-MMC and includes a composite set of moves that have been designed to ensure efficient and ergodic sampling of conformational space.16 These move sets include backbone pivots, concerted rotations, randomization of side chain torsions, perturbing torsional coordinates, rigid body translations of mobile spherical ions, and rigid body translations and rotations of the polypeptide. The frequencies for different moves were chosen based on the decision tree shown in Figure 4. These were designed to achieve efficient sampling of conformational space while preserving detailed balance. In all simulations of tethered systems, the conformational degrees of freedom of the IDR are sampled preferentially over those of the ordered domain because we are interested in the conformations that the IDR adopts in the context of being tethered to the ordered domain. Moves designed to alter the IDR degrees of freedom are proposed three times as often as moves that are designed to alter the degrees of freedom within the ordered domain. This preferential sampling of the IDR is taken into account in a modified Metropolis criterion in order to ensure against biases and preserve detailed balance.27 Each independent S-MMC simulation of a tethered system deploys a total of 8 × 107 proposed moves. In order to ensure a fair comparison between the S-MMC and HS-MMC approaches, we used identical numbers of moves for independent runs of both approaches.
Figure 4.

Decision-tree utilized that is used for the selection of S-MMC moves based on the full Hamiltonian. Each nonleaf node corresponds to a class of moves; each node is annotated with the overall probability of that move or class of moves being selected. Each edge is annotated with the probability of the decision process branching toward the child once the parent has been reached. The decision is complete once a leaf node is reached.
Additionally, we performed reference simulations, the results of which are designated with the prefixes excluded volume (EV), Flory random coil (FRC),28 and Lennard-Jones (LJ). In the EV simulations, the Wel, Wsolv (see eq 1) and the r–6 dispersive term of the Lennard-Jones potential are switched off; for the LJ simulations, the Wel and Wsolv in eq 1 are switched off. Finally, for the FRC simulations, all terms in the potential function are switched off and we generate ensembles by drawing backbone and side chain dihedral angles from a presampled library for each residue.
TREx Simulations
We used TREx MMC sampling for simulations of IDRs modeled as autonomous units. For each sequence, we created 13 replicas according to the following temperature schedule [280 K, 285 K, 294 K, 298 K, 310, K, 320 K, 330 K, 340 K, 370 K, 400 K]. The schedule was chosen based on prior results for similar types of systems7c,7d and quantification of the overlap statistics between pairs of neighboring replicas. The average swap probability for pairs of replicas was 0.4 for the prescribed schedule. For each of the IDRs shown in Figure 3, we performed three independent simulations using a total of 4.5 × 107 overall proposed moves per TREx run.
Generation of Starting Conformations
In all simulations, the starting conformations for IDRs, that is, the initial values for backbone ϕ, ψ, ω, and side chain χ dihedral angles were drawn randomly from a pre-equilibrated distribution of atomistic self-avoiding random walks. The initial coordinates for the SH3 domain were based on the model in PDB file 2JS0. To generate a starting conformation that was usable in all simulations, the coordinates extracted from the PDB file were subjected to 103 steps of standard Metropolis Monte Carlo (S-MMC) sampling at a simulation temperature of 260 K. These simulations incorporated harmonic restraints on the backbone and side chain torsion angles of the SH3 domain. The stiffness of each restraint was 0.2 kcal/(mol-deg2). After the initial 103 steps, the restraints were removed and an additional 103 steps of S-MMC sampling were performed at a simulation temperature of 298 K to converge on an equilibrated conformation for the SH3 domain that was subsequently used as the starting conformation in all simulations involving this domain. This structure had a root-mean-square deviation (RMSD) of 1.1 Å from the coordinates in the PDB file as calculated over all backbone atoms. In order to calibrate the stability of the SH3 domain with the force field of choice, we performed, multiple, long (4 × 107 steps), independent S-MMC simulations of the SH3 domain in the presence of 15 mM NaCl and in the absence of any harmonic restraints at a simulation temperature of 298 K. The results, summarized in panel B of Figure 2, demonstrate that the folded state of the SH3 domain is maintained in lengthy S-MMC simulations.
For each simulation of a tethered system, the randomly chosen IDR conformation was tethered to the N-terminus of the structure of the pre-equilibrated SH3 domain. The starting conformations for simulations of tethered systems were obtained by 103 of S-MMC simulations performed at 298 K in the presence of torsional restraints on the ordered SH3 domain. The starting conformations were further thermalized using 107 S-MMC steps of sampling after the addition of suitable numbers of Na+ and Cl– ion pairs to mimic a bulk salt concentration of 15 mM.
Introducing the HS-MMC Methodology
The Hamiltonian Switch Metropolis Monte Carlo (HS-MMC) Algorithm
The design of HS-MMC relies on the use of two Markov chains that sample conformations drawn from two distinct Boltzmann distributions. In the interest of completeness, we first introduce our notations for S-MMC sampling. We shall assume that the current coordinates of the system comprising of an IDR tethered to the OD are denoted as i. A random change is made to these coordinates and the nature of this change depends on the move set. We denote the new state proposed by virtue of the randomly chosen move to be j. The transition probability to go from state i to j is given by the Metropolis criterion29 as
| 2 |
Here, αij denotes the elements of the transition matrix that determine the probability that the prescribed move set yields a transition from i to j; ΔUij = Uj – Ui is the difference in potential energies between states j and i; β = (RT)−1 where R = 1.987 × 10–3 kcal/(mol K) is the Boltzmann constant and T is the simulation temperature in degree Kelvin. The move sets we use ensure that αij = αji; that is, the transition matrix is symmetric. Accordingly, πij can be written in compact notation in terms of the equilibrium probabilities (normalized Boltzmann weights) pi and pj associated with states i and j, respectively as
| 3 |
In HS-MMC, a majority of the states are generated using S-MMC sampling that is based on the Boltzmann distribution governed by the full Hamiltonian (FH in Figure 5), which corresponds to eq 1 in the methods section. Each S-MMC step also includes an additional test. A uniformly distributed random number rEV is drawn and compared to pEV where 0 ≤ pEV ≤ 1. If rEV < pEV, the Hamiltonian is switched for the next nEV steps of the simulation, and S-MMC sampling based on a second Markov chain is used to draw states from the Boltzmann distribution for the excluded volume (EV) Hamiltonian. The EV Hamiltonian combines two terms viz., UEV and Urestr. The term UEV is the repulsive arm of the Lennard-Jones potential and models pairwise steric repulsions between atoms within the IDR and between atoms of the IDR and those of the OD. The term Urestr refers to a set of harmonic restraints with a force constant of 0.2 kcal/(mol-deg2) that are applied over all torsional degrees of freedom of the OD in order to maintain its current configuration while the EV Hamiltonian leads to new conformations for the IDR. The EV Hamiltonian has the form:
| 4 |
Here, UEV denotes the repulsive arm of the standard 12–6 Lennard-Jones (LJ) potential and the parameters σij and εij for pairwise interactions are identical to those used for modeling LJ interactions in the full Hamiltonian.
Figure 5.

Schematic representation of the design of the HS-MMC simulation approach. Here, FH-MMC refers to a Markov chain generated using the standard Metropolis Monte Carlo simulations (MMC) based on the full Hamiltonian (FH). The simulation switches to an alternative Markov chain based on a Hamiltonian where intra-IDR interactions and those between the IDR and ordered domain are modeled using steric repulsions alone–the so-called excluded volume (EV) limit. During this EV-MMC part of the simulation, an additional term is included in the Hamiltonian that applies restraints to maintain the structure of the OD. In the schematic, the circles with letters depict distinct conformations sampled along the distinct Markov chains. The symbols pj and pi denote the Boltzmann weights associated with states j and i, respectively for the full Hamiltonian (FH) and conversely, pn′ and pm represent the Boltzmann weights associated with the EV Hamiltonian for states n and m, respectively. The acceptance ratios for the sampling that is internal to the Markov chains for the FH and EV sections are the standard Metropolis criteria. The final state of the EV-MMC part of the simulation is accepted or rejected based on the acceptance ratio proposed by Gelb that satisfies detailed balance—see eq 5
Let m denote the conformation prior to the Hamiltonian switch and start of the S-MMC sampling based on the second Markov chain while p denotes the conformation that results after S-MMC sampling for nEV steps along the second Markov chain. The conformation p is accepted or rejected according to the criterion:
 |
5 |
Here, pm′ and pp denote the equilibrium Boltzmann probabilities associated with conformations m and p, respectively for the EV Hamiltonian whereas the unprimed probabilities are those associated with the full Hamiltonian. Gelb has shown that the functional form in eq 5 satisfies detailed balance, which we further ensure by choosing moves based on symmetric transition matrices. Figures 5 shows a sketch of the design of the HS-MMC method.
Justifying the Choice of the EV Hamiltonian in HS-MMC
The design of HS-MMC is sufficiently general, and it can accommodate any approximation of the full Hamiltonian or an entirely different Hamiltonian. Our choice of the EV Hamiltonian is based on the observation that long S-MMC simulations based on this Hamiltonian generate converged distributions of thermodynamically relevant atomistic self-avoiding random walks. Importantly, the intra-IDR and IDR-OD interactions are purely short-range repulsions. Therefore, unlike other possible choices for the auxiliary Hamiltonian, the EV term ensures a systematic dilution of contacts and yields states, which if accepted, will depart from energy traps and hence improve the sampling on the rugged energy landscape encoded by the full Hamiltonian. Of course, one could just as easily apply a coupling parameter λ (0 ≤ λ ≤ 1) on the set of non-EV terms in the Hamiltonian in order to create a series of Hamiltonian replicas of the system and perform replica exchange simulations in λ-space.20b,23,30 In such methods, the improved sampling comes with the additional cost of setting up communication between the different replicas. In switching, as opposed to exchange simulations, one can use each computational core for a single HS-MMC run and this allows us to carry out multiple independent simulations in order to assess the similarity of statistics obtained across each simulation.
Instead of the UEV term, one could use the Weeks–Chandler–Andersen (WCA) potential,31 which excises a purely hard sphere term from the Lennard-Jones potential. As a result the spatial range for repulsions would be shorter for WCA when compared to the UEV terms. This might improve the acceptance ratios and allow for longer auxiliary Markov chains with the switched Hamiltonian. We have not explored the WCA model as an auxiliary potential because previous work showed that the UEV term, which belongs to the family of inverse power potentials,32 encodes significant fine structure into the energy landscape, which allows us to ensure that local conformational propensities such as the residue and context specific biases for polyproline II, α-helical, and β-strand ϕ, ψ values is preserved.18,33 This feature does not obtain with the WCA model, which eliminates all such local preferences since all sterically allowed conformations have equal weights.
Parameterization of HS-MMC
The two parameters of HS-MMC that need optimization are pEV and nEV. As noted above, the Hamiltonian switch is attempted with a probability pEV at every step along the first Markov chain. The parameter pEV needs to be low enough to guard against frequent switches to ensure efficient sampling from the distribution governed by the full Hamiltonian. It should, however, be finite enough to ensure that a sufficient number of Hamiltonian switches are attempted in a single HS-MMC run. Through a series of trials, we converged on an optimal choice of pEV = 5 × 10–4 for the systems studied here. This choice yields a percent probability of 1–5% for the acceptance criterion shown in eq 5. We deem this to be acceptable given the extent of decorrelation that is achieved by the systematic dilution of contacts engendered by the use of the EV Hamiltonian. The optimal choice for nEV can be made independently or it can be coupled to the optimization of pEV. We converged on the optimal choice for nEV by performing a series of simulations with different values for nEV and monitoring the conformational dissimilarity and energy differences between the conformations depicted as m and p in Figure 5. The goal was to prescribe a range for nEV that maximizes the conformational dissimilarity between m and p while minimizing the energy difference ΔUpm (see Figure 5). This approach resulted in the choice of nEV being in the range 25–75 steps.
It does not follow that the choices listed here can be transferred automatically to any tethered IDR-ordered domain system. The peculiarities of the system and the full Hamiltonian will mandate the necessary problem specific optimization in the choices for pEV and nEV although some level of automation seems reasonable if the problems belong to a specific category. The choices for pEV and nEV make HS-MMC fundamentally different from Gelb’s method and other variants of his NMCMC approach. In the latter, a bulk of the sampling is performed using the auxiliary Markov chain that uses the less expensive approximate Hamiltonian. This can create a severe shortage of distinct, uncorrelated conformations drawn from the ensemble for statistical analysis. Although this is not a major weakness when the application calls for a characterization of discrete points on a quantum mechanical energy surface, it becomes a major weakness for our applications because we require quantitative descriptions for a broad range of uncorrelated IDR conformations. As is inferred by the optimal choices for pEV and nEV the majority of the sampling in HS-MMC is performed using the full Hamiltonian, and the auxiliary Markov chain can be viewed as a random, unbiased interjection of an alternative set of moves that provide transitions out of energetic traps.
Results
Tests of HS-MMC for Sampling Conformational Ensembles of Tethered Systems
Figures 6–10 summarize the statistics gleaned from multiple, independent HS-MMC runs for IDRs 1–3 simulated as bristles that are tethered to the N-terminus of the SH3 domain (see Figure 3). These results are compared to those obtained from multiple, independent S-MMC simulations with identical amounts of sampling. As noted in panel B of Figure 2, the SH3 domain does not undergo large-scale conformational fluctuations under the simulation conditions used here. Accordingly, we focus our analysis on the conformational properties of the IDRs. In doing so, we obtain assessments of the improvements obtained using HS-MMC as compared to S-MMC runs. The latter, as noted above, tends to be confounded by problems of broken ergodicity. The lack of self-averaging is a classic signature of broken ergodicity and there are several approaches for diagnosing such problems.34 Here, we employ a simple criterion that quantifies the degree to which sufficiently long independent simulations produce similar statistical distributions for different conformational properties.
Figure 6.

Assessing the sampling quality for S-MMC and HS-MMC. Results are shown for IDR1 simulated in bristle mode by tethering it to the N-terminal end of the SH3 domain. Column 1 summarizes the results from ten independent S-MMC runs and these are compared to results in column 2 that summarize the results from ten independent HS-MMC runs. The histograms along the top row show results for the distributions of Rg values (in Å) extracted from each independent simulation. The histograms along the bottom row show results for the distributions of asphericity values (δ*). The results show that HS-MMC produces distributions for polymer sizes and shapes that have greater similarity from one independent run to the next (see column 2) when compared to the independent S-MMC runs (see column 1). The histograms were generated using bin widths of 0.1 Å (top row) and asphericity units of 0.01 (bottom row).
Figure 10.

Comparison of the conformational properties of IDRs modeled as autonomous units to IDRs modeled as bristles that are tethered to the N-terminus of the SH3 domain. Here, we plot the scaling of ensemble averaged spatial separations between pairs of residues i and j versus the linear sequence separation |j–i| for each IDR. In each plot, the open squares, cross marks, and open diamonds denote the expected scaling of ⟨Riij⟩ against |j–i| for a self-avoiding random walk (EV), a classic Flory random coil (FRC), and a random Lennard-Jones globule (LJ). IDR1 samples compact conformations as an autonomous unit and this feature is preserved in the bristle mode. IDR2 and IDR3, respectively sample semicompact, hairpin-like and Flory-random-coil-like conformations as autonomous units. For both sequences, their intrinsic conformational properties are maintained, although some degree of expansion is evident and is attributable to the excluded volume effect of the ordered SH3 domain.
Row A in Figure 6 shows the raw histograms obtained for the radius of gyration (Rg) values calculated over the IDR1 stretch from ten independent S-MMC and HS-MMC runs. Row B quantifies equivalent histograms for asphericity (δ*) values. We use these two parameters because Rg quantifies the overall size (density) of the IDR and δ* quantifies its shape. For each conformation, the asphericity is calculated from eigenvalues of the conformation specific gyration tensor. For spheres, δ* ≈ 0; for rods δ* ≈ 1; and for ellipsoids, δ* will have intermediate values. Visual inspection shows that the histograms obtained from the independent S-MMC runs show greater variability when compared to those obtained from HS-MMC runs (see Figure 6). We put this observation on a quantitative footing as shown in Figure 7. For each observable, viz., Rg and δ*, we calculated the overlaps between all unique pairs of histograms for S-MMC and HS-MMC runs. Panel A in Figure 7 shows the average pairwise overlap obtained for histograms of Rg values for each of the three tethered IDRs whereas panel B in Figure 7 shows equivalent results derived from histograms of δ* values. These results demonstrate that the overlap between histograms obtained from HS-MMC runs is systematically higher for equivalent amounts of sampling when compared to S-MMC runs. This is true irrespective of the observable viz., Rg or δ* and remains valid even for higher-order distributions such as the joint distributions of δ* and Rg values (Figure 8). Importantly, lengthening each of the independent simulations can increase the overlap of statistical distributions obtained among different runs. Further, the robustness of these statistical properties can be gleaned by increasing the number of independent runs by an order of magnitude because these simulations are “embarrassingly parallel” and do not require any communication between compute nodes. These simulations are efficient in that it takes ca. 250 h of wall clock time to complete a single HS-MMC run (∼108 steps for the systems studied here) on a single core of an Intel-Xeon E5-2670 2.6 GHz node with eight cores.
Figure 7.
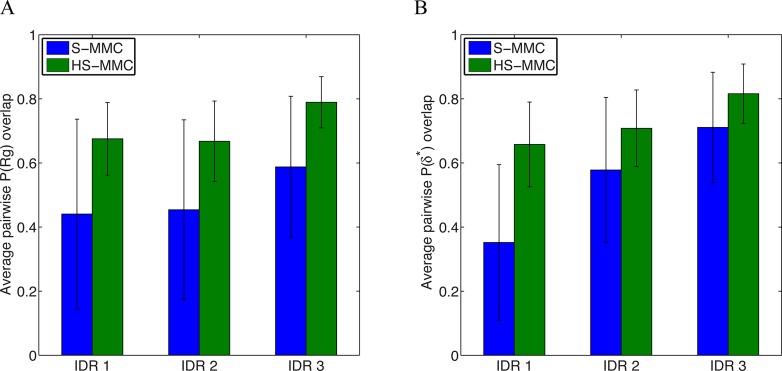
Panels A and B quantify the average overlap between pairs of distributions shown in rows 1 and 2 of Figure 6, respectively. In general, the pairwise overlap is higher for distributions generated using HS-MMC simulations (green bars in both panels) than the S-MMC simulations (blue bars). The error bars quantify the standard error in the estimate of the mean.
Figure 8.

Simulation results can also be analyzed to extract statistics for two-dimensional histograms viz., P(δ*,Rg). Panels A and B show histograms drawn from individual, independent S-MMC and HS-MMC runs, respectively, for IDR1 tethered to the SH3 domain. HS-MMC enables the sampling of a broader range of conformations for the tethered IDR. Panel C quantifies the average overlap between pairs of two-dimensional histograms P(δ*,Rg) which are extracted from pairs of independent S-MMC (blue) and HS-MMC (green) simulations, respectively. Again, the overlap between distributions obtained using HS-MMC is higher than for S-MMC runs.
HS-MMC Generates Ensembles of Increased Conformational Heterogeneity
Recently, Lyle et al.12a introduced
a measure to quantify the degree of heterogeneity within ensembles
of thermodynamically relevant conformations that are extracted from
converged simulations. This parameter, denoted as Φ, approaches
unity for a homogeneous ensemble characterized by small-scale conformational
fluctuations and approaches zero for ensembles characterized by a
broad range of diverse conformations. For a chain of N residues, each conformation c is represented as
an nd × 1 conformational vector Vc where nd = (N(N – 1)/2), Vc = {d12,d13,...,dN–1,N}, and each element dij in Vc represents the spatial distance between a unique
pair of residues, i and j. For each
pair of residues i and j, we calculate dij = 1/Zij·Σm∈iΣn∈j|rmi – rn|. Here, rmi and rnj denote the position vectors
of atoms m and n within residues i and j, respectively, and Zij is the number of unique pairwise interatomic
distances between the two residues. To compare a pair of conformations k and l, one calculates a pairwise dissimilarity
measure  where cos(Ωkl) = Vk·Vl/|Vk||Vl|. An
ensemble of nc conformations produces
an ensemble of conformational vectors, V1, V2,...,etc. These vectors are used to calculate
a distribution
where cos(Ωkl) = Vk·Vl/|Vk||Vl|. An
ensemble of nc conformations produces
an ensemble of conformational vectors, V1, V2,...,etc. These vectors are used to calculate
a distribution 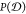 of nc(nc – 1)/2
conformational dissimilarity values. For each ensemble we actually
calculate two distributions of dissimilarities viz., the distribution of
of nc(nc – 1)/2
conformational dissimilarity values. For each ensemble we actually
calculate two distributions of dissimilarities viz., the distribution of 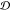 -values for
pairs of conformations within
an ensemble and a distribution of
-values for
pairs of conformations within
an ensemble and a distribution of 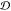 -values
comparing each conformation to an
ensemble of conformations drawn from a Flory random coil model as
described by Lyle et al.12a Averaging over
the former yields
-values
comparing each conformation to an
ensemble of conformations drawn from a Flory random coil model as
described by Lyle et al.12a Averaging over
the former yields  and averaging over the latter, which is
an ensemble of ensembles yields
and averaging over the latter, which is
an ensemble of ensembles yields  . The values of
. The values of  and
and  lead to an estimate of the degree
of heterogeneity
Φ within the ensemble. We first compute the ratio
lead to an estimate of the degree
of heterogeneity
Φ within the ensemble. We first compute the ratio 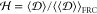 and use it to calculate
and use it to calculate 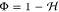 .
.
Figure 9 plots the values for Φ obtained from S-MMC versus HS-MMC runs for each of the three tethered IDRs. We find a systematic trend whereby the heterogeneity within each conformational ensemble is higher (the values of Φ are lower) for HS-MMC runs compared to S-MMC runs. This suggests that we are able to sample a broader spectrum of thermodynamically relevant conformations using HS-MMC when compared to S-MMC.
Figure 9.

Quantification of the ensemble heterogeneity Φ from ten independent S-MMC and HS-MMC simulations of the three IDRs tethered to the SH3 domain. The trends in Φ are identical between the two simulation approaches. The quantitative assessments of heterogeneity are, however, different between the two approaches and the HS-MMC method yields ensembles of higher conformational heterogeneity.
Comparative Assessments of Conformational Properties of IDRs Modeled As Autonomous Units to Tethered IDRs
Finally, Figure 10 shows a comparison between the conformational properties of each IDR modeled as an autonomous unit to that obtained for the tethered IDRs. There is no a priori reason to expect significant distortion of the intrinsic properties of IDRs given that the measured conformational properties of the stable SH3 domain, in general, remain unperturbed by the tethering of IDRs. This suggests weak coupling between the IDR and the ordered domain. As shown in Figure 10, the HS-MMC simulations yield results that support the expectation of weak coupling. Here, we plot the so-called internal scaling profiles that provide a complete summary of conformational properties for each IDR. In each plot the ordinate refers to ⟨Rij⟩, the ensemble-averaged inter-residue distances calculated for all pairs of residues that are |j–i| apart along the linear sequence. These internal scaling profiles quantify local concentrations of chain segments around each other. They are particularly useful for classifying conformational ensembles because the profiles show distinct limiting behaviors for self-avoiding random walks, Flory random coils, and random globules, respectively (see Figure 10).
In concordance with expectations based on previous reports,7c,7e,35 IDR1 forms a heterogeneous ensemble of compact, globular conformations as an autonomous unit. Conversely, IDRs 2 and 3 are expected to form semicompact hairpin-like conformations and Flory random coil-like conformations,7c respectively, and these features are reproduced for these sequences modeled as autonomous units. If the coupling between the IDRs and the SH3 domain is weak, then we expect that the intrinsic conformational properties of IDRs should be preserved even upon tethering, and Figure 10 shows that this is indeed the case. Perturbations about the intrinsic properties are attributable to the excluded volume effects of the OD. The results shown in Figure 10 are not realizable when internal scaling profiles are calculated using conformations drawn from S-MMC runs (data not shown). Instead, the results vary considerably from one run to the next as established in the analyses of Figures 6–8.
Discussion
In the preceding paragraphs, we summarized results that compare the similarities of statistics for a set of conformational properties that were obtained from multiple independent HS-MMC and S-MMC runs, respectively. These results highlight the improvements afforded by the HS-MMC algorithm in terms of (a) alleviating broken ergodicity as measured by the similarity of statistics among different runs; (b) yielding ensembles with systematically increased conformational heterogeneity; and (c) generating ensembles with conformational properties that are consistent with the expectation of weak interactions between the IDRs and the ordered SH3 domain.
The HS-MMC approach is a direct adaptation of the NMCMC method proposed by Gelb, which enables the use of approximate/alternative Hamiltonians for enhancing conformational sampling. This method bears conceptual resemblance to other Monte Carlo sampling methods such as J-walking36 and smart darting.37 One can also envisage the use of a suitable simulated tempering based approach such as Hamiltonian replica exchange,23b,30 reservoir exchange,38 general library based Monte Carlo methods,39 resolution exchange,40 or generalizations of simulated tempering that are based on Tsallis statistics.41 All of these methods offer distinct advantages. We selected a switching based method as opposed to tempering based methods in order to eliminate the need for communication between replicas and thereby leverage the use of multiple, independent “swarms” of simulations. By removing the need for interprocess communication, HS-MMC simulations can be run in a highly distributed manner, across heterogeneous hardware without the need for any form of message passing between compute nodes. A particular advantage is the ability to design direct probes for robustness by assessing the reproducibility of statistics from one run to the next. In fact, this class of approaches are well-suited to distributed computing applications, which have significant advantages such as the ability to unmask robust features of conformational landscapes as demonstrated by various investigations carried out under the auspices of the Folding@Home project.42
It is worth emphasizing that, our preliminary results aside, it does not automatically follow that all IDRs will always interact weakly with the ordered domains to which they are tethered. There exists the possibility that ordered domains can modulate the conformational properties of IDRs, especially if the compositional biases within IDR sequences are not particularly strong. It is also possible that tethered IDRs will induce local unfolding of ordered domains or secondary structure motifs as has been documented in simulations43 and experiments.44 A general framework for quantifying the synergy between IDRs and ordered domains is likely to be accessible through deployment of the HS-MMC method in conjunction with the ABSINTH force field paradigm for studying the ensembles of a large number of archetypal tethered systems.
Acknowledgments
This work was supported by grants from the National Science Foundation (MCB 1121867) and the National Institutes of Health (5R01NS056114).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- a Dunker A. K.; Brown C. J.; Lawson J. D.; Iakoucheva L. M.; Obradovic Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [DOI] [PubMed] [Google Scholar]; b Dyson H. J.; Wright P. E. Intrinsically unstructured proteins and their functions. Nature Rev. Cell. Mol. Biol. 2005, 6, 197–208. [DOI] [PubMed] [Google Scholar]
- a Dyson H. J.; Wright P. E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002, 12154–60. [DOI] [PubMed] [Google Scholar]; b Frankel A. D.; Smith C. A. Induced folding in RNA-protein recognition: More than a simple molecular handshake. Cell 1998, 922149–151. [DOI] [PubMed] [Google Scholar]; c Mucsi Z.; Hudecz F.; Hollosi M.; Tompa P.; Friedrich P. Binding-induced folding transitions in calpastatin subdomains A and C. Protein Sci. 2003, 12102327–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lacy E. R.; Filippov I.; Lewis W. S.; Otieno S.; Xiao L. M.; Weiss S.; Hengst L.; Kriwacki R. W. p27 binds cyclin–CDK complexes through a sequential mechanism involving binding-induced protein folding. Nature Struct. Mol. Biol. 2004, 114358–364. [DOI] [PubMed] [Google Scholar]; e Receveur-Brechot V.; Bourhis J. M.; Uversky V. N.; Canard B.; Longhi S. Assessing protein disorder and induced folding. Proteins-Struct. Funct. Bioinform. 2006, 62124–45. [DOI] [PubMed] [Google Scholar]; f Ganguly D.; Otieno S.; Waddell B.; Iconaru L.; Kriwacki R. W.; Chen J. Electrostatically accelerated coupled binding and folding of intrinsically disordered proteins. J. Mol. Biol. 2012, 4225674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriwacki R. W.; Hengst L.; Tennant L.; Reed S. I.; Wright P. E. Structural studies of p21(Waf1/Cip1/Sdi1) in the free and Cdk2-bound state: Conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. U.S.A. 1996, 932111504–11509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Li P.; Banjade S.; Cheng H.-C.; Kim S.; Chen B.; Guo L.; Llaguno M.; Hollingsworth J. V.; King D. S.; Banani S. F.; Russo P. S.; Jiang Q.-X.; Nixon B. T.; Rosen M. K. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 4837389336–U129. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Han T. W.; Kato M.; Xie S.; Wu L. C.; Mirzaei H.; Pei J.; Chen M.; Xie Y.; Allen J.; Xiao G.; McKnight S. L. Cell-free formation of RNA granules: Bound RNAs identify features and components of cellular assemblies. Cell 2012, 1494768–779. [DOI] [PubMed] [Google Scholar]; c Kato M.; Han T. W.; Xie S.; Shi K.; Du X.; Wu L. C.; Mirzaei H.; Goldsmith E. J.; Longgood J.; Pei J.; Grishin N. V.; Frantz D. E.; Schneider J. W.; Chen S.; Li L.; Sawaya M. R.; Eisenberg D.; Tycko R.; McKnight S. L. Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 1494753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Kwon I.; Kato M.; Xiang S.; Wu L.; Theodoropoulos P.; Mirzaei H.; Han T.; Xie S.; Corden J. L.; McKnight S. L. Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell 2013, 15551049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kwon I.; Kato M.; Xiang S.; Wu L.; Theodoropoulos P.; Mirzaei H.; Han T.; Xie S.; Corden J. L.; McKnight S. L. Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains (vol 155, pg 1049, 2013). Cell 2014, 1561–2374–374. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Milles S.; Lemke E. A. Single molecule study of the intrinsically disordered FG-repeat nucleoporin 153. Biophys. J. 2011, 10171710–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Andreeva A.; Murzin A. G. Structural classification of proteins and structural genomics: New insights into protein folding and evolution. Acta Crystall. F-Struct. Biol. Crystall. Commun. 2010, 66, 1190–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lo Conte L.; Ailey B.; Hubbard T. J. P.; Brenner S. E.; Murzin A. G.; Chothia C. SCOP: A structural classification of proteins database. Nucleic Acid. Res. 2000, 281257–259. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Nagano N.; Orengo C. A.; Thornton J. M. One fold with many functions: The evolutionary relationships between TIM barrel families based on their sequences, structures, and functions. J. Mol. Biol. 2002, 3215741–765. [DOI] [PubMed] [Google Scholar]; d Orengo C. A.; Bray J. E.; Buchan D. W. A.; Harrison A.; Lee D.; Pearl F. M. G.; Sillitoe I.; Todd A. E.; Thornton J. M. The CATH protein family database: A resource for structural and functional annotation of genomes. Proteomics 2002, 2111–21. [PubMed] [Google Scholar]
- a Toth-Petroczy A.; Simon I.; Fuxreiter M.; Levy Y. Disordered tails of homeodomains facilitate DNA recognition by providing a trade-off between folding and specific binding. J. Am. Chem. Soc. 2009, 1314215084–15085. [DOI] [PubMed] [Google Scholar]; b Vuzman D.; Levy Y. DNA search efficiency is modulated by charge composition and distribution in the intrinsically disordered tail. Proc. Natl. Acad. Sci. U.S.A. 2010, 1074921004–21009. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Khazanov N.; Levy Y. Sliding of p53 along DNA can be modulated by its oligomeric state and by cross-talks between its constituent domains. J. Mol. Biol. 2011, 4082335–355. [DOI] [PubMed] [Google Scholar]; d Vuzman D.; Levy Y. Intrinsically disordered regions as affinity tuners in protein–DNA interactions. Mol. BioSys. 2012, 8147–57. [DOI] [PubMed] [Google Scholar]; e De Sancho D.; Best R. B. Modulation of an IDP binding mechanism and rates by helix propensity and non-native interactions: Association of HIF1 α with CBP. Mol. BioSys. 2012, 81256–267. [DOI] [PubMed] [Google Scholar]; f Knott M.; Best R. B. A preformed binding interface in the unbound ensemble of an intrinsically disordered protein: Evidence from molecular simulations. PLos Comput. Biol. 2012, 87e1002605(1–10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tran H. T.; Mao A.; Pappu R. V. Role of backbone-solvent interactions in determining conformational equilibria of intrinsically disordered proteins. J. Am. Chem. Soc. 2008, 130237380–7392. [DOI] [PubMed] [Google Scholar]; b Meng W. L.; Lyle N.; Luan B. W.; Raleigh D. P.; Pappu R. V. Experiments and simulations show how long-range contacts can form in expanded unfolded proteins with negligible secondary structure. Proc. Natl. Acad. Sci. U.S.A. 2013, 11062123–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Das R. K.; Pappu R. V. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. U.S.A. 2013, 1103313392–13397. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Das R. K.; Crick S. L.; Pappu R. V. N-terminal segments modulate the α-helical propensities of the intrinsically disordered basic regions of bZIP proteins. J. Mol. Biol. 2012, 4162287–99. [DOI] [PubMed] [Google Scholar]; e Mao A. H.; Crick S. L.; Vitalis A.; Chicoine C. L.; Pappu R. V. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. U.S.A. 2010, 107188183–8188. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Zhang W. H.; Ganguly D.; Chen J. H. Residual structures, conformational fluctuations, and electrostatic interactions in the synergistic folding of two intrinsically disordered proteins. PLos Comput. Biol. 2012, 81e1002353.(1–15).. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Soranno A.; Buchli B.; Nettels D.; Cheng R. R.; Mueller-Spaeth S.; Pfeil S. H.; Hoffmann A.; Lipman E. A.; Makarov D. E.; Schuler B. Quantifying internal friction in unfolded and intrinsically disordered proteins with single-molecule spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 2012, 1094417800–17806. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schuler B.; Muller-Spath S.; Soranno A.; Nettels D. Application of confocal single-molecule FRET to intrinsically disordered proteins. Methods Mol. Biol. 2012, 896, 21–45. [DOI] [PubMed] [Google Scholar]; c Hofmann H.; Soranno A.; Borgia A.; Gast K.; Nettels D.; Schuler B. Polymer scaling laws of unfolded and intrinsically disordered proteins quantified with single-molecule spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 2012, 1094016155–16160. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Muller-Spath S.; Soranno A.; Hirschfeld V.; Hofmann H.; Ruegger S.; Reymond L.; Nettels D.; Schuler B. Charge interactions can dominate the dimensions of intrinsically disordered proteins. Proc. Natl. Acad. Sci. U.S.A. 2010, 1073314609–14614. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Ferreon A. C. M.; Gambin Y.; Lemke E. A.; Deniz A. A. Interplay of α-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc. Natl. Acad. Sci. U.S.A. 2009, 106145645–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Mukhopadhyay S.; Krishnan R.; Lemke E. A.; Lindquist S.; Deniz A. A. A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proc. Natl. Acad. Sci. U.S.A. 2007, 10482649–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Ferreon A. C. M.; Ferreon J. C.; Wright P. E.; Deniz A. A. Modulation of allostery by protein intrinsic disorder. Nature 2013, 4987454390–396. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Ferreon A. C. M.; Moosa M. M.; Gambin Y.; Deniz A. A. Counteracting chemical chaperone effects on the single-molecule α-synuclein structural landscape. Proc. Natl. Acad. Sci. U.S.A. 2012, 1094417826–17831. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Ferreon A. C. M.; Moran C. R.; Gambin Y.; Deniz A. A. Single-molecule fluorescence studies of intrinsically disordered proteins. Methods Enzymol. 2010, 472, 179–204. [DOI] [PubMed] [Google Scholar]; j Vandelinder V.; Ferreon A. C. M.; Gambin Y.; Deniz A. A.; Groisman A. High-resolution temperature-concentration diagram of α-synuclein conformation obtained from a single forster resonance energy transfer image in a microfluidic device. Analyt. Chem. 2009, 81166929–6935. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Choi U. B.; McCann J. J.; Weninger K. R.; Bowen M. E. Beyond the random coil: Stochastic conformational switching in intrinsically disordered proteins. Structure 2011, 194566–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tang X. J.; Orlicky S.; Mittag T.; Csizmok V.; Pawson T.; Forman-Kay J. D.; Sicheri F.; Tyers M. Composite low affinity interactions dictate recognition of the cyclin-dependent kinase inhibitor Sic1 by the SCFCdc4 ubiquitin ligase. Proc. Natl. Acad. Sci. U.S.A. 2012, 10993287–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Moldoveanu T.; Grace C. R.; Llambi F.; Nourse A.; Fitzgerald P.; Gehring K.; Kriwacki R. W.; Green D. R. BID-induced structural changes in BAK promote apoptosis. Nature Struct. Mol. Biol. 2013, 205589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ou L.; Waddell M. B.; Kriwacki R. W. Mechanism of Cell Cycle. Entry Mediated by the Intrinsically Disordered Protein p27(Kip1). ACS Chem. Biol. 2012, 74678–682. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ferreira M. E.; Hermann S.; Prochasson P.; Workman J. L.; Berndt K. D.; Wright A. P. H. Mechanism of transcription factor recruitment by acidic activators. J. Biol. Chem. 2005, 2802321779–21784. [DOI] [PubMed] [Google Scholar]; e Ferreon J. C.; Lee C. W.; Arai M.; Martinez-Yamout M. A.; Dyson H. J.; Wright P. E. Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2. Proc. Natl. Acad. Sci. U.S.A. 2009, 106166591–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Wojciak J. M.; Martinez-Yamout M. A.; Dyson H. J.; Wright P. E. Structural basis for recruitment of CBP/p300 coactivators by STAT1 and STAT2 transactivation domains. EMBO J. 2009, 287948–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Crick S. L.; Jayaraman M.; Frieden C.; Wetzel R.; Pappu R. V. Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proc. Natl. Acad. Sci. U.S.A. 2006, 1034516764–16769. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Marsh J. A.; Forman-Kay J. D. Sequence determinants of compaction in intrinsically disordered proteins. Biophys. J. 2010, 98102383–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao A. H.; Lyle N.; Pappu R. V. Describing sequence-ensemble relationships for intrinsically disordered proteins. Biochem. J. 2013, 449, 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lyle N.; Das R. K.; Pappu R. V. A quantitative measure for protein conformational heterogeneity. J. Chem. Phys. 2013, 13912121907(1–12). [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fisher C. K.; Ullman O.; Stultz C. M. Comparative studies of disordered proteins with similar sequences: Application to A β-40 and A β-42. Biophys. J. 2013, 10471546–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Fisher C. K.; Stultz C. M. Constructing ensembles for intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2011, 213426–431. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Fisher C. K.; Stultz C. M. Protein structure along the order-disorder continuum. J. Am. Chem. Soc. 2011, 1332610022–10025. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Fisher C. K.; Huang A.; Stultz C. M. Modeling intrinsically disordered proteins with Bayesian statistics. J. Am. Chem. Soc. 2010, 1324214919–14927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu M. M.; Kriwacki R. W.; Pappu R. V. Versatility from protein disorder. Science 2012, 337, 1460–1461. [DOI] [PubMed] [Google Scholar]
- Bright J. N.; Woolf T. B.; Hoh J. H. Predicting properties of intrinsically unstructured proteins. Prog. Biophys. Mol. Biol. 2001, 763131–173. [DOI] [PubMed] [Google Scholar]
- Papoian G. A. Proteins with weakly funneled energy landscapes challenge the classical structure-function paradigm. Proc. Natl. Acad. Sci. U.S.A. 2008, 1053814237–14238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitalis A.; Pappu R. V. Methods for Monte Carlo simulations of biomacromolecules. Annu. Rep. Comput. Chem. 2009, 5, 49–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmeier M.; Hamilton J. A.; LeGall T.; Vacic V.; Cortese M. S.; Tantos A.; Szabo B.; Tompa P.; Chen J.; Uversky V. N.; Obradovic Z.; Dunker A. K. DisProt: The database of disordered proteins. Nucleic Acid. Res. 2007, 35, D786–D793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitalis A.; Pappu R. V. ABSINTH: A new continuum solvation model for simulations of polypeptides in aqueous solutions. J. Comput. Chem. 2009, 305673–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Radhakrishnan A.; Vitalis A.; Mao A. H.; Steffen A. T.; Pappu R. V. Improved atomistic Monte Carlo simulations demonstrate that poly-l-proline adopts heterogeneous ensembles of conformations of semi-rigid segments interrupted by kinks. J. Phys. Chem. B 2012, 116236862–6871. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mao A. H.; Pappu R. V. Crystal lattice properties fully determine short-range interaction parameters for alkali and halide ions. J. Chem. Phys. 2012, 1376064104(1–9). [DOI] [PubMed] [Google Scholar]
- a Sugita Y.; Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 3141–2141–151. [Google Scholar]; b Mitsutake A.; Sugita Y.; Okamoto Y. Generalized-ensemble algorithms for molecular simulations of biopolymers. Biopolymers 2001, 60296–123. [DOI] [PubMed] [Google Scholar]
- a Vitalis A.; Caflisch A. Micelle-like architecture of the monomer ensemble of Alzheimer’s amyloid-β peptide in aqueous solution and its implications for a β aggregation. J. Mol. Biol. 2010, 4031148–165. [DOI] [PubMed] [Google Scholar]; b Vitalis A.; Lyle N.; Pappu R. V. Thermodynamics of β-sheet formation in polyglutamine. Biophys. J. 2009, 971303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gallicchio E.; Andrec M.; Felts A. K.; Levy R. M. Temperature weighted histogram analysis method, replica exchange, and transition paths. J. Phys. Chem. B 2005, 109146722–6731. [DOI] [PubMed] [Google Scholar]; b Chodera J. D.; Swope W. C.; Noe F.; Prinz J. H.; Shirts M. R.; Pande V. S. Dynamical reweighting: Improved estimates of dynamical properties from simulations at multiple temperatures. J. Chem. Phys. 2011, 13424244107(1–15). [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhang W.; Chen J. H. Efficiency of adaptive temperature-based replica exchange for sampling large-scale protein conformational transitions. J. Chem. Theory Comput. 2013, 962849–2856. [DOI] [PubMed] [Google Scholar]
- a Mitsutake A.; Sugita Y.; Okamoto Y. Replica-exchange multicanonical and multicanonical replica-exchange Monte Carlo simulations of peptides. I. Formulation and benchmark test. J. Chem. Phys. 2003, 118146664–6675. [Google Scholar]; b Sugita Y.; Kitao A.; Okamoto Y. Multidimensional replica-exchange method for free-energy calculations. J. Chem. Phys. 2000, 113156042–6051. [Google Scholar]
- Gelb L. D. Monte Carlo simulations using sampling from an approximate potential. J. Chem. Phys. 2003, 118177747–7750. [Google Scholar]
- a Bandyopadhyay P. Increasing the efficiency of Monte Carlo simulation with sampling from an approximate potential. Chem. Phys. Lett. 2013, 556, 341–345. [Google Scholar]; b Calvo F. Efficiency of nested Markov chain Monte Carlo for polarizable potentials and perturbed Hamiltonians. Int. J. Quantum Chem. 2010, 110132347–2354. [Google Scholar]; c Coe J. D.; Sewell T. D.; Shaw M. S. Nested Markov chain Monte Carlo sampling of a density functional theory potential: Equilibrium thermodynamics of dense fluid nitrogen. J. Chem. Phys. 2009, 1317074105. [DOI] [PubMed] [Google Scholar]
- Kaminski G. A.; Friesner R. A.; Tirado-Rives J.; Jorgensen W. L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105286474–6487. [Google Scholar]
- a Owicki J. C.; Scheraga H. A. Preferential sampling near solutes in Monte Carlo calculations on dilute solutions. Chem. Phys. Lett. 1977, 473600–602. [Google Scholar]; b Bigot B.; Jorgensen W. L. Sampling methods for Monte-Carlo simulations of normal-butane in dilute-solution. J. Chem. Phys. 1981, 7541944–1952. [Google Scholar]; c Mehrotra P. K.; Mezei M.; Beveridge D. L. Convergence acceleration in Monte-Carlo computer-simulation on water and aqueous solutions. J. Chem. Phys. 1983, 7863156–3166. [Google Scholar]; d Leontidis E.; Suter U. W. Monte Carlo methodologies for enhanced configurational sampling of dense systems—Motion of a spherical solute in a polymer melt as a model problem. Mol. Phys. 1994, 833489–518. [Google Scholar]; e Kentsis A.; Mezei M.; Osman R. MC-PHS: A Monte Carlo implementation of the primary hydration shell for protein folding and design. Biophys. J. 2003, 842805–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flory P. J.Statistical Mechanics of Chain Molecules; Oxford University Press: New York, 1969. [Google Scholar]
- Metropolis N.; Rosenbluth A.; Rosenbluth M.; Teller A.; Teller E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar]
- Wyczalkowski M. A.; Pappu R. V. Satisfying the fluctuation theorem in free-energy calculations with Hamiltonian replica exchange. Phys. Rev. E 2008, 772026104(1–12). [DOI] [PubMed] [Google Scholar]
- Chandler D.; Weeks J. D.; Andersen H. C. Van der Waals picture of liquids, solids, and phase transformations. Science 1983, 2204599787–794. [DOI] [PubMed] [Google Scholar]
- Hoover W. G.; Gray S. G.; Johnson K. W. Thermodynamic properties of the fluid and solid phases for inverse power potentials. J. Chem. Phys. 1971, 55, 1128–1136. [Google Scholar]
- Tran H. T.; Wang X.; Pappu R. V. Reconciling observations of sequence-specific conformational propensities with the generic polymeric behavior of denatured proteins. Biochemistry 2005, 443411369–80. [DOI] [PubMed] [Google Scholar]
- a Lu Q.; Kim J.; Straub J. E. Exploring the solid–liquid phase change of an adapted Dzugutov model using generalized replica exchange method. J. Phys. Chem. B 2012, 116298654–8661. [DOI] [PubMed] [Google Scholar]; b Straub J. E.; Rashkin A. B.; Thirumalai D. Dynamics in rugged energy landscapes with applications to the S-peptide and ribonuclease-A. J. Am. Chem. Soc. 1994, 11652049–2063. [Google Scholar]; c Straub J. E.; Thirumalai D. Exploring the energy landscape in proteins. Proc. Natl. Acad. Sci. U.S.A. 1993, 903809–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R. K.; Mao A. H.; Pappu R. V. Unmasking functional motifs within disordered regions of proteins. Sci. Signal. 2012, 5220pe17. [DOI] [PubMed] [Google Scholar]
- a Freeman D. L.; Doll J. D. Computational studies of clusters: Methods and results. Annu. Rev. Phys. Chem. 1996, 47, 43–80. [Google Scholar]; b Lopez G. E. Study of the solid–liquid transition for Ar-55 using the J-walking Monte Carlo method. J. Chem. Phys. 1996, 104176650–6653. [Google Scholar]; c Zhou R. H.; Berne B. J. Smart walking: A new method for Boltzmann sampling of protein conformations. J. Chem. Phys. 1997, 107219185–9196. [Google Scholar]; d Neirotti J. P.; Freeman D. L.; Doll J. D. Approach to ergodicity in Monte Carlo simulations. Phys. Rev. E 2000, 6257445–7461. [DOI] [PubMed] [Google Scholar]; e Mak C. H. Stochastic potential switching algorithm for Monte Carlo simulations of complex systems. J. Chem. Phys. 2005, 12221214110. [DOI] [PubMed] [Google Scholar]; f Nigra P.; Freeman D. L.; Doll J. D. Combining smart darting with parallel tempering using Eckart space: Application to Lennard-Jones clusters. J. Chem. Phys. 2005, 12211114113. [DOI] [PubMed] [Google Scholar]
- Andricioaei I.; Straub J. E.; Voter A. F. Smart darting Monte Carlo. J. Chem. Phys. 2001, 114166994–7000. [Google Scholar]
- a Chodera J. D.; Shirts M. R. Replica exchange and expanded ensemble simulations as Gibbs sampling: Simple improvements for enhanced mixing. J. Chem. Phys. 2011, 13519194110. [DOI] [PubMed] [Google Scholar]; b Li H. Z.; Li G. H.; Berg B. A.; Yang W. Finite reservoir replica exchange to enhance canonical sampling in rugged energy surfaces. J. Chem. Phys. 2006, 12514144902. [DOI] [PubMed] [Google Scholar]; c Roitberg A. E.; Okur A.; Simmerling C. Coupling of replica exchange simulations to a non-Boltzmann structure reservoir. J. Phys. Chem. B 2007, 111102415–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ruscio J. Z.; Fawzi N. L.; Head-Gordon T. How Hot? Systematic convergence of the replica exchange method using multiple reservoirs. J. Comput. Chem. 2010, 313620–627. [DOI] [PubMed] [Google Scholar]
- a Lettieri S.; Mamonov A. B.; Zuckerman D. M. Extending fragment based free energy calculations with library based Monte Carlo simulation: Annealing in interaction space. Biophys. J. 2011, 1003154–154. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mamonov A. B.; Bhatt D.; Cashman D. J.; Ding Y.; Zuckerman D. M. General library-based Monte Carlo technique enables equilibrium sampling of semi-atomistic protein models. J. Phys. Chem. B 2009, 1133110891–10904. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Mamonov A. B.; Lettieri S.; Ding Y.; Sarver J. L.; Palli R.; Cunningham T. F.; Saxena S.; Zuckerman D. M. Tunable, mixed-resolution modeling using library-based Monte Carlo and graphics processing units. J. Chem. Theory Comput. 2012, 882921–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lyman E.; Ytreberg F. M.; Zuckerman D. M. Resolution exchange simulation. Phys. Rev. Lett. 2006, 96, 2. [DOI] [PubMed] [Google Scholar]; b Lyman E.; Zuckerman D. M. Resolution exchange simulation with incremental coarsening. J. Chem. Theory Comput. 2006, 23656–666. [DOI] [PubMed] [Google Scholar]
- a Kim J.; Straub J. E. Optimal replica exchange method combined with Tsallis weight sampling. J. Chem. Phys. 2009, 13014144114. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kim J.; Straub J. E. Generalized simulated tempering for exploring strong phase transitions. J. Chem. Phys. 2010, 13315154101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Pande V. S.; Baker I.; Chapman J.; Elmer S. P.; Khaliq S.; Larson S. M.; Rhee Y. M.; Shirts M. R.; Snow C. D.; Sorin E. J.; Zagrovic B. Atomistic protein folding simulations on the submillisecond time scale using worldwide distributed computing. Biopolymers 2003, 68191–109. [DOI] [PubMed] [Google Scholar]; b Lane T. J.; Shukla D.; Beauchamp K. A.; Pande V. S. To milliseconds and beyond: Challenges in the simulation of protein folding. Curr. Opin. Struct. Biol. 2013, 23158–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson T. E.; Vitalis A.; Crick S. L.; Pappu R. V. Modulation of polyglutamine conformations and dimer formation by the N-terminus of huntingtin. J. Mol. Biol. 2010, 39651295–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrea D. M.; Kriwacki R. W. Regulated unfolding of proteins in signaling. FEBS Lett. 2013, 58781081–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey N. E.; Van Roey K.; Weatheritt R. J.; Toedt G.; Uyar B.; Altenberg B.; Budd A.; Diella F.; Dinkel H.; Gibson T. J. Attributes of short linear motifs. Mol. BioSys. 2012, 81268–281. [DOI] [PubMed] [Google Scholar]
- a Joonseong L.; Seonghoon K.; Rakwoo C.; Jayanthi L.; Gebremichael Y. Effects of molecular model, ionic strength, divalent ions, and hydrophobic interaction on human neurofilament conformation. J. Chem. Phys. 2013, 1381015103. [DOI] [PubMed] [Google Scholar]; (1–15);; b Kumar S.; Yin X. H.; Trapp B. D.; Hoh J. H.; Paulaitis M. E. Relating interactions between neurofilaments to the structure of axonal neurofilament distributions through polymer brush models. Biophys. J. 2002, 8252360–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnham C. P.; Roll-Mecak A. The chemical complexity of cellular microtubules: Tubulin post-translational modification enzymes and their roles in tuning microtubule functions. Cytoskeleton 2012, 697442–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Goryaynov A.; Ma J.; Yang W. Single-molecule studies of nucleocytoplasmic transport: From one dimension to three dimensions. Integ. Biol. 2012, 4110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Labokha A. A.; Gradmann S.; Frey S.; Huelsmann B. B.; Urlaub H.; Baldus M.; Goerlich D. Systematic analysis of barrier-forming FG hydrogels from Xenopus nuclear pore complexes. EMBO J. 2013, 322204–218. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Terry L. J.; Wente S. R. Flexible Gates: Dynamic topologies and functions for FG nucleoporins in nucleocytoplasmic Transport. Eukaryotic Cell 2009, 8121814–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14133–38. [DOI] [PubMed] [Google Scholar]
- Baker N. A.; Sept D.; Joseph S.; Holst M. J.; McCammon J. A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2001, 981810037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25131605–1612. [DOI] [PubMed] [Google Scholar]