

Abstract
Background
Bortezomib is an FDA-approved proteasome inhibitor, and oncolytic HSV-1 (oHSV) is a promising therapeutic approach for cancer. We tested the impact of combining bortezomib with oHSV for anti-tumor efficacy.
Methods
The synergistic interaction between oHSV and bortezomib was calculated using Chou-Talalay analysis. Viral replication was evaluated using plaque assay and immune fluorescence. Western-blot assays were used to evaluate induction of ER stress and unfolded protein response (UPR). Inhibitors targeting Hsp90 were utilized to investigate the mechanism of cell killing. Anti-tumor efficacy in vivo was evaluated using subcutaneous and intracranial tumor xenografts of glioma and head and neck cancer. Survival was analyzed by Kaplan-Meier curves and two-sided log rank test.
Results
Combination treatment with bortezomib and oHSV, 34.5ENVE, displayed strong synergistic interaction in ovarian cancer, head & neck cancer, glioma, and malignant peripheral nerve sheath tumor (MPNST) cells. Bortezomib treatment induced ER stress, evident by strong induction of Grp78, CHOP, PERK and IRE1α (western blot analysis) and the UPR (induction of hsp40, 70 and 90). Bortezomib treatment of cells at both sublethal and lethal doses increased viral replication (p value <0.001), but inhibition of Hsp90 ablated this response, reducing viral replication and synergistic cell killing. The combination of bortezomib and 34.5ENVE significantly enhanced anti-tumor efficacy in multiple different tumor models in vivo.
Conclusions
The dramatic synergy of bortezomib and 34.5ENVE is mediated by bortezomib- induced UPR and warrants future clinical testing in patients.
Introduction
Oncolytic herpes simplex virus-1 (oHSV) therapy utilizes viruses that are engineered to infect and replicate in cancer cells with minimal damage to non-neoplastic tissue. This therapy is currently being evaluated for safety and efficacy in multiple Phase I, II, and III clinical trials (1). The results from a phase III testing of T-Vec (an oHSV developed by Amgen) has shown promising results in tumor shrinkage. Although the overall survival data has yet to be established, there is a significant need to optimize this promising therapy in vivo. While second and third generation viruses are being created and tested in preclinical studies, drug-virus combinations can be rapidly translated to clinical trials to maximize efficacy and minimize toxicity (2).
The proteasome is a cellular organelle that controls degradation and recycling of a wide variety of proteins that regulate diverse cellular functions including cell cycle progression, cell death, gene expression, signal transduction, metabolism, morphogenesis, differentiation, antigen presentation, and neuronal function. Inhibition of the proteasome can result in cellular aggregation of unfolded proteins which induce ER stress and apoptosis. Cancer cells have increased metabolic demands and are thought to constantly be at the brink of ER stress. Thus proteasome inhibition has been investigated as a potential way to target malignant cells.
Bortezomib is a peptide-based, reversible proteasome inhibitor, which is currently Food and Drug Administration (FDA)-approved either as a single agent or in combination with other chemo-/radio- therapeutic agents for multiple myeloma. It is also used as a second line treatment for ovarian and head & neck cancers and is currently under clinical evaluation for the treatment of several other cancer types. Recent evidence indicates that most patients do not respond to this drug when it is used as a single agent, and several strategies testing its efficacy in combination with other drugs are being pursued (3, 4).
The combination of bortezomib and oHSV is intriguing because HSV-1 exploits the host proteasome during its life cycle (5, 6), but proteasome-mediated degradation of viral capsids in infected macrophages is also thought to be important for stimulating antiviral interferon (IFN) responses in these cells (7). Additionally bortezomib treatment has also been shown to induce Epstein Barr virus and Kaposi sarcoma virus lytic gene expression, suggesting that bortezomib treatment could also improve virus replication in vivo (8). These results suggest that bortezomib may have opposing effects on oHSV efficacy.
In this study, we demonstrate for the first time that the induction of the unfolded protein response after bortezomib treatment improved oHSV replication and synergistically improved cancer cell killing in vitro and in vivo. These findings demonstrate that the synergistic interaction between oHSV and bortezomib improves overall therapeutic efficacy via augmenting the cancer cell killing, providing advanced rationale for combining these agents in a clinical trial. This is the first study to demonstrate the utility of combining a clinically relevant proteasome inhibitor to augment replication of oHSV and to achieve synergistic cell killing.
Materials and methods
Cell lines, viruses, antibodies, and molecular biology reagents
All cancer cell lines - with the exception of ovarian cancer cells were cultured in Dulbecco's modified eagle's medium (DMEM; Gibco BRL, Grand Island, NY) supplemented with 10% fetal bovine serum (Gibco BRL). Ovarian cancer cell lines were maintained in RPMI medium (Gibco BRL) supplemented with 10% fetal bovine serum. Human glioma cell lines (U251T3 and LN229), Head&Neck cancer cell lines (CAL27, UMSCC11A, and UMSCC74A), ovarian cancer cell lines (PA1, A2780-CS, A2780-CR, and SKOV3) have been cultured in our laboratory and U251T3 cells were obtained as a tumorigenic clone of U251 cells by serial passage of these cells three times in mice. Normal Human Astrocyte, human umbilical vein endothelial cell (HUVEC), and Hepatocyte cells were purchased from Sciencell Research Laboratories (Carlsbad, CA, USA) and cultured according to manufacturer's instruction. U87ΔEGFR cell line expresses a truncated, constitutively active, mutant form of epidermal growth factor receptor (EGFRvIII). GBM169 are primary patient derived neurosphere cultures maintained in neurobasal medium. Human ovarian cancer patient ascites cells were harvested from patient ascites under an institutional IRB approved protocol. All cell culture media were supplemented with 2 mM L-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin. All cell lines were maintained at 37°C in a humidified atmosphere at 5% CO2. The construction and generation of 34.5ENVE (viral ICP34.5 Expressed by Nestin promotor and Vstat120 Expressing) and HSV1716 (a mutant herpes simplex virus type 1 deleted in both copies of RL1 gene which encodes the protein ICP34.5) has been previously described (9). Virus was propagated in Vero African green monkey kidney cells (American Type Culture Collection, Manassas, VA) as previously described (10).
Cytotoxicity assay
The cytotoxicity of bortezomib and 34.5ENVE in each cell line was determined by a standard MTT assay (Roche, Mannheim, Germany) as manufacturer's instructions. To measure ED50 of each bortezomib or 34.5ENVE alone, cells were plated onto 96-well plates of approximately 50% confluence on day zero and then treated with bortezomib or PBS for 16 hours, prior to drug wash-out and infection with 34.5ENVE or HBSS. Cell viability was measured 72 hours post infection. To measure synergistic cell killing, cells were treated with bortezomib and 34.5ENVE (as detailed above) at serially diluted concentrations of 0.0625, 0.125, 0.25, 0.5, 1, 2, and 4 times of their ED50 in a constant ratio. Cell viability was measured 72 hours post infection. All assays were performed in triplicate. For rescue assay with Geldanamycin and 17-AAG, cells were pretreated with either agent for 1 hour before bortezomib treatment.
In vitro viral replication assay
Cells were treated ± bortezomib (at indicated doses) for 16 hours, and following drug wash-out cells were infected with 34.5ENVE (at indicated doses) for two hours. Seventy-two hours post infection, cells and supernatant were collected, and the number of infectious particles present in the resulting supernatant was determined as previously described (9).
Western blot analysis and antibodies
Cell lysates were fractionated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. In UL30 detection experiments, cell and nuclear fractions were separated by NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, Cat# 78835, Rockford, lL). Blocked membranes were then incubated with antibodies against Anti-LC3B, Calnexin, PERK, IRE1α, PDI, Bip/GRP78, CHOP, Ero1-Lα, HSP40, HSP70, HSP90, GAPDH (Abcam, Cambridge, MA) (each diluted 1:1000); HRP-conjugated secondary anti-mouse antibody (each diluted 1:1000) (GE Healthcare, Piscataway, NJ); HRP-conjugated secondary goat anti-rabbit antibody (each diluted 1:1000) (Dako, Hamburg, Germany), and the immunoreactive bands were visualized using a using enhanced chemiluminescence (ECL) (GE Healthcare, Piscataway, NJ). UL30 antibody (diluted 1:100) used was purified rabbit IgG raised against recombinant Escherichia coli-expressed bacteriophage T7 gene 10 protein fused with the C-terminal portion of the HSV-1 pol gene (11).
Animal surgery
All mice experiments were housed and handled in accordance with the Subcommittee on Research Animal Care of the Ohio State University guidelines and adhered to the NIH Guide for the Care and Use of Laboratory Animals. Female athymic nu/nu mice (Charles River Laboratoties, Frederick, MD) were used for all in vivo experiments. Nude mice with subcutaneous tumors (100 mm3) were randomized to be treated with either intraperitoneal PBS or bortezomib (0.8 mg/kg) twice a week. For intracranial tumor studies, anesthetized nude mice were implanted with tumor cells as described (9). Three days following cell implantation, mice were randomized to receive PBS or bortezomib (0.8 mg/kg) via intra-peritoneal injection twice a week. Seven days later, mice with intracranial or subcutaneous tumors were inoculated with 34.5ENVE or Hank's Balanced Salt Solution (HBSS) (intracranial: 5 × 104 pfu or for subcutaneous tumors: 1 × 105 PFU). Intra-peritoneal bortezomib or PBS injections continued for the duration of the experiment, and animals were observed daily and euthanized at the indicated time points or when they showed signs of morbidity (hunched posture and weight loss). For subcutaneous studies, tumor volume was calculated based on tumor length and width using the following formula: volume = 0.5LW2 as described (9). For histological analysis, subcutaneous CAL27 tumors in nude mice were treated with/without bortezomib (0.8 mg/kg) twice a week. Seven days later, virus was intratumorally injected, and then injected again two days later. Three days after the second virus injection, tumors were harvested from each treatment group. Representative sections were stained with hematoxylin & Eosin (H&E) and anti-HSV-1 antibody (Dako, 1:200 dilution) and then examined by microscopy.
Statistical analysis
To compare two independent treatments for continuous endpoints, the student's t test was used. When multiple pair-wise comparisons are made, one-way ANOVA was used. Log-rank test was used to compare survival curves for survival data. When median survival was not observed for two or more groups in an experiment, survival probability on the last day of observation was calculated for each group. P values were adjusted for multiple comparisons by Holms' procedure. A P value 0.05 or less is considered significant. For all synergy analysis, cancer cells were seeded into a 96-well plate, and were treated with vehicle or bortezomib for 18 hours followed by drug wash out, and then virus infection. Three days after infection, cell viability was measured using a standard MTT assays (Roche Applied Sciences, Indianapolis IN). For synergy analysis, dose response curves and 50% effective dose (ED50) values were determined for each individual treatment (drug or virus). Fixed ratios of ED50 of drug and virus were then used to treat the cells, either with dual treatment or with individual treatments (as controls). CompuSyn software program algorithm assessed the Combination Index (CI). Combined dose-response curves were fitted to Chou-Talalay lines, which are derived from the law of mass action. CI<1 indicates synergistic interaction, whereas CI>1 is antagonistic and CI=1is considered additive (12).
Results
Interaction of Bortezomib treatment with oHSV
The sensitivity of head & neck squamous cell carcinomas, ovarian cancer, malignant peripheral nerve sheet tumors (MPNST), and glioma to bortezomib or 34.5ENVE alone in vitro was evaluated. Cells were treated with indicated doses of bortezomib and/or oHSV and cell death was measured by a standard MTT assay. Figure 1A shows that combination treatment increased cell killing compared to the predicted additive effects (dotted line), suggesting synergistic cell killing.
Figure 1. Effect of bortezomib and oHSV on cancer cell killing.

A) Treatment of ovarian cancer, head and neck cancer, and glioma cells with bortezomib and oHSV showed more than additive cell killing. Briefly, the indicated cells were treated with sub-lethal doses of bortezomib or PBS for 16 hours, followed by wash-out drug and treatment with sub-lethal dosed of 34.5ENVE or PBS. Seventy-two hours later from 34.5ENVE infection, cell viability was measured via MTT assay. Data shown are % cell death ± standard deviations of cells treated with bortezomib/PBS for 16 hours prior to media washout alone or with oHSV infection. B) Chou-Talalay analysis of combining bortezomib with 34.5ENVE in ovarian cancer, head & neck cancer, and glioma cells. Briefly, cells were treated with bortezomib for 16 hours, followed by drug wash out and treatment with 34.5ENVE at concentrations of 0.0625, 0.125, 0.25, 0.5, 1, 2, and 4 times of their ED50 in a constant ratio. Data shown as fraction affected (fa) versus combination index (CI) plots. CI < 1 indicate synergy, CI = 1 indicate additive, and CI > 1 indicate an antagonistic interaction. C) Normal Human Astrocytes (NHA), human umbilical vein derived endothelial cells (HUVEC), and Hepatocyte cells were plated and treated with/without ED50 concentration of bortezomib for 16 hours, followed by drug wash out and treatment with 34.5ENVE. 72 hours following 34.5ENVE infection, cell viability was measured by MTT assay. Data points represent the mean % cell viability relative to uninfected cells and error bars indicate ± SD for each group.
To further evaluate synergistic interaction between bortezomib and oHSV, we measured sensitivity of each cell line to bortezomib and oHSV. Briefly, cells were treated with bortezomib at concentrations ranging from 0.1 to 500 nM for 16 hours, followed by drug wash out. To test sensitivity to 34.5ENVE, cells were infected with different doses of oHSV ranging from 0.001 to 0.5 multiplicity of infection (MOI) defined as virus plaque forming units/cell (pfu/cell). 72 hours following bortezomib wash out or 34.5ENVE infection, cell viability was measured by a standard MTT assay. The 50 percent effective dose (ED50) of bortezomib and 34.5ENVE were each defined as the dosage yielding 50% cell viability at 72 hrs following treatment as compared to untreated controls. Supplementary Table S1 shows the sensitivity of each cell line to bortezomib and 34.5ENVE. To study the interaction between bortezomib and 34.5ENVE, cells were treated with bortezomib for 16 hours, followed by drug wash out and treatment with 34.5ENVE at concentrations of 0.0625, 0.125, 0.25, 0.5, 1, 2, and 4 times of their ED50 in a constant ratio. Cell viability was measured 72 hours after virus infection. The results were analyzed using the median-effect method of Chou-Talalay (COMPUSYN) (12). Fig. 1B shows combination index plots of Chou-Talalay. The plots show synergistic cell killing between bortezomib and 34.5ENVE (combination) in PA1, A2780-CS (cisplatin sensitive), SKOV3, and A2780-CR (cisplatin resistant) ovarian cancer cells CAL27, UMSCC11A, and UMSCC74A, head & neck squamous cell carcinomas and in U251T3, LN229, and U87ΔEGFR glioma cells (Fig 1B). In a separate experiment, similar synergistic cell killing was observed when clinical grade oHSV1716 was used in combination with bortezomib to treat MPNST cells (Supplementary Fig. S1) (CI values less than 1 at all fractions affected). To evaluate the effect of bortezomib on oHSV safety, we compared the cytotoxic ability of oHSV towards normal Human Astrocytes (NHA), human umbilical vein derived endothelial cells (HUVEC), and Hepatocyte cells treated with/without ED50 concentrations of bortezomib (Fig. 1C). In all three normal cells tested, no additive (dotted line) or synergy was observed between bortezomib and 34.5ENVE. These data support the conclusion that the combination of bortezomib and oHSV significantly increased sensitivity to cell death for a wide variety of solid tumor cells but not for normal cells.
Impact of Bortezomib on oHSV replication
To investigate the impact of bortezomib on 34.5ENVE replication, U251T3 glioma cells were treated with sublethal and ED50 doses of bortezomib (2 and 12 nM) followed by 34.5ENVE infection (Fig. 2A-B). Fluorescent microscopy was used to image GFP positive (infected) cells, 48 hours post infection. Figure 2A shows increased GFP positive infected cells in both concentrations of bortezomib indicating that bortezomib treatment increased viral replication in vitro. Additionally quantification of viral titers showed a significant increase in viral titer in the both sublethal and ED50 doses of bortezomib (Fig. 2B). Quantification of viral titers in cells treated with or without bortezomib revealed a significant increase in viral replication in the multiple cell lines treated with sub-lethal doses of bortezomib in all cells tested: ovarian: SKOV3 cells: 20.3 fold, head and neck cancer cells: CAL27: 1.8 fold, UMSCC74A: 1.8 fold, and glioma: U87ΔEGFR: 8.4 fold, (Fig. 2B, and supplementary Table S2). Collectively, these data demonstrate that bortezomib increased viral replication.
Figure 2. Effect of bortezomib treatment on viral replication.
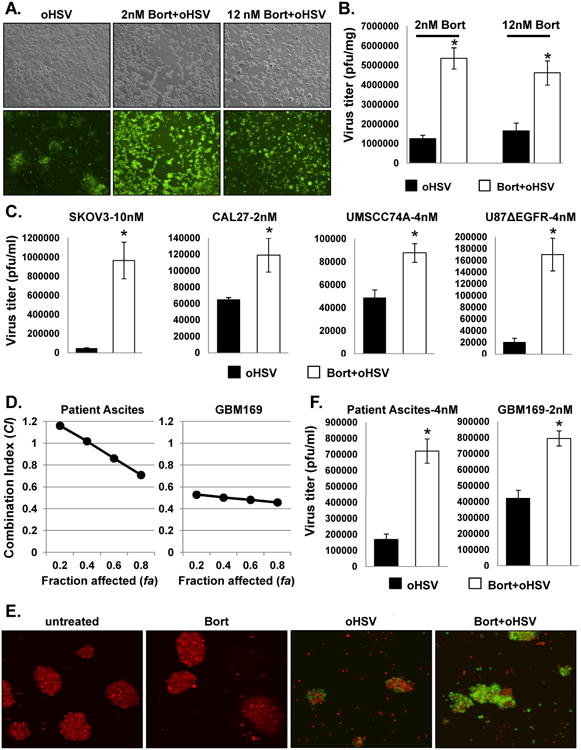
U251T3 cells were treated with 2nM or 12nM bortezomib for 16 hours prior to drug wash out and 34.5ENVE infection (MOI = 0.002 with 2nM bortezomib and 0.01 for 12 nM bortezomib) and evaluated for virus replication. A) Bright field (Top) and fluorescence microscopic images (bottom) of GFP positive infected cells 48 hours post-infection demonstrate increased virus replication following bortezomib treatment at both sublethal (2nM) and ED50 (12nM) concentrations relative to no bortezomib treated cells (magnification, × 100). B) Quantification of virus yield in the both sublethal and ED50 concentration of bortezomib treated cells. Both cells and media were harvested 48 hours (12nM) and 72 hours (2nM) after infection, and viral titers were determined by standard plaque assay. C) The indicated cell lines were treated with the indicated doses of bortezomib 16 hours prior to 34.5ENVE infection. Both cells and media were harvested 72 hours after viral infection and viral titers were determined by standard plaque assay. Data points represent the mean, and error bars indicate ± SD for each group. Asterisks indicate statistically significant differences between indicated pairs. D) Chou-Talalay analysis of combining bortezomib with 34.5ENVE in primary ovarian cancer ascites derived tumor cells and in primary GBM patient derived tumor cells maintained as undifferentiated neurospheres in vitro. Briefly primary tumor cells were treated with bortezomib for 16 hours, followed by drug wash out and treatment with 34.5ENVE at serially diluted concentrations of 0.0625, 0.125, 0.25, 0.5, 1, 2, and 4 times of their ED50 in a constant ratio. E) Patient-derived neurospheres (GBM169) treated with/without bortezomib (2nM) were stained with CellTracker CMTPX and incubated with single cell suspension of GBM 169 cells infected with 34.5ENVE (MOI = 0.1) for one hour. Twenty-four hours post-infection, bortezomib treated neurospheres showed increased GFP positive, infected cells relative to untreated cells (magnification, × 100). F) The indicated primary patient derived ovarian cancer ascites and GBM169 cells were treated with/without bortezomib (4 nM for ascites and 2 nM for GBM169) 16 hours prior to 34.5ENVE infection. Both cells and media were harvested 72 hours after viral infection and viral titers were determined by standard plaque assay. Data points represent the mean, and error bars indicate ± SD for each group. Asterisks indicate statistically significant differences between indicated pairs.
Efficacy of combination treatment in patient-derived primary tumor cells ex vivo
Next, we tested synergistic interaction of bortezomib and 34.5ENVE in ovarian cancer patient ascites derived tumor cells and GBM patient derived primary GBM neurospheres (GBM169) ex vivo (Fig. 2D). Briefly cells were treated with bortezomib for 16 hours, followed by drug wash out and treatment with 34.5ENVE at concentrations of 0.0625, 0.125, 0.25, 0.5, 1, 2, and 4 times of their ED50 in a constant ratio. Cell viability was measured 72 hours after virus infection. The results were analyzed using the median-effect method of Chou-Talalay (COMPUSYN). Synergistic cell killing was obvious in both patient derived ascites and primary GBM neurosphere cells (Fig. 2D). Patient-derived primary GBM neurosphere cells (GBM169) pre-treated with or without bortezomib were labeled with red cell tracker and treated with 34.5ENVE. Fluorescent microscopy images showed increased GFP positive infected cells in bortezomib treated cells (Figure 2E). Quantification of viral titers in cells treated with or without bortezomib revealed a significant increase in viral replication in both patient ascites and primary GBM neurospheres: Ovarian patient ascites: 4.3 fold and GBM169: 1.9 folds increase over oHSV alone (Fig. 2F).
ER stress and unfolded protein response in cells treated with bortezomib and oHSV
Bortezomib is reversible proteasome inhibitor that blocks the chymotrypsin like activity of the proteasome resulting in the accumulation of unfolded proteins. Accumulation of unfolded proteins induces ER stress leading to caspase dependent apoptosis (13-15). Treatment of both glioma and head and neck cancer cells with bortezomib showed induction of proteins indicative of ER stress in a dose dependent manner (Figure 3A and supplementary Figure S2A). In contrast oHSV is known to disarm ER stress during infection (16). To test the impact of combination treatment of oHSV and bortezomib on ER stress, we measured changes in levels of cellular proteins that are induced during ER stress. Increased expression of PERK, Calnexin, IRE1alpha, CHOP, Ero1-Lα and GRP78 proteins was observed in oHSV-infected and uninfected glioma and head and neck cancer cells after bortezomib treatment (Fig. 3B and supplementary Figure S2B).
Figure 3. Effect of bortezomib on ER stress and unfolded protein response (UPR) in infected and uninfected cancer cells.
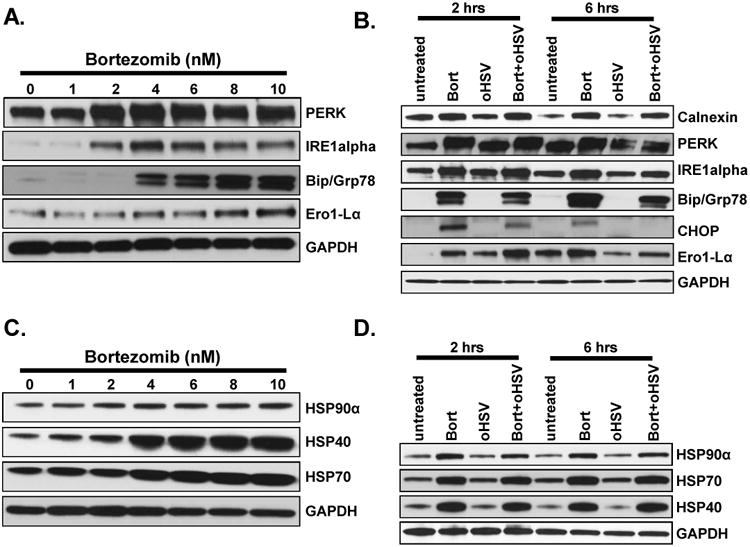
A) Dose dependent induction of ER stress in U251T3 cells. Shown are immunoblots of cell lysates from U251T3 glioma cells treated with bortezomib for 16 hours probed for expression of the indicated proteins. B) ER stress is not increased in cells treated with both bortezomib and oHSV compared to bortezomib only treated cells. Shown are immune blots of the U251T3 cells treated with/without bortezomib (12nM) for 16 hours prior to 34.5ENVE infection at an MOI of 0.01 or 1. Cells were harvested 2 and 6 hours post infection, and cell lysates were probed with antibodies against ER stress related pathway (Calnexin, PERK, IRE1alpha, Bip/GRP78, CHOP, and Ero1-Lα) GAPDH was used as a loading control. C) Dose dependent induction of HSP40, 70 and 90α in U251T3 cancer cells. Shown are immunoblots of cell lysates from the indicated cells treated with bortezomib for 16 hours probed for expression of the indicated proteins. D) Bortezomib pretreatment induced HSPs expression in uninfected and oHSV-infected U251T3 cells. U251T3 cells treated with/without bortezomib were infected with 34.5ENVE (MOI = 1) and cells were harvested 2 and 6 hours post infection. Cell lysates were probed with antibodies against HSP40, HSP70, and HSP90α. GAPDH was used as a loading control.
Along with ER stress, unfolded proteins also lead to the induction of an unfolded protein response (UPR), which constitutes increased expression of heat shock and cellular chaperone proteins. Both glioma and head and neck cancer cells treated with bortezomib showed a dose dependent induction of HSP proteins (Figure 3C and Supplementary Figure S2C). Consistent with this, we observed induction of heat shock proteins (HSPs) (HSP40, 70, and 90α) in oHSV-infected and uninfected glioma and head and neck cancer cells pre-treated with bortezomib (Fig. 3D and supplementary figure S2D). Hsp90α induction was observed at both low and high concentrations of bortezomib treatment of glioma and head and neck cancer cells. HSP90 has been previously shown to be important for localization of HSV polymerase to the nucleus (17), thus we hypothesized that bortezomib-induced HSP90 induction could be critical for the increased viral replication and cell killing following combination treatment. To test this hypothesis, we examined the impact of Geldanamycin, an HSP90 inhibitor, on combination treatment-induced tumor cell cytotoxicity. Fig. 4A shows combination index plots of Chou-Talalay of glioma cells treated with bortezomib and 34.5ENVE in the presence or absence of Geldanamycin. Treatment of glioma cells with Geldanamycin reduced synergistic cell killing interactions between 34.5ENVE and bortezomib (Fig. 4A). The synergistic interaction of the combination treatment was also reduced in cells treated with 17-AAG, a less toxic alternative HSP90 inhibitor (Fig. 4B), supporting the significance of HSP90 in oHSV and bortezomib synergistic cell killing. Consistent with the importance of HSP90, both 17-AAG and Geldanamycin treatment resulted in a loss of the increased viral replication achieved by bortezomib treatment (Fig. 4C). HSP90 is thought to increase HSV polymerase localization to the nucleus. To directly test whether bortezomib treatment affected nuclear localization of HSV polymerase (UL30), we compared the cytosolic and nuclear fractions (C.F and N.F respectively) of oHSV-infected cells treated without/with bortezomib at the indicated concentrations (Fig. 4D). Increased nuclear localization of UL30 was observed in bortezomib treated cells. Collectively, these results demonstrate that the induction of HSP90 by bortezomib is essential for the synergy with 34.5ENVE treatment.
Figure 4. Effect of bortezomib induced HSP90 on virus replication and synergistic cell killing.
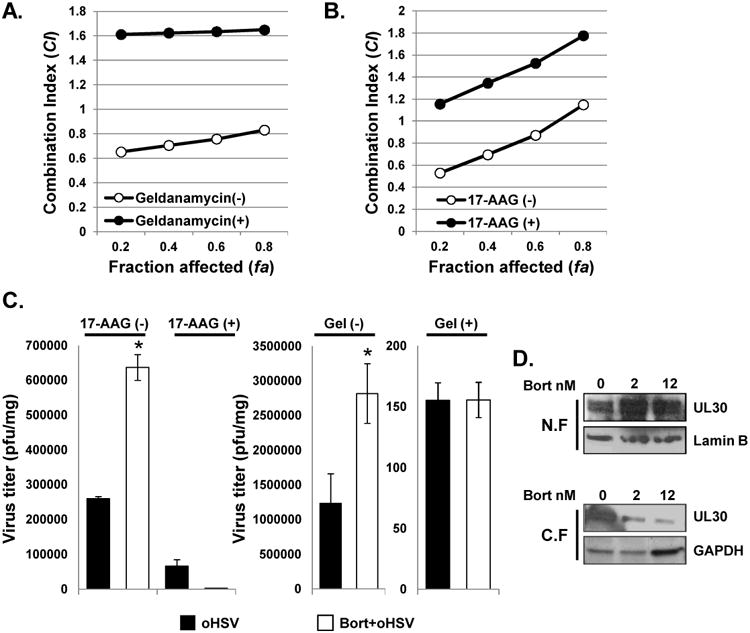
A) Chou-Talalay analysis of combining bortezomib with 34.5ENVE in ovarian cancer, head & neck cancer, and glioma cells treated with geldanamycin. Briefly, Geldanamycin (1uM) was added prior to bortezomib treatment (1 hour) and synergy between bortezomib and oHSV measured as described earlier. Data are presented as fraction affected (fa) versus combination index (CI) plots of cells with or without geldanamycin treatment. B) Effect of 17-AAG on bortezomib and oHSV interaction in cell killing. Briefly, 17-AAG (0.1uM) was added prior to bortezomib treatment (1 hour) and synergy between bortezomib and oHSV measured as described earlier. The interaction between bortezomib and oHSV on cell killing was analyzed by the median-effect method of Chou-Talalay analysis. Data are presented as fraction affected (fa) versus combination index (CI) plots of cells treated with or without 17-AAG. C) Effect of 17-AAG and Geldanamycin on increased viral replication following bortezomib treatment. U251T3 cells were pretreated with/without 0.1uM of 17-AAG or Geldanamycin (1uM) for 1 hour and then treated with 12nM bortezomib for 16 hours prior to 34.5ENVE infection (MOI = 0.01). Forty-eight hours later cells and media were harvested and viral titers were measured by standard plaque assay. Data points represent the mean, and error bars indicate ± SD for each group. Asterisks indicate statistically significant differences between indicated pairs. D) Western blot analysis for HSV polymerase (UL30). U251T3 cells were pretreated with 2nM or 12nM of bortezomib for 16 hours, followed by drug wash out and treatment with 34.5ENVE of 0.1 MOI. Four hours later, cells were harvested and proteins were fractionated for nuclear (N.F) and cytoplasmic extracts (C.F). Lamine B and GAPDH antibody was used as loading control for N.F. and C.F. respectively.
Combination treatment increased therapeutic efficacy in vivo
We next examined the therapeutic efficacy of bortezomib in subcutaneous U251T3 glioma model (Fig. 5A). Mice bearing the U251T3 glioma tumor xenograft displayed tumor growth inhibition of 91.7% in response to combinatorial treatment. Moreover, at day 23 after treatment, 6 out of 8 tumors treated with bortezomib plus 34.5ENVE had completely regressed.
Figure 5. oHSV and bortezomib treatment enhances antitumor efficacy and survival in vivo.

A) Athymic nude mice were subcutaneously implanted with U251T3 glioma cells. When tumor size reached around 100 mm3, PBS or bortezomib (0.8 mg/kg) were administered via intra-peritoneal injection twice a week for the duration of the experiment. Seven days after initiation of drug treatment mice were injected intratumorally with 5 × 104 pfu of 34.5ENVE or PBS. Data points represent the mean and 95% confidence intervals of the tumor size in each group at the indicated time points. (N=8/group) B) Athymic nude mice with intracranial GBM169 cells were treated with/without bortezomib (0.8 mg/kg administered intraperitoneally twice a week for the duration of the experiment) and were treated with intratumoral injection of HBSS or 1×105 pfu of 34.5ENVE on day 14. Data shown are Kaplan Meier survival curves of animals in each group. (N=9/ group for mice treated with PBS or bortezomib alone, and n=10/group for mice treated with bortezomib and oHSV). C) Athymic nude mice were subcutaneously implanted with CAL27 head & neck cancer cells. When tumor size reached around 100 mm3, PBS or bortezomib (0.8 mg/kg) were administered via intra-peritoneal injection twice a week for the duration of the experiment. Following one week of bortezomib treatment, animals were injected intratumorally with HBSS or 1×105 pfu of oHSV. Tumor volume was measured regularly after treatment. Data points represent the mean of the tumor size and 95% confidence intervals for each group at the indicated time points. (n=10/group) D) Kaplan Meier Survival curves of the data in (C). The percentage of surviving mice was determined by monitoring the death of mice over a period of 80 days after treatment.
Combination treatment also improved survival of mice bearing intracranial GBM169 tumors (Fig. 5B). All mice treated with PBS, bortezomib alone, or 34.5ENVE alone died with a median survival of 31 days (bortezomib alone: hazard ratio (HR) of survival = 1.18, P = 0.927, 34.5ENVE alone: hazard ratio (HR) of survival = 0.89, P = 0.797). Whereas, by day 100 following treatment, 70% (median survival > 100 days; hazard ratio (HR) of survival = 0.11, P < .001) of the animals in the bortezomib combined with 34.5ENVE group were still viable (Fig. 5B).
Similar tumor growth suppression was observed in a combination with 34.5ENVE in the CAL27 head & neck xenograft model established in nude mice (Fig. 5C-D). Untreated tumors grew rapidly, leading to a tumor volume of 1407.36 mm3 by day 33 (95% CI = 1142.36 to 1672.36 mm3), while mean tumor volumes of bortezomib, 34.5ENVE, and combination treated mice were 920.43 mm3 (difference with PBS = -486.93 mm3, P = 0.004), 217.23 (difference with PBS = -1190.13 mm3, P < 0.001), and 116.46 mm3 (difference with PBS = -1290.90 mm3, P < 0.001) respectively (Fig. 5C). These data correlate to 34.6, 84.6, and 91.7% tumor growth inhibition, respectively, compared to control PBS treated mice. Additionally, combination treatment resulted in increased survival of mice (Fig. 5D). Together these results highlight the translational significance of combining bortezomib treatment with oHSV therapy for both glioma and head and neck cancers.
Combination treatment increased viral replication and necrotic cell death in vivo
The apparent enhanced antitumor efficacy and survival benefit resulting from combinatorial treatment was further investigated by immunohistological examination of CAL27 subcutaneous tumor xenografts three days after virus treatment. As shown in Fig. 6, there was significantly increased HSV staining in tumors treated with combination treatment as compared to tumors treated with oHSV alone. A large portion of tumor sections derived from tumors treated with bortezomib and oHSV were necrotic, as evident with hematoxylin and eosin (H&E) staining (Fig. 6).
Figure 6. Histological characterization of tumor tissues.
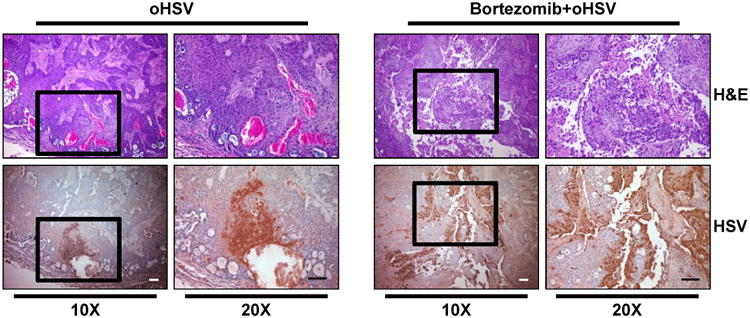
Athymic nude mice were subcutaneously implanted with CAL27 and treated as in Fig. 5. Mice were injected intra-tumorally twice with 1×105 pfu of oHSV every other day. Three days later from second virus injection, tumors were harvested and sectioned. Data shown are representative images of H&E- and HSV-stained tumor sections. Original magnification = × 10 and × 20, as indicated. Scale bar = 100 μm.
Discussion
Oncolytic herpes simplex virus-1 (oHSV) therapy has shown promise in preclinical models and several clinical studies testing its efficacy in patients are ongoing and interim analysis of a large phase three trial for a GMCSF expressing oncolytic HSV has revealed significant improvement in durable response rates (18). Proteasome inhibition has recently emerged as a promising target in cancer therapy, but its efficacy in conjunction with oHSV has not been previously investigated. Here, we report the first study to show that proteasome inhibition with bortezomib can be combined with oHSV to effectively target various solid tumors. Such combinatorial strategies hold great promise as cancer treatments because they can enhance efficacy while minimizing toxicity (19). We utilized Chou-Talalay analysis and found that bortezomib interacted synergistically with oHSV in vitro in killing various solid cancer cells including ovarian, head & neck, MPNST, and glioma. Importantly, combination treatment resulted in a greater than two- to eight-fold decrease in the dosage of either bortezomib or oHSV necessary for similar cell killing.
HSV-1 encoded ICP0 is an E3 ubiquitin ligase that utilizes cellular machinery to selectively degrade host proteins that enhance the innate antiviral immune response (20, 21). The proteasome has three distinct proteolytic activities: chymotrypsin like, trypsin like, and caspase like activities. Specifically, the chymotrypsin like activity of the proteasome has also been shown to be important for virus entry (22). Additionally, dysregulation of cellular ubiquitin-proteasome system has been shown to augment antiviral activity against HSV-1 and HSV-2 viruses (23). These results suggest that it may be counterintuitive to utilize bortezomib to increase the efficacy of oHSV. However, while ICP0 mediated modification leads to degradation of cellular antiviral proteins (21), its own proteasomal degradation by SIAH-1, a cellular ubiquitin ligase has also been shown to inhibit viral infection (24). Clinically, increased incidence of herpes zoster, Varicella zoster virus, and hepatitis B virus reactivation has also been noted in patients treated with bortezomib (25-28). Interestingly an oncolytic adenovirus was shown to synergize with bortezomib by increasing cellular apoptosis in vitro and by its immunomodulatory effects in vivo (29). Bortezomib treatment has also been found to increase apoptosis of EBV positive transformed B cells suggesting that it may be a novel strategy for the treatment of EBV-associated lymphomas (30). While our data shows no evidence of increased apoptosis, we have not examined the contribution of immunomodulatory effects on virus propagation and anti-tumor immune responses generation in vivo. Bortezomib induced activation of UPR has also been shown to induce cellular transcription factors that can activate EBV viral promoters and promote a lytic switch. Direct activation of EBV lytic cycle by bortezomib has also been reported in a variety of tumor cells lines. This effect is thought to be due to the induction of C/EBPβ, a cellular transcription factor that can then initiate the activation of EBV lytic gene expression (31). While bortezomib has been shown to increase EBV replication, other studies have also found that bortezomib inhibits VSV via activation of NF-κB, resulting in an antiviral state that ultimately inhibits VSV propagation (32). Consistent with these reports, treatment of myeloma cells with bortezomib was was found to inhibit VSV replication and show less than additive cell killing in vitro (33). Interestingly, the authors found that despite the antagonistic results in vitro bortezomib and VSV treatment improved anti-tumor efficacy in vivo. These seemingly contradictory studies make the combination of bortezomib and oHSV both promising and intriguing. Here our results demonstrate increased virus replication and cancer cell killing when cells are pre-treated with bortezomib prior to oHSV infection. In infected macrophages and dendritic cells in vivo, proteasome mediated degradation of viral capsid proteins has also been shown to release viral genomic DNA into the cytoplasm activating antiviral IFN responses in macrophages post HSV-1 and CMV infections and can aid in viral clearance (7). Immune-suppression with bortezomib may account for the increased virus reactivation observed in patients and in vivo tumor bearing mice in our experiments. In the context of our study, we have not investigated the effect of bortezomib on antiviral and anti-tumor immune responses that are generated with oHSV treatment. Future studies to investigate this interaction would be of interest.
Resistance to bortezomib remains a clinical challenge and the induction of the unfolded protein response (UPR) has been correlated with the development of resistance to apoptosis initiated by proteasome inhibition (34). The UPR leads to the induction of heat shock proteins (HSPs): a group of ubiquitously expressed chaperon proteins that ensure correct folding and prevent aggregation of specific target proteins. These chaperone proteins coordinate and rescue ER stress and are thought to contribute towards bortezomib resistance (34). Interestingly, HSP90 is also utilized by HSV-1-encoded DNA polymerase for its proper localization to the nucleus (35, 36). Our results show that the induction of HSP90 as part of the UPR activated by bortezomib is responsible for increased virus replication, and HSP90 inhibitors ablated the synergistic cell killing and increased viral replication induced by bortezomib. Together these results indicate that the combination of oHSV therapy with bortezomib is an attractive strategy to enhance therapy or deal with the development of resistance to bortezomib treatment. Here we show that the unfolded protein response induced by bortezomib increases viral replication in vitro. It is interesting to speculate if different proteasome inhibitors with different affinities for distinct proteolytic activities of the proteasome and/or their different half life of each inhibitor may account may account for the increased oHSV replication observed in bortezomib treated cells but inhibition with wild type virus observed with other proteasome inhibitors. In this study the lack of bortezomib alone to show therapeutic efficacy in mice bearing intracranial glioma is consistent with the lack of response observed in glioma patients after bortezomib treatment (37). However, the enhanced survival with combination treatment is consistent with our results which show that even sublethal doses of bortezomib can also increase viral replication and improve cell killing in vitro. While it is interesting to speculate that tumors which can induce UPR in response to bortezomib would be most sensitive to combination of bortezomib and oHSV, future studies will identify biomarkers to predict tumor types that will be most sensitive to this combination treatment.
Recently, bortezomib was found to synergize with oncolytic reovirus therapy in the treatment of multiple myeloma by induction of ER stress and NOXA-dependent cellular apoptosis (15). While reovirus induces ER stress in infected cells, HSV-1 is known to hijack cellular pathways to override ER stress signaling and maintain ER homeostasis (38). Here we show that combination of bortezomib and oHSV did not exacerbate ER stress in cells compared to cells treated with only bortezomib. To our knowledge, this is the first report showing synergy between oHSV and bortezomib. This study offers a novel therapeutic treatment strategy for cancer therapy that can be rapidly translated in patients with multiple solid tumors.
Supplementary Material
Translational Significance.
This study describes synergy between proteasome inhibition and oncolytic HSV-1 virus (oHSV). Induction of the cellular unfolded protein response (UPR) is usually associated with resistance to proteasome inhibition and other chemotherapies. Here we found that proteasome inhibition-induced UPR sensitizes cells to oncolysis. Interestingly HSV-1 is known to exploit the proteasome during its replication cycle and proteasome inhibitors have been suggested as anti-viral agents. In contrast this study shows that bortezomib treatment increases viral replication, a finding additionally supported by the clinical observation of increased incidence of latent virus reactivation in patients treated with bortezomib. Since bortezomib is FDA approved, its combination with oHSV can be rapidly translated in patients with multiple solid tumors. This study paves the way for a combination treatment strategy that can utilize suboptimal doses of bortezomib in conjunction with oHSV to maximize efficacy with minimal toxicity.
Acknowledgments
Funding: This work was supported by the National Institutes of Health grant (R01NS064607, R01CA150153, P01 CA163205, and P30NS045758) to Balveen Kaur and (R01CA155521) to JY. This work was supported by the Comprehensive Cancer Center Viral Oncology Program Award to Matthew Old and by the Pelotonia Fellowship Program to Brian S Hurwitz. Any opinions, findings, and conclusions expressed in this material are those of the authors and do not necessarily reflect those of the Pelotonia Fellowship Program.
Footnotes
Conflict of interest disclosure statement: The authors disclose no potential conflicts of interest.
References
- 1.Friedman GK, Pressey JG, Reddy AT, Markert JM, Gillespie GY. Herpes simplex virus oncolytic therapy for pediatric malignancies. Mol Ther. 2009;17:1125–35. doi: 10.1038/mt.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kanai R, Wakimoto H, Cheema T, Rabkin SD. Oncolytic herpes simplex virus vectors and chemotherapy: are combinatorial strategies more effective for cancer? Future Oncol. 2010;6:619–34. doi: 10.2217/fon.10.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dudek AZ, Lesniewski-Kmak K, Shehadeh NJ, Pandey ON, Franklin M, Kratzke RA, et al. Phase I study of bortezomib and cetuximab in patients with solid tumours expressing epidermal growth factor receptor. Br J Cancer. 2009;100:1379–84. doi: 10.1038/sj.bjc.6605043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gilbert J, Lee JW, Argiris A, Haigentz M, Jr, Feldman LE, Jang M, et al. Phase II 2-arm trial of the proteasome inhibitor, PS-341 (bortezomib) in combination with irinotecan or PS-341 alone followed by the addition of irinotecan at time of progression in patients with locally recurrent or metastatic squamous cell carcinoma of the head and neck (E1304): A trial of the Eastern Cooperative Oncology Group. Head Neck. 2012 doi: 10.1002/hed.23046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burch AD, Weller SK. Nuclear sequestration of cellular chaperone and proteasomal machinery during herpes simplex virus type 1 infection. J Virol. 2004;78:7175–85. doi: 10.1128/JVI.78.13.7175-7185.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gross S, Catez F, Masumoto H, Lomonte P. Centromere architecture breakdown induced by the viral E3 ubiquitin ligase ICP0 protein of herpes simplex virus type 1. PLoS ONE. 2012;7:e44227. doi: 10.1371/journal.pone.0044227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horan KA, Hansen K, Jakobsen MR, Holm CK, Soby S, Unterholzner L, et al. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J Immunol. 2013;190:2311–9. doi: 10.4049/jimmunol.1202749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reid EG. Bortezomib-induced Epstein-Barr virus and Kaposi sarcoma herpesvirus lytic gene expression: oncolytic strategies. Curr Opin Oncol. 2011;23:482–7. doi: 10.1097/CCO.0b013e3283499c37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoo JY, Haseley A, Bratasz A, Chiocca EA, Zhang J, Powell K, et al. Antitumor Efficacy of 34.5ENVE: A Transcriptionally Retargeted and “Vstat120”-expressing Oncolytic Virus. Mol Ther. 2011 doi: 10.1038/mt.2011.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kurozumi K, Hardcastle J, Thakur R, Yang M, Christoforidis G, Fulci G, et al. Effect of tumor microenvironment modulation on the efficacy of oncolytic virus therapy. J Natl Cancer Inst. 2007;99:1768–81. doi: 10.1093/jnci/djm229. [DOI] [PubMed] [Google Scholar]
- 11.Goodrich LD, Schaffer PA, Dorsky DI, Crumpacker CS, Parris DS. Localization of the herpes simplex virus type 1 65-kilodalton DNA-binding protein and DNA polymerase in the presence and absence of viral DNA synthesis. J Virol. 1990;64:5738–49. doi: 10.1128/jvi.64.12.5738-5749.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–81. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 13.Combaret V, Boyault S, Iacono I, Brejon S, Rousseau R, Puisieux A. Effect of bortezomib on human neuroblastoma: analysis of molecular mechanisms involved in cytotoxicity. Mol Cancer. 2008;7:50. doi: 10.1186/1476-4598-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chauhan D, Tian Z, Zhou B, Kuhn D, Orlowski R, Raje N, et al. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin Cancer Res. 2011;17:5311–21. doi: 10.1158/1078-0432.CCR-11-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelly KR, Espitia CM, Mahalingam D, Oyajobi BO, Coffey M, Giles FJ, et al. Reovirus therapy stimulates endoplasmic reticular stress, NOXA induction, and augments bortezomib-mediated apoptosis in multiple myeloma. Oncogene. 2012;31:3023–38. doi: 10.1038/onc.2011.478. [DOI] [PubMed] [Google Scholar]
- 16.Burnett HF, Audas TE, Liang G, Lu RR. Herpes simplex virus-1 disarms the unfolded protein response in the early stages of infection. Cell Stress Chaperones. 2012;17:473–83. doi: 10.1007/s12192-012-0324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathew SS, Della Selva MP, Burch AD. Modification and reorganization of the cytoprotective cellular chaperone Hsp27 during herpes simplex virus type 1 infection. J Virol. 2009;83:9304–12. doi: 10.1128/JVI.01826-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elsedawy NB, Russell SJ. Oncolytic vaccines. Expert review of vaccines. 2013;12:1155–72. doi: 10.1586/14760584.2013.836912. [DOI] [PubMed] [Google Scholar]
- 19.Balch C, Houghton A, Sober A, Soong SJ, editors. Cutaneous Melanoma. St. Louis: Quality Medical Publishing; 1998. Biologic Therapy; pp. 419–36. [Google Scholar]
- 20.Jurak I, Silverstein LB, Sharma M, Coen DM. Herpes simplex virus is equipped with RNA- and protein-based mechanisms to repress expression of ATRX, an effector of intrinsic immunity. J Virol. 2012;86:10093–102. doi: 10.1128/JVI.00930-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boutell C, Cuchet-Lourenco D, Vanni E, Orr A, Glass M, McFarlane S, et al. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog. 2011;7:e1002245. doi: 10.1371/journal.ppat.1002245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delboy MG, Nicola AV. A pre-immediate-early role for tegument ICP0 in the proteasome-dependent entry of herpes simplex virus. J Virol. 2011;85:5910–8. doi: 10.1128/JVI.00267-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiu M, Chen Y, Cheng L, Chu Y, Song HY, Wu ZW. Pyrrolidine dithiocarbamate inhibits herpes simplex virus type 1 and 2 replication and its activity may be mediated through dysregulation of ubiquitin-proteasome system. J Virol. 2013 doi: 10.1128/JVI.00869-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagel CH, Albrecht N, Milovic-Holm K, Mariyanna L, Keyser B, Abel B, et al. Herpes simplex virus immediate-early protein ICP0 is targeted by SIAH-1 for proteasomal degradation. J Virol. 2011;85:7644–57. doi: 10.1128/JVI.02207-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fukushima T, Sato T, Nakamura T, Iwao H, Nakajima A, Miki M, et al. Daily 500 mg valacyclovir is effective for prevention of Varicella zoster virus reactivation in patients with multiple myeloma treated with bortezomib. Anticancer Res. 2012;32:5437–40. [PubMed] [Google Scholar]
- 26.Tanaka H, Sakuma I, Hashimoto S, Takeda Y, Sakai S, Takagi T, et al. Hepatitis B reactivation in a multiple myeloma patient with resolved hepatitis B infection during bortezomib therapy: case report. J Clin Exp Hematop. 2012;52:67–9. doi: 10.3960/jslrt.52.67. [DOI] [PubMed] [Google Scholar]
- 27.di Meo N, Bergamo S, Dondas A, Trevisan G. Bortezomib and bilateral herpes zoster. Acta Dermatovenerol Alp Panonica Adriat. 2012;21:21–2. [PubMed] [Google Scholar]
- 28.Khalafallah AA, Woodgate M, Koshy K, Patrick A. Ophthalmic manifestations of herpes zoster virus in patients with multiple myeloma following bone marrow transplantation. BMJ Case Rep. 2013;2013 doi: 10.1136/bcr-2012-007625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boozari B, Mundt B, Woller N, Struver N, Gurlevik E, Schache P, et al. Antitumoural immunity by virus-mediated immunogenic apoptosis inhibits metastatic growth of hepatocellular carcinoma. Gut. 2010;59:1416–26. doi: 10.1136/gut.2009.196519. [DOI] [PubMed] [Google Scholar]
- 30.Zou P, Kawada J, Pesnicak L, Cohen JI. Bortezomib induces apoptosis of Epstein-Barr virus (EBV)-transformed B cells and prolongs survival of mice inoculated with EBV-transformed B cells. J Virol. 2007;81:10029–36. doi: 10.1128/JVI.02241-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shirley CM, Chen J, Shamay M, Li H, Zahnow CA, Hayward SD, et al. Bortezomib induction of C/EBPbeta mediates Epstein-Barr virus lytic activation in Burkitt lymphoma. Blood. 2011;117:6297–303. doi: 10.1182/blood-2011-01-332379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dudek SE, Luig C, Pauli EK, Schubert U, Ludwig S. The clinically approved proteasome inhibitor PS-341 efficiently blocks influenza A virus and vesicular stomatitis virus propagation by establishing an antiviral state. J Virol. 2010;84:9439–51. doi: 10.1128/JVI.00533-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yarde DN, Nace RA, Russell SJ. Oncolytic vesicular stomatitis virus and bortezomib are antagonistic against myeloma cells in vitro but have additive anti-myeloma activity in vivo. Experimental hematology. 2013;41:1038–49. doi: 10.1016/j.exphem.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roue G, Perez-Galan P, Mozos A, Lopez-Guerra M, Xargay-Torrent S, Rosich L, et al. The Hsp90 inhibitor IPI-504 overcomes bortezomib resistance in mantle cell lymphoma in vitro and in vivo by down-regulation of the prosurvival ER chaperone BiP/Grp78. Blood. 2011;117:1270–9. doi: 10.1182/blood-2010-04-278853. [DOI] [PubMed] [Google Scholar]
- 35.Burch AD, Weller SK. Herpes simplex virus type 1 DNA polymerase requires the mammalian chaperone hsp90 for proper localization to the nucleus. J Virol. 2005;79:10740–9. doi: 10.1128/JVI.79.16.10740-10749.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li YH, Tao PZ, Liu YZ, Jiang JD. Geldanamycin, a ligand of heat shock protein 90, inhibits the replication of herpes simplex virus type 1 in vitro. Antimicrob Agents Chemother. 2004;48:867–72. doi: 10.1128/AAC.48.3.867-872.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friday BB, Anderson SK, Buckner J, Yu C, Giannini C, Geoffroy F, et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. Neuro Oncol. 2012;14:215–21. doi: 10.1093/neuonc/nor198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mulvey M, Arias C, Mohr I. Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J Virol. 2007;81:3377–90. doi: 10.1128/JVI.02191-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.