

Abstract
The ability to efficiently deliver a drug to a tumor site is dependent on a wide range of physiologically imposed design constraints. Nanotechnology provides the possibility of creating delivery vehicles where these design constraints can be decoupled, allowing new approaches for reducing the unwanted side effects of systemic delivery, increasing targeting efficiency and efficacy. Here we review the design strategies of the two FDA-approved antibody-drug conjugates (Brentuximab vedotin and Trastuzumab emtansine) and the four FDA-approved nanoparticle-based drug delivery platforms (Doxil, DaunoXome, Marqibo, and Abraxane) in the context of the challenges associated with systemic targeted delivery of a drug to a solid tumor. The lessons learned from these nanomedicines provide important insight into the key challenges associated with the development of new platforms for systemic delivery of anti-cancer drugs.
Keywords: tumor targeting, active targeting, nanoparticles, liposomes, circulation, enhanced permeability and retention (EPR) effect, design rules, physiologically imposed design constraints
1. Introduction
Local delivery of a drug to a solid tumor has the potential to overcome many of the unwanted side effects of systemic delivery, however, the design of nanoparticle-based platforms for local delivery is extremely challenging and must overcome a number of physiologically imposed design constraints (Figure 1). Research on nanomedicines for in vivo diagnostics and/or therapeutics has increased dramatically over the past 10 years, and yet there are only two FDA-approved antibody-drug conjugates (Brentuximab vedotin and Trastuzumab emtansine) and four FDA-approved nanoparticle-based drug delivery platforms (Doxil, DaunoXome, Marqibo, and Abraxane) (Table 1). Here we review the design of these FDA-approved therapeutic platforms in the context of the challenges associated with systemic targeted delivery of a drug to a solid tumor.
Figure 1.
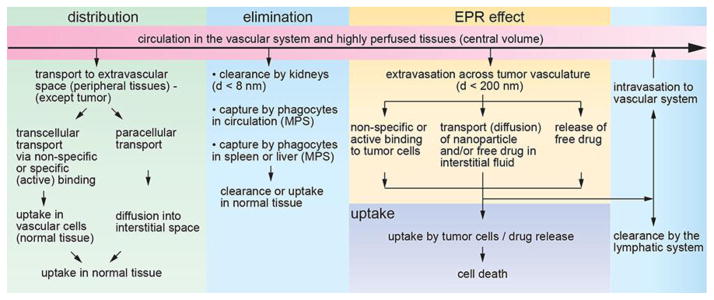
Schematic illustration of physiologically imposed design constraints for nanoparticle-based targeted drug delivery. After systemic delivery of a nanoparticle-based platform, distribution in peripheral tissues (except the tumor) can lead to uptake in normal tissues and the potential for adverse side effects. Nanoparticles can be targeted by the Mononuclear Phagocyte System (MPS) in circulation or in the liver and spleen. Small nanoparticles or nanoparticle fragments can be cleared by the kidneys. The enhanced permeability and retention (EPR) effect is usually the key to accumulation of a nanoparticle-platform in a solid tumor. A nanoparticle may be taken up by active targeting of cell surface biomarkers on the tumor cells or by passive non-specific binding. Drug release after uptake may be by passive diffusion (e.g. from a polymer nanoparticle) or by exploiting a cleavable linker. Transport of the nanoparticle platform or free drug to the tumor core usually relies on passive diffusion in the interstitial space.
Table 1.
Summary of FDA-approved antibody drug conjugates (ADCs) and nanomedicines.
| Platform | Class | Drug | d (nm) | Drug/carrier ratio | Key design feature(s) | Problem addressed |
|---|---|---|---|---|---|---|
| Brentuximab vedotin | ADC | monomethyl auristan E | ~ 10 | ≤ 8 | valine-citrulline linker cleaved by cathepsin in endosomes | monomethyl auristan E (MMAE) is too toxic to be used alone |
| Trastuzumab emtansine | ADC | mertansine | ~ 10 | ≤ 8 | non-cleavable linker; release of drug by proteolytic degradation of antibody in endosomes | mertansine is too toxic to be used alone |
| Doxil | liposome | doxorubicin | 100 | 10,000 – 15,000 | lipid encapsulation for high drug/carrier ratio, polyethylene glycol coating to evade MPS, crystallization of drug in liposome minimizes escape during circulation | drug toxicity and adverse cardiac side effects |
| DaunoXome | liposome | daunorubicin | 50 | ~ 10,000 | no polyethylene glycol coating, targeted by MPS resulting in slow release into circulation | drug toxicity and adverse cardiac side effects |
| Marqibo | liposome | vincristine | 100 | ~ 10,000 | no polyethylene glycol coating, targeted by MPS resulting in slow release into circulation | drug toxicity and adverse side effects |
| Abraxane | protein carrier | paclitaxel | 130 | >10,000 | non-specific binding of paclitaxel to albumin | overcomes very low solubility of paclitaxel |
Antibody-drug conjugates (ADCs) are a conceptually simple approach to target a drug to a tumor and reduce the toxic side effects associated with systemic delivery of a free drug. However, meeting the design criteria for ADCs has proven to be challenging. While numerous strategies for targeted drug delivery and combined theranostic nanoparticle platforms have been proposed, there have been very few systematic studies that could ultimately provide design rules for the development of new platforms. In the research community, more weight is often given to new nanoparticle drug delivery platforms, regardless of the potential for clinical translation. The unglamorous research required to fully characterize the components of a system or to contribute to the development of design rules has been largely overlooked. The quest for increasingly complex nanoparticle platforms often ignores the difficulties in overcoming the physiologically imposed constraints in accumulating a drug at therapeutic concentrations in a tumor while avoiding toxic side effects in normal tissue, the chief function of nanoparticle-based delivery.
In this review we summarize the design rationale for the six current FDA-approved nanomedicines: ADCs, liposome-based delivery platforms, and albumin-bound nanoparticles. We focus on the lessons learned from the design of these platforms in the context of the pharmacokinetics and the physiologically imposed design constraints, including circulation, the Mononuclear Phagocyte System (MPS), the Enhanced Permeability and Retention (EPR) effect, tumor transport, and toxicity. Finally, we summarize the current status of design rules for nanoparticle drug delivery platforms based on these six FDA-approved nanomedicines.
2. Chemotherapy vs targeted therapy
In the treatment of cancer, the use of one or more cytotoxic small molecules is widely used to kill highly proliferative cancer cells. However, these drugs also kill other proliferative cells in bone marrow, the gastrointestinal tract (stomach and intestines), and hair follicles, leading to common side effects such as compromised immune system (due to decreased production of leukocytes, red blood cells, and platelets), inflammation and ulceration of mucous membranes in the GI tract, and hair loss. Small molecule chemotherapeutics generally include: alkylating agents (e.g. cisplatin), anti-metabolites (e.g. gemcitabine), anti-microtubule agents (e.g. paclitaxel, vincristine), topoisomerase inhibitors (e.g. topotecan), and cytotoxic inhibitors (e.g. doxorubicin).
Targeted antibody therapies reduce the toxic side effects of anticancer drugs in normal cells and tissues by targeting a cell-surface receptor that will either directly or indirectly kill cancer cells. Indirect strategies include inducing an immune response that leads to cancer cell apoptosis or inhibiting angiogenesis.1–5 Common targets for anticancer antibodies are the B-lymphocyte antigen (CD20) expressed by lymphomas and some leukemias, vascular endothelial growth factor receptor (VEGFR) expressed by vascular endothelial cells involved in angiogenesis, and one of the epidermal growth factor receptors (e.g. HER2) upregulated in some cancer cells.5 Examples of FDA approved antibodies for cancer therapy include rituximab, trastuzumab, and bevacizumab.
The large libraries of cell surface markers overexpressed in cancer cells have provided a resource in identifying potential candidates for targeted drug delivery. However, expression levels are relative to normal cells - many of these markers are also expressed by normal endothelial cells but at lower levels. For example, two common receptors for targeting: the transferrin receptor (TfR1) and the folate receptor (FR-α) are overexpressed in many tumors but are also expressed at low levels in many normal tissues.6,7 Consequently, efficient targeting of a cell surface marker may result in delivery of a nanoparticle to both tumor and normal cells. Furthermore, a systemically delivered nanoparticle platform will be exposed to more normal cells than tumor cells during circulation.
Nanoparticle-based platforms combining a drug, biological product, and/or device (e,g. nanoparticle), are considered combination products. The path for translating new combination drug therapies is complex and the roadmap for commercialization is not well-defined. Preclinical development of a new molecular entity (NME, i.e. drug) requires assessment of Chemistry, Manufacturing, and Controls (CMC), and includes characterization of the product (e.g. physicochemical properties, pharmacokinetics, safety, toxicity, and metabolism) and evaluation of the manufacturing process.8 While there has been considerable progress, standards for CMC of nanomedicines are not well established. Therefore, platforms that are as simple as possible and use materials with established biocompatibility are more likely to be approved for clinical use.
3. Antibody-drug conjugates (ADCs), liposomes, and albumin-bound nanoparticles
3.1 Antibody-drug conjugates (ADCs)
The use of targeted antibody therapy reduces the side effects associated with potent cytotoxic drugs but is limited to antibodies that can modulate a pathway or process that results in cancer cell apoptosis. The conjugation of antibodies to anticancer drugs overcomes this limitation by separating the design requirements of targeting and treatment: the antibody is used to target a molecule that is overexpressed on cancer cells and the drug induces cell death.1–3,9,10 The antibody is usually covalently conjugated to the drug with a cleavable (e.g. peptide or disulfide) or non-cleavable (e.g. thioether) linker.11 The conjugation site on the antibody is usually a surface accessible residue with a reactive group, such as the amine side group on a lysine. Using this approach, several drug molecules (typically up to 8) can be conjugated to a single antibody. A disadvantage to random conjugation is that the linker and/or drug may block the antigen binding sites on the antibody. In addition, many drugs have limited solubility and require the addition of a polyethylene glycol unit to the linker. While this concept appears straightforward, there are currently only two FDA-approved antibody drug conjugates for cancer therapy: Brentuximab vedotin and Trastuzumab emtansine.
Brentuximab vedotin, approved by the FDA in 2011, targets CD30 overexpressed in lymphomas.10,12,13 The antibody brentuximab is conjugated to monomethyl auristan E (MMAE), an anti-mitotic drug that is too toxic to be used alone. The drug is conjugated to the thiolated antibody via a maleimide linkage, and includes a valine-citrulline peptide sequence that is cleaved by cathepsin, a protease that degrades proteins and is activated by the low pH in lysosomes.10,12,13 The valine-citrulline linker is stable in serum with only 2% of the drug released after 10 days.14
Trastuzumab emtansine, approved by the FDA in 2013 for treatment of HER2+ metastatic breast cancer, is an ADC with trastuzumab conjugated to the drug mertansine via a non-cleavable linkage.15,16 The heterobifunctional cross-linker has succinimide ester and maleimide reactive groups at either end of a cyclohexane spacer and covalently couples the sulfhydryl-terminated drug to a surface accessible amine (e.g. lysine residue) on the antibody.17 The pharmacokinetics of Brentuximab vedotin and Trastuzumab emtansine are similar (Figure 2): both have moderate AUCs, relatively low clearance, and elimination half-times of 3 – 4 days.14,15,17,18
Figure 2.

Summary of pharmacokinetic parameters for FDA-approved antibody drug conjugates (ADCs) and nanomedicines: Area Under the Curve (AUC), clearance (CL), distribution volume (Vd), and elimination half-time (t1/2). Each bar represents the range of mean values obtained from clinical trials in humans. Brentuxumab vedotin,21 Trastuzumab emtansine,18 Doxil,22 Doxorubicin,23–25 DaunoXome,26 Marqibo,27 Abraxane.28–30 Doxil has high AUC, low clearance rate, small distribution volume, and a long elimination half-time. These features are largely due to the polyethylene glycol coating that provides extended evasion of the MPS and minimizes distribution into peripheral tissues. DaunoXome and Marqibo also have a small distribution volume but are designed to have faster MPS uptake and shorter elimination half-times by having no polyethylene glycol coating. The ADCs have low clearance rates, small distribution volumes, and long elimination half-time, but relative low AUCs. Abraxane has a relatively fast clearance rate, large distribution volume, and moderate elimination half-time.
The small number of FDA-approved ADCs highlights the difficulty in coupling an antibody with a drug for cancer therapy. The key design requirements are that the ADC is stable in blood, targets tumor cells, and releases the drug to the appropriate intracellular or paracellular compartment(s) after uptake. Although the overall requirements are well understood, the design of new antibody/drug pairs remains largely empirical. The main technological challenges in developing ADCs are the difficulty in selecting antibody/drug pairs and linkers that result in selective targeting and efficient intratumoral release of the free drug.10,12,13,19,20
3.1.1 Linkers for drug ADCs and other platforms
For systemically delivered drug conjugates where the drug is covalently coupled to the delivery platform, the drug must be released at the tumor site. At the same time, the linker should be stable in circulation to avoid the cytotoxic side effects of the free drug. For example, the linker should not be degraded by endogenous proteases in the blood. Since ADCs are usually taken up by an endocytic pathway, drug release usually exploits the local biochemistry in endosomes. Linkers are generally divided into two main categories: cleavable and non-cleavable (Figure 3).31 Cleavable linkers generally exploit chemical or enzymatic cleavage and result in direct release of the drug and the remaining linker fragment. For example, the valine-citrulline peptide linkage is cleaved by cathepsin in lysosomes, and is exploited in Brentuximab vedotin. Hydrazone linkers are stable at neutral pH but are unstable in the acidic environment in lysosomes. Disulfide linkages are cleaved by reducing agents such as glutathione (a glutamic acid - cysteine - glycine peptide) that are present at much higher concentrations inside cells than in circulation,32 although the efficiency of disulfide cleavage in endosomes is relatively low.33
Figure 3.
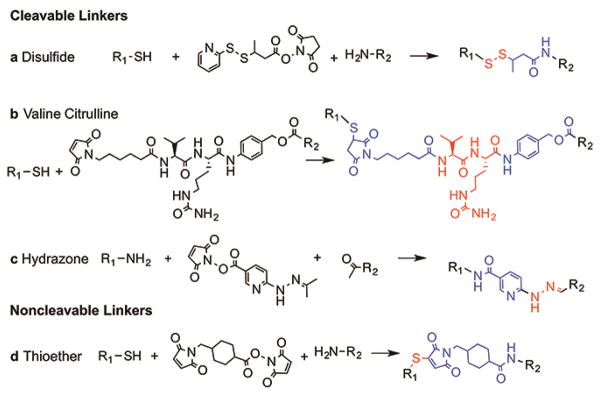
Common cleavable linkers for ADCs. (a) Disulfide bonds are formed using a cross-linking agent such as N-succinimidyl 3-(2-pyridyldithio)butyrate (SPDP) to link a thiol group to an amine. Disulfide bonds are cleavable under reducing agents. (b) The valine-citrulline bond is a common peptide linkage that can be attached to antibodies via accessible thiols. The peptide sequence cleaved by proteases, such as cathepsin, in acidic endosomes or lysosomes. (c) Hydrazone bonds can be formed using succinimidyl 4-hydrazinonicotinate acetone hydrazone (SANH) to link an amine to an aldehyde containing. Hydrazone bonds are stable at neutral pH but can be cleaved in acidic lysosomes. (d) The thiother bond is the most common non-cleavable linker, formed between a maleimide group (often on the drug) and a sulfhydryl group on the antibody using a crosslinking agent such as succinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC). In general, drug release is achieved by proteolytic degradation of the targeting antibody. The linker is shown in blue with the bond group in red.
Non-cleavable linkers, such as thioether linkers, are generally more stable in circulation than cleavable linkers. For platforms such as ADCs, non-cleavable linkers rely on intracellular proteases to degrade the antibody and release the drug and linker. The strategy of degrading the delivery platform after uptake is usually not feasible in more complex systems.
3.1.2 Drug release
Anti-cancer drugs used in chemotherapy have different mechanisms of action and hence therapeutic efficacy is dependent on drug release and delivery to the appropriate compartment within a cell.34–39 For example, anti-microtubule drugs (e.g. paclitaxel) must access microtubules in the cytoplasm whereas alkylating agents (e.g. cisplatin), anti-metabolites (e.g. gemcitabine), topoisomerase inhibitors (topotecan), and anti-tumor antibiotics (e.g. doxorubicin) must be trafficked to the nucleus. After binding to the target molecule, ADC-receptor complexes are usually internalized by endocytosis resulting in intracellular trafficking in endosomes.1,12 Therefore, cleavage of the drug from the antibody and escape from the endosome are critical final steps in drug therapy.
3.2 Liposomes
Antibody-drug conjugates typically deliver only a few drug molecules per antibody. Nanoparticle-based platforms represent an alternative approach to reducing the side effects of toxic anti-cancer drugs with very high drug/carrier ratios. There are currently four FDA-approved nanoparticle-based drug therapy platforms: liposome-based and albumin-bound drug conjugates (Table 1).
Liposomes are artificial vesicles with one or more concentric layers of phospholipids and an internal aqueous core. Small unilamellar vesicles (typically 30 – 100 nm in diameter) are less stable than their larger counterparts (100 nm – 1 μm) due to the high curvature and hence high surface tension. The combination of a lipid outer surface and aqueous core allows targeting and stability requirements to be decoupled from drug loading. The aqueous core can be loaded with drugs, DNA, siRNA, and/or contrast agents.40
Doxil and Myocet are liposomal formulations of doxorubicin, DaunoXome is a liposomal formation of daunorubicin, and Marqibo is a liposomal formulation of vincristine. Of these Doxil, DaunoXome, and Marqibo are FDA-approved. Doxil is sold as Caelyx outside the USA, and Myocet is a non-pegylated liposome-based drug sold outside the USA.
Doxil was FDA-approved for AIDS-related Kaposi’s sarcoma in 1995, for ovarian cancer in 1999, and for multiple myeloma in 2007. In 2013 the use of the generic version Lipodox was approved for treatment of ovarian cancer and Kaposi’s sarcoma.41 DaunoXome was approved by the FDA in 1996 for treatment of Kaposi’s sarcoma. Marqibo was FDA-approved in 2012 for leukemia. Myocet is sold outside the USA for treatment of breast cancer.
Doxil is formulated from a combination of fully hydrogenated soy phosphatidylcholine (HSPC), cholesterol, and a lipid with a polyethylene glycol (PEG) head group (DSPE-PEG2k) in a mole ratio of 56.4:38.3:5.3. Taking a hydrodynamic diameter of a 2k PEG of about 2 nm,42 the PEG groups correspond to an area coverage of about 60%. The DSPE-PEG provides a polymer coating that can inhibit protein adhesion and prolong evasion of the MPS.43,44 Such coatings lead to long circulation half-times (3 – 4 days in humans) and are essential to achieve significant passive accumulation at a tumor site. The Doxil liposomes are about 100 nm in diameter and have 10,000 – 15,000 doxorubicin molecules per liposome. Drug loading into preformed liposomes is achieved using a base exchange mechanism. The liposomes contain a high concentration of ammonium sulfate resulting in exchange of the drug, which is a weak base, for ammonium ions across the bilayer.45 The concentration of doxorubicin in Doxil liposomes is about 45 mM, larger than the solubility limit, resulting in precipitation of doxorubicin sulfate crystals in the liposome.
A potential limitation of liposomes is drug leakage and stability in circulation. The presence of cholesterol increases the bilayer cohesiveness and reduces leakage. In addition, the formation of a solid phase minimizes osmotic effects and is thought to contribute to stability, with more than 98% of the circulating drug remaining inside liposomes.22,46–48 The distribution volume for Doxil is close to the volume of blood indicating that the liposomes exhibit minimal uptake by normal tissues (Figure 2). The pegylated lipids in the liposomes minimize opsonization and uptake by phagocytes in the MPS system, resulting in elimination half-times of 3 – 4 days.22,49 The area under the plasma concentration curve (AUC) for Doxil is large due to the small distribution volume and long elimination half-time. In contrast, the distribution volume for free doxorubicin is very large illustrating that a significant amount of the drug is taken up in normal tissues.23–25,50 The AUC is for doxorubicin is about three orders of magnitude smaller than Doxil resulting in a clearance rate about three orders of magnitude larger.23–25,50 The elimination half-time for doxorubicin is about 20 – 25 h.
Experiments in animal models have shown that pegylated liposomes extravasate from the vascular system and accumulate at the tumor site via the EPR effect.22,45 The mechanism of drug release from the liposomes and uptake by tumor cells is not well understood. In contrast to ADCs, direct uptake of liposomes by tumor cells is thought to be negligible.45 Possible mechanisms include: disruption of the lipid bilayer by phospholipases, collapse of the ammonium salt gradient or by uptake and release by macrophages at the tumor site.22,45
DaunoXome, Marqibo, and Myocet liposomes are not pegylated and are designed to be phagocytosed by monocytes in circulation. Allowing the liposomes to be targeted by the MPS avoids high plasma concentrations and provides a reservoir from which the free drug can enter the circulation, similar to a slow infusion. This approach has been exploited for the delivery of antiparasitic or antimicrobial drugs to treat infections localized to the mononuclear phagocytic system.43 Myocet has a POPC:cholesterol mole ratio of 55.8:44.2 and is about 180 nm in diameter. DaunoXome has a DSPC:cholesterol ratio of 2:1 molar ratio and is about 50 nm in diameter.26 Marqibo has a sphingomyelin:cholesterol mole ratio of 57.4:42.6 is about 100 nm in diameter.27 DaunoXome and Marqibo have small distribution volumes indicating relatively small distribution into peripheral tissues, but relatively short elimination half-times less then 10 h highlighting the relatively fast uptake by the MPS (Figure 2).26,27,51
Liposomal drug formations have been approved for a number of indications. In many cases, clinical trials are designed for hard to treat cancers with poor prognosis and dose limiting side effects.27,52–54 Evaluating the efficacy of these therapies is not straightforward since clinical trials are usually designed to compare the liposomal formulation to the state-of-the-art drug therapy. Consequently, there are few clinical trials comparing a liposomal drug with its corresponding free drug. While many trials for Doxil and DaunoXome demonstrate comparable survival rates to the state-of-the-art drug therapy, the cardiotoxicity associated with free doxorubicin and daunorubicin is significantly reduced.26,45,55,56 Small improvements in survival rates have been reported for Doxil compared to paclitaxel in the treatment of HIV-associated Kaposi’s sarcoma57 and Doxil compared to topotecan in the treatment of recurrent or non-responsive ovarian cancer.58
Existing FDA-approved liposome technologies rely on passive accumulation through the EPR effect. Current challenges include developing platforms with improved biodistribution, pharmacokinetic properties, and active targeting. While various active targeting strategies have been explored, there are no FDA-approved platforms,43,59 highlighting the difficulties in reliably improving accumulation at the tumor site with active targeting.
3.3 Albumin-bound nanoparticles
A strategy for delivery of drugs with low aqueous solubility is to take advantage of endogenous proteins. Albumin reversibly binds hydrophobic molecules, such as vitamins and hormones, and is the most abundant protein in plasma. Albumin is a 67 kDa protein, about 4 nm in diameter and 15 nm long, similar in size to an antibody, and has a hydrodynamic radius of about 3.5 nm.60,61 Abraxane is albumin bound paclitaxel, or nab-paclitaxel (nanoparticle albumin bound), formed from lyophilized human serum albumin and paclitaxel.62 Due to its very low solubility (< 0.01 mg/mL), paclitaxel is usually mixed with the non-ionic solvent Cremophor to form an emulsion in aqueous solution. However, the solvent is toxic and can lead to a wide range of allergic reactions. Non-specific binding of paclitaxel to albumin provides an alternative solution to overcome the low solubility.
In suspension, Abraxane particles are about 130 nm in diameter, however, on injection they dissociate into smaller albumin-paclitaxel complexes or unbound paclitaxel.63 Albumin mediates endocytosis of plasma components via the albondin receptor and hence it has been suggested that albumin-paclitaxel complexes may be taken up by albumin mediated endocytosis. Abraxane was approved by the FDA in 2005 for treatment of breast, lung, pancreatic, and small cell lung cancer.62 The pharmacokinetics of Abraxane (Figure 2) and paclitaxel-Cremophor are very similar. Both exhibit an initial distribution phase in peripheral tissues, characterized by a large distribution volume, a moderate AUC, a relatively high clearance, and an elimination half-time of about 1 day (Figure 2).28–30
4. Physiologically Imposed Design Constraints
4.1 Circulation: distribution in the vascular system and peripheral tissues
Systemic delivery through the circulation is the most widely used method for drug delivery (Figure 1). In humans, vessels range in diameter from 2.5 cm in large arteries to about 7 μm in capillaries, and the volume of blood in an adult is about 4 – 5 L.64 Molecules can exit the circulation by binding to vessel walls and highly perfused tissues, transcellular transport, paracellular transport, filtration in the kidneys, the MPS, the EPR effect, or defects in the vasculature, for example due to inflammation or injury. Transcellular transport or transcytosis across the endothelium is also expected to be limited to very small particles, although its role in nanomedicine is not well understood. Paracellular transport at cell-cell adherens junctions in the endothelium is limited to molecules ≤ 2 nm in size, although particles as large as 6 nm may be able to cross the endothelium at post-capillary venules. The glomerulus in the kidneys filters particles less than about 60 – 70 kDa, or about 8 nm. Proteins in the blood can bind to particles creating a protein corona,65–67 and some these proteins are targets for receptors on phagocytic cells resulting in clearance by the MPS. Tumor neovasculature and sites of inflammation or injury are leaky and can lead to exit from circulation by the EPR effect. Finally, defects in the vasculature due to injury or disease can lead to local tissue accumulation.
Blood contains various proteins, molecules, and ions, along with red blood cells, leukocytes, and platelets. Human serum albumin, the most abundant protein in blood, is responsible for transport of a wide range of molecules in the body. The molecular weight of albumin is 67 kDa, about the threshold for glomerular filtration in the kidneys. Antibodies, complement proteins, and circulating proteins such as mannose-binding lectin are examples of opsonins, a class of proteins that promote uptake by the MPS.
4.2 The Mononuclear Phagocyte System (MPS)
The MPS consists of phagocytic cells, such as monocytes in blood and macrophages in tissue, which participate in inflammation, infection, and cancer (Figure 4).68 One of the functions of these cells is to remove pathogens and foreign matter from the body. The spleen, the largest unit in the MPS, primarily functions as a filter for blood. Macrophages in the spleen and Kupffer cells in the liver can also sequester nanoparticles resulting in accumulation in these organs.
Figure 4.

The mononuclear phagocyte system (MPS). Phagocytes, monocytes, and dendritic cells in the MPS participate in the response to inflammation and infection. Phagocytes are responsible for removing pathogens and foreign bodies such as nanoparticles from circulation. Opsonins that bind to nanoparticles initiate uptake and removal from circulation by phagocytes.
Phagocytes have receptors for molecules, termed opsonins, which can initiate binding and removal. Nanoparticles that become coated with opsonins are likely to be taken up by phagocytic cells in circulation. Examples of receptors on phagocytic cells include CR1 and Fc receptors. Therefore, an accessible Fc region of an IgG antibody on a nanoparticle platform can initiate removal from circulation by the MPS.
For most nanoparticle platforms, the circulation time should be maximized to maximize accumulation in the tumor by the EPR effect. The most successful strategy developed to date to avoid the MPS is pegylation. Pegylated liposomes (e.g. Doxil) have an elimination half-time of 2 – 4 days. However, as described previously, other strategies for drug delivery take advantage fast uptake by the MPS. Non-pegylated liposomes (e.g. Myocet) are taken up very quickly by the MPS, minimizing high plasma concentrations, and then returned to circulation over time simulating sustained delivery.
Polyethylene glycol is widely used to inhibit protein and cell adhesion on surfaces, but it does not prevent adhesion.69 An alternative approach to increasing circulation and decreasing elimination half-times is to design the delivery vehicle to appear as “self” as opposed to “non-self” to the immune system.70,71 All human cells have a unique set of “self” markers, known as the major histocompatibility complex (MHC) to prevent activation of immune cells. These molecules bind to inhibitory receptors, thereby inhibiting cell activation. A marker of “self” that regulates phagocytosis is CD47 which is a ligand for the SIRPα inhibitory receptor.72 Exploiting markers of “self” may enable the engineering of delivery vehicles to achieve longer circulation times.
4.3 Enhanced Permeability and Retention (EPR) Effect
Tumor vasculature lacks many of the features of normal blood vessels, such as well-defined smooth muscle cells and lymphatic drainage, and is inherently leaky. The preferential accumulation of a molecule or nanoparticle at locations of increased vascular permeability is known as the enhanced permeability and retention (EPR) effect (Figure 5).73–77 A growing solid tumor requires nutrients and metabolites for growth: when the tumor reaches about 1 - 2 mm3, the diffusion length and the interstitial pressure increase, restricting the supply of nutrients to the tumor core. The combination of hypoxic environment and inflammatory response leads to the formation of new vessels to supply nutrients to the tumor core, triggered by the expression of angiogenic factors such as vascular endothelial growth factor (VEGF), platelet derived growth factor (PDGF), and tumor necrosis factor α (TNFα), and downregulation of angiogenic inhibitors, such as thrombospondin-1. This process involves local removal of pericytes, the degradation of basement membrane and extracellular matrix (ECM) by matrix metalloproteinases (MMPs), and the activation of endothelial cells leading to sprouting and the formation of new vessels. Formation of the tumor neovasculature results in a rapid increase in tumor growth rate. In addition, tumor cells release vascular mediators such as bradykinin, prostaglandins, matrix metalloproteinases (MMPs), that increase the paracellular permeability at the junctions between endothelial cells.
Figure 5.

The Enhanced Permeation and Retention (EPR) effect. The accumulation of a drug delivery platform by the EPR effect requires high sustained plasma concentrations. Minimizing accumulation in peripheral tissue by transendothelial or paracellular transport is important in maintaining high plasma concentrations. Avoiding clearance by the MPS is important in increasing the elimination half-time. Activation of endothelial cells in the tumor vasculature leads to increased permeability compared to normal vasculature. The leakiness of the vasculature is dependent on the tumor type, size, and microenvironment. It is generally assumed that particles less than 200 nm in diameter are able to extravasate to the tumor site. After extravasation to the tumor site, the delivery platform must diffuse to tumor cells and induce cell death. Drug release can occur by various mechanisms and is one of the major challenges in the development of delivery platforms.
As a consequence of the recruitment of blood vessels by the tumor, the neovasculature is not hierarchically organized as in capillary beds but has an irregular architecture and heterogeneous spatial distribution.73,75 This irregular structure leads to increased resistance to blood flow and poor perfusion. In animal models, the average velocity of red blood cells in tumor neovasculature may be an order of magnitude lower than in normal tissue.78,79 Degradation of the basement membrane and the lack of smooth muscle cells and pericytes essential for constriction also contribute to leaky vessels. The obstruction and/or collapse of lymphatic vessels at the tumor core reduces drainage and results in accumulation in the local tissue.75,80 A consequence of poor lymphatic drainage is increased intratumoral pressure.
The accumulation of nanomedicines and nanoparticles in a tumor by the EPR effect has been demonstrated in animal models.73,81–83 Evidence for tumor accumulation by the EPR effect in humans is more limited. Fluorescence microscopy of patient biopsies and analysis of tumor interstitial fluid and tumor cells show significantly more doxorubicin in the tumors of patients treated with Doxil compared to free doxorubicin.46,84 Doxorubicin can undergo passive transcellular transport (lipophilicity logPoct = −0.5, and MW = 543.5)85 whereas liposomes are limited to paracellular transport at junctions where they are not size excluded. At the same time, after administration of Doxil, more than 98% of the doxorubicin in plasma is in liposomes.22 Therefore, the increased amount of doxorubicin in patients treated with Doxil supports tumor accumulation of liposomes by the EPR effect.
4.3.1 Heterogeneity
A challenge in exploiting the EPR effect for drug delivery is the inherent physical and biological heterogeneity of the tumor due to the nature of the local microenvironment, tumor type and characteristics, and degree of inflammatory activity.74 For example, pancreatic tumors are generally very poorly vascularized and hence systemic delivery via the EPR effect is thought to be inefficient. As a result of this heterogeneity, the accumulation of a nanoparticle delivery platform at a tumor site is likely to vary considerably from patient to patient. In addition, there is no well-defined size limit for transport across the leaky tumor vasculature. In animal models, the pore size cut-off for extravasation from the tumor vasculature varies from 200 nm to 1.2 μm depending on the tumor type.78,86,87 In general, a diameter of about 200 nm is usually considered an upper limit for nanoparticle delivery platforms.88
4.3.2 Enhancing the EPR effect
Various strategies have been explored to modulate the EPR effect to increase drug accumulation in the tumor.73,74 The EPR effect is involved in inflammation, and hence factors that mediate an inflammatory response can also promote leaky vasculature in tumors.73,74 For example, bradykinin mediates inflammation and induces extravasation and accumulation of fluids in inflamed tissues. Prostaglandins are upregulated by inflammatory cytokines and prevent platelet aggregation and leukocyte adhesion, thereby enhancing the EPR effect. Nitric oxide (NO) and NO-releasing factors are also important mediators of vascular permeability.
The EPR effect is usually associated with molecules that extravasate from the tumor neovasculature by paracellular transport through the relatively large pores between endothelial cells. Other pathways include passive transport across the cell membranes (usually restricted to small lipophilic molecules ≤ 500 Da), transcytosis via an endocytic or receptor mediated pathway, or transport through fenestrations in the endothelial cells.
Accumulation at a tumor site is dependent, in part, on other sinks for the nanoparticle platform. These include renal clearance, clearance by the MPS, and accumulation in peripheral tissue usually through nonspecific binding to the vasculature or highly vascularized tissues. In general, particles that avoid kidney filtration, avoid uptake by the MPS system, and have long circulation half-times are likely to result in significant accumulation in a tumor.
4.3.3 Tumor transport
After extravasation from the neovasculature, nanoparticles enter the interstitial space, usually at the perimeter of the tumor. Therefore, it is the outermost cells of the tumor mass that are first exposed to the nanoparticle platform. To reach the interior of the tumor, the nanoparticles or free drug must diffuse through the interstitial space between the cancer cells to the tumor core.75,76,87,89 Depending on the mechanism of drug release from the nanoparticle platform, transport into the tumor may depend on the rate of specific or non-specific binding to the surface of the cancer cells, the rate of uptake by the cells, and the rate of mass transport. The rate of mass transport is dependent on the density of cancer cells in the tumor and hence the effective pore size in the interstitial space. The interstitial space consists of a network of collagen fibers and other proteins and hence transport is also dependent on the size and physicochemical properties of the nanoparticle platform. The average distance from the neovasculature to the tumor core, or the diffusion length, is dependent on the vascular density and tumor size. For poorly vascularized tumors, such as pancreatic neoplasms, the diffusion length is expected to be relatively long and hence the time to reach the tumor core is also expected to be long.
4.4 Drug loading, trafficking and drug release
Drug loading is generally achieved by covalent coupling of the drug to the delivery platform (e.g. ADCs) or by drug encapsulation (e.g. liposomes). Covalent coupling usually requires a cleavable linker that will efficiently release the drug and allow delivery to the appropriate cellular compartment. The various strategies that have been developed for drug encapsulation can be classified by the drug permeability. In the simplest case, a drug can be passively loaded into a porous particle, such as a polymer (e.g. PEI) or inorganic material (e.g. mesoporous silica), where the drug release rate is dependent on the effective pore size. For these systems, the drug continuously diffuses out and hence the platform must be designed to allow sufficient drug concentration after circulation half-times of several days. Drug elution in circulation can be attenuated by engineering the delivery platform to increase the release rate after tumor or cellular uptake. In principle this could be accomplished by externally triggered release, or by exploiting the biochemical conditions in the tumor to trigger release.
The transport of a nanoparticle delivery platform into a cell usually involves binding to the cell surface, translocation across the cell membrane, and intracellular trafficking. As described previously, nanoparticle delivery platforms are usually taken up by an endocytic pathway. Moieties for active targeting, such as transferrin and folic acid, target receptors involved in endocytosis.90,91 While endocytosis is an efficient method to transport a nanoparticle delivery platform into a cell, release of the drug and escape from endosomes or lysosomes can be challenging. Late stage endosomes and lysomes have low pH and contain proteolytic enzymes, features that can be exploited for drug cleavage from a delivery vehicle. Nonetheless, design of drug delivery platforms for uptake and release remains largely empirical.
4.4 Toxicity
A key component of the development of a drug therapy is determination of the therapeutic index - the ratio of the dose that results in toxicity in 50% of patients divided by the minimum effective dose in 50% of patients. For targeted nanoparticle drug delivery and/or diagnostic platforms, an additional concern is the potential toxicity of the components of the platform other than the drug.92 Data from environmental exposure studies can be helpful in guiding design and dosing in initial development. For example, the No Observable Effect Level (NOEL), is the maximum dose with no observable adverse effects, and is usually determined in rats. The Reference Dose (RfD) is an estimate of the daily oral exposure (e.g. mg/kg/day) that is not likely to cause harmful effects during a lifetime. The RfD is usually estimated as NOEL/100. Values for NOEL and RfD for many elements can be found in the literature.
5. Targeting Efficiency
The efficacy of a drug or combination drug is often measured in animal models by the time dependence of tumor size or by the fraction of animals that survive after a candidate therapy. These parameters are particularly useful in assessing the potential therapeutic benefit of a new therapy but integrate many factors. An additional parameter that can be useful in assessing the potential efficacy of a targeted drug delivery platform is the targeting efficiency - the fraction of an intravenously administered dose that accumulates in a tumor (%ID). Despite the importance of this parameter, very few measurements are reported in the literature. Unfortunately, results are usually reported as percent of initial administered dose (ID) per gram of tumor (% ID/g), which is only useful if the tumor mass is also reported. The targeting efficiency is expected to be dependent on time post injection and the dose, and hence time-course studies are important to identify fully characterize the targeting efficiency.
Mouse models are widely used for research studies of disease progression and the development of new therapies.93 Standard models for targeting experiments include xenografts of human cell lines or explants, orthotopic xenografts, and genetically engineered mouse models.93,94 While these models are invaluable for preclinical studies of efficacy, pharmacokinetics, and biodistribution, differences in physiology can lead to differences compared to circulation and accumulation of a nanoparticle platform in humans.95 For example, differences in vascularization can lead to differences in accumulation within tumor. The difference in blood volume between mouse models and humans can also lead to large differences in dilution upon administration.
The targeting efficiency is usually measured using gamma counter, PET, HPLC, or ICP-MS (inductively coupled plasma mass spectroscopy). Methods using a gamma counter or PET require that a suitable radiolabel is conjugated to the drug delivery platform. With a gamma counter, the radioactivity in the resected tumor is measured and compared to the radioactivity of the dose. To determine the targeting efficiency from PET scans, reconstructed 3D regions of interest (ROI) are drawn around the tumor and the activity per unit mass is determined after correcting for decay and tissue density. The targeting efficiency is then determined by comparison to the activity of the initial dose. An alternative to using a radiolabel to measure the targeting efficiency is to use ICP-MS to determine the amount of one or more elemental components in the delivery platform and to compare to the initial dose.
Values of targeting efficiency per gram of tumor (%ID/g) in mouse models for different nanoparticle delivery platforms typically vary from 1 – 10% for both active and passive targeting.96–112 In some cases, the targeting efficiency per gram of tumor was greater in control experiments without the targeting molecule. However, as described above, it is difficult to make detailed comparisons since the tumor mass may be considerably different.
The lack of consistency in experimental approach makes it very difficult to draw any conclusions from these targeting studies. The difficulties in achieving highly efficient targeting suggest that distribution and elimination elsewhere in the body may be faster than accumulation by the EPR effect. Assessment of the kinetic parameters is difficult, as described previously, due to the heterogeneity of the tumor neovasculature that depends on tumor size and location in the body. In animal models, additional variables include the cell line and/or animal model.
The development of general guidelines for animal studies of nanoparticle delivery platforms would greatly increase the value of research in this field. Key parameters are: amount of initial dose in the tumor, the amount cleared from the body, and the amount in organs (especially liver and spleen) and normal tissue. The time and dose dependencies of these parameters as would be performed in standard pharmacokinetic studies are also important.
6. Design rules for targeted nanoparticle drug delivery platforms
The development of nanoparticle-based delivery platforms to overcome the side effects of systemic delivery is challenging. The inherent difficulty in controlling variables makes it difficult to perform experiments in a way that that allows direct comparison, and as a result, many studies are largely empirical in their design. This is often compounded by the lack of awareness of both the physiological and engineering constraints in the development of nanoparticle delivery platforms. The very few FDA-approved cancer nanomedicines suggest that there is a need for improving our understanding of the physiologically imposed design constraints and the structure-property relations of the engineered nanoparticle platforms.
The general guidelines for nanoparticle delivery platforms are summarized below and in Table 2. These represent general guidelines – for specific applications, some conditions may not be applicable.
Table 2.
Summary of design criteria for nanoparticle drug delivery platforms.
| Function | Design Requirements | Possible Strategies |
|---|---|---|
| circulation |
|
|
| distribution |
|
|
| elimination |
|
|
| tumor accumulation |
|
|
| tumor cell uptake |
|
|
| drug release |
|
|
Stability in circulation
The nanoparticle platform should be stable in circulation and should not be degraded or destabilized under flow and at physiological temperature. In addition, the particle should not bind with components of blood that could lead to aggregation, non-specific binding to the endothelium, or uptake by the MPS (see below). To avoid rapid clearance by the kidneys, the delivery platform should be > 8 nm.
Minimize tissue (or peripheral) volume
The distribution of the nanoparticle platform should be limited to the central volume consisting of the blood vessels and tissues highly perfused by blood. Distribution of the nanoparticle platform to normal tissue (peripheral volume) should be minimized. To minimize the peripheral volume, the particle should be designed to minimize specific and non-specific binding to the endothelium that could lead to uptake in normal endothelial cells or trafficking to normal tissue, and to avoid paracellular transport across the endothelium into normal tissue.
To minimize non-specific binding to the endothelium, it is generally thought that particles should have a neutral or slightly negative zeta potential, and should be hydrophilic. Particles should also be designed to avoid binding of components in blood, and in particular, opsonins that promote uptake by phagocytes (see Evading the MPS). Binding of components in blood can also lead to aggregation and enhanced non-specific binding to the endothelium.
A potential disadvantage of active targeting platforms is that the target molecule may be expressed by the normal endothelium. Even if the target molecule is expressed at low levels, the large area of the normal endothelium, compared to the neovasculature or tumor, may result in significant elimination by uptake in normal tissue.
Evading the MPS
To increase the half-time, the particle should be designed to minimize recognition and clearance by the MPS. Modification of the surface with polyethylene glycol (PEG) is commonly used for this purpose. Minimizing clearance by the MPS will increase the elimination half-time and increase active or passive tumor accumulation by the EPR effect.
Maximizing tumor accumulation
Tumor accumulation is usually dependent on extravasation of the delivery platform across the tumor vasculature by the EPR effect. To exploit the EPR effect, the nanoparticle diameter should be less than about 200 nm. The rate of accumulation is related to the plasma concentration and the extravasation rate constant. To maintain a high plasma concentration, the nanoparticle platform should be (1) stable in circulation, (2) not accumulate in normal tissue (small peripheral volume), (3) not be cleared by the kidneys, and (4) should evade the MPS. The extravasation rate constant is expected to be highly dependent on the tumor type, location, and the architecture of the tumor neovasculature. One strategy for increasing the extravasation rate constant is to transiently enhance the EPR effect.
Drug loading
The method of drug loading imposes numerous design constraints on the delivery platform. For example, the continuous drug diffusion from passively loaded delivery platforms makes it more difficult to deliver a therapeutic dose to the tumor and increases toxic side effects in normal tissue. In contrast, drugs that are covalently coupled to the delivery platform must be released in the tumor. A further consideration is the number of drug molecules per delivery platform, which influences the dose. The drug / delivery platform ratio can vary over many orders of magnitude: from 1 to 104 or higher. While a high drug / delivery platform ratio can reduce the dose of the delivery platform to achieve a fixed drug dose, this is only effective if a single delivery platform can induce death in many tumor cells.
Uptake and trafficking
The mechanism of drug action is an important design constraint in defining where the drug must ultimately be delivered. Whilst many anti-cancer drugs target microtubules or DNA in the cell, there are other direct or indirect mechanisms to induce tumor cell death. For many platforms designed for intracellular delivery, uptake occurs by an endocytic pathway. Although endocytic pathways can be very efficient in internalizing these platforms, drug release and endosomal escape are key challenges in achieving high therapeutic efficacy.
Future Perspectives
The FDA-approved ADCs, liposomal and protein drug delivery platforms overcome a variety of problems associated with very high drug toxicity (Brentuximab vedotin and Trastuzumab emtansine), low solubility (Abraxane), and side effects associated with high doses of the free drug (Doxil, DaunoXome, Marqibo). The strategies to overcome these problems differ significantly. Doxil liposomes have a polyethylene glycol coating to achieve extended evasion of the MPS, leading to low clearance rates and long elimination half-time. Precipitation of the drug in the liposome increases stability resulting in relatively little free drug in circulation. These features allow accumulation of the liposomes at the tumor site. In contrast DaunoXome, Marqibo, and Abraxane modulate the release of free drug in circulation by phagocytosis (DaunoXome, Marqibo) or dissociation (Abraxane). The antibody-drug conjugates provide active targeting to which promotes efficient uptake by tumor cells after extravasation, however, escape from endosomes requires an efficient release strategy.
DaunoXome, Marqibo, and Abraxane are not designed for long circulation half-times, and hence liposomes modified to evade the MPS (e.g. Doxil) and incorporating a targeting moiety represent a logical evolution of current technologies. Nonetheless, active targeting with liposomes, or immunoliposomes, has proven to be extremely difficult. Improved methods for efficient conjugation of targeting moieties that remain active is a critical need in the field.
The FDA-approved ADCs, liposomal and protein drug delivery platforms all rely on the EPR effect for extravasation from the circulation and accumulation at the tumor site. Despite its importance, there remain many gaps in our understanding of the tumor vasculature and methods for maximizing tumor accumulation. Similarly, opportunities for locally enhancing the EPR effect at the site of a tumor or inflammation have not been fully explored.
While the release of the free drug from antibody drug conjugates is thought to occur in endosomes after endocytosis in tumor cells, the mechanism of drug release by liposomes is not well understood. This is important since the diffusion of large nanoparticles, such a liposomes, in the tumor extracellular space is likely to be much slower than the free drug. Relatively little is known about the release profile for optimum therapeutic efficacy.
In summary, advancing our understanding of the design rules for achieving long circulation times, efficient extravasation and tumor accumulation, and optimum release profiles is likely to be key to developing the next generation of targeted cancer drug therapies.
Acknowledgments
This work was supported by the National Institutes of Health (U54CA151838) and the Sol Goldman Pancreatic Cancer Research Center at Johns Hopkins University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nature Reviews Drug Discovery. 2006;5:147–159. doi: 10.1038/nrd1957. [DOI] [PubMed] [Google Scholar]
- 2.Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23:1147–1157. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- 3.Scott AM, Allison JP, Wolchok JD. Monoclonal antibodies in cancer therapy. Cancer Immun. 2012;12:14. [PMC free article] [PubMed] [Google Scholar]
- 4.Galluzzi L, Vacchelli E, Fridman WH, Galon J, Sautes-Fridman C, Tartour E, Zucman-Rossi J, Zitvogel L, Kroemer G. Trial Watch: Monoclonal antibodies in cancer therapy. Oncoimmunology. 2012;1:28–37. doi: 10.4161/onci.1.1.17938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Firer MA, Gellerman G. Targeted drug delivery for cancer therapy: the other side of antibodies. J Hematol Oncol. 2012;5:70. doi: 10.1186/1756-8722-5-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daniels TR, Bernabeu E, Rodriguez JA, Patel S, Kozman M, Chiappetta DA, Holler E, Ljubimova JY, Helguera G, Penichet ML. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim Biophys Acta. 2012;1820:291–317. doi: 10.1016/j.bbagen.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parker N, Turk MJ, Westrick E, Lewis JD, Low PS, Leamon CP. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Analytical Biochemistry. 2005;338:284–293. doi: 10.1016/j.ab.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 8.Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nature Reviews Drug Discovery. 2010;9:203–214. doi: 10.1038/nrd3078. [DOI] [PubMed] [Google Scholar]
- 9.Chari RV, Miller ML, Widdison WC. Antibody-drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed Engl. 2014;53:3796–3827. doi: 10.1002/anie.201307628. [DOI] [PubMed] [Google Scholar]
- 10.Mullard A. Maturing antibody-drug conjugate pipeline hits 30. Nature Reviews Drug Discovery. 2013;12:329–332. doi: 10.1038/nrd4009. [DOI] [PubMed] [Google Scholar]
- 11.Nolting B. Linker technologies for antibody-drug conjugates. Methods Mol Biol. 2013;1045:71–100. doi: 10.1007/978-1-62703-541-5_5. [DOI] [PubMed] [Google Scholar]
- 12.Sievers EL, Senter PD. Antibody-drug conjugates in cancer therapy. Annu Rev Med. 2013;64:15–29. doi: 10.1146/annurev-med-050311-201823. [DOI] [PubMed] [Google Scholar]
- 13.Sassoon I, Blanc V. Antibody-drug conjugate (ADC) clinical pipeline: a review. Methods Mol Biol. 2013;1045:1–27. doi: 10.1007/978-1-62703-541-5_1. [DOI] [PubMed] [Google Scholar]
- 14.Bradley AM, Devine M, DeRemer D. Brentuximab vedotin: An anti-CD30 antibody-drug conjugate. American Journal of Health-System Pharmacy. 2013;70:589–597. doi: 10.2146/ajhp110608. [DOI] [PubMed] [Google Scholar]
- 15.Lu D, Burris HA, Wang B, Dees EC, Cortes J, Joshi A, Gupta M, Yi JH, Chu YW, Shih T, Fang L, Girish S. Drug interaction potential of trastuzumab emtansine (T-DM1) combined with pertuzumab in patients with HER2-positive metastatic breast cancer. Curr Drug Metab. 2012;13:911–922. doi: 10.2174/138920012802138688. [DOI] [PubMed] [Google Scholar]
- 16.Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh DY, Dieras V, Guardino E, Fang L, Lu MW, Olsen S, Blackwell K, Group ES. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783–1791. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.LoRusso PM, Weiss D, Guardino E, Girish S, Sliwkowski MX. Trastuzumab Emtansine: A Unique Antibody-Drug Conjugate in Development for Human Epidermal Growth Factor Receptor 2-Positive Cancer. Clinical Cancer Research. 2011;17:6437–6447. doi: 10.1158/1078-0432.CCR-11-0762. [DOI] [PubMed] [Google Scholar]
- 18.Girish S, Gupta M, Wang B, Lu D, Krop IE, Vogel CL, Burris HA, LoRusso PM, Yi JH, Saad O, Tong B, Chu YW, Holden S, Joshi A. Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother Pharmacol. 2012;69:1229–1240. doi: 10.1007/s00280-011-1817-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trail PA. Antibody Drug Conjugates as Cancer Therapeutics. Antibodies. 2013;2:113–129. [Google Scholar]
- 20.Shen BQ, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu SF, Mai E, Li D, Tibbitts J, Baudys J, Saad OM, Scales SJ, McDonald PJ, Hass PE, Eigenbrot C, Nguyen T, Solis WA, Fuji RN, Flagella KM, Patel D, Spencer SD, Khawli LA, Ebens A, Wong WL, Vandlen R, Kaur S, Sliwkowski MX, Scheller RH, Polakis P, Junutula JR. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol. 2012;30:184–189. doi: 10.1038/nbt.2108. [DOI] [PubMed] [Google Scholar]
- 21.Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL, Forero-Torres A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med. 2010;363:1812–1821. doi: 10.1056/NEJMoa1002965. [DOI] [PubMed] [Google Scholar]
- 22.Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clin Pharmacokinet. 2003;42:419–436. doi: 10.2165/00003088-200342050-00002. [DOI] [PubMed] [Google Scholar]
- 23.Erttmann R, Erb N, Steinhoff A, Landbeck G. Pharmacokinetics of doxorubicin in man: dose and schedule dependence. J Cancer Res Clin Oncol. 1988;114:509–513. doi: 10.1007/BF00391502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacquet JM, Bressolle F, Galtier M, Bourrier M, Donadio D, Jourdan J, Rossi JF. Doxorubicin and doxorubicinol: intra- and inter-individual variations of pharmacokinetic parameters. Cancer Chemother Pharmacol. 1990;27:219–225. doi: 10.1007/BF00685716. [DOI] [PubMed] [Google Scholar]
- 25.Piscitelli SC, Rodvold KA, Rushing DA, Tewksbury DA. Pharmacokinetics and pharmacodynamics of doxorubicin in patients with small cell lung cancer. Clin Pharmacol Ther. 1993;53:555–561. doi: 10.1038/clpt.1993.69. [DOI] [PubMed] [Google Scholar]
- 26.Gill PS, Wernz J, Scadden DT, Cohen P, Mukwaya GM, von Roenn JH, Jacobs M, Kempin S, Silverberg I, Gonzales G, Rarick MU, Myers AM, Shepherd F, Sawka C, Pike MC, Ross ME. Randomized phase III trial of liposomal daunorubicin versus doxorubicin, bleomycin, and vincristine in AIDS-related Kaposi’s sarcoma. J Clin Oncol. 1996;14:2353–2364. doi: 10.1200/JCO.1996.14.8.2353. [DOI] [PubMed] [Google Scholar]
- 27.Silverman JA, Deitcher SR. Marqibo(R) (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother Pharmacol. 2013;71:555–564. doi: 10.1007/s00280-012-2042-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sparreboom A, Scripture CD, Trieu V, Williams PJ, De T, Yang A, Beals B, Figg WD, Hawkins M, Desai N. Comparative preclinical and clinical pharmacokinetics of a cremophor-free, nanoparticle albumin-bound paclitaxel (ABI-007) and paclitaxel formulated in Cremophor (Taxol) Clin Cancer Res. 2005;11:4136–4143. doi: 10.1158/1078-0432.CCR-04-2291. [DOI] [PubMed] [Google Scholar]
- 29.Ando M, Yonemori K, Katsumata N, Shimizu C, Hirata T, Yamamoto H, Hashimoto K, Yunokawa M, Tamura K, Fujiwara Y. Phase I and pharmacokinetic study of nab-paclitaxel, nanoparticle albumin-bound paclitaxel, administered weekly to Japanese patients with solid tumors and metastatic breast cancer. Cancer Chemother Pharmacol. 2012;69:457–465. doi: 10.1007/s00280-011-1726-5. [DOI] [PubMed] [Google Scholar]
- 30.Stinchcombe TE, Socinski MA, Walko CM, O’Neil BH, Collichio FA, Ivanova A, Mu H, Hawkins MJ, Goldberg RM, Lindley C, Claire Dees E. Phase I and pharmacokinetic trial of carboplatin and albumin-bound paclitaxel, ABI-007 (Abraxane) on three treatment schedules in patients with solid tumors. Cancer Chemother Pharmacol. 2007;60:759–766. doi: 10.1007/s00280-007-0423-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leriche G, Chisholm L, Wagner A. Cleavable linkers in chemical biology. Bioorg Med Chem. 2012;20:571–582. doi: 10.1016/j.bmc.2011.07.048. [DOI] [PubMed] [Google Scholar]
- 32.Mills BJ, Lang CA. Differential distribution of free and bound glutathione and cyst(e)ine in human blood. Biochem Pharmacol. 1996;52:401–406. doi: 10.1016/0006-2952(96)00241-9. [DOI] [PubMed] [Google Scholar]
- 33.Austin CD, Wen XH, Gazzard L, Nelson C, Scheller RH, Scales SJ. Oxidizing potential of endosomes and lysosomes limits intracellular cleavage of disulfide-based antibody-drug conjugates. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:17987–17992. doi: 10.1073/pnas.0509035102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chabner BA, Roberts TG. Timeline - Chemotherapy and the war on cancer. Nature Reviews Cancer. 2005;5:65–72. doi: 10.1038/nrc1529. [DOI] [PubMed] [Google Scholar]
- 35.DeVita VT, Chu E. A History of Cancer Chemotherapy. Cancer Res. 2008;68:8643–8653. doi: 10.1158/0008-5472.CAN-07-6611. [DOI] [PubMed] [Google Scholar]
- 36.Kim R, Tanabe K, Uchida Y, Emi M, Inoue H, Toge T. Current status of the molecular mechanisms of anticancer drug-induced apoptosis - The contribution of molecular-level analysis to cancer chemotherapy. Cancer Chemother Pharmacol. 2002;50:343–352. doi: 10.1007/s00280-002-0522-7. [DOI] [PubMed] [Google Scholar]
- 37.Gascoigne KE, Taylor SS. How do anti-mitotic drugs kill cancer cells? Journal of Cell Science. 2009;122:2579–2585. doi: 10.1242/jcs.039719. [DOI] [PubMed] [Google Scholar]
- 38.Hannon MJ. Metal-based anticancer drugs: From a past anchored in platinum post-genomic future and biology. Pure and Applied Chemistry. 2007;79:2243–2261. [Google Scholar]
- 39.Nussbaumer S, Bonnabry P, Veuthey JL, Fleury-Souverain S. Analysis of anticancer drugs: A review. Talanta. 2011;85:2265–2289. doi: 10.1016/j.talanta.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 40.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nature Reviews Drug Discovery. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 41.Rose PG. Pegylated liposomal doxorubicin: optimizing the dosing schedule in ovarian cancer. Oncologist. 2005;10:205–214. doi: 10.1634/theoncologist.10-3-205. [DOI] [PubMed] [Google Scholar]
- 42.Galloway JF, Winter A, Lee KH, Park JH, Dvoracek CM, Devreotes P, Searson PC. Quantitative characterization of the lipid encapsulation of quantum dots for biomedical applications. Nanomedicine. 2012;8:1190–1199. doi: 10.1016/j.nano.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine. 2006;1:297–315. [PMC free article] [PubMed] [Google Scholar]
- 44.Vllasaliu D, Fowler R, Stolnik S. PEGylated nanomedicines: recent progress and remaining concerns. Expert Opin Drug Deliv. 2013 doi: 10.1517/17425247.2014.866651. [DOI] [PubMed] [Google Scholar]
- 45.Barenholz Y. Doxil(R)--the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160:117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 46.Gabizon A, Catane R, Uziely B, Kaufman B, Safra T, Cohen R, Martin F, Huang A, Barenholz Y. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994;54:987–992. [PubMed] [Google Scholar]
- 47.Lasic DD, Frederik PM, Stuart MC, Barenholz Y, McIntosh TJ. Gelation of liposome interior. A novel method for drug encapsulation. FEBS Lett. 1992;312:255–258. doi: 10.1016/0014-5793(92)80947-f. [DOI] [PubMed] [Google Scholar]
- 48.Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993;1151:201–215. doi: 10.1016/0005-2736(93)90105-9. [DOI] [PubMed] [Google Scholar]
- 49.Mross K, Niemann B, Massing U, Drevs J, Unger C, Bhamra R, Swenson CE. Pharmacokinetics of liposomal doxorubicin (TLC-D99; Myocet) in patients with solid tumors: an open-label, single-dose study. Cancer Chemother Pharmacol. 2004;54:514–524. doi: 10.1007/s00280-004-0825-y. [DOI] [PubMed] [Google Scholar]
- 50.Gustafson DL, Rastatter JC, Colombo T, Long ME. Doxorubicin pharmacokinetics: Macromolecule binding, metabolism, and excretion in the context of a physiologic model. J Pharm Sci. 2002;91:1488–1501. doi: 10.1002/jps.10161. [DOI] [PubMed] [Google Scholar]
- 51.Bellott R, Auvrignon A, Leblanc T, Perel Y, Gandemer V, Bertrand Y, Mechinaud F, Bellenger P, Vernois J, Leverger G, Baruchel A, Robert J. Pharmacokinetics of liposomal daunorubicin [DaunoXome] during a phase I-II study in children with relapsed acute lymphoblastic leukaemia. Cancer Chemother Pharmacol. 2001;47:15–21. doi: 10.1007/s002800000206. [DOI] [PubMed] [Google Scholar]
- 52.Solomon R, Gabizon AA. Clinical pharmacology of liposomal anthracyclines: Focus on pegylated liposomal doxorubicin. Clinical Lymphoma & Myeloma. 2008;8:21–32. doi: 10.3816/clm.2008.n.001. [DOI] [PubMed] [Google Scholar]
- 53.Sarris AH, Hagemeister F, Romaguera J, Rodriguez MA, McLaughlin P, Tsimberidou AM, Medeiros LJ, Samuels B, Pate O, Oholendt M, Kantarjian H, Burge C, Cabanillas F. Liposomal vincristine in relapsed non-Hodgkin’s lymphomas: Early results of an ongoing phase II trial. Annals of Oncology. 2000;11:69–72. doi: 10.1023/a:1008348010437. [DOI] [PubMed] [Google Scholar]
- 54.Skubitz KM. Phase II trial of pegylated-liposomal doxorubicin (Doxil (TM)) in sarcoma. Cancer Investigation. 2003;21:167–176. doi: 10.1081/cnv-120016412. [DOI] [PubMed] [Google Scholar]
- 55.Safra T, Muggia F, Jeffers S, Tsao-Wei DD, Groshen S, Lyass O, Henderson R, Berry G, Gabizon A. Pegylated liposomal doxorubicin (doxil): Reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m(2) Annals of Oncology. 2000;11:1029–1033. doi: 10.1023/a:1008365716693. [DOI] [PubMed] [Google Scholar]
- 56.O’Brien ME, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, Catane R, Kieback DG, Tomczak P, Ackland SP, Orlandi F, Mellars L, Alland L, Tendler C. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Annals of Oncology. 2004;15:440–449. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]
- 57.Cianfrocca M, Lee S, Von Roenn J, Tulpule A, Dezube BJ, Aboulafia DM, Ambinder RF, Lee JY, Krown SE, Sparano JA. Randomized trial of paclitaxel versus pegylated liposomal doxorubicin for advanced human immunodeficiency virus-associated Kaposi sarcoma: evidence of symptom palliation from chemotherapy. Cancer. 2010;116:3969–3977. doi: 10.1002/cncr.25362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ. Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol. 2001;19:3312–3322. doi: 10.1200/JCO.2001.19.14.3312. [DOI] [PubMed] [Google Scholar]
- 59.Paszko E, Senge MO. Immunoliposomes. Curr Med Chem. 2012;19:5239–5277. doi: 10.2174/092986712803833362. [DOI] [PubMed] [Google Scholar]
- 60.Tojo A, Kinugasa S. Mechanisms of glomerular albumin filtration and tubular reabsorption. Int J Nephrol. 2012;2012:481520. doi: 10.1155/2012/481520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peters T. All about albumin: biochemistry, genetics, and medical applications. Academic Press; San Diego: 1996. [Google Scholar]
- 62.Miele E, Spinelli GP, Miele E, Tomao F, Tomao S. Albumin-bound formulation of paclitaxel (Abraxane ABI-007) in the treatment of breast cancer. Int J Nanomedicine. 2009;4:99–105. doi: 10.2147/ijn.s3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yardley DA. nab-Paclitaxel mechanisms of action and delivery. J Control Release. 2013;170:365–372. doi: 10.1016/j.jconrel.2013.05.041. [DOI] [PubMed] [Google Scholar]
- 64.Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007;100:158–173. doi: 10.1161/01.RES.0000255691.76142.4a. [DOI] [PubMed] [Google Scholar]
- 65.Lundqvist M, Stigler J, Cedervall T, Berggard T, Flanagan MB, Lynch I, Elia G, Dawson K. The Evolution of the Protein Corona around Nanoparticles: A Test Study. ACS Nano. 2011;5:7503–7509. doi: 10.1021/nn202458g. [DOI] [PubMed] [Google Scholar]
- 66.Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:14265–14270. doi: 10.1073/pnas.0805135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tenzer S, Docter D, Rosfa S, Wlodarski A, Kuharev J, Rekik A, Knauer SK, Bantz C, Nawroth T, Bier C, Sirirattanapan J, Mann W, Treuel L, Zellner R, Maskos M, Schild H, Stauber RH. Nanoparticle Size Is a Critical Physicochemical Determinant of the Human Blood Plasma Corona: A Comprehensive Quantitative Proteomic Analysis. ACS Nano. 2011;5:7155–7167. doi: 10.1021/nn201950e. [DOI] [PubMed] [Google Scholar]
- 68.Hume DA. The mononuclear phagocyte system. Curr Opin Immunol. 2006;18:49–53. doi: 10.1016/j.coi.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 69.Groth T, Liu ZM, Niepel M, Peschel D, Kirchhof K, Altankov G, Faucheux N. Chemical and Physical Modifications of Biomaterial Surfaces to Control Adhesion of Cells. Advances in Regenerative Medicine: Role of Nanotechnology and Engineering Principles. 2010:253–284. [Google Scholar]
- 70.Medzhitov R, Janeway CA., Jr Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296:298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- 71.Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 72.Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK, Discher DE. Minimal “Self” Peptides That Inhibit Phagocytic Clearance and Enhance Delivery of Nanoparticles. Science. 2013;339:971–975. doi: 10.1126/science.1229568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63:136–151. doi: 10.1016/j.addr.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 74.Maeda H. Macromolecular therapeutics in cancer treatment: the EPR effect and beyond. J Control Release. 2012;164:138–144. doi: 10.1016/j.jconrel.2012.04.038. [DOI] [PubMed] [Google Scholar]
- 75.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nature Reviews Clinical Oncology. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66C:2–25. doi: 10.1016/j.addr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63:131–135. doi: 10.1016/j.addr.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 78.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK. Vascular-Permeability in a Human Tumor Xenograft - Molecular-Size Dependence and Cutoff Size. Cancer Res. 1995;55:3752–3756. [PubMed] [Google Scholar]
- 79.Yuan F, Salehi HA, Boucher Y, Vasthare US, Tuma RF, Jain RK. Vascular permeability and microcirculation of gliomas and mammary carcinomas transplanted in rat and mouse cranial windows. Cancer Res. 1994;54:4564–4568. [PubMed] [Google Scholar]
- 80.Matsumura Y. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent SMANCS. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 81.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55:3752–3756. [PubMed] [Google Scholar]
- 82.Jain RK. Transport of molecules across tumor vasculature. Cancer Metastasis Rev. 1987;6:559–593. doi: 10.1007/BF00047468. [DOI] [PubMed] [Google Scholar]
- 83.Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71:409–419. doi: 10.1016/j.ejpb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 84.Symon Z, Peyser A, Tzemach D, Lyass O, Sucher E, Shezen E, Gabizon A. Selective delivery of doxorubicin to patients with breast carcinoma metastases by stealth liposomes. Cancer. 1999;86:72–78. [PubMed] [Google Scholar]
- 85.Pyka A, Babuska M, Zachariasz M. A comparison of theoretical methods of calculation of partition coefficients for selected drugs. Acta Pol Pharm. 2006;63:159–167. [PubMed] [Google Scholar]
- 86.Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, Jain RK. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:4607–4612. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jain RK. Determinants of Tumor Blood-Flow - A Review. Cancer Res. 1988;48:2641–2658. [PubMed] [Google Scholar]
- 88.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nature Nanotechnology. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 89.Jain RK. Delivery of molecular and cellular medicine to solid tumors. Adv Drug Deliv Rev. 1997;26:71–90. doi: 10.1016/s0169-409x(97)00027-6. [DOI] [PubMed] [Google Scholar]
- 90.Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 91.Paulos CM, Reddy JA, Leamon CP, Turk MJ, Low PS. Ligand binding and kinetics of folate receptor recycling in vivo: impact on receptor-mediated drug delivery. Mol Pharmacol. 2004;66:1406–1414. doi: 10.1124/mol.104.003723. [DOI] [PubMed] [Google Scholar]
- 92.Nel A, Xia T, Madler L, Li N. Toxic potential of materials at the nanolevel. Science. 2006;311:622–627. doi: 10.1126/science.1114397. [DOI] [PubMed] [Google Scholar]
- 93.Frese KK, Tuveson DA. Maximizing mouse cancer models. Nature Reviews Cancer. 2007;7:645–658. doi: 10.1038/nrc2192. [DOI] [PubMed] [Google Scholar]
- 94.Liu M, Hicklin D. Human Tumor Xenograft Efficacy Models. In: Teicher BA, editor. Tumor Models in Cancer Research. Springer: 2011. pp. 99–124. [Google Scholar]
- 95.Steichen SD, Caldorera-Moore M, Peppas NA. A review of current nanoparticle and targeting moieties for the delivery of cancer therapeutics. Eur J Pharm Sci. 2012;48:416–427. doi: 10.1016/j.ejps.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang G, Yang Z, Lu W, Zhang R, Huang Q, Tian M, Li L, Liang D, Li C. Influence of anchoring ligands and particle size on the colloidal stability and in vivo biodistribution of polyethylene glycol-coated gold nanoparticles in tumor-xenografted mice. Biomaterials. 2009;30:1928–1936. doi: 10.1016/j.biomaterials.2008.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.von Maltzahn G, Park JH, Agrawal A, Bandaru NK, Das SK, Sailor MJ, Bhatia SN. Computationally guided photothermal tumor therapy using long-circulating gold nanorod antennas. Cancer Res. 2009;69:3892–3900. doi: 10.1158/0008-5472.CAN-08-4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rossin R, Pan DPJ, Qi K, Turner JL, Sun XK, Wooley KL, Welch MJ. Cu-64-labeled folate-conjugated shell cross-linked nanoparticles for tumor imaging and radiotherapy: Synthesis, radiolabeling, and biologic evaluation. Journal of Nuclear Medicine. 2005;46:1210–1218. [PubMed] [Google Scholar]
- 99.Oyewumi MO, Yokel RA, Jay M, Coakley T, Mumper RJ. Comparison of cell uptake, biodistribution and tumor retention of folate-coated and PEG-coated gadolinium nanoparticles in tumor-bearing mice. J Control Release. 2004;95:613–626. doi: 10.1016/j.jconrel.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 100.Yang X, Hong H, Grailer JJ, Rowland IJ, Javadi A, Hurley SA, Xiao Y, Yang Y, Zhang Y, Nickles RJ, Cai W, Steeber DA, Gong S. cRGD-functionalized, DOX-conjugated, and (6)(4)Cu-labeled superparamagnetic iron oxide nanoparticles for targeted anticancer drug delivery and PET/MR imaging. Biomaterials. 2011;32:4151–4160. doi: 10.1016/j.biomaterials.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Benezra M, Penate-Medina O, Zanzonico PB, Schaer D, Ow H, Burns A, DeStanchina E, Longo V, Herz E, Iyer S, Wolchok J, Larson SM, Wiesner U, Bradbury MS. Multimodal silica nanoparticles are effective cancer-targeted probes in a model of human melanoma. Journal of Clinical Investigation. 2011;121:2768–2780. doi: 10.1172/JCI45600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Karmani L, Labar D, Valembois V, Bouchat V, Nagaswaran PG, Bol A, Gillart J, Leveque P, Bouzin C, Bonifazi D, Michiels C, Feron O, Gregoire V, Lucas S, Borght TV, Gallez B. Antibody-functionalized nanoparticles for imaging cancer: influence of conjugation to gold nanoparticles on the biodistribution of 89Zr-labeled cetuximab in mice. Contrast Media & Molecular Imaging. 2013;8:402–408. doi: 10.1002/cmmi.1539. [DOI] [PubMed] [Google Scholar]
- 103.Chattopadhyay N, Fonge H, Cai Z, Scollard D, Lechtman E, Done SJ, Pignol JP, Reilly RM. Role of Antibody-Mediated Tumor Targeting and Route of Administration in Nanoparticle Tumor Accumulation in Vivo. Molecular Pharmaceutics. 2012;9:2168–2179. doi: 10.1021/mp300016p. [DOI] [PubMed] [Google Scholar]
- 104.Natarajan A, Gruettner C, Ivkov R, DeNardo GL, Mirick G, Yuan A, Foreman A, DeNardo SJ. NanoFerrite particle based radioimmunonanoparticles: Binding affinity and in vivo pharmacokinetics. Bioconjugate Chemistry. 2008;19:1211–1218. doi: 10.1021/bc800015n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cornelissen B, Able S, Kersemans V, Waghorn PA, Myhra S, Jurkshat K, Crossley A, Vallis KA. Nanographene oxide-based radioimmunoconstructs for in vivo targeting and SPECT imaging of HER2-positive tumors. Biomaterials. 2013;34:1146–1154. doi: 10.1016/j.biomaterials.2012.10.054. [DOI] [PubMed] [Google Scholar]
- 106.Melancon MP, Lu W, Yang Z, Zhang R, Cheng Z, Elliot AM, Stafford J, Olson T, Zhang JZ, Li C. In vitro and in vivo targeting of hollow gold nanoshells directed at epidermal growth factor receptor for photothermal ablation therapy. Molecular Cancer Therapeutics. 2008;7:1730–1739. doi: 10.1158/1535-7163.MCT-08-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hong H, Zhang Y, Engle JW, Nayak TR, Theuer CP, Nickles RJ, Barnhart TE, Cai W. In vivo targeting and positron emission tomography imaging of tumor vasculature with Ga-66-labeled nano-graphene. Biomaterials. 2012;33:4147–4156. doi: 10.1016/j.biomaterials.2012.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hicke BJ, Stephens AW, Gould T, Chang YF, Lynott CK, Heil J, Borkowski S, Hilger CS, Cook G, Warren S, Schmidt PG. Tumor targeting by an aptamer. Journal of Nuclear Medicine. 2006;47:668–678. [PubMed] [Google Scholar]
- 109.Lim EK, Kim B, Choi Y, Ro Y, Cho EJ, Lee J, Ryu SH, Suh JS, Haam S, Huh YM. Aptamer-conjugated magnetic nanoparticles enable ef cient targeted detection of integrin avb3 via magnetic resonance imaging. J Biomed Mater Res Part A. 2013;102:45–49. doi: 10.1002/jbm.a.34678. [DOI] [PubMed] [Google Scholar]
- 110.Kumar M, Yigit M, Dai GP, Moore A, Medarova Z. Image-Guided Breast Tumor Therapy Using a Small Interfering RNA Nanodrug. Cancer Res. 2010;70:7553–7561. doi: 10.1158/0008-5472.CAN-10-2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chanda N, Kattumuri V, Shukla R, Zambre A, Katti K, Upendran A, Kulkarni RR, Kan P, Fent GM, Casteel SW, Smith CJ, Boote E, Robertson JD, Cutler C, Lever JR, Katti KV, Kannan R. Bombesin functionalized gold nanoparticles show in vitro and in vivo cancer receptor specificity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:8760–8765. doi: 10.1073/pnas.1002143107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Choi CHJ, Alabi CA, Webster P, Davis ME. Mechanism of active targeting in solid tumors with transferrin-containing gold nanoparticles. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1235–1240. doi: 10.1073/pnas.0914140107. [DOI] [PMC free article] [PubMed] [Google Scholar]