

Abstract
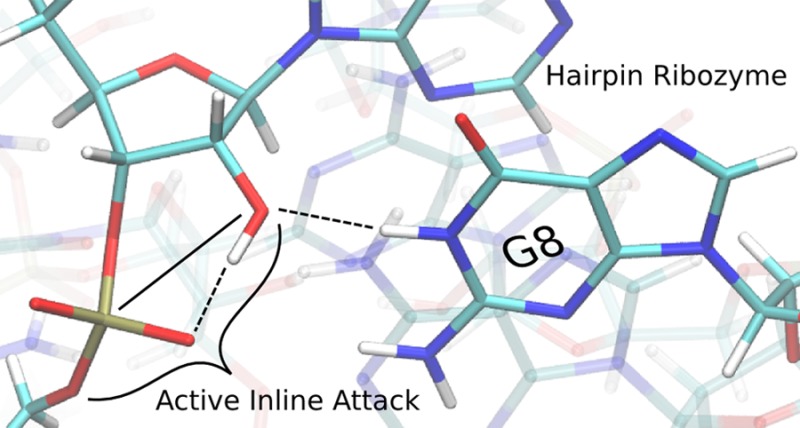
The hairpin ribozyme accelerates a phosphoryl transfer reaction without catalytic participation of divalent metal ions. Residues A38 and G8 have been implicated as playing roles in general acid and base catalysis, respectively. Here we explore the structure and dynamics of key active site residues using more than 1 μs of molecular dynamics simulations of the hairpin ribozyme at different stages along the catalytic pathway. Analysis of results indicates hydrogen bond interactions between the nucleophile and proR nonbridging oxygen are correlated with active inline attack conformations. Further, the simulation results suggest a possible alternative role for G8 to promote inline fitness and facilitate activation of the nucleophile by hydrogen bonding, although this does not necessarily exclude an additional role as a general base. Finally, we suggest that substitution of G8 with N7- or N3-deazaguanosine which have elevated pKa values, both with and without thio modifications at the 5′ leaving group position, would provide valuable insight into the specific role of G8 in catalysis.
The discovery of ribozymes has led to speculation about how these RNA molecules are able to catalyze reactions with their limited repertoire of functional groups.1,2 The hairpin ribozyme (HPr) is a small, self-cleaving RNA that catalyzes both scission and ligation of the phosphodiester backbone, and has been the focus of extensive experimental1−3 and theoretical4−6 study. HPr is intriguing in that its activity does not require divalent metal ions,7 implying catalytic roles for nucleobases (Figure 1). This discovery was interesting since free nucleobases are fairly inert to proton transfer and have pKa values that differ from the typical amino acid values in protein enzymes. Nonetheless, an early crystal structure suggested an active site containing nucleobases aligned for acid–base catalysis, with A38 positioned to donate a proton to the 5′ oxygen leaving group and G8 poised to remove a proton from the O2′ nucleophile.8
Figure 1.
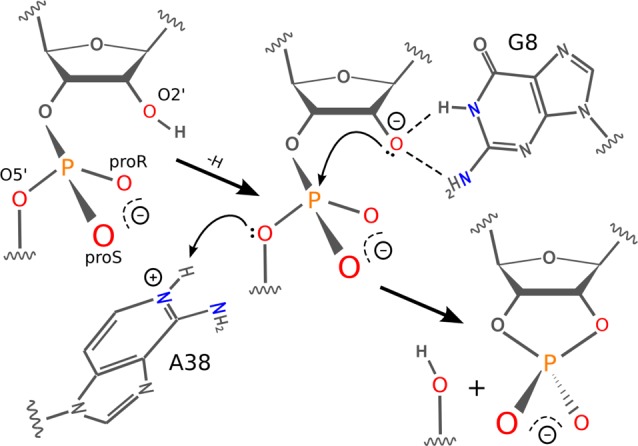
General reaction scheme for the hairpin ribozyme. The nucleophile is activated by a general or specific base. G8, in neutral form, stabilizes the activated nucleophile by dual hydrogen bond donation. A38 acts as a general acid to donate a proton to the leaving group facilitating cleavage.
Experimental evidence supports a mechanism where A38 acts as the general acid. Substitution of A38 with an abasic residue leads to a 14 000-fold reduction in the cleavage rate and a shift toward the basic in the pH–rate profile of several pH units;9 similarly, substitution of A38 with an N1-deazaadenosine, which replaces the N1 nitrogen with a carbon, abolishes catalysis.10 Reduction of the catalytic rate due to substitution of A38 with purine can be rescued by introduction of an enhanced leaving group,11 and direct pKa measurements of A38 in precatalytic10 and transition state mimic12 structures show the N1 imino shifts toward neutrality, and are in close agreement with apparent pKa’s derived from the pH–rate profile, which has been shown to reflect ionization of A38.13
Evidence suggesting the role of G8 as a general base is somewhat less compelling. Substitution of G8 with an abasic residue has an 850-fold reduction in catalysis and no shift in the pH–rate profile.14 Measurement of microscopic pKa values for 8-azaguanine were approximately 3 units higher than the apparent pKa values determined from the kinetic pH–rate profile.15 The simplest interpretation of the observed lack of correlation of activity with deprotonation at this position was that G8 functions in the protonated form. However, substitution of G8 with an imidazole (pKa ≈ 7) results in a bell-shaped pH–rate profile,16 and substitution with diaminopurine (pKa 5.1) in the presence of an enhanced leaving group results in a pH–rate profile that is log-linear over most of the pH-range, indicative of diaminopurine acting as a general base in this context.11 Also, G8 is specifically alkylated at the N1 position when the 2′-OH nucleophile is replaced by a 2′-bromoacetamide group, which is consistent with the hypothesis that it acts as a general base.17
We report molecular dynamics simulation results for the HPr at different stages along the reaction path. The goal is to characterize the structure of plausible states along the reaction path in order to help reconcile the functional roles of A38 and G8 and provide a departure point for further study of transition state barriers using quantum methods. Simulations of the precatalytic reactant (R) state (i.e., having the nucleophile protonated) were performed for 500 ns based on the crystal structure18 (PDB ID 2OUE) with A38 in both the neutral (R-A38°) and protonated (R-A38+) forms. In addition, we completed a 150 ns activated precursor (AP) simulation with the A-1:O2′ deprotonated, the scissile phosphate protonated on the proR oxygen (G+1:O2P), and A38 protonated (AP-A38+-O2P+). One 6 ns simulation of the reactant state with a thio substitution at the proR position was performed, along with quantum chemical calculations, in order to assess the probability of the proR position being protonated. Two 150 ns transition state (TS) mimic simulations were performed (both having A38 protonated), the first (TS-P(V)-A38+) based on the vanadate TS mimic crystal structure19 (PDB ID 2P7E), and the second (TS-2′-5′-A38+) based on the crystal structure of a TS mimic containing a 2′ to 5′ linkage to the scissile phosphate20 (PDB ID 3CQS). Finally, we have performed an 85 ns simulation of the vanadate TS mimic crystal19 (TS-P(V)-A38+-X) with 12 HPr monomers modeled explicitly and arranged according to the experimental crystal space group symmetry.
Simulations were performed with NAMD version 2.921 (TS-P(V)-A38+-X was performed with Amber12 CUDA22) in the NPT ensemble using the AMBER parm99 force field with the corrected α/γ torsional parameters23 and sodium and chloride ions parametrized24 for use with the TIP4P-ew25 water model. We developed parameters for nonstandard residues according to a protocol based on the development of the original AMBER parameter set.26 Simulation temperature was maintained at 300 K using Langevin dynamics, with a damping coefficient of 1 ps–1. Pressure was controlled using a Langevin piston, with a target pressure of 1 atm, period of 100 fs, and decay time of 50 fs. Full details are provided in the Supporting Information.
Interactions between the Nucleophile and the proR Nonbridging Oxygen Promote Active Inline Conformations. Active inline attack conformations27 are important requirements for RNA transphosphorylation reactions. Although these conformations are often rare, the free energy required to bring the nucleophile inline has been predicted, at least in some cases, to be only modest and likely not a dominant factor on the overall catalytic rate.28 Here we define an “active” inline attack geometry to be one that has an O2′–P′O5′ angle (θinl) of more than 125° and O2′–P distance (Dinl) of less than 3.5 Å (Figure 2A). Figure 3 shows the distribution of active inline attack conformations for the reactant and activated precursor state simulations. Active conformations are observed for 7.7% and 1.7% of the configurations in the R-A38° and R-A38+ simulations, respectively, and in both cases are highly correlated with hydrogen bonds between the 2′OH of A-1 and the proR nonbridging oxygen (G+1:O2P) on the scissile phosphate (see Supporting Information). In the case of the AP simulation, a hydrogen bond between the protonated proR oxygen and the 2′O– is similarly correlated with active inline conformations (Figure 2A). These results predict that hydrogen bond interactions between the nucleophile and proR of the scissile phosphate are important for formation of active inline attack conformations. Thio substitution experiments on a minimal sequence self-cleaving HPr domain indicate that there is only a very small (roughly 4-fold) stereospecific thio effect at the proR position.29 Quantum chemical calculations of the HPr active site suggest that protonation of the proR position is unlikely in either the native or thio-substituted ribozyme (Figures S5–S7). MD simulations of the reactant state with proR thio substitution suggest that hydrogen bonding is slightly weakened with respect to the native ribozyme, but is still correlated with inline attack conformations (see Supporting Information for full details).
Figure 2.
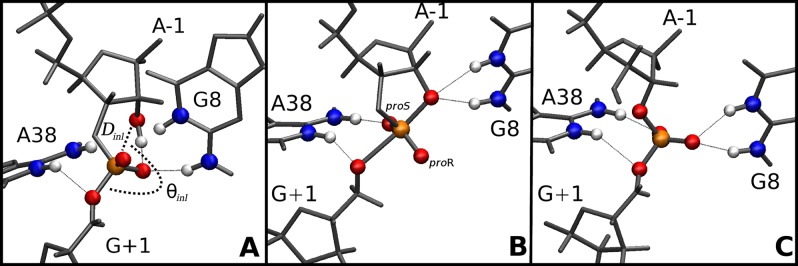
Average active site structure for (A) R-A38+ simulation (inline attack distance, Dinl, and angle, θinl, are labeled), (B) TS-P(V)-A38+ simulation, and (C) TS-2′-5′-A38+ simulation. Labels are shown for the nonbridging proR (O2P) and proS (O1P) oxygens.
Figure 3.
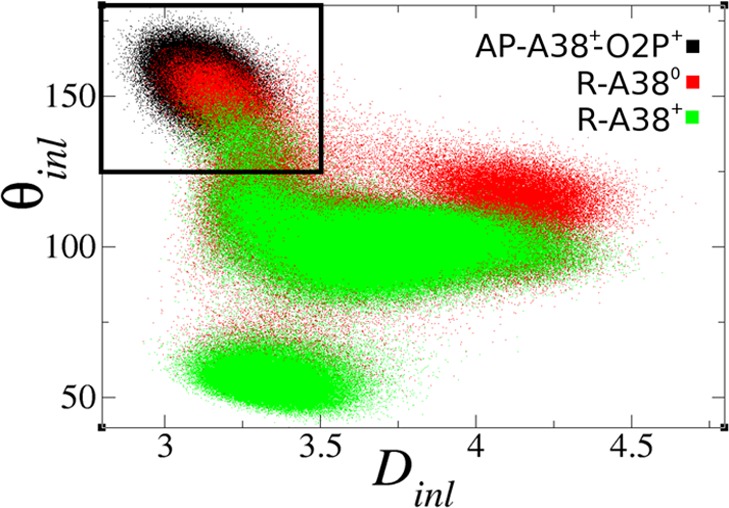
Inline attack angles (θinl in degrees) vs distances (Dinl in Å) for reactant and active precursor state simulations (sampled every 1 ps). The black outline indicates active values that favor catalysis.
G8 Hydrogen Bonds with the Nucleophile, Facilitating Its Activation and Promoting Inline Attack Geometry. In the R-A38° simulation, active inline conformations were correlated with hydrogen bonding between G8:N1/N2 and the A-1:O2′ nucleophile (Figure S1), as indicated by the shorter average distance of the active versus inactive conformations (2.95 and 3.53 Å, respectively). This interaction is also present throughout the TS-P(V)-A38+ simulation (Figure 2B). These interactions raise the possibility that a role for G8 in the catalytic mechanism is to donate hydrogen bonds to the 2′ position, facilitating deprotonation and stabilizing the negative charge of the activated precursor while maintaining an inline attack conformation. This proposed role for G8 is not dependent on its status as a general base, an additional role that others have suggested.11,17
A38 is poised to act as the general acid in the TS mimic simulations as suggested by crystallographic data. The structure of the active site in the TS-2′-5′-A38+ simulation overall resembles that of the TS-P(V)-A38+ simulation, with a few notable differences (Figure 2B and C). A common feature is that A38 is poised to act as a general acid in both simulations: the N6 exocyclic amine hydrogen bonds the G+1:O2P nonbridging oxygen, positioning N1 to donate a hydrogen bond to G+1:O5′, which is consistent with its role as the general acid. However, G8 in the TS-2′-5′-A38+ simulation donates a hydrogen bond to the G+1:O1P nonbridging oxygen rather than the A-1:O2′ as it does in the TS-P(V)-A38+ simulation. This behavior is consistent with what is observed in the crystal structures corresponding to these constructs.19
To provide support for our solution structure predictions and help ascertain to what degree our results are influenced by artifacts in our models or simulation protocol, we have carried out crystal simulations (TS-P(V)-A38+X) to compare directly with crystallographic data. Overall, the structures from the crystal simulation were much closer to the experimental crystal structure than were the structures from the corresponding solution simulations (Figure 4). The average structure from the crystal simulation was very close to the crystal structure, having a root-mean-square deviation (RMSD) of 0.89 Å (all heavy atom) and 0.58 Å (active site heavy atoms), whereas the corresponding value from the solution simulation was 2.97 and 1.54 Å, respectively. In the crystal simulation, G8:N1 maintains a tight interaction with the A-1:O2′ nucleophile in all 12 simulated monomers lending further evidence for its role in stabilizing the activated nucleophile. Also, A38:N1 remains poised to act as a general acid in a majority of the simulated monomers, although in the simulated ensemble it also sometimes observed to donate a hydrogen bond to the G+1:O1P nonbridging oxygen as observed in the 2′-5′ TS mimic crystal.19
Figure 4.
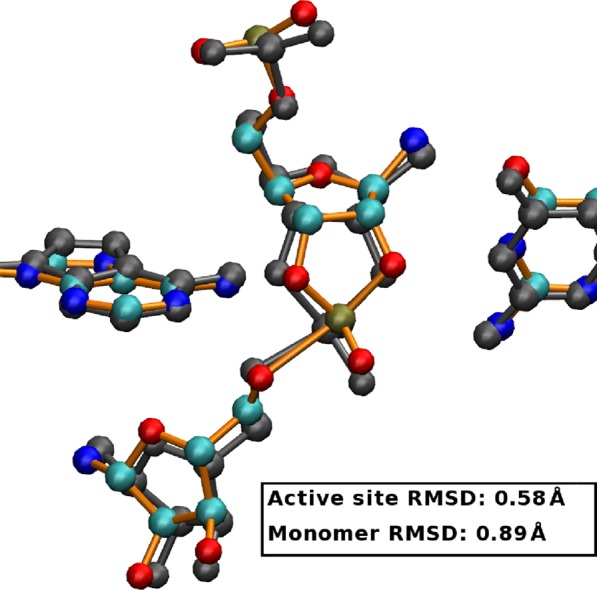
Comparison of experimental (PDB: 2p7e, gray) and average crystal simulation (TS-P(V)-A38+-X, colored) active sites. RMSD is 0.890 (all heavy atom) and 0.581 (active site heavy atom).
Chemical modification of G8 and the 5′ leaving group may provide insight into the mechanism. Our simulations predict that G8 donates two hydrogen bonds to the nucleophile, and this may facilitate its activation and positioning for inline attack. This brings into question whether G8 may also act as a general base. Experimentally, one could test this role by substitution of G8 with N7- or N3-deazaguanosine, in conjunction with a thio substitution at the 5′ leaving group position. The N7- and N3-deaza modifications preserve the hydrogen bonding groups at positions 1 and 2 but shift the pKa at the N1 position to higher values. Substitution of sulfur at the 5′ position creates an enhanced leaving group that should eliminate the general acid step as rate-controlling. If the catalytic rate and pH–rate profile upon G8 modification remain largely unchanged, then this is consistent with the hypothesis that G8 does not act as a general base but provides only electrostatic stabilization through hydrogen bonding. A similar supposition has been suggested previously based on determination of microscopic pKa values with 8-azaguanosine substitution.15 If G8 proves not to act as a general base, it is possible that activation of the nucleophile occurs through a specific base mechanism.18
Acknowledgments
The authors are grateful for financial support provided by the National Institutes of Health (GM62248 to D.M.Y., GM63162 to J.E.W.). This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation Grant Number OCI-1053575.
Supporting Information Available
Molecular dynamics simulation setup. Development of partial charges of nonstandard residues. Overlays of the average active in-line attack and inactive active site structures from the R-A38° and R-A38+ simulations. Overlays of 12 randomly selected active structures from each of the three precatalytic simulations. Comparison of solution (TS-P(V)-A38+) and crystal (TS-P(V)-A38+-X) simulation RMSD values. Impact of the proR-thio substitution at the scissile phosphate on the active conformation of the reactant state. Potential energy surfaces of the proton transfer between the nucleophile and the nonbridging oxygen or sulfur and between nucleophile and the deprotonated Guanine N1. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Doherty E. A.; Doudna J. A. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 457–475. [DOI] [PubMed] [Google Scholar]
- Bevilacqua P. C.; Yajima R. Curr. Opin. Chem. Biol. 2006, 10, 455–464. [DOI] [PubMed] [Google Scholar]
- Fedor M. J. Annu. Rev. Biophys. 2009, 38, 271–299. [DOI] [PubMed] [Google Scholar]
- Nam K.; Gao J.; York D. M. J. Am. Chem. Soc. 2008, 130, 4680–4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam K.; Gao J.; York D. RNA 2008, 14, 1501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlýnský V.; Banás P.; Hollas D.; Réblová K.; Walter N. G.; Sponer J.; Otyepka M. J. Phys. Chem. B 2010, 114, 6642–6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray J. B.; Seyhan A. A.; Walter N. G.; Burke J. M.; Scott W. G. Chem. Biol. 1998, 5, 587–595. [DOI] [PubMed] [Google Scholar]
- Rupert P. B.; Ferré-D’Amaré A. R. Nature 2001, 410, 780–786. [DOI] [PubMed] [Google Scholar]
- Kuzmin Y. I.; Costa C. P. D.; Cottrell J. W.; Fedor M. J. J. Mol. Biol. 2005, 349, 989–1010. [DOI] [PubMed] [Google Scholar]
- Guo M.; Spitale R. C.; Volpini R.; Krucinska J.; Cristalli G.; Carey P. R.; Wedekind J. E. J. Am. Chem. Soc. 2009, 131, 12908–12909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kath-Schorr S.; Wilson T. J.; Li N.-S.; Lu J.; Piccirilli J. A.; Lilley D. M. J. J. Am. Chem. Soc. 2012, 134, 16717–16724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman J. A.; Guo M.; Jenkins J. L.; Krucinska J.; Chen Y.; Carey P. R.; Wedekind J. E. J. Am. Chem. Soc. 2012, 134, 16933–16936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell J. W.; Scott L. G.; Fedor M. J. J. Biol. Chem. 2011, 286, 17658–17664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmin Y. I.; Da Costa C. P.; Fedor M. J. J. Mol. Biol. 2004, 340, 233–251. [DOI] [PubMed] [Google Scholar]
- Liu L.; Cottrell J. W.; Scott L. G.; Fedor M. J. Nat. Chem. Biol. 2009, 5, 351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson T. J.; Ouellet J.; Zhao Z.-y.; Harusawa S.; Araki L.; Kurihara T.; Lilley D. M. RNA 2006, 12, 980–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J. M.; Perrin D. M. J. Am. Chem. Soc. 2006, 128, 16540–16545. [DOI] [PubMed] [Google Scholar]
- Salter J.; Krucinska J.; Alam S.; Grum-Tokars V.; Wedekind J. E. Biochemistry 2006, 45, 686–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torelli A. T.; Krucinska J.; Wedekind J. E. RNA 2007, 13, 1052–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torelli A. T.; Spitale R. C.; Krucinska J.; Wedekind J. E. Biochem. Biophys. Res. Commun. 2008, 371, 154–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kaleé L.; Schulten K. J. Comput. Chem. 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.et al. AMBER 12; University of California, San Francisco: San Francisco, CA, 2012.
- Pérez A.; Marchán I.; Svozil D.; Šponer J.; Cheatham T. E. III; Laughton C. A.; Orozco M. Biophys. J. 2007, 92, 3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung I. S.; Cheatham T. E. III. J. Phys. Chem. B 2008, 112, 9020–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn H. W.; Swope W. C.; Pitera J. W.; Madura J. D.; Dick T. J.; Hura G. L.; Head-Gordon T. J. Chem. Phys. 2004, 120, 9665–9678. [DOI] [PubMed] [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar]
- Soukup G. A.; Breaker R. R. RNA 1999, 5, 1308–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min D.; Xue S.; Li H.; Yang W. Nucleic Acids Res. 2007, 35, 4001–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbitt S.; Hegg L. A.; Fedor M. J. Chem. Biol. 1997, 4, 619–630. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.