

Abstract
The mechanisms by which mucosal homeostasis is maintained are of central importance to inflammatory bowel disease. Critical to these processes is the intestinal epithelial cell (IEC), which regulates immune responses at the interface between the commensal microbiota and the host1,2. CD1d presents self and microbial lipid antigens to natural killer T (NKT) cells, which are involved in the pathogenesis of colitis in animal models and human inflammatory bowel disease3–8. As CD1d crosslinking on model IECs results in the production of the important regulatory cytokine interleukin (IL)-10 (ref.9), decreased epithelial CD1d expression—as observed in inflammatory bowel disease10,11—may contribute substantially to intestinal inflammation. Here we show in mice that whereas bone-marrow-derived CD1d signals contribute to NKT-cell-mediated intestinal inflammation, engagement of epithelial CD1d elicits protective effects through the activation of STAT3 and STAT3-dependent transcription of IL-10, heat shock protein 110 (HSP110; also known as HSP105), and CD1d itself. All of these epithelial elements are critically involved in controlling CD1d-mediated intestinal inflammation. This is demonstrated by severe NKT-cell-mediated colitis upon IEC-specific deletion of IL-10, CD1d, and its critical regulator microsomal triglyceride transfer protein (MTP)12,13, as well as deletion of HSP110 in the radioresistant compartment. Our studies thus uncover a novel pathway of IEC-dependent regulation of mucosal homeostasis and highlight a critical role of IL-10 in the intestinal epithelium, with broad implications for diseases such as inflammatory bowel disease.
To examine the function of CD1d in the intestinal epithelium, we generated mice with tamoxifen-inducible IEC-specific deletion of MTP, a lipid transfer protein encoded by Mttp and required for CD1d function12,13 (Villin-CreERT2 (ref. 14) × Mttpfl/fl (ref. 15); hereafter referred to as MttpΔIEC). MttpΔIEC mice exhibited no evidence of epithelial lipid accumulation (Extended Data Fig. 2a) and endoplasmic reticulum (ER) stress (Extended Data Fig. 2b), but showed decreased IEC-mediated, CD1d-but not major histocompatibility complex (MHC)-class-I-dependent antigen presentation despite unimpaired CD1d expression (Extended Data Fig. 3a, b, c left). MttpΔIEC mice exhibited increased sensitivity to oxazolone-induced colitis, a CD1d-and NKT-cell-dependent colitis model4, as shown by increased mortality (Fig. 1a, left), weight loss (Fig. 1a, right) and histological injury (Fig. 1b and Extended Data Fig. 3d), in comparison with wild-type littermates (Villin-CreERT2-negative Mttpfl/fl mice). Increased severity of colitis was not caused by primary epithelial barrier defects (Extended Data Fig. 4a) but was due to loss of function of intestinal epithelial CD1d. Thus, bone marrow chimaeras with selective deficiency of CD1d in the radioresistant compartment exhibited severe mortality and morbidity in the oxazolone model (Fig. 1c, d), increased production of pathogenic IL-13 and IL-1β in colon explants (Fig. 1e)and purified CD11b+lamina propria cells (Extended Data Fig. 4b), and reduced secretion of protective IL-10 (Fig. 1e). In accordance with a protective role of intestinal epithelial CD1d, its expression was upregulated in response to oxazolone colitis in wild-type, but not MttpΔIEC, mice (Extended Data Fig. 3c, right). Intriguingly, pathogenic signals were similarly CD1d-dependent but emanated from non-epithelial cells in a manner dependent on adaptive immune cells. Thus, MttpΔIEC mice on a Rag1−/−or Cd1d1−/−Cd1d2−/−(CD1d-knockout) background were protected from oxazolone colitis (Extended Data Fig. 4c, d). Consistent with a role of non-epithelial cells in the delivery of pathogenic CD1d signals, antibody-mediated neutralization of (non-epithelial) CD1d in MttpΔIECmice reduced the severity of oxazolone-induced colitis (Fig. 1f,g). Thus, intestinal epithelial CD1d- and MTP-dependent lipid antigen presentation protects from oxazolone colitis, whereas CD1d- and NKT-cell-mediated inflammation emanates from non-epithelial cells, presumably bone-marrow-derived lamina propria cells.
Figure 1. Intestinal epithelial MTP and CD1d protect from oxazolone colitis.

a–k, Survival (a, left, c, left, f, left, h, left), body weight (a, right, c, right, f, right, h, right), histopathology (b, d, g, i), cytokine secretion (enzyme-linked immunosorbent assay (ELISA)) in colon explant cultures (e, j), and cytokine expression (quantitative polymerase chain reaction (qPCR)) of sorted colonic iNKT cells combined from intraepithelial and lamina propria compartments (k) of the indicated mice upon rectal challenge with oxazolone (ox., a–k) or vehicle (ethanol (EtOH, a–e, h–j)). c–e, Wild-type (WT) and CD1d-knockout (CD1d-KO) mice were reconstituted with wild-type bone marrow. f, g, 19G11 (anti-CD1d) or isotype control antibody (IgG2b) were administered. Results representative of three independent experiments are shown. Mean ± standard error of the mean (s.e.m.) of the indicated number of mice is shown. j, Right, Mann–Whitney U-test was applied. In all other panels, Student’s t-test or log-rank test (survival) were applied.
To investigate directly the role of epithelial CD1d, we developed mice with inducible, IEC-specific deletion of CD1d (Villin-CreERT2×Cd1d1fl/fl; hereafter referred to as Cd1d1ΔIEC) (Extended Data Fig. 5a). Tamoxifen-treated Cd1d1ΔIEC mice exhibited normal numbers of invariant (i)NKT cells and lacked intestinal epithelial expression of CD1d and CD1d- but not MHC-class-I-mediated antigen presentation by IECs (Extended Data Fig. 5b–e). Oxazolone-challenged Cd1d1ΔIEC mice, like MttpΔIEC mice, exhibited increased mortality (Fig. 1h, left), weight loss (Fig. 1h, right), histopathological injury (Fig. 1i), colon shortening (Extended Data Fig. 5f) and elevated secretion of IL-13 and IL-1β, as well as impaired secretion of IL-10 from colon explant cultures (Fig. 1j). Moreover, colonic iNKT cells from Cd1d1ΔIEC mice showed increased IL-13 and IFN-γ expression in oxazolone colitis (Fig. 1k). Thus, intestinal epithelial CD1d protects from intestinal inflammation.
To gain insight into the mechanisms underlying protection by intestinal epithelial CD1d, we performed transcriptional profiling of intestinal mucosal scrapings from wild-type and MttpΔIEC mice in the oxazolone model (Extended Data Table 1 and data not shown). HSP110, encoded by Hsph1, exhibited decreased expression in MttpΔIEC mice (Fig. 2a), which was of interest given its known role in induction of epithelial CD1d expression16. In line with a potential MTP- and CD1d-dependent role of HSP110 in intestinal inflammation, HSP110 messenger RNA (Fig. 2b, left) and protein expression (Extended Data Fig. 6a) were increased in IECs from wild-type but not MttpΔIEC mice upon oxazolone challenge. Similar observations were made for Cd1d1ΔIEC mice (Fig. 2b, right). In addition, intestinal epithelial but not lamina propria HSP110 expression was reduced in active and inactive human ulcerative colitis compared with healthy controls (Fig. 2c and Extended Data Fig. 6b), which is in accordance with impaired epithelial CD1d expression in inflammatory bowel disease, as previously reported10. Furthermore, consistent with regulation of CD1d expression by HSP110 (ref. 16), oxazolone colitis was associated with induction of CD1d expression in wild-type but not MttpΔIEC IECs (Extended Data Fig. 3c, right). Together, these data indicate that IEC CD1d and MTP are required for epithelial HSP110 upregulation during oxazolone-induced colitis and that alterations in this pathway are observed in human ulcerative colitis.
Figure 2. HSP110 elicits protective effects downstream of intestinal epithelial CD1d.

a, b, Hsph1 expression (qPCR) in purified IECs of the indicated mouse strains at baseline (a) or on day 5 after rectal oxazolone (Ox.) or ethanol (EtOH) (b). c, HSP110 immunohistochemistry in colon of human ulcerative colitis and controls. d–i, Survival (d, bottom, h, left), body weight (d, top, h, right), histopathology (e, i), and cytokine secretion in colon explants (ELISA) (f) in the oxazolone model. d, f, Wild-type (WT) and HSP110-KO recipients of wild-type bone marrow received 19G11 anti-CD1d or isotype control antibody. g–i, Mice received adenoviruses expressing HSP110 (AdHSP110) or β-galactosidase (AdLacZ). g, Immunofluorescence of colonic GFP after AdHSP110-IRES2-eGFP administration. DAPI, 4′,6-diamidino-2-phenylindole. j–l, MODE-K Hsph1 and Il10 (qPCR) after antibody-mediated CD1d crosslinking (1B1) (j–k) or α-GalCer presentation to iNKT cells (24.7 hybridoma) in the presence/ absence of neutralizing CD1d antibodies (19G11) (l). mAb, monoclonal antibody. m, Hsph1 and Il10 (qPCR) in purified small intestinal IECs from wild-type and TD-CD1d recipients of wild-type bone marrow after injection with vehicle or α-GalCer (6 mice per group). c, g, Scale bars, 50 μm. Results representative of two or three independent experiments are shown. Means ±s.e.m. of the indicated number of mice or triplicate cultures (l) are indicated. Student’s t-test or log-rank test (survival) were applied.
To understand better the functional consequences of epithelial HSP110 deficiency observed in MttpΔIEC and Cd1d1ΔIEC mice, bone marrow chimaeras of HSP110-knockout (HSP110-KO) or wild-type mice receiving wild-type bone marrow were studied. Compared with wild-type recipients, HSP110-knockout recipients of wild-type bone marrow exhibited increased mortality and weight loss (Fig. 2d), microscopic injury (Fig. 2e) and increased IL-13 and IL-1β secretion (Fig. 2f) in the oxazolone model. Morbidity and mortality in HSP110-knockout recipients of wild-type bone marrow reflected that observed for conventional HSP110-knockout mice, suggesting critical, protective effects of HSP110 in the radioresistant compartment, including IECs (Extended Data Fig. 6c). Antibody-mediated neutralization of CD1d reduced the severity of colitis in HSP110-knockout bone marrow chimaeras, in line with pathogenic CD1d-dependent signals originating from non-epithelial lamina propria cells (Fig. 2d, f). Together, these studies indicate that reduced HSP110 expression in the absence of functional epithelial CD1d may contribute to severe oxazolone colitis. Consistent with this hypothesis, reconstitution of HSP110 expression by adenoviruses targeting IECs (Fig. 2g) ameliorated oxazolone-induced colitis in MttpΔIEC mice, as shown by decreased mortality (Fig. 2h, left), weight loss (Fig. 2h, right) and microscopic injury (Fig. 2i). It is noteworthy that adenoviruses also targeted lamina propria cells (Fig. 2g), which may have contributed to protection from intestinal inflammation.
As mice with an IEC-specific deficiency in MTP and CD1d exhibited impaired epithelial expression of HSP110, we tested whether engagement of epithelial CD1d supports HSP110 expression. To this end, we investigated HSP110 in purified (Extended Data Fig. 7) colonic IECs after antibody-mediated CD1d crosslinking, a treatment previously shown to induce production of barrier-protective IEC-derived IL-10 (ref. 9). CD1d crosslinking led to increased expression of Il10 (Fig. 2j) and Hsph1 (Fig. 2k) in IECs from wild-type but not MttpΔIECmice. Similar observations were made upon in vitro co-culture of NKT cells with a mouse IEC line (Fig. 2l). Previous studies in a cultured human IEC line suggested that epithelial IL-10 production upon CD1d crosslinking is elicited through retrograde signalling dependent on the cytoplasmic tail of CD1d9. We therefore investigated whether bone marrow chimaeras with selective deletion of the cytoplasmic CD1d tail (hereafter referred to as tail-deleted (TD)-CD1d) in radioresistant cells exhibit impaired expression of Il10 and Hsph1 in purified IECs. Indeed, IECs obtained from these mice showed impaired expression of Il10 and Hsph1 under constitutive conditions and reduced expression after administration of the CD1d-binding, iNKT-cell-activating lipid α-galactosylceramide (α-GalCer) (Fig. 2m). These data demonstrate that engagement of epithelial CD1d induces expression of HSP110 and IL-10, presumably through retrograde CD1d signalling. Additional effects of the CD1d cytoplasmic tail, such as regulation of intracellular CD1d trafficking17 and potential effects on NKT cell homing5, may further contribute to protection from intestinal inflammation.
As the Il10, Hsph1 and Cd1d1 genes contain STAT sites in their promoters (ref. 18 and T.O. et al., unpublished observations), and as Hsph1 and Cd1d1 exhibit reduced expression in Stat3-deficient IECs (Gene Expression Omnibus accession number GSE15955)19, we reasoned that STAT3 may contribute to Il10 and Hsph1 expression downstream of CD1d. Indeed, epithelial STAT3 became phosphorylated in response to CD1d engagement by antibody crosslinking (Fig. 3a, left) and iNKT-cell co-culture (Fig. 3a, right, and Extended Data Fig. 8a). Moreover, impaired epithelial STAT3 phosphorylation was observed in Cd1d1ΔIEC mice (Fig. 3b) and TD-CD1d recipients of wild-type bone marrow (Fig. 3c). Furthermore, short interfering RNA (siRNA)-mediated knockdown of Stat3 in the IEC line MODE-K (Extended Data Fig. 8b) led to reduced expression of Cd1d1, Hsph1 and Il10 (Fig. 3d). Together, these data demonstrate that STAT3 acts downstream of epithelial CD1d to induce expression of Il10, Hsph1 and Cd1d1.
Figure 3. Epithelial CD1d induces STAT3-dependent expression of IL-10 and HSP110.

a, MODE-K pSTAT3 after CD1d-crosslinking (1B1 monoclonal antibody clone) or iNKT (24.7 hybridoma) co-culture. b, Colonic pSTAT3 immunohistochemistry 5 days after rectal oxazolone (Ox.) or ethanol (EtOH). Scale bar, 50 μm. c, pSTAT3 in purified small intestinal IECs from the indicated bone marrow chimaeras (IECs from one mouse per lane). d–f, Cd1d1, Hsph1 and Il10 (qPCR) in MODE-K cells after treatment with recombinant HSP110 (e) or Stat3/ control siRNA knockdown (d, f) followed by viral transduction at a multiplicity of infection (MOI) of 5 (f). g, Il10, Hsph1 and Tgfb in purified IECs (qPCR) 6 h after rectal oxazolone in the presence or absence of viral infection. The fold change compared to oxazolone-treated Cd1d1+/+ mice without virus is indicated. h, Western blot of IECs 24 h after rectal oxazolone. i, Western blot of purified CD11b+ lamina propria cells 5 days after rectal oxazolone. j–l, Survival (j, left), body weight (j, right), histopathology (k) and cytokine secretion by colon explants (ELISA; l) of the indicated oxazolone-challenged mice with or without viral infection and antibody treatment as indicated. Results representative of three independent experiments are shown. Means ±s.e.m. of the indicated number of mice or triplicate cultures (d–f) are indicated. Student’s t-test or log-rank test (survival) were applied. NS, not significant.
IL-10 and HSP110 do not only exhibit STAT3-dependent expression but also signal via STAT3 (refs 18, 20) and may thus further contribute to sustained activation of epithelial STAT3 and STAT3-dependent expression of Cd1d1, Il10 and Hsph1. Consistent with this concept, both recombinant and adenoviral HSP110 induced epithelial expression of Il10 and Cd1d1 in MODE-K cells, which was abrogated upon siRNA-mediated knockdown of Stat3 (Fig. 3e, f). Similar findings were made in vivo, as adenoviruses expressing HSP110, but not LacZ, restored epithelial expression of IL-10 in Cd1d1ΔIEC mice in the oxazolone model (Fig. 3g, h). These data demonstrate that HSP110 acts in a STAT3-dependent manner to support the expression of IL-10 and CD1d by IECs.
Given HSP110-dependent regulation of epithelial IL-10 expression, we investigated whether the protective effects of HSP110 are dependent on IL-10. CD11b+ lamina propria cells from Cd1d1ΔIEC mice showed decreased phosphorylated (p)STAT3 expression in oxazolone colitis, consistent with decreased IL-10 production by IECs and IL-10 receptor (IL-10R) signalling by lamina propria cells (Fig. 3i). Moreover, antibody-mediated IL-10R blocking prevented adenoviral HSP110-dependent amelioration of colitis (Fig. 3j–l). To investigate further the role of epithelial IL-10, we generated Il10−/− bone marrow chimaeras. Il10 deletion in the radioresistant compartment resulted in increased mortality (Fig. 4a) and colonic IL-13 production (Fig. 4b) in the oxazolone model. Moreover, a similar trend was observed in mice with Il10 deletion in the radiosensitive compartment (Fig. 4a, b). These data thus reveal that, in addition to the established role of bone-marrow-derived cells in IL-10-mediated control of intestinal inflammation21,22, IL-10 derived from radioresistant cells provides an additional, non-redundant pathway required for regulation of intestinal inflammation. To address whether protective IL-10 originates from the intestinal epithelium, we investigated oxazolone colitis in mice with inducible, epithelial-specific deletion of Il10 (Villin-CreERT2 × Il10fl/fl; hereafter referred to as Il10ΔIEC). Similar to mice with IEC-specific deletion of Mttp and Cd1d1 and mice with deletion of Hsph1 and Il10 in the radioresistant compartment, Il10ΔIEC mice exhibited increased mortality (Fig. 4c), weight loss (Fig. 4d) and histopathological inflammation (Fig. 4e) in the oxazolone model. Furthermore, in accordance with STAT3-mediated signalling by IL-10 (ref. 18) and STAT3-dependent expression of Hsph1 and Cd1d1 (Fig. 3d), IEC-specific Il10 deletion was associated with reduced epithelial expression of Hsph1 and a trend towards a reduction in Cd1d1 expression in the oxazolone model (Fig. 4f). These results demonstrate that IEC-derived IL-10, downstream of epithelial CD1d, HSP110 and STAT3 is critical for regulation of intestinal inflammation.
Figure 4. Epithelial IL-10 is critical for control of intestinal inflammation.

a–f, Survival (a, c), IL-13 production in colon explants on day 1 after rectal oxazolone (Ox.) or ethanol (EtOH) (b), body weight (d), histopathology (e), and Il10 (left), Hsph1 (middle) and Cd1d1 (right) expression (qPCR; f) in colonic IECs 5 days after rectal oxazolone challenge in the indicated mouse strains. Results representative of three (a, b) or two (c–f) independent experiments are shown. Means ±s.e.m. of the indicated number of mice are shown. Student’s t-test or log-rank test (survival) were applied.
NKT cells are central mediators of intestinal inflammation3,4. In studies aiming to identify the cellular origin of pathogenic lipid antigen presentation in oxazolone colitis, we made the unanticipated observation that intestinal epithelial CD1d protects from intestinal inflammation in a manner dependent on STAT3, IL-10 and HSP110, whereas pathogenic NKT-cell activation emanates from bone-marrow-derived cells in a CD1d-restricted fashion (Extended Data Fig. 1a). As further outlined in the Supplementary Discussion and Extended Data Fig. 9, such opposing roles of CD1d-restricted presentation by bone-marrow-derived and epithelial cells are at least partially dependent on the expression of co-stimulatory molecules such as CD40. Our findings highlight the role of the intestinal epithelium as a key regulator of mucosal immunity and underscore the important role of intestinal epithelial CD1d and IL-10 in determining the host’s response to environmental stimuli, which activate CD1d-restricted NKT cells5.
METHODS
Mice
Mice (C57BL/6J unless indicated otherwise) were housed in a specific pathogen-free (SPF) barrier facility. Cd1d1−/− Cd1d2−/−(ref. 24), TD-CD1d (ref. 17), Il10−/− (ref. 25), Villin-CreERT2 (ref. 14), Il10fl/fl (ref. 26), Rag1−/−(ref. 27), HSP110-KO (ref. 23) (C57BL/6N) and Mttpfl/fl (ref. 15) (C57BL/6J/129) mice have been described. To generate the floxed/neo Cd1d1 allele, a LoxP (L83) site at 10310 and a FNFL (Frt-Neo-Frt-LoxP) cassette at 12190 was inserted to flank exon 2, 3 and 4 (about 1.9 kb) of the Cd1d1 gene. A gene-targeting vector was constructed by retrieving one 5 kb long homology arm (5′ to L83), one 1.9 kb sequence containing exon 2/3/4, FNFL cassette, and one 2 kb short homology arm (end of FNFL to 3′). The FNFL cassette conferred G418 resistance during gene targeting in PTL1 (129B6 hybrid) embryonic stem (ES) cells. The Cd1d1 allele was PCR amplified and sequenced to confirm the targeted C57BL/6J allele based on the C57BL/6J-specific mutation in Cd1d2 allele28. Three targeted ES cells with targeted C57BL/6J allele were injected into C57BL/6J blastocysts to generate chimaeras. Male chimaeras were bred to bACTFlpe females or EIIa-Cre females to transmit the floxed Cd1d1 allele (Cd1d1L/+) (with neo cassette removed by Fple recombinase) and Cd1d1-null allele (Cd1d1+/−) (with exon 2/3/4 and neo cassette removed by Cre recombinase) through the germ line. Mice carrying floxed Cd1d1 alleles were crossed to Villin-CreERT2 mice (ref. 14) expressing Cre recombinase.
To activate Cre recombinase, 4–5-week-old Villin-CreERT2 mice were orally gavaged daily with 1 mg tamoxifen for a total of 5 days. For bone marrow chimaeras, recipients received total body irradiation of 1,100 rad in two separate doses 4 h apart. The next day, 0.5 × 106 bone marrow cells were delivered intravenously. For α-GalCer studies, mice were injected with 2 μg of α-GalCer (KRN7000, Avanti Polar Lipids) intraperitoneally (i.p.) 8 weeks after bone marrow reconstitution and RNA was isolated from Percoll-purified IECs (see later) 12 h after injection. For oxazolone colitis, mice were pre-sensitized by abdominal skin application of 3% (w/v) oxazolone in 100% ethanol, followed by rectal administration of 1% oxazolone in 50% ethanol after 5 days. Where indicated, mice received 1 mg of neutralizing antibodies against IL-10R (R&D), CD1d (19G11, BioXcell) or isotype control i.p., or 5 × 108 adenoviruses in PBS i.p., each before skin sensitization and before and 2 days after rectal oxazolone challenge. Body weight, rectal bleeding and stool consistency were analysed on a daily basis. Tissues were examined for evidence of colitis by five established criteria for colitis activity in a blinded fashion by a pathologist (J.G.): hypervascularization, presence of mononuclear cells, epithelial hyperplasia, epithelial injury, and presence of granulocytes. For a schematic overview of procedures please refer to Extended Data Fig. 1b.
Animal studies were conducted in a gender- and age-matched manner using littermates for each experiment. Both male and female mice were used and were 8 weeks of age at the time of experiments (11–16 weeks of age for bone marrow chimaera studies). The number of animals used per group was based on previous experimental results and observed variability. Histopathology analysis was performed in a blinded manner. For all other in vitro and in vivo analyses, investigators were not blinded to treatment allocation. Animal studies were performed in compliance with ethical regulations and were approved by the ethics committees of Harvard Medical School and the Christian Albrechts University.
Patients
Human studies were approved by the ethics committee of the Ludwig Maximilians University. All subjects provided written informed consent. Immunohistochemical staining was performed on intestinal biopsies from ten healthy controls (patients undergoing screening colonoscopy without detection of malignancy), four patients with active ulcerative colitis, and five patients with inactive ulcerative colitis.
Isolation of IECs and lymphocytes, flow cytometry, and sorting
To isolate IECs, the intestines were opened, washed in cold PBS, cut into pieces, and transferred into tubes containing HBSS without calcium and magnesium. After the addition of dithiothreitol (DTT) at 1 mM (Sigma), intestines were shaken at 250 r.p.m. for 10 min at room temperature to remove mucus. After washing in PBS, tissues were incubated in RPMI-1640 with 1 U ml−1 Dispase (Roche) for 30–40 min at 37 °C at 250 r.p.m. After filtering through a 100 μm strainer and centrifugation for 5 min at 1,500 r.p.m., the pellet was resuspended in 5 ml of 100% Percoll and placed beneath 8 ml of 40% Percoll. After centrifugation for 20 min at 1,500 r.p.m. the top layer was removed, washed three times in PBS and used as IECs.
To isolate intraepithelial (IEL) and lamina propria lymphocytes, large intestines were collected, fat tissue removed, the intestines cut longitudinally and washed in PBS in order to remove faecal content, cut into 30 mm pieces, and shaken in RPMI-1640 containing 20 mM HEPES, 1 mM DTT and 5 mM EDTA for 30 min at 37 °C. To isolate IELs, cells derived from the epithelial compartment were resupended in 4 ml of 40% Percoll and layered on top of 2 ml of 75% Percoll. After centrifugation for 20 min at 2,000 r.p.m. the middle layer was removed, washed in 2% FBS in PBS and the IELs were obtained. For lamina propria lymphocytes, the non-epithelial compartment was washed in PBS, cut into smaller pieces and incubated in RPMI-1640 containing 5% FBS, 1.5 mg ml−1 collagenase type II and 0.5 mg ml−1 dispase (GIBCO) for 1 h at 37 °C under constant horizontal shaking (250 r.p.m.). The digested tissues were filtered through a 40 μm strainer and, after Percoll gradient centrifugation and washing as described earlier, were used as lamina propria lymphocytes in flow cytometry. Where indicated, CD11b+ lamina propria cells were purified using magnetic beads according to the manufacturer’s instructions (Miltenyi Biotec). Liver lymphocytes were prepared as described before29.
Primary iNKT cells were sorted by double staining with PBS57-loaded CD1d tetramers and CD3ε using a BD FACSAria II SORP UV sorter. RNA samples were prepared using an RNeasy Micro Kit and cDNAs were synthesized using the Omniscript RT Kit (Qiagen).
PBS57-loaded or -unloaded CD1d tetramers were obtained from the NIH Tetramer Core Facility. Flow cytometry antibodies were obtained from eBiosciences. Flow cytometry was performed using a MACSQuant (Miltenyi Biotec) and a BD FACSVerse (BD Biosciences) and data were analysed with FlowJo software (TreeStar).
Antigen presentation assays
Freshly isolated IECs, MODE-K cells30, and CD11c-magnetic-bead-purified (Miltenyi Biotec) splenic dendritic cells were incubated with 100 ng ml−1 α-GalCer for 4 h, washed three times with PBS, and aliquoted into 96-well plates at 1 × 105 cells per well. NKT-cell hybridomas (24.7, DN32.D3 and 14S.6 (ref. 31)) were added at a 1:1 ratio. Where indicated, soluble, stimulatory CD28 antibody was added (1 μg ml−1; clone 37.51; eBioscience). In addition, where indicated, cells were transfected with expression plasmids or siRNA 24 h and 72 h, respectively, before co-culture with NKT cells. Mouse IL-2, IL-10, IL-12p70, IL-13 and IL-1β production were assessed by ELISA (OptEIA; BD Biosciences) in supernatants after 24 h of co-culture.
Cell stimulation by crosslinking
CD1d crosslinking on freshly isolated colonic IECs was achieved as described previously32 using plate-bound anti-mCD1d antibody (1B1, BD). In the case of MODE-K cells, surface CD1d was crosslinked by addition of soluble anti-mCD1d antibody (1B1, 10 μg ml−1, 60 min) followed by washing and addition of secondary goat anti-rat IgG (Biolegend, 10 μg ml−1) for the indicated time as well as subsequent cell lysis (see later). MODE-K STAT3 phosphorylation in response to iNKT-cell engagement was investigated after co-culture with the iNKT-cell hybridoma 24.7 for the indicated time, removal of non-adherent iNKT cells by thorough washing, and cell lysis. Where indicated, MODE-K cells were pre-treated with 19G11 anti-CD1d antibody for blocking of CD1d (10 μg ml−1, BioXcell).
Adenoviruses
Type 5, E1/E3-deleted adenoviruses expressing murine HSP110 (AdHSP110) or Escherichia coli β-galactosidase (AdLacZ) under the control of the cytomegalovirus promoter, were constructed according to the in vitro ligation method33,34. Briefly, murine Hsph1 (NM-013559) was isolated and amplified by PCR from mouse intestinal cDNA using the following primers, cloned into NheI and EcoRI sites of pIRES2-eGFP (Clontech), sequenced, and the resulting plasmid was called pHSP110-IRES2-eGFP. Forward primer: ATAAAGCTAGCGCCACCatgtc ggtggttgggctag, containing an NheI site and a Kozak like sequence; reverse primer: AATTAGAATTCTTAgtccaggtccatgttgacagagc, containing an EcoRI site (the sequence complementary to Hsph1 cDNA is shown in lower case). An NheI-HSP110-KpnI or NheI-HSP110-IRES2-eGFP-NotI fragment from pHSP110-IRES2-eGFP was transferred into pHMCMV6 (pHMCMV6-HSP110 and pHMCMV6-HSP110-IRES2-eGFP, respectively). Subsequently, pAdHM15RGD-CMV6-HSP110 and pAdHM15RGD-CMV6-HSP110-IRES2-eGFP were constructed by ligating I-CeuI/PI-SceI-digested pAdHM15-RGD and pHMCMV6-HSP110-IRES2-eGFPorpHMCMV6-HSP110. To prepare the virus, a PacI fragment of pAdHM15-RGD-CMV-HSP110 or pAdHM15-RGD-CMV-HSP110 was transfected into 293 cells. The adenoviruses were purified by two rounds of caesium chloride density centrifugation and dialysed against Tris-HCl (pH 7.5). The concentrations of plaque-forming units (p.f.u.) of individual stocks were determined from the tissue culture infectious dose 50 titre.
Colon organ culture
Standardized segments (1 cm × 1 cm) of the transverse colon were washed in cold PBS supplemented with penicillin and streptomycin (GIBCO) and cultured in 24-well flat-bottom culture plates (Falcon) in RPMI 1640 media (GIBCO) for 24 h at 37 °C. Supernatants were analysed for cytokines IL-10, IL-13 and IL-1β by ELISA (all BD).
Western blotting
Protein extraction and western blotting were performed as described previously35. The following antibodies were used: anti-IL10 (R&D), anti-HSP110 (BD), anti-STAT3 (Cell signaling), anti-pSTAT3 (Cell signaling) and anti-β-actin (Cell Signaling).
Real-time RT–PCR
RNA samples were prepared using an RNeasy Mini Kit and cDNAs were synthesized using the Omniscript RT Kit (Qiagen). Real-time RT–PCR was performed using SYBR Green I Master Mix (Roche) and a CFX96 Real-Time System (Bio-Rad). Values were normalized to β-actin. The following primer sets were used: β-actin, 5′-GATGCTCCCCGGGCTGTATT-3′ and 5′-GGGGTA CTTCAGGGTCAGGA-3′; Hsph1, 5′-CAGGTACAAACTGATGGTCAACA-3′ and 5′-TGAGGTAAGTTCAGGTGAAGGG-3′; Il10, 5′-GAGAGCTGCAGGGC CCTTTGC-3′ and 5′-CTCCCTGGTTTCTCTTCCCAAGACC-3′; Tgfb, 5′-TGT ACGGCAGTGGCTGAACCA-3′ and 5′-TGTCACAAGAGCAGTGAGCGCT-3′, Cd1d1, 5′-GCAGCCAGTACGCTCTTTTC-3′ and 5′-ACAGCTTGTTTCTGGC AGGT-3′; Stat3, 5′-CCCGTACCTGAAGACCAAGT-3′ and 5′-ACACTCCGA GGTCAGATCCA-3′; Il13, 5′-AGCATGGTATGGAGTGTGGACCTG-3′ and 5′-CAGTTGCTTTGTGTAGCTGAGCAG-3′, Ifng 5′-TCAGCAACAGCAAGG CGAAAAAGG-3′ and 5′-CCACCCCGAATCAGCAGCGA-3′.
Transfection, siRNA treatment and recombinant HSP110
Where indicated, MODE-K cells were transfected with mouse CD40 or CD1d (in pSRα-neo), with SignalSilence Stat3 siRNA II or with SignalSilence Control siRNA (Cell Signaling Technology) using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s instructions. Il10 siRNA was obtained from Invitrogen and transduced by Amaxa Nucleofector technology (Lonza) according to the manufacturer’s instructions. Cells were used for antigen presentation assays after 72 h or were incubated with the indicated adenoviruses after 48 h at a MOI of 5 before RNA preparation after another 24 h as described earlier. Where indicated, recombinant HSP110 was added at a final concentration of 6 μg ml−1 to MODE-K cells 24 h before RNA extraction.
XBP1 splicing assay
The assay was performed as described previously36.
In vivo permeability assay
In vivo intestinal permeability was measured using the FITC-labelled dextran (FD-4) method as described previously37,38 with modifications. Mice were administered 150 μl of FD-4 (50 mg ml−1) by oral gavage before and 18 h after rectal challenge with oxazolone. Serial dilutions of FD-4 were made to generate a standard curve and serum concentrations of FD-4 were determined using a BioTek FLx800 Fluorescence Microplate Reader (BioTek) with an excitation wavelength of 490 nm and emission wavelength of 530 nm.
Immunohistochemistry and immunofluorescence
A polyclonal rabbit anti-HSP110 antibody (anti-mouse 1:250, anti-human 1:500; Sigma-Aldrich), an anti-STAT3 (anti-mouse 1:900) and anti-pSTAT3 antibody (anti-mouse 1:350), both from Cell Signaling, as well as a rabbit polyclonal anti-GFP antibody conjugated to FITC (1:1,000) (Novus Biologicals) were used. All images were taken by a digital camera on a Nikon Eclipse Ti microscope.
Osmium tetroxide staining of intracellular lipid droplets
Intestinal tissue was fixed in 10% neutral buffered formalin and transferred into 1% osmium tetroxide with periodic shaking. The tissue was rinsed with distilled water and incubated in 0.5% periodic acid, washed and counterstained with haematoxylin and eosin.
Microarray analysis
MttpΔIEC and littermate wild-type mice were skin sensitized with oxazolone, and IECs were harvested via colonic epithelial scraping as described previously35, either before administration of rectal oxazolone (n =2 per group) or 6 h thereafter (n =4 per group). Isolated RNAs were pooled for each condition and subjected to transcriptomic analysis with mouse genome 430 2.0 array (Affymetrix) at the Biopolymers Core Facility (Harvard Medical School). Data analysis was performed with Agilent GeneSpring GX and Affymetrix GCOS software under default parameter settings with GC-RMA normalization.
Statistical analysis
Statistical testing was performed using the unpaired Student’s t-test unless otherwise indicated. The Mann–Whitney U-test was applied when data were demonstrated to not follow a Gaussian distribution, which is indicated in the respective figures legends. Comparisons of mortality were made by analysing Kaplan–Meier survival curves, and the log-rank test was used to assess differences in survival. All P values were two-tailed, and statistical significance was assumed at P < 0.05.
Extended Data
Extended Data Figure 1. Proposed model of CD1d signalling in polarized intestinal epithelia and overview of experimental procedures.

a, The proposed model of protective (blue) and pathogenic (red) effects of lipid antigen presentation in intestinal inflammation. Bone-marrow-derived antigen-presenting cells (APCs) contribute to oxazolone colitis in a CD1d- and iNKT-cell-dependent manner. By contrast, engagement of intestinal epithelial CD1d elicits protective functions through cytoplasmic CD1d tail-dependent activation of STAT3, and STAT3-dependent transcription of Cd1d1, Il10 and Hsph1. Epithelial IL-10 and HSP110 support this protective self-reinforcing pathway through STAT3-dependent signalling. Interference with any of the elements involved in this regulatory pathway (MTP, CD1d, IL-10 or HSP110) is associated with uncontrolled intestinal inflammation, thus highlighting a critical role of this pathway in the control of intestinal inflammation. Conv., conventional. b, Overview of experimental procedures.
Extended Data Figure 2. Absence of enterocyte lipid accumulation and ER stress in MttpΔIEC mice.

a, Absence of lipid accumulation in MttpΔIEC mice as shown in representative haematoxylin and eosin and rare osmium tetroxide (arrows) staining in the caecum, proximal, and distal colon. Scale bar, 25 μm. b, Xbp1 splicing in small intestinal (SI) and colonic IECs of the indicated mice before (left) (n = 5 mice per group) and 6 h after rectal challenge with oxazolone (Ox., right) (n =5 mice per group). MODE-K cells were treated with either thapsigargin (Tg) or vehicle (DMSO) as positive and negative controls, respectively. Results representative of three independent experiments are shown.
Extended Data Figure 3. Impaired CD1d- but not MHC-class I-restricted antigen presentation in MttpΔIEC mice.

a, CD1d-mediated presentation of the exogenous lipid antigen α-GalCer to the invariant NKT-cell hybridomas 24.7 and DN32.D3 and of endogenous lipid antigens (autoreactivity) to the non-invariant NKT-cell hybridoma 14S.6 by IECs from CD1d-knockout (CD1d-KO) mice, MttpΔIEC mice and wild-type littermates (Mttp+/+) (n = 5 mice per group). b, Presentation of H2-Kb-restricted SIINFEKL by the indicated IECs (see a) to the SIINFEKL-responsive hybridoma RF33.70. c, Representative histograms of CD1d cell surface expression as determined by flow cytometry of colonic IECs of the indicated mouse strains before (left, n = 5 mice per group) and 6 h after (right, n = 5 mice per group) rectal oxazolone challenge. d, Representative haematoxylin and eosin stainings of the indicated mouse strains upon rectal challenge with oxazolone (Ox.) or vehicle (ethanol (EtOH)). Scale bar, 40 μm. Results representative of three independent experiments are shown. Mean ±s.e.m. of triplicate cultures are shown. Student’s t-test was applied.
Extended Data Figure 4. Increased morbidity and mortality in oxazolone-challenged MttpΔIEC mice is due to CD1d-restricted components of the adaptive immune system.

a, Intestinal permeability as determined by FD-4 before and 18 h after rectal challenge with oxazolone (ox.) in the indicated mouse strains. Each symbol represents a single mouse. b, IL-1β secretion by CD11b+ cells from colonic lamina propria of the indicated bone marrow chimaeras. CD11b+cells were isolated using magnetic microbeads 24 h after rectal challenge with oxazolone or ethanol and cultured for 24 h before measurement of IL-1β in culture supernatants by ELISA. Mean ±s.e.m. of triplicate cultures are shown. c, d, Survival and body weight of the indicated mouse strains at the indicated days after rectal oxazolone challenge. Results representative of three independent experiments are shown. Mean ±s.e.m. of the indicated number of mice are shown. Student’s t-test was applied.
Extended Data Figure 5. Development and characterization of mice with IEC-specific Cd1d1 deletion.

a, Schematic map of the targeting strategy for generation of Cd1d1fl/fl mice. A LoxP (L83) site was inserted at 10310 and a FNFL (Frt-Neo-Frt-LoxP) cassette at 12190 to flank exons 2, 3, and 4 (about 1.9 kb) of the Cd1d1 gene to generate the ‘floxed/neo’ Cd1d1 allele. A gene-targeting vector was constructed by retrieving one 5 kb long homology arm (5′ to L83), one 1.9 kb sequence containing exon 2/3/4, FNFL cassette, and one 2 kb short homology arm (end of FNFL to 3′). The FNFL cassette conferred G418 resistance during gene targeting in PTL1 (129B6 hybrid) embryonic stem (ES) cells. The targeted Cd1d1 allele was PCR amplified and sequenced to confirm the targeted C57BL/6 allele based on the C57BL/6-specific mutation in the Cd1d2 allele. Three targeted ES cells with targeted C57BL/6 allele were injected into C57BL/6 blastocysts to generate chimaeric mice. Male chimaeras were bred to bACTFlpe females or EIIa-Cre females to transmit the floxed Cd1d1 allele (Cd1d1L/+) (with neo cassette removed by Fple recombinase) through the germ line. Mice carrying the floxed Cd1d1 allele were crossed to tissue-specific VillinCre-ERT2 and exons 2, 3 and 4 and Frt-LoxP were removed by Cre recombinase in intestinal epithelial cells. b–e, Impaired CD1d- but not MHC-class I-restricted antigen presentation in Cd1d1ΔIEC mice. b, Cd1d1ΔIEC mice exhibit normal numbers of invariant NKT cells in liver, spleen and colonic lamina propria as determined by flow cytometry using α-GalCer/CD1d tetramers (n = 4 mice per group). c, Representative histograms of CD1d cell surface expression on colonic IECs of the indicated mouse strains before (left, n =5 mice per group) and 6 h after (right, n =5 mice per group) rectal oxazolone challenge. d, CD1d-mediated antigen presentation by colonic IECs of the indicated mouse strains (n = 5 mice per group). Presentation of the model glycolipid antigen α-GalCer to invariant NKT-cell hybridomas 24.7 and DN32.D3 and of endogenous antigens (autoreactivity) to the non-invariant NKT-cell hybridoma 14S.6 is shown. e, Presentation of H2-Kb-restricted SIINFEKL presentation by the indicated IECs (n = 5 mice per group) to the SIINFEKL-responsive hybridoma RF33.70. Results representative of three independent experiments are shown b–e, Mean ±s.e.m. of triplicate cultures are shown. Student’s t-test was applied. f, Intestinal epithelial CD1d is protective in oxazolone colitis. Representative macroscopic colon images of Cd1d1ΔIEC and wild-type littermates upon rectal challenge with oxazolone (Ox., n =5 mice per group) or vehicle (ethanol (EtOH), n =4 mice per group).
Extended Data Figure 6. Epithelial HSP110 expression is decreased in MttpΔIEC mice and in human ulcerative colitis.

a, HSP110 immunohistochemistry in mice with IEC Mttp deletion (bottom) as compared with wild-type littermates (top) 6 h after rectal oxazolone challenge. b, HSP110 immunohistochemistry in the colonic intestinal epithelium and the lamina propria of healthy controls and patients with active and inactive ulcerative colitis. Signal intensity was scored on a scale from 0 to 3 in a blinded fashion. Each symbol represents a single patient. The median and significance level as determined by the Mann–Whitney U-test are shown. c, Mortality in conventional HSP110-knockout mice in the oxazolone colitis model. Results representative of three independent experiments are shown. Mean ±s.e.m. of the indicated number of mice is shown. Student’s t-test and the log-rank test (survival) were applied. NS, not significant.
Extended Data Figure 7. Purity of isolated IECs.

After isolation of colonic IECs, potential contamination with haematopoetic cells was investigated by flow cytometry. The major population of cells was gated (upper left), was shown to contain viable cells (upper middle), and to contain largely EpCAM-positive epithelial cells that do not stain with leukocyte markers (middle and bottom). The upper right panel shows isotype control staining. Results representative of three independent experiments (n = 5 mice per experiment) are shown.
Extended Data Figure 8. CD1d-dependent STAT3 phosphorylation upon co-culture of IECs and iNKT cells.
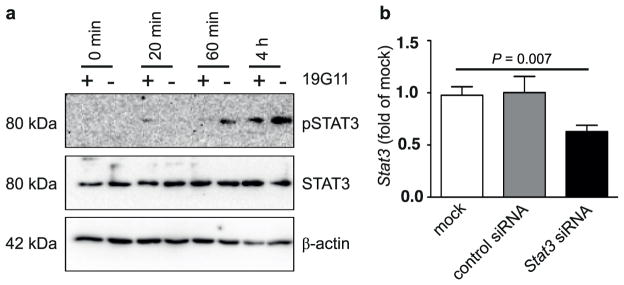
a, STAT3 phosphorylation in IECs upon co-culture with iNKT cells is CD1d-dependent. Expression of pSTAT3, STAT3 and β-actin as determined by western blotting of MODE-K cells at the indicated time after addition of the iNKT-cell hybridoma 24.7. Where indicated, MODE-K cells were pre-incubated with a monoclonal blocking antibody directed against CD1d (19G11; +) or an isotype control (−). b, siRNA-mediated knockdown of Stat3 in MODE-K cells as determined 68 h after siRNA transfection. Results representative of three independent experiments are shown. Means ±s.e.m. of triplicates are shown. Student’s t-test was applied.
Extended Data Figure 9. CD1d-mediated cytokine production is dependent on the APC and expression of co-stimulatory molecules.

a–f, IL-10 (a, c–f) and IL-12p70 (b) secretion of co-cultures of the IEC line MODE-K or splenic dendritic cells (DCs) together with the iNKT-cell hybridoma 24.7. APCs were loaded with α-GalCer (αGC; 100 ng ml−1) before washing and co-culture with iNKT cells. c, MODE-K cells (IEC) were transfected with control siRNA or siRNA directed against Il10 before co-culture with iNKT cells. d, f, MODE-K cells were transfected with CD40 (d) and CD1d (f) as indicated. Histograms demonstrated increased expression of CD40 (d) and CD1d (f) after transfection. e, Stimulatory anti-CD28 antibody was added during co-culture of MODE-K and iNKT cells. Means ±s.e.m. of quadruplicate cultures are shown. Student’s t-test was applied. Results are representative of two independent experiments.
Extended Data Table 1.
Genes downregulated in expression in oxazolone-challenged MttpΔIEC mice
| Fold Change* | GeneID | Description |
|---|---|---|
| 8.721 | Ifi202b | interferon activated gene 202B |
| 3.878 | Eya3 | eyes absent 3 homolog (Drosophila) |
| 3.7 | Sipa1l1 | signal-induced proliferation-associated 1 like 1 |
| 3.661 | Gna14 | AV230778 RIKEN full-length enriched, 0 day neonate skin Mus musculus cDNA clone 4632401H08 3′, mRNA sequence. |
| 3.534 | Cd79b | CD79B antigen |
| 3.347 | Mapk14 | mitogen activated protein kinase 14 |
| 3.347 | Aasdhppt | aminoadipate-semialdehyde dehydrogenase-phosphopantetheinyl transferase |
| 3.259 | Marcks | myristoylated alanine rich protein kinase C substrate |
| 3.216 | Tgtp | T-cell specific GTPase |
| 3.216 | Rbmx | RNA binding motif protein, X chromosome |
| 3.178 | Tcl1b3 | T-cell leukemia/lymphoma 1B, 3 |
| 3.162 | Clu | clusterin |
| 3.134 | Indo | indoleamine-pyrrole 2,3 dioxygenase |
| 3.12 | Ncl | uy94h11.x1 NCI_CGAP_Mam5 Mus musculus cDNA clone IMAGE:3667269 3′ similar to SW:NUCL_MOUSE P09405 NUCLEOLIN ; mRNA sequence. |
| 3.101 | Hspa4 | heat shock protein 4 |
| 3.068 | Gbp2 | guanylate nucleotide binding protein 2 |
| 3.057 | Ncbp2 | nuclear cap binding protein subunit 2 |
| 3.034 | Mcm5 | minichromosome maintenance deficient 5, cell division cycle 46 (S. cerevisiae) |
| 3.024 | Hsph1 | heat shock protein 110 |
Fold change: Mttp+/+ / Mttp IEC
Acknowledgments
The authors thank H.-C. Hung for technical assistance with microinjection, Y. Xie for performing osmium staining, A. Bedynek and M. Friedrich for performing immunohistochemistry of the human biopsies, F. A. Zhu for assistance with antigen presentation assays, D. Shouval, M. Sablon and D. Perez for animal care and husbandry, K. Tashiro for technical assistance with adenovirus preparation, V. M. Thiele for technical assistance, J. Cusick for help with manuscript preparation, and S. E. Plevy for discussions and reagents. This work was supported by: National Institutes of Health (NIH) (grants DK044319, DK051362, DK053056, DK088199) and the Harvard Digestive Diseases Center (DK0034854) (R.S.B.); the European Research Council (ERC Starting Grant agreement no. 336528), the Deutsche Forschungsgemeinschaft (DFG) (ZE 814/4-1, ZE 814/5-1, ZE 814/6-1), the Crohn’s and Colitis Foundation of America (Postdoctoral Fellowship Award), the European Commission (Marie Curie International Reintegration Grant no. 256363) and the DFG Excellence Cluster “Inflammation at Interfaces” (S.Z.); the DFG (OL 324/1-1) (T.O.); HL38180, DK56260, Washington University DDRCC P30DK52574 (morphology core) (N.O.D.); HDDC Pilot and Feasibility Grant (K.B.); NCI P30CA013696 (C.-S.L.), the DFG (BR 1912/6-1) and the Else Kroener-Fresenius-Stiftung (Else Kroener-Exzellenzstipendium 2010_EKES.32) (S.B.); Grant-in-Aid for Challenging Exploratory Research 24659823 from Japan Society for Promotion of Science (K.W.); the ERC under the European Community’s Seventh Framework Programme (FP7/2007-2013/ERC Grant agreement no. 260961), the National Institute for Health Research Cambridge Biomedical Research Centre, the Austrian Science Fund and Ministry of Science P21530-B18 and START Y446-B18, Innsbruck Medical University (MFI 2007-407) and the Addenbrooke’s Charitable Trust, CiCRA (A.K.); the European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement SysmedIBD (no. 305564) (W.M., S.S.); the NIH (grants HL59561, DK034854, AI50950), the Helmsley Charitable Trust and the Wolpow Family Chair in IBD Treatment and Research (S.B.S.). PBS57-loaded and unloaded mouse CD1d tetramer was obtained through the NIH Tetramer Facility. The authors thank M. A. Exley and S. P. Colgan for discussions.
Footnotes
Online Content
Any additional Methods, Extended Data display items and Source Data are available in the online version of the paper; references unique to these sections appear only in the online paper.
Supplementary Information is available in the online version of the paper.
Author Contributions T.O., J.F.N., C.M.D. and K.B. performed in vitro and in vivo experiments and analysed the results; N.O.D. performed osmium tetroxide staining; J.G. obtained and scored histopathologies; C.-S.L. generated Cd1d1fl/fl mice; C.J. contributed to the analysis of CD1dΔIEC mice; S.B. and K.S. contributed to the immunohistochemical analysis of ulcerative colitis patients; K.W., K. Katayama, A.N. and H.M. generated adenoviruses; K. Kawasaki and K.N. provided HSP110-KO mice; W.M. and S.B.S. provided and participated in the analysis of the Il10ΔIEC mice; S.S. contributed to the coordination of experimental studies; A.K. contributed to MttpΔIEC studies and to the analysis of microarray data; R.S.B. and S.Z. designed the study, coordinated the experimental work and wrote the manuscript with input from co-authors. All authors discussed the results and commented on the manuscript.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
References
- 1.Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saenz SA, Taylor BC, Artis D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol Rev. 2008;226:172–190. doi: 10.1111/j.1600-065X.2008.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fuss IJ, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113:1490–1497. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heller F, Fuss IJ, Nieuwenhuis EE, Blumberg RS, Strober W. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity. 2002;17:629–638. doi: 10.1016/s1074-7613(02)00453-3. [DOI] [PubMed] [Google Scholar]
- 5.Olszak T, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336:489–493. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wingender G, Kronenberg M. Role of NKT cells in the digestive system. IV The role of canonical natural killer T cells in mucosal immunity and inflammation. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1–G8. doi: 10.1152/ajpgi.00437.2007. [DOI] [PubMed] [Google Scholar]
- 7.Heller F, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Liao CM, et al. dysregulation of CD1d-restricted type II natural killer T cells leads to spontaneous development of colitis in mice. Gastroenterology. 2012;142:326–334. doi: 10.1053/j.gastro.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colgan SP, Hershberg RM, Furuta GT, Blumberg RS. Ligation of intestinal epithelial CD1d induces bioactive IL-10: critical role of the cytoplasmic tail in autocrine signaling. Proc Natl Acad Sci USA. 1999;96:13938–13943. doi: 10.1073/pnas.96.24.13938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perera L, et al. Expression of nonclassical class I molecules by intestinal epithelial cells. Inflamm Bowel Dis. 2007;13:298–307. doi: 10.1002/ibd.20026. [DOI] [PubMed] [Google Scholar]
- 11.Blumberg RS, et al. Expression of a nonpolymorphic MHC class I-like molecule, CD1D, by human intestinal epithelial cells. J Immunol. 1991;147:2518–2524. [PubMed] [Google Scholar]
- 12.Brozovic S, et al. CD1d function is regulated by microsomal triglyceride transfer protein. Nature Med. 2004;10:535–539. doi: 10.1038/nm1043. [DOI] [PubMed] [Google Scholar]
- 13.Zeissig S, et al. Primary deficiency of microsomal triglyceride transfer protein in human abetalipoproteinemia is associated with loss of CD1 function. J Clin Invest. 2010;120:2889–2899. doi: 10.1172/JCI42703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El Marjou F, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 15.Raabe M, et al. Analysis of the role of microsomal triglyceride transfer protein in the liver of tissue-specific knockout mice. J Clin Invest. 1999;103:1287–1298. doi: 10.1172/JCI6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Colgan SP, et al. Intestinal heat shock protein 110 regulates expression of CD1d on intestinal epithelial cells. J Clin Invest. 2003;112:745–754. doi: 10.1172/JCI17241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiu YH, et al. Multiple defects in antigen presentation and T cell development by mice expressing cytoplasmic tail-truncated CD1d. Nature Immunol. 2002;3:55–60. doi: 10.1038/ni740. [DOI] [PubMed] [Google Scholar]
- 18.Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nature Rev Immunol. 2010;10:170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- 19.Pickert G, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamagishi N, Fujii H, Saito Y, Hatayama T. Hsp105β upregulates hsp70 gene expression through signal transducer and activator of transcription-3. FEBS J. 2009;276:5870–5880. doi: 10.1111/j.1742-4658.2009.07311.x. [DOI] [PubMed] [Google Scholar]
- 21.Chaudhry A, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity. 2011;34:566–578. doi: 10.1016/j.immuni.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubtsov YP, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 23.Nakamura J, et al. Targeted disruption of Hsp110/105 gene protects against ischemic stress. Stroke. 2008;39:2853–2859. doi: 10.1161/STROKEAHA.107.506188. [DOI] [PubMed] [Google Scholar]
- 24.Smiley ST, Kaplan MH, Grusby MJ. Immunoglobulin E production in the absence of interleukin-4-secreting CD1-dependent cells. Science. 1997;275:977–979. doi: 10.1126/science.275.5302.977. [DOI] [PubMed] [Google Scholar]
- 25.Kühn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 26.Roers A, et al. T cell-specific inactivation of the interleukin 10 gene in mice results in enhanced T cell responses but normal innate responses to lipopolysaccharide or skin irritation. J Exp Med. 2004;200:1289–1297. doi: 10.1084/jem.20041789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mombaerts P, et al. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 28.Park SH, Roark JH, Bendelac A. Tissue-specific recognition of mouse CD1 molecules. J Immunol. 1998;160:3128–3134. [PubMed] [Google Scholar]
- 29.Zeissig S, et al. Hepatitis B virus-induced lipid alterations contribute to natural killer T cell-dependent protective immunity. Nature Med. 2012;18:1060–1068. doi: 10.1038/nm.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vidal K, Grosjean I, Revillard JP, Gespach C, Kaiserlian D. Immortalization of mouse intestinal epithelial cells by the SV40-large T gene. Phenotypic and immune characterization of the MODE-K cell line. J Immunol Methods. 1993;166:63–73. doi: 10.1016/0022-1759(93)90329-6. [DOI] [PubMed] [Google Scholar]
- 31.Behar SM, Podrebarac TA, Roy CJ, Wang CR, Brenner MB. Diverse TCRs recognize murine CD1. J Immunol. 1999;162:161–167. [PubMed] [Google Scholar]
- 32.Yue SC, Shaulov A, Wang R, Balk SP, Exley MA. CD1d ligation on human monocytes directly signals rapid NF-κB activation and production of bioactive IL-12. Proc Natl Acad Sci USA. 2005;102:11811–11816. doi: 10.1073/pnas.0503366102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katayama K, et al. A novel PPARγ gene therapy to control inflammation associated with inflammatory bowel disease in a murine model. Gastroenterology. 2003;124:1315–1324. doi: 10.1016/s0016-5085(03)00262-2. [DOI] [PubMed] [Google Scholar]
- 34.Mizuguchi H, Kay MA. A simple method for constructing E1- and E1/E4-deleted recombinant adenoviral vectors. Hum Gene Ther. 1999;10:2013–2017. doi: 10.1089/10430349950017374. [DOI] [PubMed] [Google Scholar]
- 35.Chen Z, Chen L, Qiao SW, Nagaishi T, Blumberg RS. Carcinoembryonic antigen-related cell adhesion molecule 1 inhibits proximal TCR signaling by targeting ZAP-70. J Immunol. 2008;180:6085–6093. doi: 10.4049/jimmunol.180.9.6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaser A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Furuta GT, et al. Eosinophils alter colonic epithelial barrier function: role for major basic protein. Am J Physiol Gastrointest Liver Physiol. 2005;289:G890–G897. doi: 10.1152/ajpgi.00015.2005. [DOI] [PubMed] [Google Scholar]
- 38.Furuta GT, et al. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med. 2001;193:1027–1034. doi: 10.1084/jem.193.9.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]