

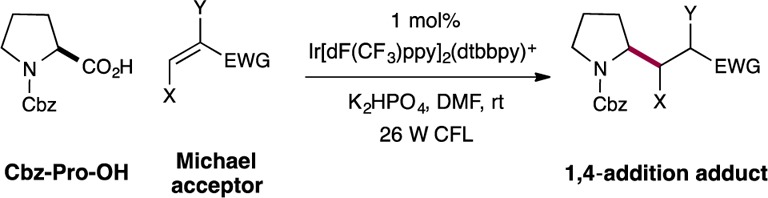
Abstract
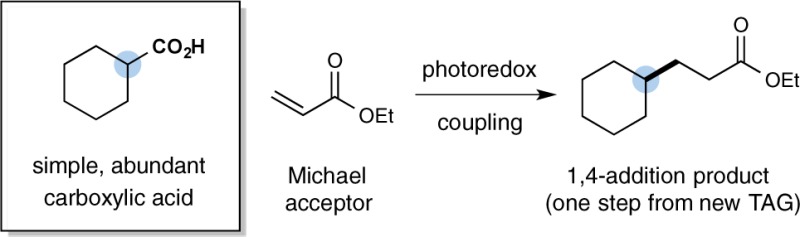
The direct application of carboxylic acids as a traceless activation group for radical Michael additions has been accomplished via visible light-mediated photoredox catalysis. Photon-induced oxidation of a broad series of carboxylic acids, including hydrocarbon-substituted, α-oxy, and α-amino acids, provides a versatile CO2-extrusion platform to generate Michael donors without the requirement for organometallic activation or propagation. A diverse array of Michael acceptors is amenable to this new conjugate addition strategy. An application of this technology to a three-step synthesis of the medicinal agent pregabalin (commercialized by Pfizer under the trade name Lyrica) is also presented.
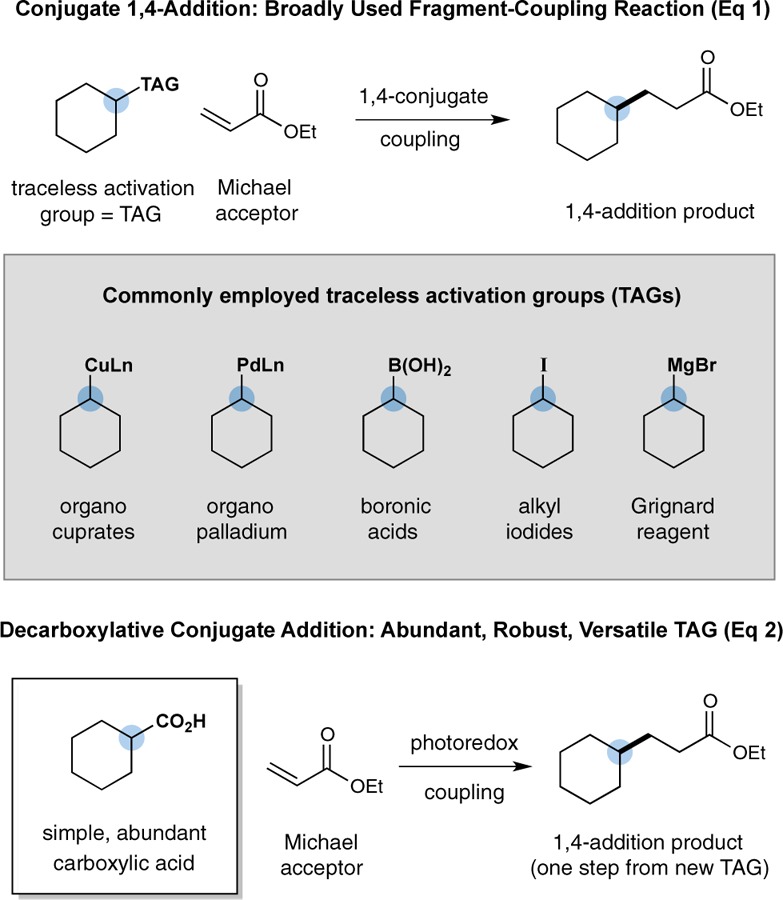
Since the discovery of the Michael reaction in 1887,1 1,4-conjugate additions have become a central bond construction within the field of organic synthesis.2 This broadly employed fragment-coupling mechanism is founded upon the use of a series of electrophilic olefins (now referred to as Michael acceptors) that combine with nucleophiles3 or SOMO-activated partners4 in a regioselective 1,4-addition pathway (eq 1). Over the past four decades, significant advances have been made in the development of traceless activation groups (TAGs) such as cuprates, boronic acids, halides, and Grignard reagents that enable a broad range of organic structures to participate as nucleophiles or donor components in this 1,4-coupling pathway.2,3 Expanding upon this theme, we recently questioned whether simple and abundant carboxylic acids might be generically5 employed as a traceless activation group for radical 1,4-conjugate additions via the use of a photoredox-mediated CO2-extrusion mechanism.6,7 Herein, we describe the successful execution of these ideals and present a new, decarboxylative 1,4-addition reaction that enables a broad array of organic molecules to participate as Michael donors without the need for organometallic activation or propagation (eq 2). We envision that the use of carboxylic acids as a generic TAG for Michael chemistry will (i) reduce operational complexity, (ii) greatly expand the scope, and (iii) lower the costs associated with performing 1,4-conjugate addition reactions.
 |
1 |
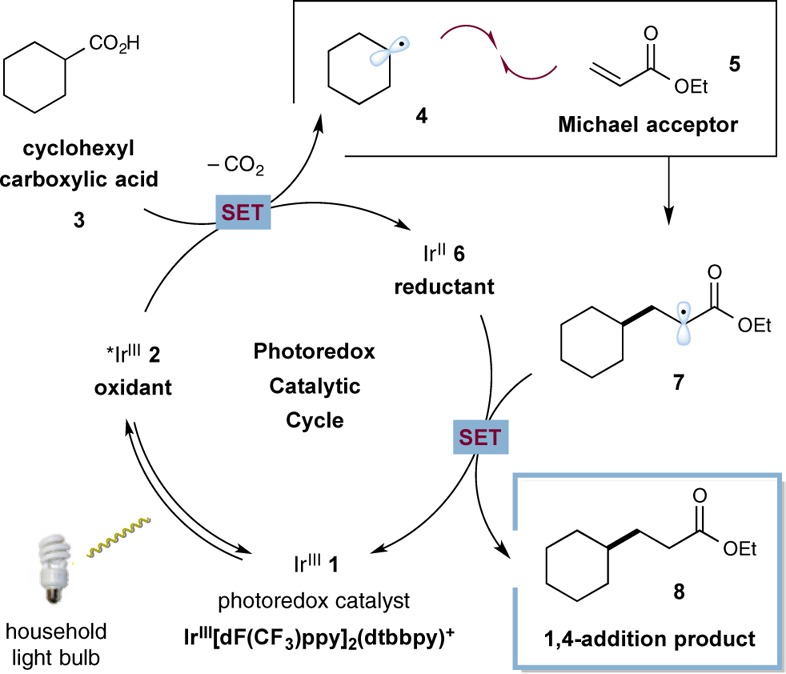
Recently, our laboratory introduced a new protocol for the direct construction of benzylic amines from α-amino acids using a previously unknown photoredox-catalyzed decarboxylation–aryl coupling mechanism.8 A critical feature of this new Csp3-arylation reaction is the capacity for aliphatic carboxylic acids to undergo CO2-extrusion under mild conditions (room temperature, household light bulb) to generate primary, secondary, and tertiary radicals, which subsequently participate in hetero radical–radical couplings. Seeking to take advantage of this new oxidative decarboxylation pathway, we recognized that simple carboxylic acids should therefore be suitable and generic precursors for radical conjugate addition reactions under similar photoredox conditions (given the long established success of SOMO-activated fragments in Michael additions).4 As described more specifically in Scheme 1, it is well-known that photoredox catalysts, such as Ir[dF(CF3)ppy]2(dtbbpy)+1, can readily accept photons from visible light to generate the strongly oxidizing excited state *Ir[dF(CF3)ppy]2(dtbbpy)+2, (E1/2*III/II = +1.21 V vs saturated calomel electrode (SCE) in CH3CN).9 We assumed that base-promoted deprotonation of a carboxylic acid substrate (cyclohexyl carboxylic acid (3) is depicted) and subsequent single-electron transfer (SET) oxidation of the resulting carboxylate functionality (hexanoate ion, E1/2 = +1.16 V vs SCE)10 by the visible-light-excited photocatalyst *Ir[dF(CF3)ppy]2(dtbbpy)+2 should be thermodynamically favorable, to generate the reduced Ir[dF(CF3)ppy]2(dtbbpy) 6 and the carboxyl radical species which would immediately extrude CO2 to give the SOMO species 4. We anticipated that the alkyl radical 4 would be highly nucleophilic and thereby readily undergo conjugate addition with electron-deficient olefin 5 to forge a new C–C bond with concomitant formation of the alkyl radical 7.4 Facile SET reduction of the α-acyl radical 7 (E1/2red = −0.60 V vs SCE)11 by the strongly reducing Ir[dF(CF3)ppy]2(dtbbpy) complex 6 (E1/2 = −1.37 V vs SCE)9 would deliver the desired 1,4-conjugate addition product (upon enolate protonation), while simultaneously regenerating photocatalyst 1.
Scheme 1. Proposed Mechanism for Decarboxylative Alkylation.
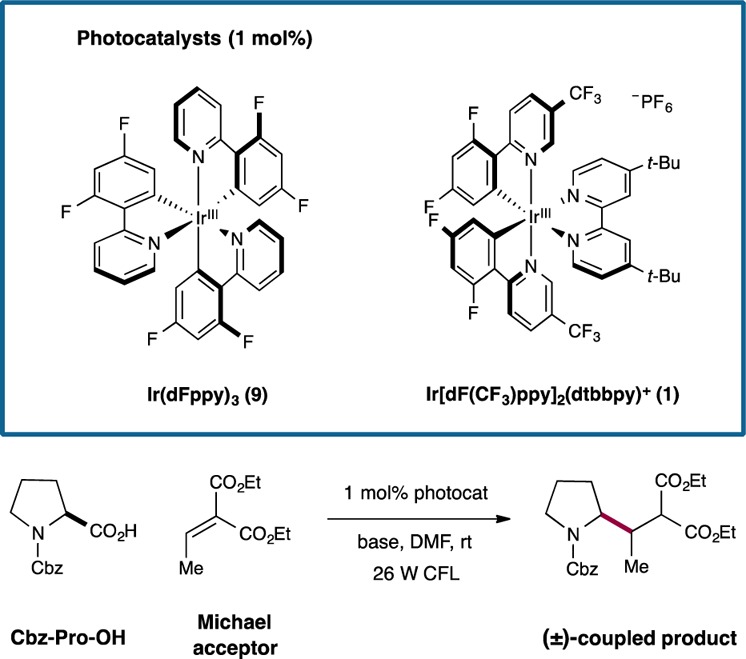
Evaluation of this CO2-extrusion/conjugate addition strategy was first examined with Cbz-protected proline, diethyl ethylidenemalonate as the Michael acceptor, and a series of Ir-based photocatalysts in the presence of CsF (base) and a household 26 W fluorescent light bulb (Table 1). We were delighted to find that the proposed decarboxylative alkylation was indeed feasible in the presence of photocatalysts that have readily reduced excited states, such as Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1) (entry 1, 67% yield); however, less oxidizing systems (Ir(dFppy)3 and Ir(ppy)2(dtbbpy)PF6) resulted in dramatic decreases in efficiency (entries 2 and 3). A survey of inorganic bases revealed that K2HPO4 is the preferred carboxylic acid deprotonation system, providing the desired pyrrolidine Michael adduct in the highest yield (entry 9, 92% yield). It is important to note that control experiments have demonstrated that both photocatalyst and visible light are essential for the desired transformation to occur (entries 4 and 5).
Table 1. Decarboxylative Conjugate Addition: Catalyst Evaluation.
| entry | photocatalyst | base | yield (%)a |
|---|---|---|---|
| 1 | Ir[dF(CF3)ppy]2(dtbbpy)+ | CsF | 67 |
| 2 | Ir(dFppy)3 | CsF | <5 |
| 3 | Ir(ppy)2(dtbbpy)+ | CsF | 10 |
| 4 | none | CsF | 0 |
| 5b | Ir[dF(CF3)ppy]2(dtbbpy)+ | CsF | 0 |
| 6 | Ir[dF(CF3)ppy]2(dtbbpy)+ | CsOAc | 73 |
| 7 | Ir[dF(CF3)ppy]2(dtbbpy)+ | Cs2CO3 | 65 |
| 8 | Ir[dF(CF3)ppy]2(dtbbpy)+ | K2CO3 | 31 |
| 9 | Ir[dF(CF3)ppy]2(dtbbpy)+ | K2HPO4 | 92 |
Isolated yields.
Reaction performed in the absence of visible light. Abbreviations: ppy, 2-phenylpyridyl; dtbbpy, 4,4′-di-tert-butyl-2,2′-bipyridyl; Cbz, benzyl carbamoyl; CFL, compact fluorescent light.
With optimal reaction conditions in hand, we next evaluated the scope of Michael acceptors that can participate in this new CO2-extrusion, conjugate addition protocol. As revealed in Table 2, these mild reaction conditions are compatible with a wide range of functional groups, such as esters, bromides, ketones, aldehydes, sulfones, imides, and carboxylic acids, providing a versatile platform for further synthetic manipulations. Unsaturated ketones are well tolerated in both cyclic and acyclic form (products 10 and 11, 81–88% yield). Moreover, acyclic enals undergo the desired radical conjugate addition with excellent efficiency (product 12, 92% yield). The resulting aldehyde adducts are versatile precursors to a wide range of bicyclic systems including pyrrolizidine12 and indolizidine13 alkaloids, which are large classes of natural products showing a variety of interesting biological activities (see Supporting Information (SI) for a demonstration of a pyrrolizidine alkaloid synthesis). Moreover, this protocol could be further applied to other electrophilic olefins, including α,β-unsaturated imides, sulfones, and malonates, as well as acrylates and maleates, to furnish a variety of alkylated amines in good to excellent yields (products 14–24, 69–93% yield). In terms of the scope of acrylate coupling partners, unsubstituted as well as α-aryl and α-alkyl groups are well tolerated (products 17–22, 69–90% yield). Interestingly, variation of the electronic nature of the aromatic ring at the α-position of the acrylates has little influence on the reaction efficiency (products 19–22, 76–90% yield). With respect to the alkylidene malonate substrates, linear and sterically demanding substituents (R = CH2CH2Ph, cyclohexyl) can be attached at the β-olefin position without loss in overall yield (products 23 and 24, 87–92% yield). Intriguingly, this new protocol provides a strategy for direct access to γ-amino acids from naturally occurring α-amino acids. As demonstrated in Table 2, the direct decarboxylative coupling of Cbz-Pro-OH with 2-benzylacrylic acid provides the corresponding γ-amino acid with useful reaction efficiency (product 13, 57% yield).
Table 2. Decarboxylative Addition: Michael Acceptor Scopea.
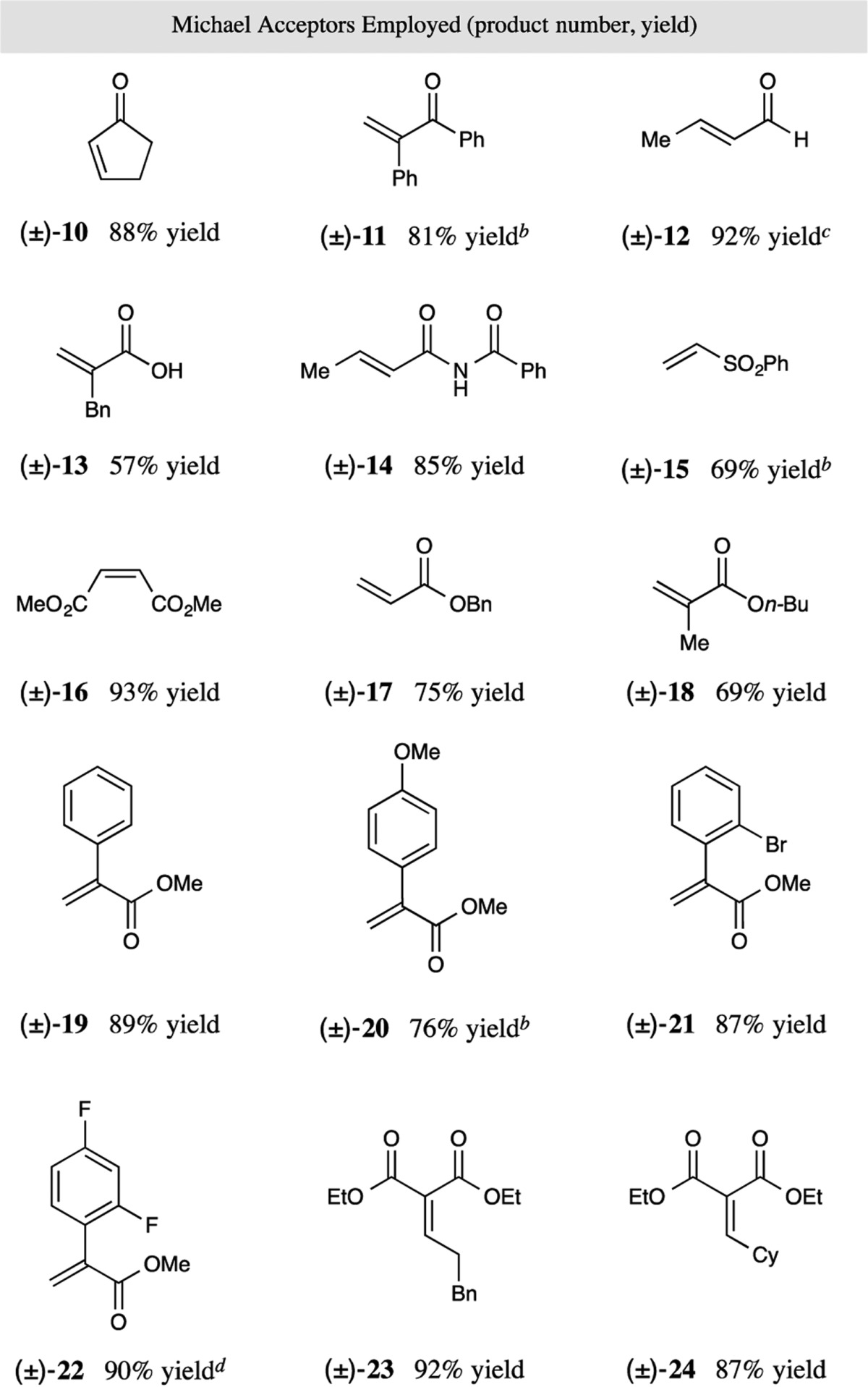
Reaction performed using the optimized conditions from Table 1 (see SI). All cited yields are isolated. Ratios of diastereoisomers determined by 1H NMR analysis are between 1 and 1.5:1.
Performed with 1.2 equiv of CsF instead of K2HPO4.
Performed on a 1.0 mmol scale.
Performed with 1.5 equiv of Cbz-Pro-OH.
We next turned our attention to the scope of carboxylic acids that can participate in this new conjugate addition protocol. As revealed in Table 3, a variety of cyclic (e.g., cyclohexyl, cyclopentyl, and cyclobutyl) and acyclic (e.g., 2-hexyldecanoic acid) hydrocarbon radical precursors can be readily employed with good levels of efficiency (products 27–30, 53–97% yield). Moreover, this transformation appears to be tolerant of a broad range of substituents at the carboxylic α-position. Indeed a series of cyclic systems that incorporate α-oxy groups (e.g., tetrahydro-2H-pyran-2-carboxylic and 2-tetrahydrofuranoic acid), provide the corresponding conjugate adduct in high yield (products 25 and 26, ≥90% yield). Importantly, a variety of natural α-amino acids (e.g., Boc-protected Phe, Pro, Trp, Ser, Gly) readily participate in this decarboxylative conjugate addition to generate α-alkylated amines in a highly efficient fashion (products 31, 35, 36, 40, and 44, 57–97% yield). Notably, α-amino acids bearing an unprotected amide or indole ring were also tolerated under these standard conditions (products 32 and 36, 57–84% yield). Not surprisingly, this decarboxylative protocol is also amenable to the use of unnatural α-amino acids such as N-Boc-2-piperidinecarboxylic acid and N-Boc-morpholine-3-carboxylic acid (products 42 and 43, 94–95% yield). Perhaps most remarkably, we have found that the amino acid component can be successfully extended to dipeptides, providing the corresponding coupled products in excellent yields (products 37 and 38, 88–90% yield). It is important to note that this highly efficient conjugate addition strategy employs a 1:1 ratio of carboxylic acid and electron-deficient olefin at room temperature, without the need for stoichiometric oxidants and/or forcing reaction conditions.
Table 3. Decarboxylative Conjugate Addition: Scope of Carboxylic Acid Fragmenta.
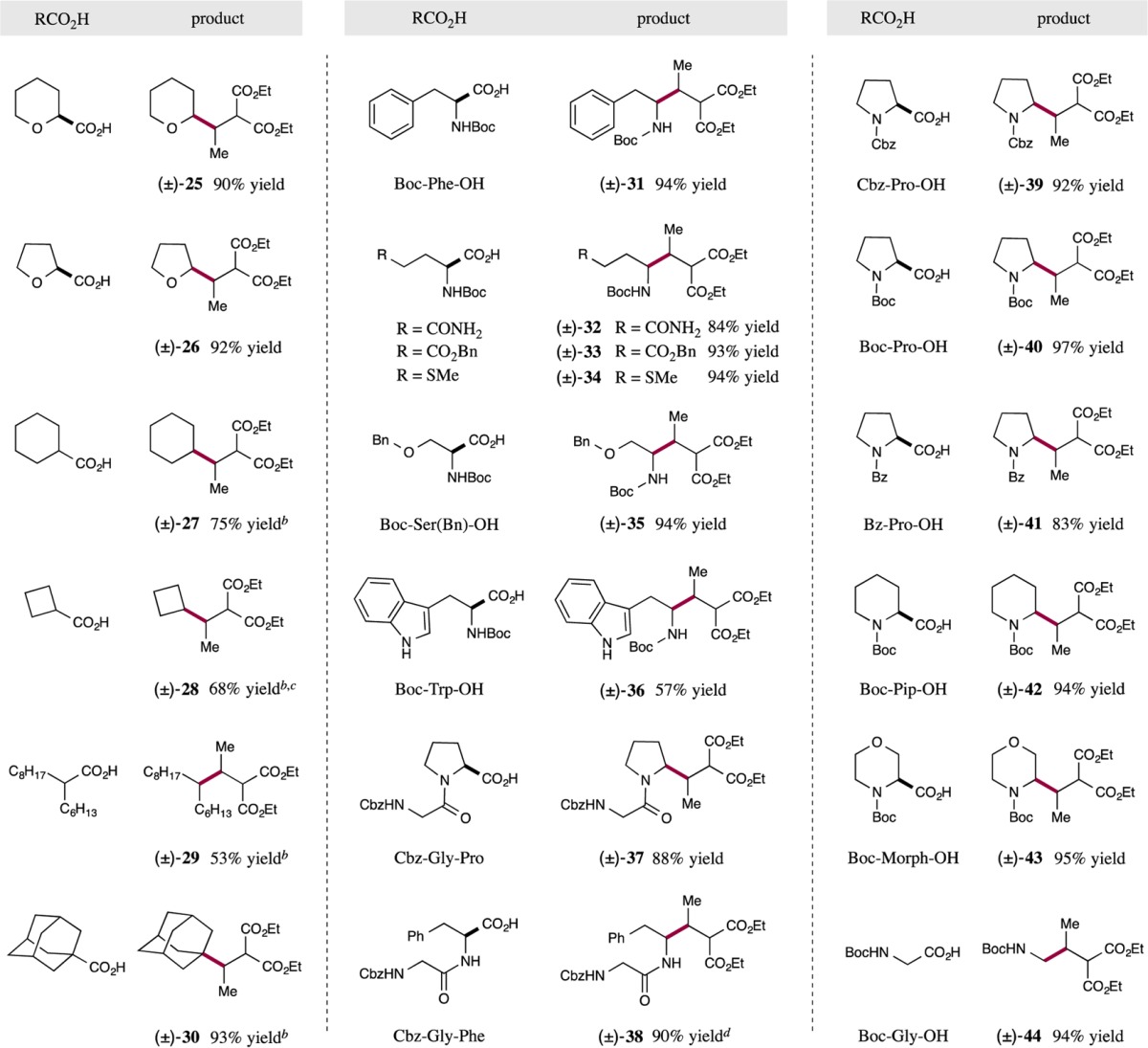
Reaction performed using the optimized conditions from Table 1 (see SI). The cited yields are isolated. Ratios of diastereoisomers determined by 1H NMR analysis are between 1 and 1.5:1.
Reaction performed using 34 W blue LED, instead of 26 W fluorescent light.
Cyclopentanecarboxylic acid and octanoic acid also worked under the reaction conditions, affording the corresponding products in ≥58% yield with 1,4-dioxane as a solvent.
Reaction concentration is 0.2 M.
To further demonstrate the operational simplicity and generality of this new Michael addition protocol, we present a three-step racemic synthesis of pregabalin, an anticonvulsant drug that has been commercialized by Pfizer under the trade name Lyrica (eq 3).14
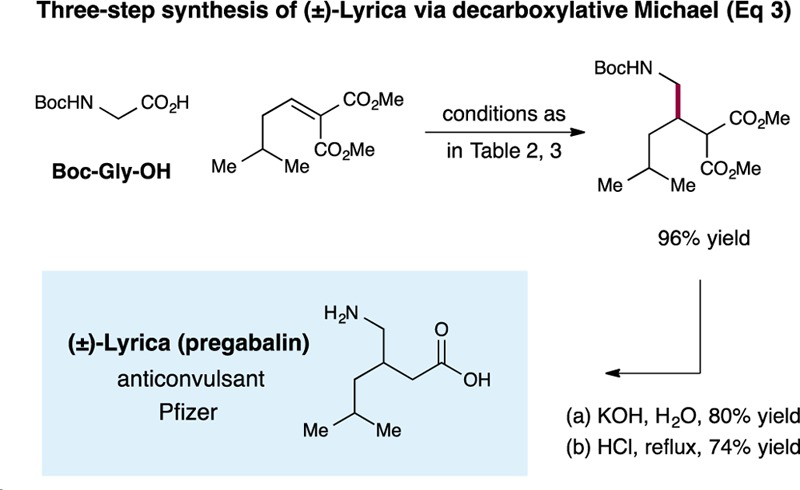 |
3 |
At the present time, pregabalin is produced on scale via a process that involves a cyanide conjugate addition reaction. As revealed in eq 3, exposure of Boc-protected glycine and 3-methylbutylidene malonate to our decarboxylative alkylation conditions provided the corresponding malonate with excellent efficiency (96% yield). Hydrolysis under basic conditions followed by treatment with acid promoted decarboxylation to afford racemic pregabalin in only three steps.
In conclusion, we have demonstrated the utility of carboxylic acids as a traceless activation group for radical conjugate addition via visible light-mediated photoredox catalysis. The versatile method tolerates a wide range of functional groups and shows broad scope with regard to both the carboxylic acid and Michael acceptor components. More importantly, this new process provides an alternative to generating Michael donors without the requirement of organometallic activation or propagation.
Acknowledgments
Financial support was provided by the NIH General Medical Sciences (NIHGMS Grant R01 GM103558-03) and gifts from Merck and Amgen. Z.Z. and L.C. are grateful for postdoctoral fellowships from the Shanghai Institute of Organic Chemistry.
Supporting Information Available
Experimental procedures and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Michael A. J. Prakt. Chem. 1887, 35, 349. [Google Scholar]; b Michael A. J. Prakt. Chem. 1894, 49, 20. [Google Scholar]
- For selected reviews on 1,4-conjugate additions, see:; a Perlmutter P.Conjugate Addition Reactions in Organic Synthesis; Pergamon: Oxford, 1992. [Google Scholar]; b Schmalz H.-G. In Comprehensive Organic Synthesis; Trost B. M., Fleming I., Eds.; Pergamon Press: Oxford, 1991; Vol. 4, Chapter 1.5. [Google Scholar]
- For selected reviews on nucleophilic conjugate additions, see:; a Krause N.; Gerold A. Angew. Chem., Int. Ed. 1997, 36, 186. [Google Scholar]; b Rossiter B. E.; Swingle N. M. Chem. Rev. 1992, 92, 771. [Google Scholar]; c Leonard J.; Diez-Barra E.; Merino S. Eur. J. Org. Chem. 1998, 2051. [Google Scholar]; d Sibi M. P.; Manyem S. Tetrahedron 2000, 56, 8033. [Google Scholar]; e Vicario J. L.; Badia D.; Carrillo L. Synthesis 2007, 2065. [Google Scholar]; f Alexakis A.; Bäckvall J. E.; Krause N.; Pàmies O.; Diéguez M. Chem. Rev. 2008, 108, 2796. [DOI] [PubMed] [Google Scholar]; g Harutyunyan S. R.; den Hartog T.; Geurts K.; Minnaard A. J.; Feringa B. L. Chem. Rev. 2008, 108, 2824. [DOI] [PubMed] [Google Scholar]
- For selected reviews on radical conjugate additions, see:; a Renaud P.; Gerster M. Angew. Chem., Int. Ed. 1998, 37, 2562. [DOI] [PubMed] [Google Scholar]; b Sibi M. P.; Porter N. A. Acc. Chem. Res. 1999, 32, 163. [Google Scholar]; c Sibi M. P.; Manyem S.; Zimmerman J. Chem. Rev. 2003, 103, 3263. [DOI] [PubMed] [Google Scholar]; d Srikanth G. S. C.; Castle S. L. Tetrahedron 2005, 61, 10377. [Google Scholar]
- The use of carboxylic acids as radical precursors for conjugate additions has previously been accomplished in specific cases; however, a generic system has not been developed to our knowledge. See:; a Yoshimi Y.; Masuda M.; Mizunashi T.; Nishikawa K.; Maeda K.; Koshida N.; Itou T.; Morita T.; Hatanaka M. Org. Lett. 2009, 11, 4652. [DOI] [PubMed] [Google Scholar]; b Chen L.; Chao C. S.; Pan Y.; Dong S.; Teo Y. C.; Wang J.; Tan C.-H. Org. Biomol. Chem. 2013, 11, 5922. [DOI] [PubMed] [Google Scholar]; c Miyake Y.; Nakajima K.; Nishibayashi Y. Chem. Commun. 2013, 49, 7854. [DOI] [PubMed] [Google Scholar]
- For specifically tailored carboxylate derivatives that have been developed to enable radical conjugate addition see:; a Barton D. H. R.; Crich D.; Kretzschmar G. Tetrahedron Lett. 1984, 25, 1055. [Google Scholar]; b Barton D. H. R.; Crich D.; Kretzschmar G. J. Chem. Soc., Perkin Trans. 1 1986, 39. [Google Scholar]; c Barton D. H. R.; Sas W. Tetrahedron 1990, 46, 3419. [Google Scholar]; d Ahmad-Junan S. A.; Walkington A. J.; Whiting D. A. J. Chem. Soc., Perkin Trans. 1 1992, 2313. [Google Scholar]; e Barton D. H. R.; Chern C.-Y.; Jaszberenyi J. C. Tetrahedron Lett. 1992, 33, 5013. [Google Scholar]; f Barton D. H. R.; Chern C.-Y.; Jaszberenyi J. C. Tetrahedron 1995, 51, 1867. [Google Scholar]; g Garner P. P.; Cox P. B.; Klippenstein S. J. J. Am. Chem. Soc. 1995, 117, 4183. [Google Scholar]; h Barton D. H. R.; Liu W. Tetrahedron Lett. 1997, 38, 2431. [Google Scholar]; i Zhu X.; Ganesan A. J. Comb. Chem. 1999, 1, 157. [Google Scholar]; j Garner P.; Anderson J. T.; Cox P. B.; Klippenstein S. J.; Leslie R.; Scardovi N. J. Org. Chem. 2002, 67, 6195. [DOI] [PubMed] [Google Scholar]; k Schnermann M. J.; Overman L. E. Angew. Chem., Int. Ed. 2012, 51, 9576. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Lackner G. L.; Quasdorf K. W.; Overman L. E. J. Am. Chem. Soc. 2013, 135, 15342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reviews on decarboxylations, see:; a Johnson R. G.; Ingham R. K. Chem. Rev. 1956, 56, 219. [Google Scholar]; b Vijh A. K.; Conway B. E. Chem. Rev. 1967, 67, 623. [Google Scholar]; c Griesbeck A. G.; Kramer W.; Oelgemoller M. Synlett 1999, 1169. [Google Scholar]; d Gooβen L. J.; Rodriguez N.; Gooβen K. Angew. Chem., Int. Ed. 2008, 47, 3100. [Google Scholar]; e Cornella J.; Larrosa I. Synthesis 2012, 653. [Google Scholar]
- Zuo Z.; MacMillan D. W. C. J. Am. Chem. Soc. 2014, 136, 5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry M. S.; Goldsmith J. I.; Slinker J. D.; Rohl R.; Pascal R. A.; Malliaras G. G.; Bernhard S. Chem. Mater. 2005, 17, 5712. [Google Scholar]
- Galicia M.; Gonzalez F. J. J. Electrochem. Soc. 2002, 149, D46. [Google Scholar]
- Bortolamei N.; Isse A. A.; Gennaro A. Electrochim. Acta 2010, 55, 8312. [Google Scholar]
- a Wrbbel J. T. In The Alkaloids: Chemistry and Pharmacology; Brossi A., Ed.; Academic Press: New York, 1985; Vol. 26, Chapter 7. [Google Scholar]; b Rizk A. F. H.Naturally Occurring Pyrrolizidine Alkaloids; CRC Press: Boston, 1991. [Google Scholar]; c Takahata H.; Momose T. In The Alkaloids; Cordell G. A., Ed.; Academic Press: New York, 1993; Vol. 26, p 327. [Google Scholar]; d Liddell J. R. Nat. Prod. Rep. 1998, 15, 363.9736995 [Google Scholar]; e Kim H.-Y.; Stermitz F. R.; Li J. K.-K.; Coulombe R. A. Jr. Food Chem. Toxicol. 1999, 37, 619. [DOI] [PubMed] [Google Scholar]; f Tepe J. J.; William R. M. J. Am. Chem. Soc. 1999, 121, 2951. [Google Scholar]; g Liddell J. R. Nat. Prod. Rep. 2000, 17, 455. [DOI] [PubMed] [Google Scholar]
- a Howard A. S.; Michael J. P. In The Alkaloids: Chemistry and Pharmacology; Brossi A., Ed.; Academic Press: New York, 1986; Vol. 28, Chapter 3. [Google Scholar]; b Daly J. W.; Spande T. F.; Garraffo H. M. J. Nat. Prod. 2005, 68, 1556. [DOI] [PubMed] [Google Scholar]
- a Martinez C. A.; Hu S.; Dumond Y.; Tao J.; Kelleher P.; Tully L. Org. Process Res. Dev. 2008, 12, 392. [Google Scholar]; b Mujahid M.; Muthukrishnan M. Chirality 2013, 25, 965. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.