

Abstract
The nitrogen mustard Chlorambucil (Chl) generates covalent adducts with double-helical DNA and inhibits cell proliferation. Among these adducts, interstrand cross-links (ICLs) are the most toxic, as they stall replication by generating DNA double strand breaks (DSBs). Conversely, intrastrand cross-links generated by Chl are efficiently repaired by a dedicated Nucleotide Excision Repair (NER) enzyme. We synthesized a novel cross-linking agent that combines Chl with the G-quadruplex (G4) ligand PDS (PDS-Chl). We demonstrated that PDS-Chl alkylates G4 structures at low μM concentrations, without reactivity toward double- or single-stranded DNA. Since intramolecular G4s arise from a single DNA strand, we reasoned that preferential alkylation of such structures might prevent the generation of ICLs, while favoring intrastrand cross-links. We observed that PDS-Chl selectively impairs growth in cells genetically deficient in NER, but did not show any sensitivity to the repair gene BRCA2, involved in double-stranded break repair. Our findings suggest that G4 targeting of this clinically important alkylating agent alters the overall mechanism of action. These insights may inspire new opportunities for intervention in diseases specifically characterized by genetic impairment of NER, such as skin and testicular cancers.
The nitrogen mustard Chlorambucil (Chl; Figure 1) is a drug employed in the treatment of various types of malignancies.1 Chl possess two electrophilic sites and generates covalent adducts with double-helical DNA, especially via reaction with N7 of guanines, either within the same strand (intrastrand) or between the two opposite DNA strands (interstrand).2 Interstrand cross-links (ICLs) are the most toxic adducts generated by Chl, since they block DNA replication and elicit DSB formation leading to growth inhibition in highly proliferating cells, but accompanied by significant toxicity in healthy cells that can cause secondary malignacies.3 Moreover, Chl has poor DNA recognition properties that can cause off target alkylation. Substantial improvements in the molecular recognition properties have been reported by conjugating Chl to the A-T specific minor groove binder Distamycin4 or to highly sequence-specific DNA binding hairpin polyamides.5 The enhanced DNA recognition properties of these compounds enables treatment at lower doses. However, the drug’s mechanism of action is not altered in these analogues, and some of the side effects associated with ICL mediated toxicity persist.
Figure 1.

PDS and Chl structures.
Herein, we describe an investigation of the DNA cross-linking properties and the mechanism of action of a novel Chl analogue in which the alkylator is coupled with the potent G4 ligand pyridostatin6 (PDS, Figure 1).
G4s are four-stranded nucleic acid secondary structures that can exist within guanine-rich DNA and RNA sequences via stacking of hydrogen-bonded G-tetrads.7 G4 nucleic acids are the subject of widespread investigation as targets for small molecule intervention in biological studies.8 We have demonstrated that G4s exist in human cells and can be recognized and trapped by the small molecule ligands PDS and carboxy-PDS.9 Given that G4s are preferentially formed during DNA replication,9a cross-linking of these structures may improve the specificity of alkylating agents toward highly proliferative cancer cells.10 Selective cross-linking of G4s within double-helical DNA can be achieved in vitro by combining G4 ligands with different alkylating agents.11 However, the therapeutic advantages offered by covalent G4 targeting have yet to be explored. We aimed to investigate the cellular responses stimulated by selective G4 alkylation.
To address this aim, we synthesized a novel G4 alkylating agent by combination of Chl with the potent G4 ligand PDS (PDS-Chl). We previously described the synthesis of a PDS analogue bearing an alkyne functional group (1) and demonstrated that such chemical modification and its further chemical functionalization do not impair the G4 recognition properties of the PDS scaffold.12 We took advantage of this to generate a hybrid PDS-Chl by means of copper-catalyzed 1,3-dipolar cycloaddition, reacting (1) with an azido Chl analogue (2) (Scheme 1).
Scheme 1. Synthesis of PDS-Chl.

Conditions: (i) CuSO4·5H2O, sodium ascorbate, H2O/t-BuOH (71%); (ii) CH2Cl2, TFA (99%).
Compound (2) was prepared by chlorination of Chl using thionyl chloride followed by coupling with 2-azidoethanamine (see Supporting Information (SI)). Removal of the Boc groups using TFA followed by HPLC purification afforded PDS-Chl in good yields (Scheme 1).
We investigated the reactivity of PDS-Chl toward different DNA structures using synthetic oligonucleotides fluorescently labeled at both the 5′- and 3′-ends, with 6-FAM and TAMRA, respectively. The tested oligonucleotides included the G4-forming sequences (c-MYC and h-Telo), a double-stranded DNA (ds-DNA) and a mutated c-MYC sequence (c-MYC-MUT) no longer able to fold into a G4 conformation (SI). Oligonucleotides were annealed at a final concentration of 200 nM in the presence of 100 mM KCl, buffered at pH 7.4 to promote G4 formation. Annealed oligonucleotides were then incubated with increasing doses of PDS-Chl (0.2–5 μM) for 1 h at 37 °C. The alkylated adducts were separated by polyacrylamide gel electrophoresis (PAGE) under denaturing conditions (8 M urea) and detected by fluorescence (6-FAM). We observed formation of a new band only when PDS-Chl was incubated with G4 forming sequences, with no alkylation observed for ds-DNA and the mutated c-MYC sequence (Figure 2).
Figure 2.

G4-specific alkylation. PDS-Chl (0–5 μM) was incubated with 0.2 μM oligonucleotides, for 1 h at 37 °C. Alkylated DNA (ALKY) was separated from unreacted oligonucleotides on a 15% denaturing PAGE. Oligonucleotides were detected by recording the 6-FAM fluorescence signal (λex= 488 nm).
The faster migrating band observed for the alkylated adduct suggests the formation of a covalently locked secondary structure that cannot be denatured. Compressed DNA secondary structures occupy a smaller hydrodynamic volume compared to their single-stranded counterparts, causing bands to migrate faster than expected from their molecular weight.13 Imaging the same gel by exciting the second fluorescent tag (TAMRA, λex= 583 nm) attached to the opposite 3′-end of the oligonucleotides gave identical banding, which indicates that strand cleavage is not the cause of the new band (Figure S6). We explored the alkylation target sites of unlabeled h-Telo and c-MYC, after 1 h incubation with PDS-Chl at 37 °C, by nuclease digestion of the alkylated oligonucleotides to single nucleobases that were purified and analyzed by LC-MS to detect alkylated bases (SI). The MS fragmentation profile of Chl adducts with nucleobases enabled the identification of the alkylated sites after incubation of PDS-Chl with c-MYC.14 As expected, PDS-Chl mainly reacts with water under these conditions, as the bis-hydrated product was the most abundant in the reaction mixture (Figure 3). The detected alkylated nucleobases were the following: mono-adenine adducts at N1 and the exocyclic NH2, mono-guanine adducts at N7 and N1 (minor product), in addition to a bis-adduct with both adenine and guanine (Figure 3).
Figure 3.

Alkylated adducts with adenine and guanine detected by LC-MS analysis of nuclease digested c-MYC incubated 1 h with PDS-Chl.
Analysis of the relative base composition in the mixture before and after treatment with different doses of PDS-Chl (0.2–5 μM) confirmed alkylation sites. Incubation of c-MYC with PDS-Chl confirmed dose-dependent alkylation of both adenine and guanine (Figure S7). h-Telo showed alkylation of adenine only; thus, the adenines in the loops of the telomeric G4 are the only accessible sites for the alkylating agent (Figure S8). This is in agreement with previous reports11c and is consistent with the hydrogen-bonding of the guanine N7 lone pair in G-tetrads leading to protection from alkylation.7 Importantly, no alkylation was observed upon incubation of ds-DNA with up to 100 μM of PDS-Chl (Figure S9), confirming a structure-specific reaction.
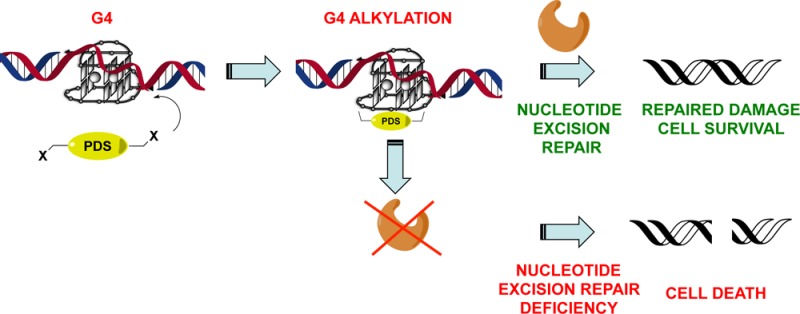
Finally, we sought to explore the therapeutic potential of PDS-Chl within this overall strategy. We reasoned that selective cross-linking of an intramolecular G4 structure would predominantly cause intrastrand cross-linking. Intrastrand cross-links are commonly formed in DNA after exposure to UV light, as this triggers [2 + 2] cyclo-addition of adjacent thymidine bases.15 Nature has evolved nucleotide excision repair (NER) enzymes to overcome intrastrand cross-links and repair T-T dimers.15 NER recognizes the damaged DNA strand, excises it, and replaces it with freshly synthesized DNA.15 Because of its importance to DNA repair after UV exposure, deficiency in NER is responsible for several photosensitivity diseases and is a significant cancer risk factor and a common genetic signature of skin cancers.15 NER-deficient cells are sensitized to the chemotherapeutic drug cisplatin in which the cis-geometry of the drug imposes the formation of covalent bonds with adjacent purine bases to produce mainly 1,2- or 1,3-intrastrand cross-links.16 Similarly, we postulated that our G4-specific alkylator PDS-Chl might show selectivity toward NER-deficient cells by preferentially inducing intrastrand cross-links via G4 structural recognition. To test this hypothesis, we applied PDS-Chl to SV-40 mutated human fibroblasts genetically deficient in the expression of a vital NER component XPA (XPA–, GM04312D) and compared this to the wild type (WT) counterpart that expresses XPA (XPA+, GM00637).17 These cells have been widely used to characterize NER deficient sensitization to drugs, since they comprise an identical cell line pair derived from the same tissue of different patients (respectively, with and without XPA deficiency) and differ only for the expression of XPA.18 Using an impedance-based continuous cell-monitoring approach (SI), we examined, in real time, the effect on cell growth of PDS-Chl, PDS, and Chl to these cell lines. We found that PDS-Chl impaired the cell growth of the XPA– cells but showed no activity against XPA+ fibroblasts up to 100 μM, the highest testable dose compatible with the solubility of the compound (Table 1). Thus, the absence of NER activity sensitizes the cells to PDS-Chl, which become >5-fold more sensitive to this compound compared to their WT counterpart that expresses NER enzymes. By analogy with the preferential growth inhibition exhibited by PDS-Chl for XPA− cells, cisplatin showed a GI50 of ∼44 μM for XPA+ and ∼4.1 μM for the XPA–, therefore also showing a preference (10-fold) for NER deficient cells.18 Conversely, both PDS and Chl alone showed no preference for XPA– cells, and in fact, Chl actually showed a preference for XPA+ cells (Table 1).
Table 1. GI50 Values of PDS, Chl, and PDS-Chl Measured in XPA+ and XPA– Human Fibroblasts.
| GI50(XPA+) (μM) | GI50 (XPA−) (μM) | |
|---|---|---|
| PDS | 1.7 ± 0.6 | 0.8 ± 0.1 |
| Chl | 3.7 ± 1.8 | 22 ± 4 |
| PDS-Chl | >100 | 21 ± 5 |
To rule out the possibility that differences in cell permeability may bias the outcomes for cell lines derived from different patients, we further evaluated PDS-Chl in GM04312D cells that were supplemented with exogenous XPA cDNA, which restores XPA expression and activity in the same cell line. We found that PDS-Chl is no longer able to induce any growth inhibition in GM04312D cells for doses as high as 100 μM (GI50 > 100 μM) when XPA activity is restored, further supporting the hypothesis that the activity measured is NER-dependent. We next investigated whether the cross-linking properties of PDS-Chl were responsible for the growth inhibition activity measured in XPA– cells. We generated the hydrolyzed analogue of PDS-Chl by incubation of the compound in water at 37 °C overnight; full conversion was ensured by HPLC analysis (Figure S10). The hydrolyzed PDS-Chl derivative did not impair XPA+ and XPA– growth for doses up to 100 μM, supporting the hypothesis that the cross-linking activity of PDS-Chl is essential to impair growth in XPA– cells. Given these experiments suggest a preference for intrastrand cross-linking by PDS-Chl, we next investigated whether the agent generates ICLs. Since Chl generates DNA DSBs via ICLs, we postulated that cells deficient in BRCA2 expression would be sensitized to this compound, given the crucial relevance of this gene in the DSB repair machinery, homologous recombination.8c We applied PDS-Chl, PDS, and Chl to HCT116 colon carcinoma cells carrying either the WT BRCA2+/+ gene, or where both copies have been removed (BRCA2–/–). We found that Chl is 40-fold more active in BRCA2–/– cell lines (GI50 1.25 ± 0.7 μM) as compared to BRCA2+/+ WT cells (GI50 50 ± 2 μM). PDS is also more active against BRCA2–/– cells, as previously reported.8c Importantly, we found that PDS-Chl shows negligible activity against both BRCA2 positive and negative cells up to a 100 μM dose (GI50 > 100 μM), supporting the hypothesis that ICLs do not contribute to the mechanism of action of this compound.
Our findings suggest that tethering the nitrogen mustard Chl to the G4 ligand PDS reprograms the mechanism of action of this drug, preventing the generation of ICLs by selective cross-linking of single-stranded DNA G4s. This alters the mechanism of action of the nitrogen mustard moiety in PDS-Chl, resulting in an agent that preferentially generates intrastrand cross-links via selective G4 recognition, thus sensitizing NER-deficient cells lines to this G4-selective alkylating agent.
Reprogramming a DNA cross-linker by means of G4-directed reactivity may be a promising strategy to consider for diseases where genetic impairment of NER is relevant, such as skin, testicular, and drug-resistant cancers.
Acknowledgments
We thank Cancer Research U.K. for program funding and core support. We thank Dr. Raphaël Rodriguez and Dr. David Tannahill for insightful discussions, Dr. Mike Booth for HPLC support, Dr. Dan Le and Dr. Chris Lowe for proofreading of the manuscript.
Supporting Information Available
All experimental details and supplementary tables and figures cited above. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Faguet G. B. J. Clin. Oncol. 1994, 12, 1974. [DOI] [PubMed] [Google Scholar]
- a Mattes W. B.; Hartley J. A.; Kohn K. W. Nucleic Acids Res. 1986, 14, 2971. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hartley J. A.; Bingham J. P.; Souhami R. L. Nucleic Acids Res. 1992, 20, 3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson B. J.; Shield A. J. Mutat. Res. 1996, 355, 41. [DOI] [PubMed] [Google Scholar]
- Pezzoni G.; Grandi M.; Biasoli G.; Capolongo L.; Ballinari D.; Giuliani F. C.; Barbieri B.; Pastori A.; Pensenti E.; Mongelli N.; Spreafico F. Br. J. Cancer 1991, 64, 1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. D.; Dziegielewski J.; Wurtz N. R.; Dziegielewska B.; Dervan P. B.; Beerman T. A. Nucleic Acids Res. 2003, 31, 1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez R.; Muller S.; Yeoman J. A.; Trentesaux C.; Riou J. F.; Balasubramanian S. J. Am. Chem. Soc. 2008, 130, 15758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sen D.; Gilbert W. Nature 1988, 334, 364. [DOI] [PubMed] [Google Scholar]; b Davis J. T. Angew. Chem., Int. Ed. 2004, 43, 668. [DOI] [PubMed] [Google Scholar]
- a Balasubramanian S.; Hurley L. H.; Neidle S. Nat. Rev. Drug Discov. 2011, 10, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rodriguez R.; Miller K. M.; Forment J. V.; Bradshaw C. R.; Nikan M.; Britton S.; Oelschlaegel T.; Xhemalce B.; Balasubramanian S.; Jackson S. P. Nat. Chem. Biol. 2012, 8, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]; c McLuckie K. I.; Di Antonio M.; Zecchini H.; Xian J.; Caldas C.; Krippendorff B. F.; Tannahill D.; Lowe C.; Balasubramanian S. J. Am. Chem. Soc. 2013, 135, 9640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Biffi G.; Tannahill D.; McCafferty J.; Balasubramanian S. Nat. Chem. 2013, 5, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Biffi G.; Di Antonio M.; Tannahill D.; Balasubramanian S. Nat. Chem. 2014, 6, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doria F.; Nadai M.; Folini M.; Di Antonio M.; Germani L.; Percivalle C.; Sissi C.; Zaffaroni N.; Alcaro S.; Artese A.; Richter S. N.; Freccero M. Org. Biomol. Chem. 2012, 10, 2798. [DOI] [PubMed] [Google Scholar]
- a Bertrand H.; Bombard S.; Monchaud D.; Teulade-Fichou M.-P. J. Biol. Inorg. Chem. 2007, 12, 1003. [DOI] [PubMed] [Google Scholar]; b Di Antonio M.; Doria F.; Richter S. N.; Bertipaglia C.; Mella M.; Sissi C.; Palumbo M.; Freccero M. J. Am. Chem. Soc. 2009, 131, 13132. [DOI] [PubMed] [Google Scholar]; c Doria F.; Nadai M.; Folini M.; Scalabrin M.; Germani L.; Sattin G.; Mella M.; Palumbo M.; Zaffaroni N.; Fabris D.; Freccero M.; Richter S. N. Chemistry 2013, 19, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Verga D.; Hamon F.; Poyer F.; Bombard S.; Teulade-Fichou M. P. Angew. Chem., Int. Ed. 2014, 53, 994. [DOI] [PubMed] [Google Scholar]
- a Muller S.; Sanders D. A.; Di Antonio M.; Matsis S.; Riou J. F.; Rodriguez R.; Balasubramanian S. Org. Biomol. Chem. 2012, 10, 6537. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Di Antonio M.; Biffi G.; Mariani A.; Raiber E. A.; Rodriguez R.; Balasubramanian S. Angew. Chem., Int. Ed. 2012, 51, 11073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques J. P.; Susskind M. M. Nucleic Acids Res. 1991, 19, 2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Haapala E.; Hakala K.; Jokipelto E.; Vilpo J.; Hovinen J. Chem. Res. Toxicol. 2001, 14, 988. [DOI] [PubMed] [Google Scholar]; b Florea-Wang D.; Haapala E.; Mattinen J.; Hakala K.; Vilpo J.; Hovinen J. Chem. Res. Toxicol. 2003, 16, 403. [DOI] [PubMed] [Google Scholar]
- Li L. In DNA Repair, Genetic Instability, and Cancer; Wei Q., Li L., Chen D. J., Eds.; World Scientific Publishing Co Pte. Ltd.: Singapore, 2007. [Google Scholar]
- a Koberle B.; Masters J. R.; Hartley J. A.; Wood R. D. Curr. Biol. 1999, 9, 273. [DOI] [PubMed] [Google Scholar]; b McKay B. C.; Becerril C.; Ljungman M. Oncogene 2001, 20, 6805. [DOI] [PubMed] [Google Scholar]
- Enoiu M.; Jiricny J.; Scharer O. D. Nucleic Acids Res. 2012, 40, 8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens E. V.; Nishizuka S.; Antony S.; Reimers M.; Varma S.; Young L.; Munson P. J.; Weinstein J. N.; Kohn E. C.; Pommier Y. Mol. Cancer Ther. 2008, 7, 10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.