

Abstract
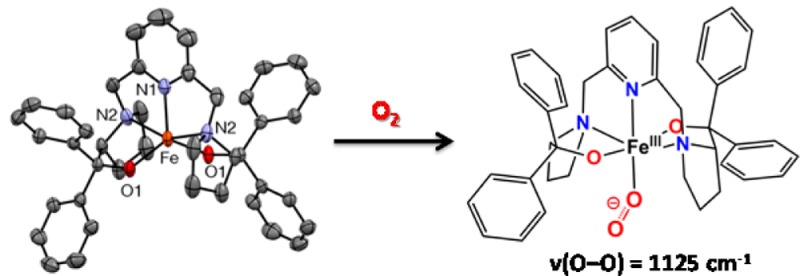
O2 bubbling into a THF solution of FeII(BDPP) (1) at −80 °C generates a reversible bright yellow adduct 2. Characterization by resonance Raman and Mössbauer spectroscopy provides complementary insights into the nature of 2. The former shows a resonance-enhanced vibration at 1125 cm–1, which can be assigned to the ν(O–O) of a bound superoxide, while the latter reveals the presence of a high-spin iron(III) center that is exchange-coupled to the superoxo ligand, like the FeIII–O2– pair found for the O2 adduct of 4-nitrocatechol-bound homoprotocatechuate 2,3-dioxygenase. Lastly, 2 oxidizes dihydroanthracene to anthracene, supporting the notion that FeIII–O2– species can carry out H atom abstraction from a C–H bond to initiate the 4-electron oxidation of substrates proposed for some nonheme iron enzymes.
The formation of an iron(III)-superoxo species upon O2 binding to an iron(II) center is invariably the first step proposed for the activation of O2 by iron oxygenases.1−3 Although such species are well-characterized for enzymatic and synthetic heme centers,4,5 only recently have iron-superoxo intermediates of nonheme iron enzymes been reported. The first example is the O2 adduct of the diiron(II,III) myo-inositol oxygenase, which has been spectroscopically assigned to be an FeIIIFeIII–O2– species.6 O2 adducts for two aromatic ring-cleaving dioxygenases have been observed in crystallo;7,8 corresponding EPR and Mössbauer studies of homoprotocatechuate 2,3-dioxygenase (HPCD) in frozen solution show its O2 adduct to be an antiferromagnetically coupled high-spin iron(III)-superoxo species.9 For the above enzymes and related iron enzymes that catalyze 4-e– substrate oxidations, the iron(III)-superoxo species must carry out the first 1-e– oxidation step.10 There are thus far only two reports of synthetic nonheme iron-superoxo complexes, both of which derive from diiron(II) precursors.11,12 No monoiron-superoxo species has yet been reported. Based on the chemistry some of us developed to synthesize a stable five-coordinate nickel(III) complex, [NiIII(BDPP)](PF6) (see Figure 1 for the structure of the BDPP ligand),13 we have prepared its iron(II) derivative to take advantage of its two anionic alkoxide donors to promote O2 binding. Herein we present spectroscopic evidence for the formation of a paramagnetic mononuclear nonheme iron(III)-superoxo complex.
Figure 1.
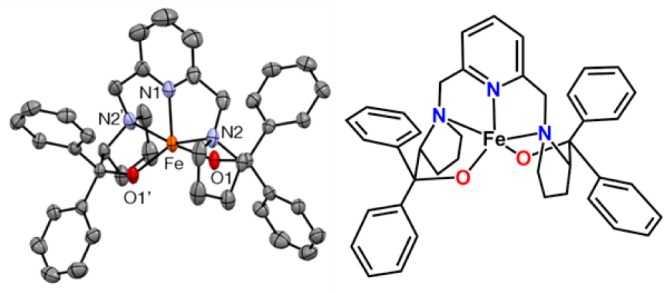
X-ray structure (left) and a schematic drawing (right) of Fe(BDPP) (1); hydrogen atoms not shown. Selected bond lengths (Å) and angles (deg): Fe1–O1 1.9275(18), Fe1–N1 2.100(3), Fe1–N2 2.271(2), O1–Fe1–O1′ 123.04(12), N2–Fe1–N2′ 151.78(14).
Treatment of H2BDPP with NaH in CH3CN and then with FeCl2 forms a red solution of FeII(BDPP) (1). Evaporation of the solvent affords a solid that yields dark red crystals of 1 upon recrystallization from CH2Cl2/pentane. Complex 1 exhibits two UV–vis absorption bands at 325 (sh, εM 1500) and 525 nm (εM 570) in THF and gives rise to a quasi-reversible cyclic voltammogram (ΔE = 80 mV) with an E1/2 value at 122 mV vs Ag/AgCl in CH3CN (Figure S1). X-ray crystallography of 1 (Figure 1) reveals a mononuclear iron(II) complex with a distorted square pyramidal geometry (τ = 0.48).14
The five-coordinate iron(II) center of 1 would appear to be well set up to bind O2. Indeed, bubbling of O2 through a THF solution of 1 at −80 °C generates a bright yellow solution with an intense absorption band at 330 nm (εM 9400) (Figure 2); an isosbestic point at 465 nm is observed in the conversion of 1 to 2 (Figure 2 inset). For comparison, the O2 adduct of [FeII2(μ–OH)2(6-Me3TPA)2]2+ (6-Me3TPA = tris(6-methylpyridyl-2-methyl)amine) observed in CH2Cl2 at −80 °C exhibits a similarly intense UV band at 325 nm (εM 10 300) and has been characterized to be an iron(II)iron(III)-superoxo complex.11 Interestingly, bubbling of N2 through the THF solution of 2 for 5 min at −80 °C regenerates 1, showing that 2 is a reversible Fe–O2 adduct; several cycles of alternating N2/O2 purges can be achieved (Figure S2).
Figure 2.
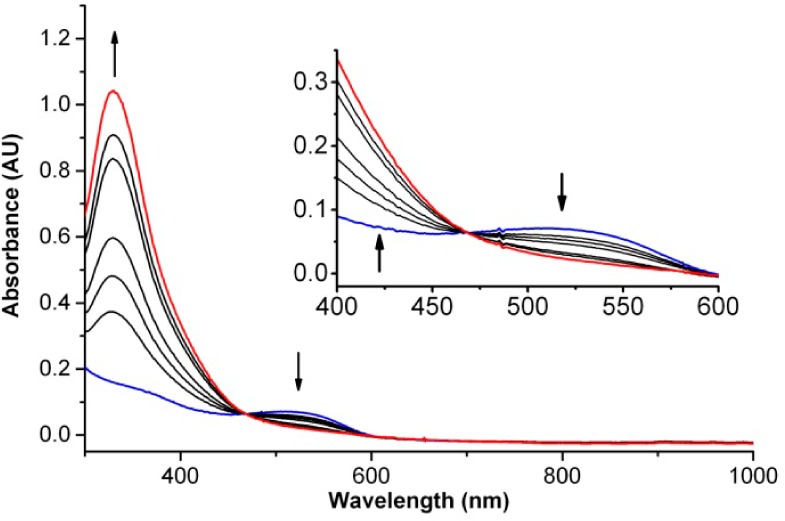
Formation of 2 (red line) at −80 °C by bubbling O2 through a THF solution of 1 (0.1 mM) (blue line). The spectra shown were taken over a 15 s time frame.
Excitation of 2 obtained with 413.1 nm irradiation reveals a resonance-enhanced vibration at 1125 cm–1 (Figure 3), presumably arising from the bound O2. Its frequency falls within the 1100–1200 cm–1 range found for the ν(O–O) features of other mononuclear metal-superoxo complexes (Table 1).15 Hooke’s Law predicts a downshift of 64 cm–1 for the ν(O–O) of the 18O2 isotopologue. This downshift moves the ν(O–O) for 2 into a region obscured by a THF mode, but the corresponding experiment in THF-d8 shows a peak at 1062 cm–1. Taken together, the observed frequency and 63 cm–1 downshift support a superoxo ligand for 2. Given the pentadentate nature of BDPP and the reversibility of O2 binding, it is likely that the superoxide is bound end-on, as deduced for the other complexes listed in Table 1.
Figure 3.
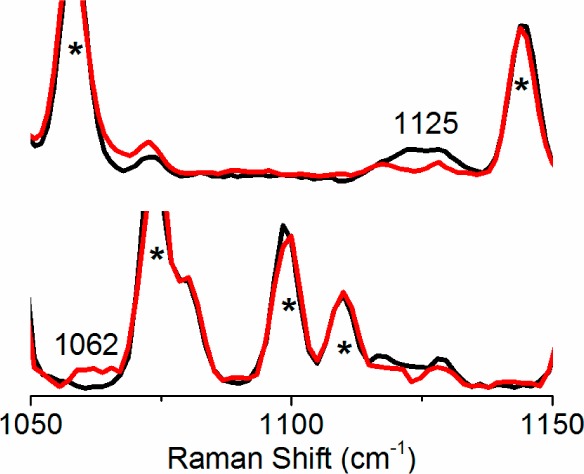
Resonance Raman spectra of 2 (λex 413.1 nm, 30 mW, 77 K) prepared by bubbling O2 into 1 (5 mM) in THF or THF-d8 at −80 °C. Top: black, 16O2; red, 18O2 in THF. Bottom: black, 16O2; red, 18O2 in THF-d8. Asterisks denote solvent peaks.
Table 1. Raman Data for Metal-Superoxo Complexesa.
| superoxo complexes | ν(16O–16O), cm–1 | ν(18O–18O), cm–1 | ref |
|---|---|---|---|
| 2 | 1125 | 1062 | this work |
| [FeII2(μ-OH)2(6-Me3TPA)2]2+ + O2 | 1310 | 1239 | (11) |
| [CuII(TMG3tren)(η1-O2)]+ | 1117 | 1059 | (15a) |
| [CuII(6-pivTPA)O2]+ | 1130 | 1067 | (15b) |
| [NiII(TMC)O2]+ | 1131 | 1067 | (15c) |
| [CrIII(TMC)(η1-O2)(Cl)]+ | 1170 | 1104 | (15d) |
Abbreviations: 6-Me3TPA = tris(6-methylpyridyl-2-methyl)amine; 6-pivTPA = (6-pivaloylamidopyridyl-2-methyl)-bis(6-methylpyridyl-2-methyl)amine; TMC = 1,4,8,11-tetramethylcyclam; TMG3tren =1,1,1-tris{2-[N2-(1,1,3,3-tetramethylguanidino)]ethyl}amine).
Figure 4 shows 4.2 K Mössbauer spectra of 2 recorded in zero field (A) and parallel applied fields B = 2.2 mT (B) and B = 45 mT (C). The spectrum of Figure 4C is typical of those observed for high-spin (S1 = 5/2) FeIII with a ground Kramers doublet that is magnetically uniaxial (such as the MS = ±5/2 doublet of a mononuclear FeIII which has gz ≫ gx,gy). Indeed, the isomer shift, δ = 0.58(3) mm/s, derived from analyzing the magnetic hyperfine pattern is strongly indicative of high-spin FeIII. If 2 would represent an exchange-coupled FeIII–O2– pair like that for the HPCD superoxo intermediate,9 the ground state would have integer spin S = 2 or 3, corresponding to antiferromagnetic or ferromagnetic coupling, respectively. In either case, we would expect to observe for B = 0 a quadrupole doublet. Instead, the zero field spectrum (ZFS) of 2 (Figure 4A) displays two spectral components, namely, a majority paramagnetic component with similar features as the spectrum of Figure 4C plus a doublet (ca. 10% of Fe) with δ = 0.58 mm/s and quadrupole splitting ΔEQ ≈ 1.7 mm/s that is not present in Figure 4C. The features of the zero field spectrum are highly unusual but can be understood using the ideas developed in our analysis of the Pox state of the nitrogenase P clusters17 (where the two lowest spin levels are so closely spaced that they are mixed by 57Fe magnetic hyperfine interactions; see Supporting Information).
Figure 4.
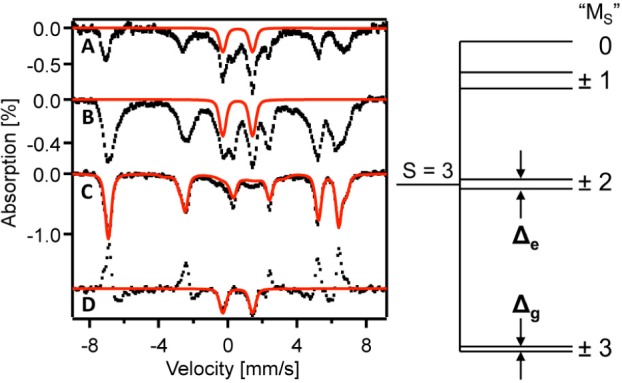
Left: 4.2 K Mössbauer spectra of 2 recorded in zero field (A) parallel applied fields of 2.2 mT (B) and 45 mT (C). (D) Difference spectrum “2.2 mT minus 45 mT”. Red lines in A, B, and D outline a quadrupole doublet that represents ≈10% of the Fe attributed to 2. Red line in C is a spectral simulation for 2 based on eq 1 and represents 89% of the Fe. Red line represents two similar S = 3 species, 2a and 2b (parameters of 2b are given in italics): % Fe 62(27), D = −1.2(−1.2) cm–1, E/D = 0.08(0.08), δ = 0.58(0.58), ΔEQ = −1.65(−1.65) mm/s, η ≈ 0.0(0.6), Az/gnβn = −13.8(−14.4) T (because ⟨Sx,y⟩ ≈ 0, the spectra are insensitive Ax and Ay). Right: Splittings of the S = 3 multiplet; for labels, see ref (18).
The proof that 2 contains an exchange-coupled FeIII–O2– pair is independent of whether the coupling is ferromagnetic or antiferromagnetic. We have analyzed our data using the S = 3 spin Hamiltonian
| 1 |
with
| 2 |
For the S = 3 multiplet, the ZFS parameters D, (E/D), and the 57Fe magnetic hyperfine tensor, A, are related to the corresponding local quantities of the S1 = 5/2 FeIII site by D = (2/3)D1, (E/D) = (E/D)1 and A = (5/6)A1.9 For D < 0, the S = 3 multiplet has two low-lying non-Kramers doublets (Figure 4), whose properties are fundamental to understanding the unusual spectroscopic properties of 2. For B = 0, the MS = ±3 ground doublet18 is split by Δg = 3D(E/D)3, while the first excited, MS = ±2, doublet is split by Δe = 15D(E/D)2.
The observation of magnetic hyperfine structure even in zero field (Figure 4A) shows that Δg of 2 must be exceptionally small, namely, Δg < 0.003 cm–1. For such a small value of Δg, the ground doublet develops a large expectation value of the electronic spin along z (Figure S6A), even in weak applied fields. The magnetic splitting of the spectrum of Figure 4C is determined by the internal magnetic field Bint,z = −⟨Sz⟩Az/gnβn ≈ ±3(Az/gnβn), where the + and – refer to the lower and upper members of the ground doublet, respectively, and Bint,x,y vanishes for βB ≪ |D|. A uniaxial electronic doublet yields a characteristic 6-line Mössbauer pattern like that of Figure 4C. The observation that Bint,z approaches the maximum value obtainable for an isolated doublet at B = 2.2 mT also shows Δg < 0.003 cm–1. For B = 45 mT, ⟨Sz⟩ is saturated at ⟨Sz⟩ = ±3, which implies a value Az/gnβn ≈ −14 T for the z component of the magnetic hyperfine tensor.
Interestingly, the central part of Figure 4A,B contains a doublet (red solid lines) that is absent in Figure 4C. This quadrupole doublet originates from the excited states of the S = 3 manifold, mainly from the MS = ±2 doublet, and represents the same molecular species as the magnetic feature of the ground state (explained in Supporting Information). Consider now the difference spectrum shown in Figure 4D, obtained by subtracting Figure 4C from Figure 4B. The difference spectrum shows a quadrupole doublet (red solid line) with ΔEQ ≈ 1.65 mm/s and δ = 0.58 mm/s, values that are the same as those obtained from analysis of the 6-line pattern of Figure 4C (as they must be if both spectra represent 2). This quadrupole doublet disappears for B = 45 mT. At 2.2 mT and 4.2 K, it contributes ∼10% of the absorption of species 2, a population that fits to an S = 3 level splitting corresponding to D ≈ −1.2 cm–1 (the same D value was obtained from level mixing at B = 3.0 T; see Figure S5).
The red lines in Figures 4C and S5 are spectral simulations based on eq 1 using the parameters given in the figure caption. For our final simulations, we assumed that 2 appears as two conformers, 2a and 2b, representing 62 and 27% of the total absorption (explanation given in Supporting Information), possibly due to slight differences in the Fe–O2– moiety arising from interactions with the frozen THF solution. The remaining ∼11% of the absorption belongs to a broad and shallow background that has not yet been identified.
The HPCD-superoxo complex9 has an S = 2 ground multiplet with D < 0, E/D = 0.20, and exhibits an EPR signal near g = 8.2. We have searched for a parallel mode EPR signal for 2 dissolved in THF, acetone, and dichloromethane, but no signal attributable to 2 was found. For D < 0, the EPR intensities of S = 3 and 2 ground doublets are proportional to Δg2, that is, proportional to (E/D)6 and (E/D)4, respectively.17 Analyzing our data assuming S = 3 and 2 yielded (E/D)S=3 = 0.08 and (E/D)S=2 = 0.02 (see Supporting Information). Thus, in either case, the expected signal intensity of the ground doublet is expected to be 4 orders of magnitude smaller than that found for the HPCD intermediate.
Finally, the spectra of Figures 4 and S5 do not reveal whether the ground state of 2 has S = 3 or 2. In principle, this information can be obtained from Mössbauer spectroscopy. Suppose the electronic spin system is in the slow relaxation regime at 11 K. At this temperature, the excited MS = ±2 doublet would be sufficiently populated to be detected and would yield at B = 100 mT a 6-line Mössbauer spectrum with features completely determined by the parameters of the 45 mT ground state spectrum of Figure 4C. (The excited MS = ±1 states of an S = 2 system would not yield a 6-line spectrum because Δe would be too large for (E/D)S=2 = 0.02.) Preliminary data hint at the presence of this 6-line spectrum (i.e., at S = 3), but the onset of intermediate relaxation at 11 K cautions us to reserve final judgment. While the above analysis could have been presented with minor modifications for S = 2 (see SI), our preliminary 11 K data suggested to us to describe the spectra for an S = 3 system. Further Mössbauer experiments in strong applied fields at different temperatures as well as EPR studies in different solvents including glassing solvents should shed further light on the nature of the coupling.
The low-temperature stability of 2 has led us to test the notion that an iron(III)-superoxo moiety can abstract a H atom from a substrate C–H bond. This question was examined by adding an excess of 9,10-dihydroanthracene (DHA, DC–H 78 kcal/mol16) to a THF solution of 2 at −70 °C, which resulted in the exponential decay of its characteristic 330 nm band. Anthracene was formed in 90% yield relative to the amount of 2, and neither anthrone nor anthraquinone was found as byproducts. The reaction followed first-order kinetics in the presence of excess DHA, and a plot of the pseudo-first-order rate constants against the concentration of DHA gave a straight line, from which a second-order rate constant k2 of 0.005 M–1 s–1 was obtained at −70 °C (Figure S3). When DHA-d4 was used as substrate, a kinetic isotope effect of 7 was observed, showing that C–H bond cleavage is involved in the rate-determining step. Our data can be compared with those of the two other metal-superoxo complexes for which the kinetics of intermolecular C–H bond cleavage have been studied. After adjustment for differences in temperature and substrate DC–H values, it appears that 2 has a C–H bond cleaving rate comparable to that of [CrIII(TMC)O2(Cl)]+ (k2 = 0.17 M–1 s–1 at −10 °C for DHA oxidation)15d but slower than that of [CuII(6-pivTPA)O2]+ (k2 = 0.19 M–1 s–1 at −125 °C for 1-benzyl-1,4-dihydronicotinamide (DC–H 71 kcal/mol) oxidation).15b In addition, the latter two complexes were found to exhibit KIE values for C–H bond cleavage of 5015d and 12,15b respectively, that may implicate hydrogen tunneling. What the differences among the KIE values indicate about the nature of the superoxo ligand and how that affects the H atom abstraction mechanism should be an interesting topic for future work. In addition, as observed for [CrIII(TMC)O2(Cl)]+,15d we also found a 1:1 reaction stoichiometry of 2 consumed and anthracene formed, indicating that 2 provided two oxidizing equivalents for the oxidation of DHA. This result suggests that the FeIII–OOH species, presumably formed upon H atom abstraction of DHA by 2, must react further with the nascent DHA· radical. The nature of these iron byproducts in the DHA oxidation and related reactions is under investigation and will be reported in a subsequent publication.
To summarize, we have generated the first synthetic example of a mononuclear iron(III)-superoxo complex in a nonheme ligand environment, providing a model complex with which to compare corresponding complexes that have been trapped, or are likely to occur, in the catalytic cycles of nonheme iron oxygenases.6−10 For 2, resonance Raman and Mössbauer spectroscopy provide complementary information. Thus, the former reveals a vibration at 1125 cm–1 that arises from the superoxo ligand, while the Mössbauer spectra demonstrate that 2, observed as two related conformers 2a and 2b, contains a S = 5/2 FeIII center that is exchange-coupled to a radical which, of course, is the superoxo moiety. The demonstration that 2 can oxidize dihydroanthracene at −70 °C supports the mechanistic notion that FeIII–O2– species can carry out the H atom abstraction from a substrate C–H bond that is necessary for the initiation of the 4-e– oxidation of substrates by nonheme iron enzymes such as myo-inositol oxygenase.6
Acknowledgments
This work was supported by grants from the Ministry of Science and Technology of Taiwan (102-2113M-003-007-MY3 to W.Z.L.) and the U.S. National Science Foundation (CHE-1058248 and CHE-1361773 to L.Q. and CHE-1305111 to E.M.).
Supporting Information Available
Experimental details, figures showing the cyclic voltammogram of 1, the reversibility of O2 binding to 1, a plot of kobs vs DHA concentration for the reaction of 2 with DHA and DHA-d4, and Mössbauer spectra obtained at B = 0 and 3.0 T, comments on the Mössbauer spectra of 2, magnetization curves for MS = ±2 and ±3 doublets, and a crystallographic file in CIF format for 1. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bertini I.; Gray H. B.; Stiefel E. I.; Valentine S. J.. Biological Inorganic Chemistry: Structure & Reactivity; University Science Books, Sausalito, CA, 2007. [Google Scholar]
- Sono M.; Roach M. P.; Coulter E. D.; Dawson J. H. Chem. Rev. 1996, 96, 2841. [DOI] [PubMed] [Google Scholar]
- Costas M.; Mehn M. P.; Jensen M. P.; Que L. Jr. Chem. Rev. 2004, 104, 939. [DOI] [PubMed] [Google Scholar]
- Meunier B.; de Visser S. P.; Shaik S. Chem. Rev. 2004, 104, 3947. [DOI] [PubMed] [Google Scholar]
- Momenteau M.; Reed C. A. Chem. Rev. 1994, 94, 659. [Google Scholar]
- Xing G.; Diao Y.; Hoffart L. M.; Barr E. W.; Prabhu K. S.; Arner R. J.; Reddy C. C.; Krebs C.; Bollinger J. M. Jr. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovaleva E. G.; Lipscomb J. D. Science 2007, 316, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeoung J.-H.; Bommer M.; Lin T.-Y.; Dobbek H. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 12625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbughuni M. M.; Chakrabarti M.; Hayden J. A.; Bominaar E. L.; Hendrich M. P.; Münck E.; Lipscomb J. D. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 16788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Donk W.; Krebs C.; Bollinger J. M. Jr. Curr. Opin. Struct. Biol. 2010, 20, 673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan X.; Que L. Jr. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M.; Helms B.; Slonkina E.; Friedle S.; Lee D.; DuBois J.; Hedman B.; Hodgson K. O.; Fréchet J. M. J.; Lippard S. J. J. Am. Chem. Soc. 2008, 130, 4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.-Z.; Chiang C.-W.; Lin T.-H.; Kuo T.-S. Chem.—Eur. J. 2012, 18, 50. [DOI] [PubMed] [Google Scholar]
- Addison A. W.; Rao T. N.; Reedijk J.; van Rijn J.; Verschoor G. C. J. Chem. Soc., Dalton Trans. 1984, 1349. [Google Scholar]
- a Schatz M.; Raab V.; Foxon S. P.; Brehm G.; Schneider S.; Reiher M.; Holthausen M. C.; Sundermeyer J.; Schindler S. Angew. Chem., Int. Ed. 2004, 43, 4360. [DOI] [PubMed] [Google Scholar]; b Peterson R. L.; Himes R. A.; Kotani H.; Suenobu T.; Tian L.; Siegler M. A.; Solomon E. I.; Fukuzumi S.; Karlin K. D. J. Am. Chem. Soc. 2011, 133, 1702. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cho J.; Kang H. Y.; Liu L. V.; Sarangi R.; Solomon E. I.; Nam W. Chem. Sci. 2013, 4, 1502. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Cho J.; Woo J.; Nam W. J. Am. Chem. Soc. 2010, 132, 5958. [DOI] [PubMed] [Google Scholar]
- a Bordwell F. G.; Cheng J. P.; Ji G.; Satish A. V.; Zhang X. J. Am. Chem. Soc. 1991, 113, 9790. [Google Scholar]; b Roth J. P.; Mayer J. M. Inorg. Chem. 1999, 38, 2760. [DOI] [PubMed] [Google Scholar]
- Surerus K. K.; Hendrich M. P.; Christie P. D.; Rottgardt D.; Orme-Johnson W. H.; Münck E. J. Am. Chem. Soc. 1992, 114, 8579. [Google Scholar]
- For B = 0, the true eigenstates are “nonmagnetic” states composed of symmetric and antisymmetric combinations of |+MS⟩ and |−MS⟩ states; see eq 2 of ref (17). For B > 2 mT (for the ground doublet) and B > 100 mT (for the excited doublet), the levels are better described by the magnetic |±MS⟩ states.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.