

Abstract
The p53-binding protein 1 (53BP1) is a well-known DNA damage response (DDR) factor, which is recruited to nuclear structures at the site of DNA damage and forms readily visualized ionizing radiation (IR) induced foci. Depletion of 53BP1 results in cell cycle arrest in G2/M phase as well as genomic instability in human as well as mouse cells. Within the DNA damage response mechanism, 53BP1 is classified as an adaptor/mediator, required for processing of the DNA damage response signal and as a platform for recruitment of other repair factors. More recently, specific 53BP1 contributions to DSB repair pathway choice have been recognized and are being characterized. In this review, we have summarized recent advances in understanding the role of 53BP1 in regulating DNA DSBs repair pathway choice, variable diversity joining [V(D)J] recombination and class-switch recombination (CSR).
INTRODUCTION
Double-strand breaks (DSBs) in DNA are a significant threat to genomic stability and cellular viability (1–6). Double-strand breaks arise from both endogenous and exogenous sources, including oxygen radicals, replication errors, chemical mutagens and ionizing radiation (IR). Timely signaling to recognize damage and initiate cellular repair of DSBs with appropriate fidelity is critical for genome maintenance (7) as unrepaired DSBs can lead to cancer, accelerated aging and immune deficiency (3, 8, 9). Two distinct pathways, non-homologous end-joining (NHEJ) and homologous recombination (HR), have evolved to repair DSBs. The non-homologous end-joining repair ligates DNA ends together with little or no requirement for intrastrand homology while HR uses the homologous sequence from an undamaged sister chromatid/chromosome as a template to synthesize a new strand of DNA during repair. The requirement for a sister chromatid or homologous chromosome during HR (10) means these repair pathways have some cell cycle specificity (3) with HR repair mostly limited to S and G2 phase cells (13). While the NHEJ repair pathway can function throughout the cell cycle and is the predominant pathway in G1 cells (11, 12). The HR pathway is also the primary means for repair of spontaneous DSBs that arise due to collapsed DNA replication forks (14, 15). The DNA damage response (DDR) pathways are signal transduction pathways that are initiated by DNA damage sensors followed by mediator and effector activation (16, 17). Known DDR mediator/adaptor proteins include MDC1 (mediator of DNA Damage Checkpoint 1), 53BP1, BRCA1 (Breast Cancer 1, early onset), TOPBP1 (Topoisomerase II-binding protein 1) and Claspin (17).
Domain Structure and Functions of 53BP1
53BP1 is a large (350 kD) multi-domain protein (Fig. 1) that was initially identified by a yeast two-hybrid screen using p53 as the bait protein. The protein binds to p53 through its tandem COOH-terminal BRCT (Brca1 carboxyl-terminus) repeats (18), which are DDR specific domains. Other 53BP1 domains that have been identified and characterized are the chromatin-binding Tudor domain, an OLIG (oligomerization) domain, GAR (glycine-arginine rich) domain, two tandem BRCT domains and an N-terminal domain containing 28 SQ/TQ elements (Fig. 1) (19). The N-terminal S/T-Q residues of 53BP1 are ATM dependent phosphorylation sites required for RIF1 (Rap1-interacting factor 1) and PTIP (Pax transactivation domain-interacting protein) recruitment to DNA DSB sites (20–23).
FIG. 1.
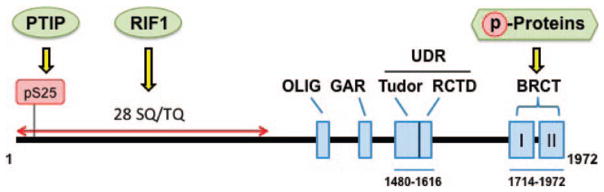
Domain structure of 53BP1. The protein has 1972 amino acids within which four major domains have been identified: OLIG, GAR, UDR and BRCT. The UDR domain ranges from amino acids 1480 to 1616 and has two subdomains tudor and RCTD. The BRCT domain ranges from amino acid 1714 to 1972 and interacts with specific phospho-proteins (p-Proteins). There are 28 SQ/TQ phosphorylation sites within the N-terminal region and PTIP interacts with pS25 while RIF1 interacts with multiple phosphorylated SQ/TQ amino acids.
The 53BP1 tudor domain specifically binds histone H4 dimethylated lysine-20 (H4K20me2), for localization to damage sites (24–27). The specificity of 53BP1-H4K20me2 binding was confirmed by both nuclear magnetic resonance (NMR) and X-ray crystallography spectroscopy studies (28). Moreover, a W1494A substitution within the tudor domain abolishes IR-induced 53BP1 focus formation (24). While the tudor domain is necessary for IR-induced focus formation, it is not sufficient for efficient 53BP1 recruitment to DSB sites, as it has been demonstrated that the OLIG (oligomerization) and RCTD (Region C to terminal of tudor domain) domains also facilitate DSB recognition (28). Region C to terminal of tudor domain (RCTD) is a 15 amino acid long C-terminal extension of the tudor domain. Chromatin histone H4K20me2 levels are unaltered in response to DNA damage (29), suggesting that the higher-order changes in chromatin structure induced by DSBs expose embedded H4K20me2 sites enabling 53BP1 recruitment to DSBs (24, 28, 30). Recent studies have demonstrated that RNF168 (RING finger protein 168) dependent ubiquitination of histones H2A and H2AX also facilitates 53BP1 recruitment to DNA damage sites (31). Thus, 53BP1 probably simultaneously recognizes mono-nucleosomes containing H2A ubiquitinated on lysine 15 (H2A K15Ub) and dimethylated H4K20 (H4K20me2). 53BP1 binds to nucleosomes as a dimer using its methyl-lysine-binding tudor domain and a carboxy-terminal extension (31). The tudor and RCTD domains are both defined as the ubiquitination dependent recruitment (UDR) motif. 53BP1 is described as a bivalent histone modification reader that recognizes a histone ‘code’ (32) produced by DSB signaling (31). 53BP1 also binds to methylated histone H3K79, but binding to H3K79me or H4K20me2 is mutually exclusive during DDR (28). The 53BP1 glycine-arginine rich (GAR) domain is a site for protein arginine methyltransferase (PRMT) – dependent methylation (33). BRCA1 carboxy-terminus (BRCT) domains are phospho-group binding motifs that mediate protein-protein interactions. The BRCT domains of 53BP1 are not required for 53BP1 recruitment to DNA DSB sites nor do they bind to phosphorylated histone H2AX (γ-H2AX) (34), an early marker of DSBs. However, phospho-H2AX is required for the recruitment of 53BP1 to DNA DSB sites (35, 36). The 53BP1–BRCT domains mediate the interaction between 53BP1 and EXPAND1, a protein shown to promote chromatin changes after DNA damage and facilitate DSB repair (37).
53BP1 Determines DSB Repair Pathway Choice
53BP1 is a key DNA repair factor since the association of 53BP1 with DSBs in G1 promotes NHEJ (Fig. 2) (12, 38–40) and plays a pivotal role in defining DSB repair pathway choice in the G1 and S/G2 phases cell cycle (33). The NHEJ is the major repair pathway in G1 cells due to lack of sister chromatids for HR (3, 4). Double-strand break repair by NHEJ promotes a direct ligation of the two broken DNA ends. The initial end recognizing components of the NHEJ pathway, the Ku70–Ku80/86 (Ku) heterodimer and the DNA dependent protein kinase catalytic subunit (DNA-PKcs), act in concert with ataxia telangiectasia mutated (ATM), Mre11, Rad50 and NBS1 (MRN), MDC1, RNF8 (ring finger protein 8, E3 ubiquitin-protein ligase) and XRCC4 (X-ray repair cross-complementing protein 4) –LIG4 (ligase 4) complex. 53BP1 does not affect the initial processing of DNA ends by Ku heterodimer, H2AX phosphorylation by ATM/DNA-PKcs or the loading of MDC1 on γ-H2AX. Rather, 53BP1 acts as a mediator for the loading of other subsequent repair factors. The association of 53BP1 with DSBs in G1 promotes NHEJ, V(D)J and CSR (Fig. 2) (12, 38–40).
FIG. 2.

Role of 53BP1 during the cell cycle. 53BP1 mediates the recruitment of factors in the cell cycle G1 phase that play a role in NHEJ, V(D)J and CSR. During S phase after exposure to DNA damaging agents, ATM/MOF-dependent phosphorylated 53BP1 interacts with RIF1, which is critical for intra-S-phase checkpoint activation. Release of 53BP1 from DNA DSB sites allows BRCA1 during S/G2 phase recruitment of the HR-related proteins required for DNA end resection.
The HR for DSB repair pathway requires a homology template and is initiated by DNA 5′-end resection. The initial processing of the DSB ends is a key determinant of DSB repair pathway choice and is tightly regulated during the cell cycle (41). End resection is initiated by MRN complex and facilitated by CtBP-interacting protein (CtIP) (42, 43). A more extensive end resection is subsequently carried out by DNA replication ATP-dependent helicase-like homolog (DNA2); Exonuclease 1(EXO1); and Bloom Syndrome gene product a RecQ helicase family member (BLM) that generates longer single-stranded DNA (ssDNA) stretches (44, 45). Binding of replication protein A (RPA) to the exposed ssDNA protects against nuclease cleavage and hairpin formation. Two critical proteins involved in HR are BRCA1 and BRCA2 (breast cancer gene 1/2) (46). BRCA1 is a critical component of the HR-mediated DNA repair pathway. Tumors with BRCA1-mutations show evidence of genomic instability and defective DNA DSB repair while BRCA1 deficiency leads to impaired HR (46–51). BRCA1 facilitates the initiation of 5′-end resection and recruitment of PALB2 (partner and localizer of BRCA2 gene) into a BRCA2/PALB2 complex (43) that initiates RAD51 filament formation on ssDNA to catalyze homology search, strand invasion and strand exchange (52–54).
53BP1 does not block MRN functions, exonuclease 1, RPA or RAD51, but has an antagonistic relationship with BRCA1 (55), acting as an inhibitor of BRCA1 accumulation at DSB sites, specifically in the G1 phase of the cell cycle (56). Tumor predisposition, embryonic lethality and the HR defect in Brca1-deficient mice can be rescued by deleting 53BP1 (57, 58). CtBP-interacting protein associates with BRCA1 and the MRN complex to play an essential role in 5′-end resection (42, 59). CtIP and BRCA1 interaction is cyclin-dependent kinase (CDK)-dependent and formation of a functional MRN-CtIP-BRCA1 complex loaded onto DSB ends initiates HR-mediated DSB repair (43, 60). In S and G2-phase cells, CtIP-phosphorylation at S327 and T847 by CDK promotes end resection (61). CDK-dependent phosphorylation of S327 also mediates CtIP association with the C-terminal BRCT domains of BRCA1 as well as with MRN (62). CDK-dependent phosphorylation of CtIP at T847 is required for DSB resection and subsequent S and G2 phase HR (60).
Non-homologous end joining repair of DSBs in G1 phase cells is promoted by 53BP1-RIF1 complexes that protect the DSB ends from exonuclease processing (39, 56, 63). RIF1 accumulation at DSB sites is strongly antagonized by BRCA1-CtIP complex. Resection at the 5′ end of DSBs is not only a requirement for the initiation of repair by the HR pathway but simultaneously blocks KU70/80 complex-mediated classic NHEJ as Ku proteins cannot bind to resected ssDNA. Thus, competition between 53BP1 and BRCA1 during the critical initial stages of DSB repair determines repair pathway choice, either NHEJ or HR (Fig. 2). However, Kakarougkas and coworkers have demonstrated a requirement for 53BP1 in heterochromatin associated DSB repair by HR in G2 phase (64). For DSBs located in heterochromatic (HC) regions, repair requires ATM, H2AX, MRN, ring finger containing nuclear factor 8 (RNF8), RNF168 and the 53BP1 protein (65–67). The proposed role of 53BP1 is to tether ATM to DSBs, promoting concentration of pKAP-1 at HC–DSBs. The resulting pKAP-1 foci cause release of the large isoform of the chromatin remodeling protein (CHD3) and HC relaxation (67, 68). Thus, in HC–DSB repair, 53BP1 is proposed to promote phosphorylated KAP-1 foci formation that favors HR.
53BP1 has no known catalytic activity, but acts, instead, as an adaptor/mediator for DNA damage signaling and responds to IR-induced DNA damage by redistributing to DSB sites to form discrete nuclear foci (69, 70). It has been demonstrated that 53BP1 acts as a transducer of the DNA damage checkpoint signal through regulation of p53 accumulation and the G2/M and intra-S checkpoint in response to IR. The 53BP1 N-terminus contains 28 SQ/TQ motifs phosphorylated by the DSB-responsive kinase ATM following IR (71), however, accumulation of 53BP1 at DNA DSB sites is independent of ATM (72). Instead, N-terminal 53BP1 phosphorylation leads to interaction with other DSB response proteins e.g., Paxip1/PTIP (Pax transactivation domain interacting protein-1) and RIF1 (Fig. 1). A ubiquitously expressed nuclear PAX-transactivation domain-interacting protein, PTIP, associates constitutively with two of the known histone methyltransferases - MLL3 (mixed lineage leukemia) and MLL4-that catalyze trimethylation of histone H3 at lysine 4 (H3K4me3) (73, 74). PTIP acts as a gene transcription regulator by controlling histone H3 methylation and participates in cellular responses to DNA damage (71). Human PTIP (hPTIP) was recognized as a DDR protein in a screening of proteins capable of interacting with SQ/TQ peptides, phosphorylation sites for the ATM/ATR (ataxia telangiectasia-related) protein kinases (75). Human PTIP has three pairs of tandem BRCT domains required for its recruitment to damage site (75, 76) as hPTIP specifically interacts with phosphorylated serine-25 of 53BP1 (pSer-25) residue (Fig. 3) (71) and this interaction increases in response specifically to IR but not other DNA-damaging agents (72). ATM dependent phosphorylation of 53BP1 at serine-25 is required for PTIP interaction but not for 53BP1 localization to IR-induced DSBs. Gene knockout of PTIP is embryonic lethal and PTIP depleted cells have extensive unrepaired DNA ends (73). Structural maintenance chromosome 1 (SMC1) protein, also an ATM substrate, could not be phosphorylated at DNA damage sites in the absence of PTIP (77). Thus, PTIP can be considered as a transducer of 53BP1-dependent DNA damage signaling (77). PA1 (PTIP-associated protein1) is a PTIP binding partner, also recruited to DSBs through PTIP and contributes to repair of IR-induced DSB. PTIP and PA1 are components of a Set1-like histone methyltransferase complex that contributes to the G2/M IR checkpoint and cell survival (73, 78). Depletion of either does not impact RPA or RAD51 focus formation, but their role lies mostly in NHEJ (78).
FIG. 3.
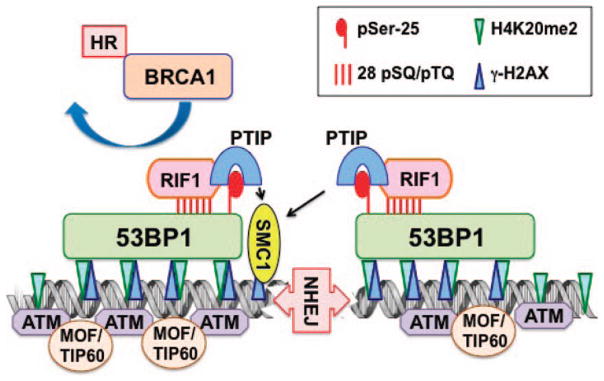
53BP1 interacts with chromatin to enhance NHEJ and suppress HR. Chromatin associated proteins like ATM, MOF and TIP60 facilitate the recruitment of proteins involved in DNA DSB repair through modifications of 53BP1, which is bound to chromatin through direct interaction with H4K20me2 sites. ATM dependent phosphorylation of N-terminal 53BP1 sites is required for RIF1 (28 pSQ/TQ) and PTIP (pSer-25) recruitment to DNA DSB sites. RIF1 accumulation at DSB sites antagonizes BRCA1-CtIP complex recruitment, thus suppresses HR.
Rap1-Interacting Factor 1 (RIF1)
RIF1, a highly conserved protein present from yeast to mammals, was originally discovered in budding yeast as a protein that associates with the telomeric DNA-binding protein Rap1 and negatively regulates telomere length (79). In mammals, RIF1 does not regulate telomere length (80–82) but localizes to DNA damage sites and its depletion results in cellular sensitivity to IR, reduced HR-dependent DSB repair and defective intra-S-phase checkpoint (80–82). Most recently, a study of Rif1-knockout mice suggested that it functions during repair of stalled replication forks (82).
RIF1 and 53BP1 act in the same DNA damage response pathway to promote DSB repair (23). ATM-dependent 53BP1 N-terminal SQ/TQ phosphorylation acts as a platform for RIF1 interaction at DSB sites (56) (Fig. 3), suggesting that RIF1 functions downstream of 53BP1 during DSB repair in G1 cells (Fig. 2). RIF1 interacts with phosphorylated 53BP1 (Fig. 3) through its amino-terminal HEAT-like (Huntington, elongation factor 3, 4, a regulatory subunit of protein phosphatase 2A and Tor1, a target of rapamycin) repeats (56) and this interaction blocks DSB end resection to promote NHEJ (Figs. 2 and 3) (23, 56, 83–85). RIF1 also physically interacts with the BLM complex through a conserved C-terminal domain and both are recruited with similar kinetics to stalled replication forks suggesting that BLM and RIF1 work in a common pathway to promote recovery of stalled forks (82, 86, 87). It has been shown that 53BP1 interacts with BLM and 53BP1 is recruited to sites of aberrant fork structures where it functions to suppress HR. Like 53BP1, RIF1 is also essential for class switch recombination (CSR) (23, 56, 85) where 53BP1 plays a role in productive CSR and suppression of mutagenic DNA repair through distinct phospho-dependent interactions with RIF1 and PTIP (Fig. 3) (88).
53BP1 is Critical for Variable, Diversity and Joining (V(D)J) Recombination and Class Switch Recombination
Non-homologous end joining is important not only for the DSB repair but also for rejoining of the DNA ends generated during V(D)J recombination of immunoglobulin and T-cell receptor loci, which occurs specifically during Band T-cell differentiation (3). V(D)J recombination involves the assembly of Ig and T-cell receptor variable region exons from variable (V), diversity (D) and joining (J) gene segments. Defective NHEJ results in immunodeficiency and hypersensitivity to IR as reported in multiple mouse models (3). Similarly, antibody diversification by CSR, such as IgM to IgG class switching, is initiated by activation-induced cytidine deaminase, an enzyme that produces multiple DNA DSBs within highly repetitive DNA switch regions (89). Switch regions are then re-joined, with varying amounts of DNA loss, by a mechanism that requires an intact DDR and the classical NHEJ (C-NHEJ) or alternative-NHEJ (A-NHEJ) pathways. Among the DDR factors, 53BP1 has the most profound effect on V(D)J and CSR (Fig. 2) (90). Mice deficient for 53BP1 are viable but radiosensitive and immune-deficient (22, 36, 89), having a decrease in CSR-dependent antibody classes with a 50–80% reduction in B and T lineage cells in bone marrow and thymus (22, 89). Loss of CSR-dependent antibody and lymphocytes are due to defective V(D)J and CSR pathways. However, ATM deficiency results in only modest V(D)J recombination or lymphocyte developmental defects; whereas H2AX deficiency alone leads to no obvious defects in these processes (22, 89). Deficiency for DSB response factors also leads to general genomic instability and impairs CSR to significant, but varying, degrees. Deficiency of 53BP1 impairs CSR far more severely than ATM or H2AX deficiency, suggesting additional roles for 53BP1 in CSR (89, 91).
An inherited NHEJ defect was identified in fibroblasts isolated from a patient with a novel severe combined immunodeficiency that displayed high sensitivity to IR and defective V(D)J recombination (92). The defective NHEJ factor was later characterized as XLF (XRCC4 like factor) (93), deficiency of which causes growth retardation and immunodeficiency (94), due to defective NHEJ dependent V(D)J recombination that impacts antibody diversity. XLF interacts with the XRCC4-DNA Ligase IV complex to promote DNA end joining in NHEJ and down regulation of XLF results in radiosensitivity to IR and defective DSB repair (93). Although XLF-deficient mice have moderately impaired lymphocyte development, embryonic stem cells and fibroblasts from XLF−/− mice are IR sensitive and have defective for V(D)J recombination on episomal substrates (95). Overlapping functions of XLF with 53BP1 in V(D)J recombination and DNA repair have been identified (91) as either XLF or 53BP1 deficiency alone led to only a partial reduction in peripheral lymphocyte numbers. However, double deficiency of both XLF and 53BP1 in mice severely impairs C-NHEJ, V(D)J recombination, lymphocyte development, general genomic instability and growth. 53BP1/XLF double deficiency blocks lymphocyte development at early progenitor stages, owing to severe defects in end joining during chromosomal V(D)J recombination. Thus 53BP1 and XLF have some common functions and one may partially compensate for the other’s deficiency during V(D)J recombination pathway.
Future Prospects
Recent studies have made considerable advances in understanding the role of 53BP1 in regulating DSBs repair, specifically in the identification of key DNA repair factors that enhance or block the recruitment of factors at the DSB sites to facilitate DSB repair pathway selection. Although the factors that interact with 53BP1 to regulate NHEJ or HR are becoming defined, what we expect in the near future is a better understanding of how these factors themselves are regulated during pathway selection. For instance, is it chromatin structure (heterochromatin vs. euchromatin) (4–6) or the status of DNA substrate proximity (sister chromatid or homologous chromosome) or the cell cycle phase or position of the DNA DSB or the combination of a few or all of these processes that determines the function of 53BP1 in choosing the DSB repair pathway. However, answering these questions may not completely define the role of 53BP1 within the physiological contexts of V(D)J recombination and CSR, processes that employ overlapping but distinct NHEJ pathways to repair endogenous DSB intermediates. Because the repair of programmed DSBs is also essential for the production of a full immune repertoire by V(D)J and CSR (96), further exploration is necessary before endeavoring to inhibit 53BP1 pharmacologically, which could inhibit breast cancer invasion and metastasis (97) or suppress tumor growth and promote ovarian cancer cell apoptosis (98), since untoward immunological outcomes may also be produced. On a more fundamental level, 53BP1 interacts with specific modified histones, suggesting that it may modulate chromatin structure (Fig. 3), but a detailed understanding for how chromatin modifications and structure determine recruitment of proteins specific for individual DSB repair pathways requires further study. Similarly, how chromatin structures influence the function of the many oncogenes known to regulate tumorigenesis, needs further elucidation. In the near future, as in other areas of basic research, a finer understanding of the relationship of 53BP1 interactions to tumor development and treatment resistance will be of increasing importance to further advances in patient specific targeted therapies.
Acknowledgments
The authors thank the members of our laboratory for the discussions and their suggestions. The research in authors’ laboratory is supported by NIH grants CA129537, CA154320 and U19A1091175 (TK Pandita).
References
- 1.Pandita TK, Hittelman WN. The contribution of DNA and chromosome repair deficiencies to the radiosensitivity of ataxia-telangiectasia. Radiat Res. 1992;131(2):214–23. [PubMed] [Google Scholar]
- 2.Pandita TK, Hittelman WN. Initial chromosome damage but not DNA damage is greater in ataxia telangiectasia cells. Radiat Res. 1992;130(1):94–103. [PubMed] [Google Scholar]
- 3.Scott SP, Pandita TK. The cellular control of DNA double-strand breaks. J Cell Biochem. 2006;99(6):1463–75. doi: 10.1002/jcb.21067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pandita TK, Richardson C. Chromatin remodeling finds its place in the DNA double-strand break response. Nucleic Acids Res. 2009;37(5):1363–77. doi: 10.1093/nar/gkn1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar R, Horikoshi N, Singh M, Gupta A, Misra HS, Albuquerque K, et al. Chromatin modifications and the DNA damage response to ionizing radiation. Front Oncol. 2012;2:214. doi: 10.3389/fonc.2012.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hunt CR, Ramnarain D, Horikoshi N, Iyenger P, Pandita RK, Shay JW, et al. Histone Modifications and DNA Double-Strand Break Repair after Exposure to Ionizing Radiations. Radiat Res. 2013 doi: 10.1667/RR3308.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pfeiffer P, Goedecke W, Obe G. Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Mutagenesis. 2000;15(4):289–302. doi: 10.1093/mutage/15.4.289. [DOI] [PubMed] [Google Scholar]
- 8.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKinnon PJ. DNA repair deficiency and neurological disease. Nat Rev Neurosci. 2009;10(2):100–12. doi: 10.1038/nrn2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hong S, Sung Y, Yu M, Lee M, Kleckner N, Kim KP. The logic and mechanism of homologous recombination partner choice. Molecular Cell. 2013;51(4):440–53. doi: 10.1016/j.molcel.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elliott B, Jasin M. Double-strand breaks and translocations in cancer. Cell Mol Life Sci. 2002;59(2):373–85. doi: 10.1007/s00018-002-8429-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.West SC. Molecular views of recombination proteins and their control. Nature reviews Molecular cell Biology. 2003;4(6):435–45. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 14.Sonoda E, Hochegger H, Saberi A, Taniguchi Y, Takeda S. Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair (Amst) 2006;5(9–10):1021–9. doi: 10.1016/j.dnarep.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 15.Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, et al. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J. 1998;17(2):598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 17.Li L, Zou L. Sensing, signaling, and responding to DNA damage: organization of the checkpoint pathways in mammalian cells. J Cell Biochem. 2005;94(2):298–306. doi: 10.1002/jcb.20355. [DOI] [PubMed] [Google Scholar]
- 18.Iwabuchi K, Bartel PL, Li B, Marraccino R, Fields S. Two cellular proteins that bind to wild-type but not mutant p53. Proc Natl Acad Sci U S A. 1994;91(13):6098–102. doi: 10.1073/pnas.91.13.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adams MM, Carpenter PB. Tying the loose ends together in DNA double strand break repair with 53BP1. Cell Div. 2006;1:19. doi: 10.1186/1747-1028-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DiTullio RA, Jr, Mochan TA, Venere M, Bartkova J, Sehested M, Bartek J, et al. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nature Cell Biol. 2002;4(12):998–1002. doi: 10.1038/ncb892. [DOI] [PubMed] [Google Scholar]
- 21.Morales JC, Xia Z, Lu T, Aldrich MB, Wang B, Rosales C, et al. Role for the BRCA1 C-terminal repeats (BRCT) protein 53BP1 in maintaining genomic stability. J Biol Chem. 2003;278(17):14971–7. doi: 10.1074/jbc.M212484200. [DOI] [PubMed] [Google Scholar]
- 22.Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, et al. 53BP1 is required for class switch recombination. J Cell Biol. 2004;165(4):459–64. doi: 10.1083/jcb.200403021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Molecular Cell. 2013;49(5):858–71. doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127(7):1361–73. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Charier G, Couprie J, Alpha-Bazin B, Meyer V, Quemeneur E, Guerois R, et al. The Tudor tandem of 53BP1: a new structural motif involved in DNA and RG-rich peptide binding. Structure. 2004;12(9):1551–62. doi: 10.1016/j.str.2004.06.014. Epub 2004/09/03. [DOI] [PubMed] [Google Scholar]
- 26.Greeson NT, Sengupta R, Arida AR, Jenuwein T, Sanders SL. Di-methyl H4 lysine 20 targets the checkpoint protein Crb2 to sites of DNA damage. J Biol Chem. 2008;283(48):33168–74. doi: 10.1074/jbc.M806857200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, et al. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO reports. 2006;7(4):397–403. doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zgheib O, Pataky K, Brugger J, Halazonetis TD. An oligomerized 53BP1 tudor domain suffices for recognition of DNA double-strand breaks. Molecular Cellular Biology. 2009;29(4):1050–8. doi: 10.1128/MCB.01011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, et al. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Development. 2004;18(11):1251–62. doi: 10.1101/gad.300704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iwabuchi K, Basu BP, Kysela B, Kurihara T, Shibata M, Guan D, et al. Potential role for 53BP1 in DNA end-joining repair through direct interaction with DNA. J Biol Chem. 2003;278(38):36487–95. doi: 10.1074/jbc.M304066200. [DOI] [PubMed] [Google Scholar]
- 31.Fradet-Turcotte A, Canny MD, Escribano-Diaz C, Orthwein A, Leung CC, Huang H, et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499(7456):50–4. doi: 10.1038/nature12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Misri S, Pandita S, Kumar R, Pandita TK. Telomeres, histone code, and DNA damage response. Cytogenet Genome Res. 2008;122(3–4):297–307. doi: 10.1159/000167816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Molecular Cell. 2012;47(4):497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 34.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123(7):1213–26. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 35.Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, et al. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nature Cell Biol. 2002;4(12):993–7. doi: 10.1038/ncb884. [DOI] [PubMed] [Google Scholar]
- 36.Ward IM, Minn K, van Deursen J, Chen J. p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Molecular Cell Biol. 2003;23(7):2556–63. doi: 10.1128/MCB.23.7.2556-2563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huen MS, Huang J, Leung JW, Sy SM, Leung KM, Ching YP, et al. Regulation of chromatin architecture by the PWWP domain-containing DNA damage-responsive factor EXPAND1/MUM1. Molecular Cell. 2010;37(6):854–64. doi: 10.1016/j.molcel.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, et al. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Molecular Cell. 2011;42(3):319–29. doi: 10.1016/j.molcel.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med. 2010;207(4):855–65. doi: 10.1084/jem.20100244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009;417(3):639–50. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–71. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 42.Yu X, Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol Cell Biol. 2004;24(21):9478–86. doi: 10.1128/MCB.24.21.9478-9486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459(7245):460–3. doi: 10.1038/nature07955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gravel S, Chapman JR, Magill C, Jackson SP. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22(20):2767–72. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Development. 2011;25(4):350–62. doi: 10.1101/gad.2003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Molecular Cell. 2001;7(2):263–72. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 47.Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88(2):265–75. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- 48.Snouwaert JN, Gowen LC, Latour AM, Mohn AR, Xiao A, DiBiase L, et al. BRCA1 deficient embryonic stem cells display a decreased homologous recombination frequency and an increased frequency of non-homologous recombination that is corrected by expression of a brca1 transgene. Oncogene. 1999;18(55):7900–7. doi: 10.1038/sj.onc.1203334. [DOI] [PubMed] [Google Scholar]
- 49.Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Molecular Cell. 1999;4(4):511–8. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 50.Moynahan ME, Cui TY, Jasin M. Homology-directed dna repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Research. 2001;61(12):4842–50. [PubMed] [Google Scholar]
- 51.Jasin M. Homologous repair of DNA damage and tumorigenesis: the BRCA connection. Oncogene. 2002;21(58):8981–93. doi: 10.1038/sj.onc.1206176. [DOI] [PubMed] [Google Scholar]
- 52.Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Molecular Cell. 2006;22(6):719–29. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 53.Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci U S A. 2009;106(17):7155–60. doi: 10.1073/pnas.0811159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu J, Doty T, Gibson B, Heyer WD. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat Struct Mol Biol. 2010;17(10):1260–2. doi: 10.1038/nsmb.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang J, Cho NW, Cui G, Manion EM, Shanbhag NM, Botuyan MV, et al. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol. 2013;20(3):317–25. doi: 10.1038/nsmb.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Molecular cell. 2013;49(5):872–83. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 57.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141(2):243–54. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17(6):688–95. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, et al. Human CtIP promotes DNA end resection. Nature. 2007;450(7169):509–14. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huertas P, Jackson SP. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem. 2009;284(14):9558–65. doi: 10.1074/jbc.M808906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kass EM, Jasin M. Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett. 2010;584(17):3703–8. doi: 10.1016/j.febslet.2010.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen L, Nievera CJ, Lee AY, Wu X. Cell cycle-dependent complex formation of BRCA1.CtIP. MRN is important for DNA double-strand break repair. J Biol Chem. 2008;283(12):7713–20. doi: 10.1074/jbc.M710245200. [DOI] [PubMed] [Google Scholar]
- 63.Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456(7221):529–33. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kakarougkas A, Ismail A, Klement K, Goodarzi AA, Conrad S, Freire R, et al. Opposing roles for 53BP1 during homologous recombination. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Lobrich M, et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Molecular Cell. 2008;31(2):167–77. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 66.Goodarzi AA, Noon AT, Jeggo PA. The impact of heterochromatin on DSB repair. Biochem Soc Trans. 2009;37(Pt 3):569–76. doi: 10.1042/BST0370569. [DOI] [PubMed] [Google Scholar]
- 67.Noon AT, Shibata A, Rief N, Lobrich M, Stewart GS, Jeggo PA, et al. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nature Cell Biology. 2010;12(2):177–84. doi: 10.1038/ncb2017. [DOI] [PubMed] [Google Scholar]
- 68.Goodarzi AA, Kurka T, Jeggo PA. KAP-1 phosphorylation regulates CHD3 nucleosome remodeling during the DNA double-strand break response. Nat Struct Mol Biol. 2011;18(7):831–9. doi: 10.1038/nsmb.2077. [DOI] [PubMed] [Google Scholar]
- 69.Bork P, Hofmann K, Bucher P, Neuwald AF, Altschul SF, Koonin EV. A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. Faseb J. 1997;11(1):68–76. [PubMed] [Google Scholar]
- 70.Derbyshire DJ, Basu BP, Serpell LC, Joo WS, Date T, Iwabuchi K, et al. Crystal structure of human 53BP1 BRCT domains bound to p53 tumour suppressor. EMBO. 2002;21(14):3863–72. doi: 10.1093/emboj/cdf383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Munoz IM, Jowsey PA, Toth R, Rouse J. Phospho-epitope binding by the BRCT domains of hPTIP controls multiple aspects of the cellular response to DNA damage. Nucleic Acids Res. 2007;35(16):5312–22. doi: 10.1093/nar/gkm493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jowsey PA, Doherty AJ, Rouse J. Human PTIP facilitates ATM-mediated activation of p53 and promotes cellular resistance to ionizing radiation. J Biol Chem. 2004;279(53):55562–9. doi: 10.1074/jbc.M411021200. [DOI] [PubMed] [Google Scholar]
- 73.Cho YW, Hong T, Hong S, Guo H, Yu H, Kim D, et al. PTIP associates with MLL3-and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem. 2007;282(28):20395–406. doi: 10.1074/jbc.M701574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Patel SR, Kim D, Levitan I, Dressler GR. The BRCT-domain containing protein PTIP links PAX2 to a histone H3, lysine 4 methyltransferase complex. Dev Cell. 2007;13(4):580–92. doi: 10.1016/j.devcel.2007.09.004. Epub 2007/10/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Manke IA, Lowery DM, Nguyen A, Yaffe MB. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003;302(5645):636–9. doi: 10.1126/science.1088877. [DOI] [PubMed] [Google Scholar]
- 76.Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302(5645):639–42. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 77.Wu J, Prindle MJ, Dressler GR, Yu X. PTIP regulates 53BP1 and SMC1 at the DNA damage sites. J Biol Chem. 2009;284(27):18078–84. doi: 10.1074/jbc.M109.002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gong Z, Cho YW, Kim JE, Ge K, Chen J. Accumulation of Pax2 transactivation domain interaction protein (PTIP) at sites of DNA breaks via RNF8-dependent pathway is required for cell survival after DNA damage. J Biol Chem. 2009;284(11):7284–93. doi: 10.1074/jbc.M809158200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hardy CF, Sussel L, Shore D. A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev. 1992;6(5):801–14. doi: 10.1101/gad.6.5.801. [DOI] [PubMed] [Google Scholar]
- 80.Silverman J, Takai H, Buonomo SB, Eisenhaber F, de Lange T. Human Rif1, ortholog of a yeast telomeric protein, is regulated by ATM and 53BP1 and functions in the S-phase checkpoint. Genes Development. 2004;18(17):2108–19. doi: 10.1101/gad.1216004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xu L, Blackburn EH. Human Rif1 protein binds aberrant telomeres and aligns along anaphase midzone microtubules. J Cell Biol. 2004;167(5):819–30. doi: 10.1083/jcb.200408181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Buonomo SB, Wu Y, Ferguson D, de Lange T. Mammalian Rif1 contributes to replication stress survival and homology-directed repair. J Cell Biol. 2009;187(3):385–98. doi: 10.1083/jcb.200902039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science. 2013;339(6120):700–4. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Feng L, Fong KW, Wang J, Wang W, Chen J. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J Biol Chem. 2013;288(16):11135–43. doi: 10.1074/jbc.M113.457440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339(6120):711–5. doi: 10.1126/science.1230624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yamazaki S, Ishii A, Kanoh Y, Oda M, Nishito Y, Masai H. Rif1 regulates the replication timing domains on the human genome. EMBO. 2012;31(18):3667–77. doi: 10.1038/emboj.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cornacchia D, Dileep V, Quivy JP, Foti R, Tili F, Santarella-Mellwig R, et al. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO. 2012;31(18):3678–90. doi: 10.1038/emboj.2012.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Callen E, Di Virgilio M, Kruhlak MJ, Nieto-Soler M, Wong N, Chen HT, et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell. 2013;153(6):1266–80. doi: 10.1016/j.cell.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol. 2004;5(5):481–7. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- 90.Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456(7221):524–8. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Oksenych V, Alt FW, Kumar V, Schwer B, Wesemann DR, Hansen E, et al. Functional redundancy between repair factor XLF and damage response mediator 53BP1 in V(D)J recombination and DNA repair. Proc Natl Acad Sci U S A. 2012;109(7):2455–60. doi: 10.1073/pnas.1121458109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dai Y, Kysela B, Hanakahi LA, Manolis K, Riballo E, Stumm M, et al. Nonhomologous end joining and V(D)J recombination require an additional factor. Proc Natl Acad Sci U S A. 2003;100(5):2462–7. doi: 10.1073/pnas.0437964100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124(2):301–13. doi: 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 94.Buck D, Malivert L, de Chasseval R, Barraud A, Fondaneche MC, Sanal O, et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124(2):287–99. doi: 10.1016/j.cell.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 95.Zha S, Alt FW, Cheng HL, Brush JW, Li G. Defective DNA repair and increased genomic instability in Cernunnos-XLF-deficient murine ES cells. Proc Natl Acad Sci U S A. 2007;104(11):4518–23. doi: 10.1073/pnas.0611734104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261–92. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li X, Xu B, Moran MS, Zhao Y, Su P, Haffty BG, et al. 53BP1 functions as a tumor suppressor in breast cancer via the inhibition of NF-kappaB through miR-146a. Carcinogenesis. 2012;33(12):2593–600. doi: 10.1093/carcin/bgs298. [DOI] [PubMed] [Google Scholar]
- 98.Hong S, Li X, Zhao Y, Yang Q, Kong B. 53BP1 suppresses tumor growth and promotes susceptibility to apoptosis of ovarian cancer cells through modulation of the Akt pathway. Oncol Rep. 2012;27(4):1251–7. doi: 10.3892/or.2012.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]