

Abstract
Eosinophils function contributes to human allergic and autoimmune diseases, many of which currently lack curative treatment. Development of more effective treatments for eosinophil-related diseases requires expanded understanding of eosinophil signaling and biology. Cell signaling requires integration of extracellular signals with intracellular responses, and is organized in part by cholesterol rich membrane microdomains (CRMMs), commonly referred to as lipid rafts. Formation of these organizational membrane domains is in turn dependent upon the amount of available cholesterol, which can fluctuate widely with a variety of disease states. We tested the hypothesis that manipulating membrane cholesterol content in primary human peripheral blood eosinophils (PBEos) would selectively alter signaling pathways that depend upon membrane-anchored signaling proteins localized within CRMMs (e.g., mitogen activated protein kinase [MAPK] pathway), while not affecting pathways that signal through soluble proteins, like the Janus Kinase/Signal Transducer and Activator of Transcription [JAK/STAT] pathway. Cholesterol levels were increased or decreased utilizing cholesterol-chelating methyl-β-cyclodextrin (MβCD), which can either extract membrane cholesterol or add exogenous membrane cholesterol depending on whether MβCD is preloaded with cholesterol. Human PBEos were pretreated with MβCD (cholesterol removal) or MβCD+Cholesterol (MβCD+Chol; cholesterol delivery); subsequent IL-5-stimulated signaling and physiological endpoints were assessed. MβCD reduced membrane cholesterol in PBEos, and attenuated an IL-5-stimulated p38 and extracellular-regulated kinase 1/2 phosphorylation (p-p38, p-ERK1/2), and an IL-5-dependent increase in interleukin-1β (IL-1β) mRNA levels. In contrast, MβCD+Chol treatment elevated PBEos membrane cholesterol levels and basal p-p38, but did not alter IL-5-stimulated phosphorylation of ERK1/2, STAT5, or STAT3. Furthermore, MβCD+Chol pretreatment attenuated an IL-5-induced increase in cell survival at 48 hours, measured as total cellular metabolism. The reduction in cell survival following cholesterol addition despite unaltered STAT phosphorylation contradicts the current dogma in which JAK/STAT activation is sufficient to promote eosinophil survival, and suggests an additional, unidentified mechanism critically regulates IL-5-mediated human PBEos survival.
Introduction
Human eosinophils are terminally differentiated granular leukocytes that contribute to inflammation and asthmatic airway remodeling, coronary artery disease, and Alzheimer's disease [1]–[10]. Eosinophils are activated as part of the immune system inflammatory response by cytokines released from mast and T helper 2 cells. The binding of extracellular interleukin (IL) 5 family cytokines (IL-5, IL-3, and Granulocyte Macrophage-Colony Stimulating Factor [GM-CSF]) activates inflammatory hallmarks in eosinophils, including prolonged survival, adherence, degranulation (release of cytotoxic proteins), and secretion of pro-inflammatory cytokines and chemokines [11]–[16]. In particular, IL-5 binding to its cognate receptor activates the mitogen activated protein kinase kinase/extracellular-regulated kinase (MEK/ERK), p38 mitogen activated protein kinase (MAPK), and Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) pathways [17]–[22] by inducing phosphorylation of the respective signaling proteins. Enhanced eosinophil survival promoted by these pathways is in turn characterized by up-regulation of pro-survival factors [23], [24], as well as down-regulation of pro-apoptotic factors like caspase 3 activation (cleavage) [25]–[27]. Cyclin family proteins, traditionally defined by their role in regulating cell division, interact with caspases [28] and may also influence survival, as cyclin D3 protein levels parallel eosinophil survival [19]. Eosinophil inflammatory response to stimuli requires proper synchronization of these numerous signaling events.
Eosinophil activation is ultimately the culmination of coordinating extracellular signals with downstream intracellular responses. One of the ways cells ensure specificity in coupling signal receipt with responses is by clustering signaling proteins based on cell membrane architecture, which is in turn determined by lipid distribution. Specificity can be achieved both by the inclusion and exclusion of signaling proteins to/from cholesterol-rich membrane microdomains (CRMMs), which are classically characterized by their resistance to detergents, and often referred to as lipid rafts. CRMMs cluster proteins that regulate signaling in most cells, including T cells, macrophages, and neutrophils [29]–[33], and specifically serve as docking and localization sites for membrane-anchored Src family kinases, which regulate MAPK signal-transduction [34]. These domains are fluid, allowing dynamic regulation, and their structure is dependent upon available cholesterol. As circulating cells, eosinophils are exposed to plasma cholesterol, and are likely to undergo cell membrane reorganization in response to disease states like hypercholesterolemia.
While studies have linked eosinophil-mediated inflammation with diseases caused or affected by abnormal cholesterol levels, including atherosclerosis and Alzheimer's, and recent studies have assessed the relationship between hypercholesterolemia and asthma [1], [5]–[8], [10], [35]–[41], little is known about how cholesterol directly regulates inflammatory signaling in eosinophils. The potential for CRMM disruption to provide separation of distinct intracellular responses to the same extracellular signal is highlighted by recent studies demonstrating that while the IL-5 receptors localize to both CRMMs and detergent-soluble membrane regions in eosinophils, the localization determines which intracellular signaling proteins are directly associated with the receptor [42], [43]. Lei and colleagues found that IL-5Rs localized to CRMMs co-immunoprecipitate with the membrane-anchored MAPK signaling cascade initiator, Lyn tyrosine kinase. These CRMM-localized IL-5Rs do not, however, co-immunoprecipitate with JAK2 [43], a soluble (cytosolic) tyrosine kinase initiator of the JAK/STAT pathway. Conversely, the IL-5Rs in the detergent-soluble portions of the membrane bind JAK2 but not Lyn [43]. These data predict that altering membrane composition by depleting or enhancing cholesterol levels would selectively disrupt pathways like MAPK which are dependent upon membrane-anchored signals, while leaving pathways like JAK/STAT, which utilize soluble proteins, intact following IL-5 binding to its cognate receptor. Manipulating cholesterol levels to selectively alter CRMM-dependent pathways downstream of IL-5 stimulation in primary human eosinophils may allow mechanistic separation of signaling pathways, mimic a variety of human disease states associated with both low and high serum cholesterol levels, and will enhance understanding of eosinophilic inflammation.
This study defines the consequences of membrane cholesterol depletion and addition on specific signaling proteins and inflammatory hallmarks in human PBEos. We tested the hypothesis that reducing cellular membrane cholesterol attenuates MAPK signaling and downstream physiological responses, but not JAK/STAT signaling. We found that reducing membrane cholesterol selectively down-regulated a subset of responses to IL-5, including reduced ERK1/2 and p38 phosphorylation, along with IL-1β mRNA induction. In contrast, depleting membrane cholesterol did not alter STAT3, STAT5, or cyclin D3 phosphorylation nor total cell metabolic activity following IL-5 stimulation. Furthermore, the addition of exogenous cholesterol basally activated p-p38 and led to reduced cellular metabolic activity 48 hours after IL-5 stimulation, despite maintained STAT phosphorylation. These data suggests an additional, unidentified cholesterol-dependent mechanism regulates IL-5-mediated human PBEos survival.
Methods
Human Eosinophil Isolation
Eosinophils were purified from peripheral human blood obtained from atopic patients under informed written consent. The University of Wisconsin-Madison Center for Health Sciences Human Subjects Committee approved this protocol.
Unless specified, all laboratory reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA) or Fisher Scientific (Itasca, IL, USA). Heparinized blood was diluted with HBSS (Mediatech, Manassas, VA, USA) without Ca2+, and a granulocyte mixture was obtained from the leukocyte buffy coat after centrifugation, 20 min at 700× g, through a Percoll monolayer (1.090 g/ml) (Pharmacia, Piscataway, NJ, USA). Eosinophils were purified by negative selection as previously described [44]; recovered eosinophils were at least 97% pure and 98% viable as evaluated by Giemsa's-based Diff-Quik stain (Baxter Scientific Products, McGaw Park, IL, USA) and trypan blue exclusion, respectively.
Cholesterol Treatments
Freshly isolated PBEos (106 cells/100 µL) were pre-incubated in media (RPMI-1640 [ATCC; Manassas, VA, USA] supplemented with 25 mM HEPES and 0.1% human serum albumin [Irvine Scientific; Santa Ana, CA, USA]) for 30 min followed by a 1-hour treatment with media, the cholesterol chelator MβCD (5 mg/mL), or soluble cholesterol (MβCD+Chol, 5 mg/mL), which is MβCD pre-loaded with cholesterol (or concentrations indicated in Figure 1 legend). All incubations were at 37°C.
Figure 1. MβCD reduced cholesterol dose-wise in human peripheral blood eosinophils, while MβCD+Chol (2% cholesterol) elevated cholesterol.

Three to five-hundred thousand eosinophils per treatment were treated 1 hour with media, MβCD, or MβCD+2%Chol (A–C) then fixed in 2% paraformaldehyde, stained with 50 µg/mL Filipin III (cholesterol stain), and analyzed via flow cytometry. (A) Pooled data from dose-wise treatment (n = 5). (B) Representative histogram from n = 16 experiments of cells stained with FIII after treated with i. media, ii. 5 mg/mL MβCD, iii. or 5 mg/mL MβCD+2%Chol, as well as unstained eosinophils (iv.). (C) Quantification of 16 independent experiments of FIII-stained eosinophils after treatment with 5 mg/mL MβCD or 5 mg/mL MβCD+2%Chol. Unmarked comparisons were non-significant.
Eicosapentaenoic Acid Treatment
EPA (Sigma E7006, cis-5,8,11,14,17-Eicosapentaenoic acid) was supplied as a liquid, and diluted to final concentration in culture media. Freshly isolated PBEos were cultured in (RPMI-1640, 10 mM HEPES, 2 mM L-glutamine, 1% penicillin/streptomycin, 1% ciprofloxacin, 10% FBS) for 2 hours prior to the addition of 33 uM EPA or control-media, and overnight culture (18 hours).
Flow Cytometry
To measure cell membrane cholesterol, human PBEos treated +/− MβCD or MβCD+Chol were fixed with 2% paraformaldehyde, then, stained with 50 µg/mL filipin III (FIII) for 1 hour. To compare receptor expression, MβCD or MβCD+Chol treated PBEos were immediately stained with 10 µg/mL PE-conjugated anti-IL-5Rα (BD Biosciences, Franklin Lakes, NJ, USA) or 2.5 µg/mL PE-conjugated anti-IL-5Rβ (eBiosciences, San Diego, CA, USA), then fixed with 2% paraformaldehyde in media. Cells were suspended in 1× PBS (pH 7.4) supplemented with 0.25% BSA, 1 mM Na2EDTA. Ten thousand events within the PBEos-defined forward and side-scatter gate were collected using a BD Biosciences (San Jose, CA, USA) LSRII flow cytometer (355 nm laser [525/50 filter] for FIII, and 561 nm laser [582/15 filter] for PE). Data analyses were performed using CellQuest software (BD Biosciences; Franklin Lakes, NJ, USA) and Flowing Software 2 (Turku Centre for Biotechnology, University of Turku, Finland). Flow data are summarized as the median fluorescence intensity (MFI).
Immunoblotting
Human PBEos were pretreated for 1 hour with media +/− indicated concentrations of MβCD, MβCD+Chol, or EPA, then stimulated with media +/− 1 nM IL-5 (R&D Systems; Minneapolis, MN, USA) for indicated times at 37°C. Samples were diluted in ice-cold buffer A (20 mM Tris, pH 7.4, 137 mM NaCl, 1 mM EDTA, 1 mM Na3VO4, 20 mM β-glycerophosphate, 10 mM NaF, and [mammalian] protease inhibitor cocktail from Sigma), pelleted by centrifugation at 4°C, and lysed via vortexing in buffer A plus detergents (1% Triton X-100, 0.25% deoxycholate, and 0.1% SDS) at a concentration of 4×107 cells/mL.
For cyclin D3 and caspase 3, treated and stimulated PBEos were placed on ice, pelleted by centrifugation, and lysed in buffer B (1% NaDodSO4 [SDS]), 10 mM DTT, 0.5 mM Na3VO4, 1 mM EDTA, 10% glycerol, 10 mM Tris, pH 8.0) at a concentration of 4×107 cells/mL. Lysates were boiled for 5 minutes, sonicated 4 times, and held at −80°C for 15 minutes.
Following either lysis protocol, Western blotting was performed as previously reported [44]. Briefly, lysate insoluble material was removed via centrifugation, and lysates were loaded at 0.6–1×106 cells per well on 10%, 12.4%, or 15% SDS-PAGE gels as appropriate. Blots were incubated according to manufacturer instructions with appropriate antibodies: phospho-specific ERK1/2 [pY185/187], phospho-specific p38 [pTpY180/182] (Invitrogen; Eugene, OR, USA), phospho-specific STAT5 [pY694], phospho-specific STAT3 [pY705] (Cell Signaling; Billerica, MA), anti-C4/Actin (BD Biosciences; Franklin Lakes, NJ, USA), anti-cyclin D3 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), or anti-caspase 3 (Cell Signaling; Billerica, MA, USA) followed by appropriate HRP-conjugated secondary antibodies (Santa Cruz Biotechnology; Santa Cruz, CA, USA). Band quantification was performed using ImageJ software (National Institutes of Health, USA). Due to tight binding of phospho-specific antibodies and subsequent removal of protein upon stripping, actin was used as a loading control for all Western blots.
Cell Viability and Metabolic Activity
Short-term cellular viability was measured via trypan blue exclusion. Briefly, following 4 or 24 hour continuous treatment with MβCD or MβCD+Chol, a cell suspension was mixed 1∶1 with trypan blue and mixed gently via pipetting 20 times. Live cells (those excluding trypan blue) were counted using a hemocytometer and used to calculate percent viable compared to total cell count.
To assess metabolic activity, human PBEos pretreated 1 hour with media, MβCD, or MβCD+Chol, washed, and cultured in triplicated +/−100 pM IL-5 for 48 hours. Afterwards, Phenazine methosulfate (PMS) and MTS (3-[4,5-dimethylthiazol-2-yl]-5-[3-carboxymethoxyphenyl]-2-[4-sulfophenyl]-2H-tetrazolium [Promega; Madison, WI, USA]) were added (final concentrations 7.3 µg/mL and 0.23 mg/mL, respectively). The metabolism of MTS to formazan was quantified by measuring the OD at 490 nm after 1-hour incubation.
Isolation of RNA and subsequent qPCR
Purified PBEos were cultured 1 hour +/− MβCD or MβCD+Chol, then, stimulated with media +/−1 nM IL-5 for 4 hours. Total RNA was extracted (RNeasy Mini Kit [Qiagen; Valencia, CA, USA]), and the reverse transcription reaction was performed using the Superscript III system (Invitrogen/Life Technologies; Grand Island, NY, USA). mRNA expression was determined by qPCR using SYBR Green Master Mix (SABiosciences; Frederick, MD, USA). Human IL-1β specific primers (forward primer: TCGAGGCACAAGGCACAACAGG; reverse primer: CCATGGCTGCTTCAGACACTTGAGC) were used to quantify IL-1β mRNA levels, using the reference gene, β-glucuronidase to normalize. Data are expressed as fold change using the comparative cycle threshold (ΔΔCT) method as described previously [45], and values given are fold change (2-ΔΔCt) compared to the level in media-pretreated, non-stimulated eosinophils, which was set at 1 (n = 5).
Statistical Analysis
Mean fluorescence intensity, protein band densitometry, and OD measurements were compared among treatment groups (media, MβCD, MβCD+Chol) and stimulants (media +/− IL-5). Where appropriate, measures were log or square root-transformed to achieve normal distributions and homogeneity of variance for ANOVA. A two-sided p-value<0.05 was regarded as statistically significant.
Results
MβCD and MβCD+Chol alter eosinophil membrane cholesterol levels in a dose-dependent manner
To test the hypothesis that the level of membrane-bound cholesterol alters signaling in primary human PBEos, we manipulated cholesterol content in vitro utilizing MβCD and MβCD+2% Cholesterol (w/w) (MβCD+2%Chol). Empty MβCD is a cholesterol chelator and depletes membrane cholesterol from the cells, whereas MβCD preloaded with cholesterol can deposit exogenous cholesterol into the cell membrane. Filipin III (FIII) is a fluorescent polyene antibiotic that stoichiometrically binds membrane-integrated cholesterol in a 1∶1 ratio [46], and is detectable via flow cytometry. Therefore, FIII serves as powerful tool for rapidly comparing relative membrane cholesterol levels in cell populations [47]–[50]. To define dose-dependent changes in cellular membrane cholesterol, FIII fluorescence was measured in purified human PBEos treated with step-wise doses of MβCD or MβCD+2%Chol. MβCD-treated human PBEos displayed dose-dependent decreases of FIII fluorescence relative to media-controls, reflecting a loss of membrane cholesterol. Treating PBEos with increasing concentrations of MβCD+2%Chol saturated cellular membrane cholesterol levels, reflected by a plateau in FIII fluorescence (Figure 1A). FIII concentration was not a limiting factor since doubling the FIII concentration did not further increase the FIII fluorescent signal after any of the treatment conditions (data not shown). Relative to media control, treatment with 5 mg/mL MβCD significantly decreased FIII median fluorescent intensity (MFI) by ∼25 (p<0.01, n = 15; Figure 1B and C,). Conversely, 5 mg/mL MβCD+2%Chol treatment significantly increased FIII MFI ∼2× (Figure 1B, C) (p<0.001, n = 15). These data indicate MβCD treatment quantitatively alters membrane cholesterol level in PBEos. Neither 5 mg/mL MβCD nor MβCD+2%Chol treatment altered PBEos size or density as indicated by forward and side scatter, respectively (Figure S1). Based on trypan blue viability measurements, neither 5 mg/mL MβCD or MβCD+2%Chol significantly altered cell viability 4 and 24 hours post-treatment (data not shown), and these doses were therefore selected as general working concentrations for the remainder of the study.
IL-5 receptor subunit surface expression was independent of membrane cholesterol content
Altering receptor surface expression levels is one way in which cells dynamically alter sensitivity to extracellular ligands, and hence subsequent intracellular signaling. To determine whether altered membrane cholesterol levels influence PBEos surface expression of the IL-5-specific receptor subunit α (IL-5Rα) and the IL-5 family common β chain subunit (IL-5Rβ), which dimerize to form the functional IL-5 receptor, respective surface expression levels were measured via flow cytometry in unpermeabilized cells following treatment with MβCD or MβCD+2%Chol (Figure 2A and B respectively). No significant changes in MFI were observed across treatments for IL-5Rα (Figure 1C, n = 9, p>0.05) or IL-5Rβ (Figure 1C, n = 3, p>0.05). These data indicate that neither MβCD nor MβCD+2%Chol treatment altered IL-5Rα or IL-5Rβ surface expression.
Figure 2. Membrane cholesterol manipulation did not alter IL-5Rα or β surface expression.
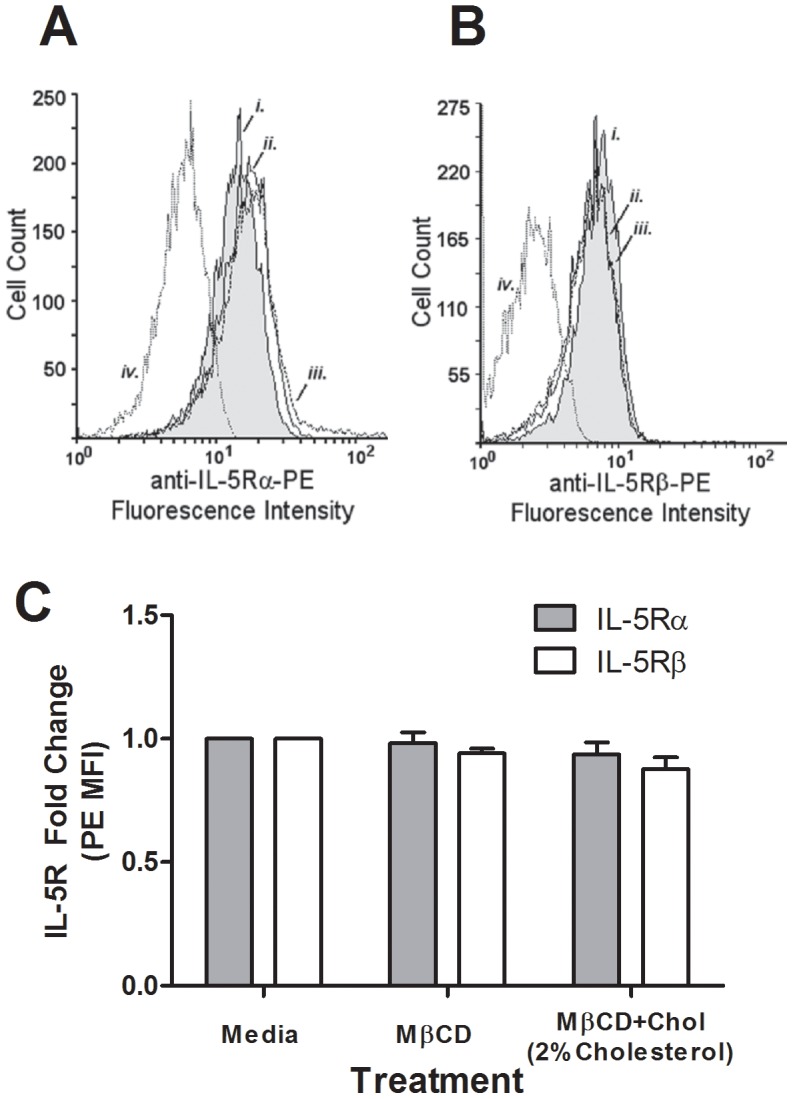
Representative histograms of PBEos treated 1 hour with media, MβCD, or MβCD+2%Chol (3–5×105 per treatment) then stained with (A) PE-conjugated anti-IL-5Rα (n = 9) or (B) PE-conjugated anti-IL-5Rβ (n = 3). (C) Pooled data from receptor staining: grey bars, IL-5Rα (n = 9), open bars, IL-5Rβ (n = 3). Error bars indicate SEM, p-values from one-way ANOVA. Unmarked comparisons were non-significant.
IL-5 induced STAT phosphorylation was independent of cellular membrane cholesterol
Given unchanged IL-5Rα and IL-5Rβ surface expression in human PBEos following cholesterol manipulation, alterations in downstream IL-5-induced signaling would likely be due to the effects cellular cholesterol levels have on proteins that transduce signal from the receptor through intracellular pathways rather than total receptor expression. We therefore assessed cholesterol-mediated changes in activation of the JAK/STAT, MEK/ERK, and p38-MAPK pathways by quantifying the phosphorylated (activated) state of representative signaling proteins from each pathway after cholesterol deposition or depletion. Unlike MAPK pathways, JAK/STAT signal transduction is mediated by soluble proteins, and functions independently of membrane-anchored signaling proteins localized to CRMMs. We therefore predicted that cholesterol modulation would not alter JAK/STAT signaling. As indicated in Figure 3A–C, neither MβCD (cholesterol depletion) nor MβCD+2%Chol (cholesterol addition) altered IL-5-stimulated phosphorylation of STAT5 (p-STAT5, n = 15) or STAT3 (p-STAT3, n = 5). These data indicate that signaling through the JAK/STAT pathway in PBEos was independent of membrane cholesterol levels.
Figure 3. Membrane cholesterol manipulation did not affect IL-5-induced p-STAT5 or p-STAT3.
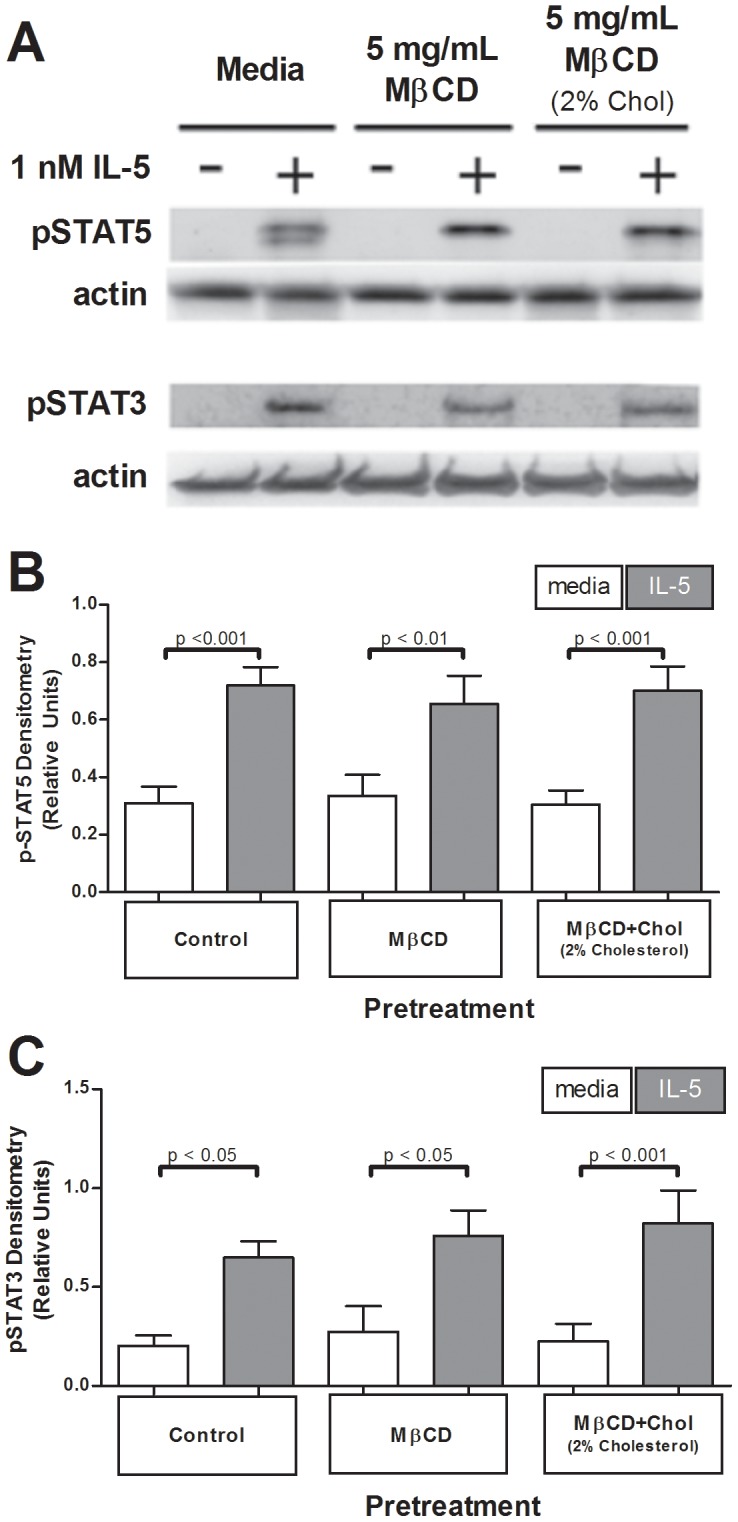
One million eosinophils per treatment pretreated 1 hour with media, 5 mg/mL MβCD, or 5 mg/mL MβCD+2%Chol, were stimulated +/− IL-5 for 15 min. (A) Samples were immunoblotted for active STAT5 or STAT3. (B) Data were pooled from 19 experiments (p-STAT5) or (C) 5 experiments (p-STAT3) and normalized to actin loading; pooled data were square root transformation for normalcy. Error bars indicate SEM, p-values from one-way ANOVA. Unmarked comparisons were non-significant.
IL-5-induced ERK and p38 phosphorylation are selectively disrupted by altered membrane cholesterol levels in human eosinophils
Our model predicted cholesterol depletion would attenuate IL-5-induced MAPK signaling (e.g., MEK/ERK and p38), because both pathways are activated by membrane-anchored kinases. Human PBEos pretreated with media, MβCD, or MβCD+2%Chol were stimulated with IL-5 for 15 min, and lysates were immunoblotted for phosphorylated ERK1/2 and p38. Figure 4A and B demonstrate MβCD pretreatment attenuated IL-5-induced ERK1/2 phosphorylation compared to media-pretreated, IL-5-stimulated controls (p<0.001, n = 13; Figure 4B). MβCD pretreatment similarly attenuated an IL-5-induced increase in p-p38 levels (p<0.05, n = 16; Figure 4A and C). Cholesterol treatments did not alter total ERK1/2 or p38 protein expression levels (data not shown). These data demonstrate that cholesterol depletion decreased IL-5-induced activation of MEK/ERK and p38 pathways.
Figure 4. Cholesterol depletion attenuated IL-5-induced phosphorylated ERK1/2 and p38, while cholesterol addition enhanced basal p38 phosphorylation.
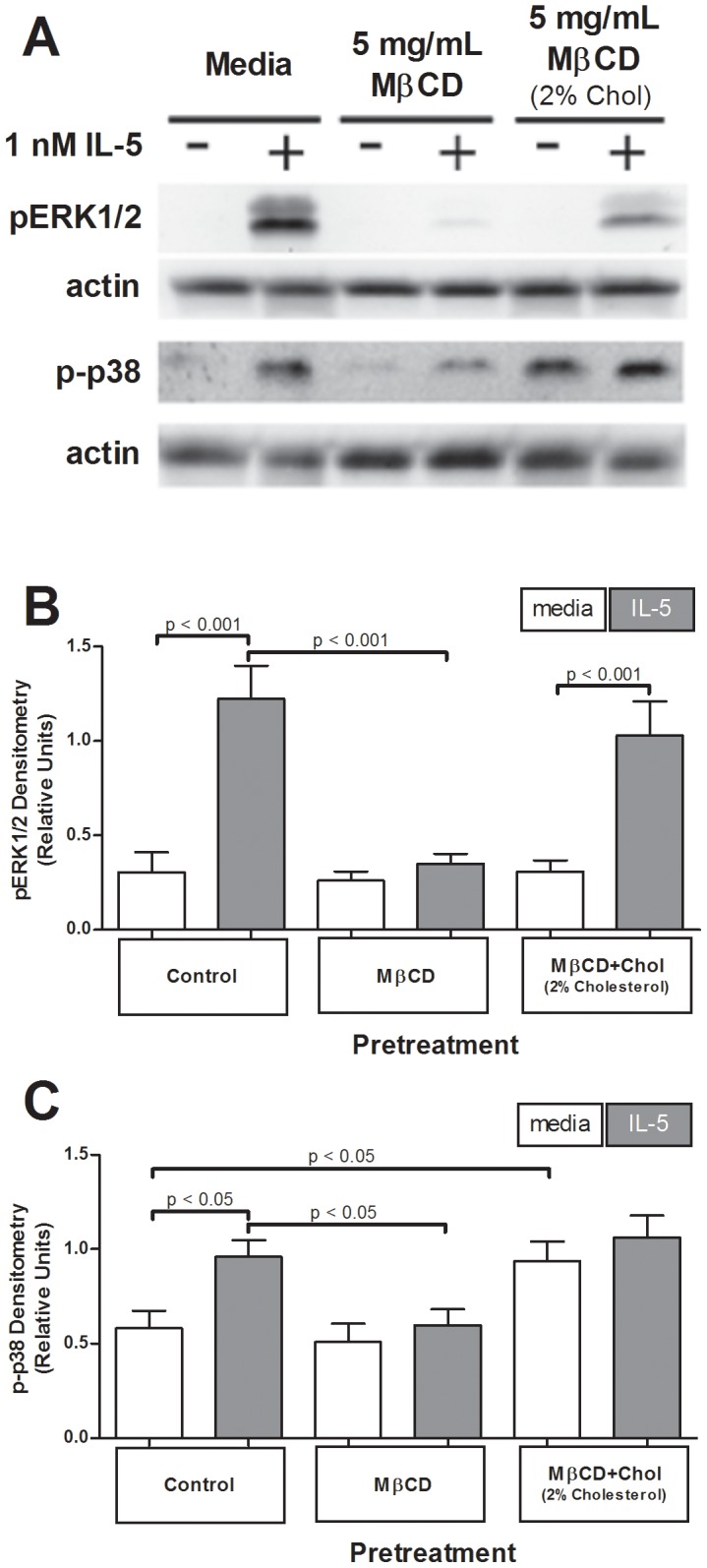
One million eosinophils per treatment pretreated 1 hour with media, 5 mg/mL MβCD, or 5 mg/mL MβCD+2%Chol, were stimulated +/− IL-5 for 15 min. (A) Samples were immunoblotted for active ERK1/2 or p38. (B) 14 experiments (ERK1/2) or (C) 16 experiments (p38) normalized to actin loading; pooled data were square root transformation for normalcy. Error bars indicate SEM, p-values from one-way ANOVA. Unmarked comparisons were non-significant.
To determine whether adding exogenous cholesterol, predicted to enhance CRMM formation, would have the opposite effect and increase ERK/p38 phosphorylation, PBEos were treated with MβCD+2%Chol and lysates assessed for p-ERK/p38. Pretreatment with MβCD+2%Chol did not significantly change total levels of IL-5-induced p-ERK1/2 or p-p38 compared to media-treated, IL-5 stimulated controls (p>0.05, n = 13, n = 16, respectively; Figure 4A–C). Interestingly, the addition of exogenous cholesterol increased basal p38 phosphorylation levels relative to media-pretreated, media-stimulated control eosinophils (p<0.05, n = 16; Figure 4A and C). Furthermore, while IL-5 continued to potentiate p-ERK1/2 relative media-control after MβCD+2%Chol (3× increase, p<0.001, n = 13, Figure 4A and B), cholesterol addition limited the dynamic range of the p-p38 response. The increase in basal p38 phosphorylation eliminated the IL-5-induced increase in p-p38 in MβCD+2%Chol-treated cells, (p>0.05, n = 16; Figure 4A and C). Therefore the fact that PBEos receiving exogenous cholesterol exhibited the same levels of p38 phosphorylation as control cells post-IL-5 was due to elevated basal p38 phosphorylation, rather than IL-5 stimulation. These data indicate that increasing membrane cholesterol levels did not alter overall phosphorylation of MEK/ERK pathway proteins following IL-5 stimulation, but increased basal p38 phosphorylation and eliminated the p38 response to IL-5.
To control for effects of exposing cells to MβCD, PBEos were treated with MβCD+1% Cholesterol (MβCD+1%Chol), which resulted in a no-net change in eosinophil membrane cholesterol levels as quantified via FIII signal in flow cytometry (n = 5; Figure S2A and B). PBEos pretreated with MβCD+1%Chol exhibited no differences in basal or IL-5-stimulated p-p38 levels compared with media pretreated, IL-5-stimulated controls (n = 5; Figure S2C and D). MβCD+1%Chol treatment similarly did not alter total STAT3 or STAT5 protein expression levels (data not shown).
While p38 phosphorylation was clearly regulated by membrane cholesterol, MβCD treatment alone cannot distinguish whether the phosphorylation events are specifically dependent upon lipid raft organization, or total cholesterol content. To begin to separate these mechanisms, we treated cells with eicosapentaenoic acid (EPA), a polyunsaturated fatty acid which can redistribute cholesterol from lipid rafts across the membrane, and disrupts nascent lipid raft architecture [51]–[54]. If the regulation of p38 phosphorylation was solely dependent upon total membrane cholesterol, EPA treatment was predicted to have no effect. Changes in p38 phosphorylation following EPA treatment, however, would indicate the signaling event is dependent upon lipid raft organization. PBEos were cultured overnight (18 hours) in the presence or absence of 33 uM EPA, followed by 15 min. stimulation with 1 nM IL-5. Western blots of lysates probed with p-p38 antibodies revealed that EPA treatment increased basal p38 phosphorylation in a manner parallel to MβCD+2%Chol (Fig. 4), such that there was no IL-5 induced increase, and total p-p38 levels were similar to media/IL-5 treated cells (Fig. S3A, B, n = 3). As a control, lysates were also probed for p-STAT5; EPA did not change IL-5-induced increases in p-STAT5 (Fig. S3A, C, n = 3), consistent with the fact this pathway is regulated by soluble proteins. Baseline levels of p-STAT5 were undetectable in these experiments, and therefore not included in quantification (Figs. S3A, C). These data demonstrate that p38 phosphorylation was dependent upon lipid raft architecture of the plasma membrane, independently of total cholesterol content.
Reduced cellular membrane cholesterol attenuated IL-5-induced increase in IL-1β mRNA
Eosinophils can be stimulated to produce and release IL-1β in a MAPK-dependent manner [55], [56]. IL1β mRNA contains known AU-rich elements (ARE) that are well-defined cis-elements in the 3′ untranslated region of mRNAs that are controlled by ERK1/2 in eosinophils and responsible for mRNA stabilization and accumulation [57], [58]. As cholesterol depletion decreased p-ERK1/2 levels (MAPK signaling, Fig. 4), we tested the hypothesis MβCD would cause a concomitant reduction in IL-1β mRNA expression induced by IL-5. PBEos pretreated with MβCD expressed significantly less IL-5-stimulated IL-1β mRNA relative to media pretreated, IL-5-stimulated controls (p<0.05, n = 5; Figure 5). Cells treated with MβCD +1%Chol for a no net cholesterol change responded to IL-5 stimulation with increases in IL-1β mRNA levels (p<0.01 for IL-5 stimulation) similar to media-treated controls (no difference with p>0.05, n = 5; Figure 5). Pretreatment with MβCD +2%Chol to increase membrane cholesterol similarly did not alter basal levels or IL-5 induced IL-1β mRNA compared with control (p<0.05 for IL-5 induction). These data are consistent with the reduction in p-ERK1/2 and p-p38 following cholesterol depletion (4), and a model in which IL-1β mRNA production is regulated by MAPK signaling.
Figure 5. Cholesterol depletion decreased IL-5-induced IL-1β mRNA.

One million eosinophils per treatment were pretreated 1 hour with media, 5 mg/mL MβCD, 5 mg/mL MβCD+1%Chol, or 5 mg/mL MβCD+2%Chol, then stimulated +/−1 nM IL-5 for 4 hours. Sample lysates were assessed for IL-1β mRNA. Pooled data from 5 experiments display fold change from media-pretreated, media-stimulated; error bars indicate SEM, p-values from one-way ANOVA. Unmarked comparisons were non-significant.
Elevated cellular membrane cholesterol down-regulated IL-5-dependent cyclin-D3 expression
As the human cyclin D3 gene has upstream consensus sequences for both STAT and MEK/ERK-regulation activating protein 1 promoter regions [59], we tested the hypothesis that cyclin D3 expression would be sensitive to cholesterol manipulation. Cholesterol depletion (MβCD) did not significantly alter IL-5-stimulated increase in cyclin D3 protein expression compared to media-pretreated controls (Figure 6A and B), though protein levels trended upward. The addition of membrane cholesterol via MβCD+2%Chol-pretreatment, however, dramatically reduced IL-5-induced cyclin D3 protein expression to almost undetectable levels (p<0.001, n = 5; Figure 6A and B). MβCD+1%Chol (no net cholesterol change) had no effect on IL-5-induced expression of cyclin D3 compared to media pretreated controls (p>0.05, n = 5), resulting in increased cyclin D3 protein expression following IL-5 stimulation (p<0.05, n = 5; Figure 6A and B) similar to the trend seen in control cells.
Figure 6. Cholesterol addition reduced IL-5-induced cyclin D3 expression.
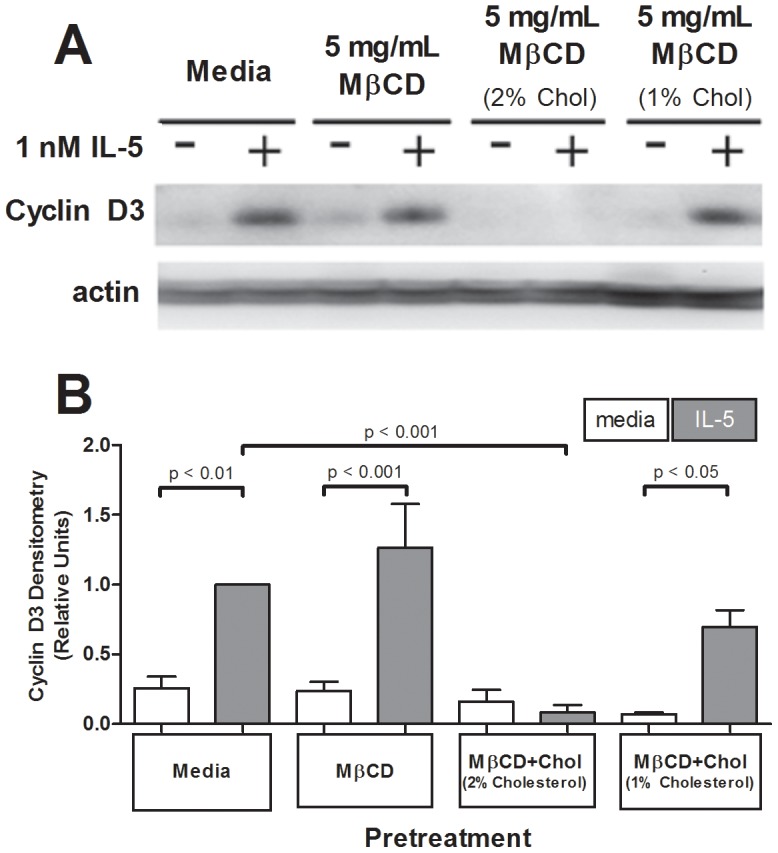
One million eosinophils per treatment were pretreated 1 hour with media, 5 mg/mL MβCD, 5 mg/mL MβCD+2%Chol, or 5 mg/mL MβCD+1%Chol, washed, then stimulated with media or 1 nM IL-5 for 16 hours. (A) Sample lysates were immunoblotted for cyclin D3. (B) Pooled data from 5 experiments represented as ratio relative to media pretreated (normalized to actin loading), IL-5-stimulated. Error bars indicate SEM, p-values from one-way ANOVA. Unmarked comparisons were non-significant.
Elevated cellular membrane cholesterol attenuated IL-5-induced metabolic activity and enhanced caspase 3 activation despite IL-5-stimulation
To determine how membrane cholesterol manipulation regulates eosinophil survival, we used MTS assays to quantify metabolic activity as a survival indicator. As shown in Figure 7A, neither cholesterol reduction (MβCD pretreatment) nor no-cholesterol change (MβCD+1%Chol pretreatment) altered IL-5-induced increase in metabolic activity at 48 hours post-treatment (p<0.001 each, n = 4). In contrast, elevated membrane cholesterol (MβCD+2%Chol pretreatment) attenuated IL-5-induced 48-hour survival relative to media pretreatment (p<0.001, n = 4; Figure 7A). Loss of IL-5-induced cellular metabolism, despite consistent STAT phosphorylation (Fig. 3 above), suggests an alternate, unidentified mechanism exists regulating the otherwise classic IL-5-induced eosinophil survival.
Figure 7. Cholesterol addition reduced IL-5-induced eosinophil survival and enhanced caspase 3 cleavage.

One million eosinophils per treatment were pretreated 1 hour with media, 5 mg/mL MβCD, 5 mg/mL MβCD+1%Chol or 5 mg/mL MβCD+2%Chol, washed, then stimulated with media or 100 pM IL-5. (A) Viability was assessed via MTS assay after 48 hours. Pooled data from 4 experiments represented as fold change from media pretreated, IL-5-stimulated. (B) After 24 hours, pre-treated and stimulated PBEos were lysed and immunoblotted for caspase 3 (pro- and active/cleaved form). (C) Pooled data from 5 experiments represented as a ratio of pro-caspase 3 to cleaved caspase 3. Error bars indicate SEM, p-values from one-way ANOVA. Unmarked comparisons were non-significant.
To determine whether this loss of survival corresponded with activation of apoptotic pathways, we quantified caspase 3 cleavage (activation) as a ratio of pro- to cleaved-caspase 3 in lysates from PBEos harvested 24 hours post-cholesterol treatment and subsequent IL-5 stimulation. In media pre-treated cells, 24 hour IL-5 stimulation significantly elevated the pro∶cleaved caspase 3 ratio compared non-stimulated-controls (p<0.05, n = 5; Figure 7B and C), indicating decreased caspase 3 cleavage, consistent with an IL-5 induced increase in cell survival. IL-5 stimulation likewise elevated the pro∶cleaved caspase 3 ratio after MβCD pretreatment (p<0.05, n = 5; Figure 7C) or MβCD+1%Chol pretreatment (p<0.001, n = 5; Figure 7C). Pretreatment with MβCD+2%Chol to increase membrane cholesterol, however, resulted in nearly complete cleavage of pro-caspase 3 in both IL-5 stimulated and unstimulated conditions (Figure 7B), which significantly decreased pro∶cleaved ratio relative to media pretreated control cell lysates (p<0.05, n = 5; Figure 7C). These data indicate cholesterol addition stimulated caspase 3 activation, suggesting exogenous cholesterol increases PBEos cell death regardless of the generally pro-survival IL-5 stimulus and retained STAT activation.
Discussion
To determine whether eosinophil inflammatory responses are sensitive to cholesterol regulation, we defined the effects that altering cell membrane cholesterol content has on specific eosinophil signaling pathways. Depleting cellular cholesterol in vitro attenuated IL-5-induced p38 and MEK/ERK phosphorylation, and IL-1β mRNA increases in human eosinophils without altering surface expression levels of the IL-5 receptor. Exogenous cholesterol supplementation elevated basal p38 activation, and attenuated IL-5-induced increases in cyclin D3 protein expression and total cellular metabolic activity. Neither manipulation altered IL-5-induced JAK/STAT signaling, as assayed by STAT3 and STAT5 phosphorylation, importantly demonstrating there was not a global downregulation of eosinophil signaling. These data suggest membrane cholesterol composition selectively regulates IL-5-induced signaling events that are dependent upon membrane-anchored signaling proteins, with further specificity highlighted by the differential responses between MEK/ERK and p38. Future studies will identify the proteins that confer cholesterol sensitivity to the MAPK pathways, with likely candidates including membrane-anchored Raf and Lyn, which act upstream of p38 and ERK1/2.
Selective, cholesterol-dependent sensitivity of the MEK/ERK pathway in response to IL-5 contrasts cholesterol-independent JAK/STAT signaling, and is consistent with the study by Lei et al demonstrating the localization of IL-5Rs to membrane microdomains defines which intracellular signaling proteins are bound to the receptor [43]. Completely unexpected, however, were our findings that the addition of exogenous cholesterol dramatically increased basal p38 phosphorylation to levels equivalent to IL-5 stimulation. These data suggest that eosinophils exhibiting elevated membrane cholesterol as part of a disease state will functionally express constitutively active p38. In addition, such eosinophils are predicted to manifest decreased responsiveness to IL-5 as the pre-phosphorylated p38 precluded further IL-5 activation.
IL-5 stimulation triggers events that promote long-term survival of eosinophils, which otherwise undergo significant levels of apoptosis within 48 hours of in vitro culture [60]. Survival contributes to overall eosinophil inflammatory capacity. Our data demonstrate, however, that cholesterol supplementation increased caspase 3 cleavage despite IL-5 stimulation, suggesting an additional, unidentified protein(s) or pathway(s) regulates eosinophil apoptotic signaling in a cholesterol-dependent manner. It is possible that cholesterol (non-cytokine)-induced phospho-p38 (active p38) is responsible for attenuating IL-5-mediated eosinophil survival, as previous studies demonstrate that p38 promotes caspase 3 cleavage under specific conditions, leading to apoptosis [61]. Our data predict a model in which p38 activated by a non-cytokine source triggers apoptosis, regardless of IL-5 stimulation and persistent JAK/STAT survival signals. This model further predicts that inhibiting basal p38 phosphorylation in the presence of exogenous cholesterol would restore IL-5-induced cell survival.
Phosphorylated p38 may also be responsible for the decrease in cyclin D3 protein levels observed in cells treated with exogenous cholesterol. While the functional role of cyclin D3 in terminally differentiated and tissue human eosinophils is unclear, recent studies demonstrate function likely extends beyond cell cycle control [19], [28]. Future experiments may determine whether cholesterol-induced phospho-p38 promotes cyclin D3 protein degradation or prevents IL-5 mediated increased production, and may provide a mechanism for fine-tuning IL-5-induced cyclin D3 protein expression.
Additional studies will also be required to determine the mechanism by which exogenous cholesterol increased basal p38 phosphorylation. One potential mechanism may be cytokine-independent interactions between p38 and the scaffold protein TAB1, which can activate p38 in PBEos [62]. The cellular consequences of increasing baseline levels of p-p38 in huPBEos similarly remains to be determined, but a genome study in mouse embryonic kidney cells found that p38 activation up-regulates a distinct array of genes such that each subset of downstream effectors is specific to the upstream p38-activation source (i.e., osmostress, cytokine, or protein synthesis) [63]. The genomic study suggests cholesterol-based p38 activation regulates an array of genes distinct from IL-5 induced p38 activation.
IL-1β is a key inflammatory cytokine, traditionally viewed as a product of monocytes and macrophages, but recent studies have demonstrated human eosinophils also have the capacity to release active IL-1β that increases IL-17 expression by CD4+ T lymphocytes [45]. IL-1-β production and release are controlled at many different levels. IL-1β mRNA accumulation depends on its stability and the MAP kinase activity [64]. Our data support that reduced MAP kinase activation due to depletion of cholesterol subsequently decreases the level of IL-1β mRNA. Synthesis and extracellular release of mature IL-1β requires two coordinate signals in monocytes and macrophages. The second signal to complement IL-5 for the production and the release of a mature IL-1β protein from eosinophils remains unknown. Controlled cholesterol depletion may provide an excellent tool for dissecting the mechanism of IL-1β expression and secretion in eosinophils, without altering other key cellular aspects, including viability.
One limitation of the current approach is that while MβCD is considered a cholesterol chelator, particularly given the dose and incubation times used in the study, MβCD treatment may deplete cholesterol from the entire cell membrane rather than solely cholesterol-rich lipid rafts. At high doses (typically >10 mM), MβCD may also extract phospholipids and membrane proteins [65]. In a complimentary approach, we treated cells overnight with eicosapentaenoic acid (EPA), a polyunsaturated fatty acid that alters lipid raft structure and composition without depleting total cellular cholesterol. Treating cells with EPA therefore distinguishes whether MβCD -induced changes in cell signaling are due to alterations in total membrane cholesterol or membrane organization. EPA treatment resulted in increased basal p38 phosphorylation and a concomitant loss of IL-5 responsiveness in eosinophils, mimicking the effects of increasing cholesterol with MβCD +2%Chol. Whether EPA treatment redistributes cholesterol across the membrane, eliminating lipid rafts, or in fact forms larger, more ordered lipid rafts appears to be condition- and cell type-dependent [51]–[54], [66]. Since cholesterol addition enhances the formation of cholesterol-rich lipid rafts [67], [68], these data suggest that 33 µM, EPA likely increases the size and organization of lipid rafts in human eosinophils, a hypothesis that can be tested in future studies. The data furthermore demonstrate that the changes in p38 phosphorylation induced by cholesterol manipulation are due to altered membrane structure, rather than being dependent solely upon total cholesterol concentration. Future studies may refine the specific role of membrane architecture in eosinophil signaling, and potentially distinguish various classes of cell membrane microdomains including sphingolipid-rich, caveolae, tetraspanin-rich, and galectin lattices [69]–[72].
The cholesterol-mediated regulation of eosinophil inflammatory capacity may reflect physiological conditions in which there is (1) enhanced HDL-packaging and cholesterol export, (2) hypercholesterolemia, or (3) hypercholesterolemia treated utilizing fish oil or cholesterol-lowering drugs like stating. Statins can reduce high serum cholesterol levels (clinically defined as >240 mg/dl) to levels as low as 70 mg/dl, and circulating eosinophils are directly exposed to those serum cholesterol concentrations. Though there is not currently a defined stoichiometry between circulating and cell membrane cholesterol levels, tumor cells isolated from mice fed a high-cholesterol diet for 4 weeks had a cholesterol/protein ratio (ug/mg) twice that of cells from animals fed a normal diet [73]. In the same study, cultured cells treated with 10 µM Simvastatin had a 3× reduction in the cholesterol/protein ratio. The 25% reduction in eosinophil membrane cholesterol achieved with 5 mg/mL MβCD, and the 2× increase with our 2% exogenous cholesterol treatment therefore likely fall within a clinically-relevant physiological range. It is reasonable to predict that eosinophils isolated from human subjects over a range of normal and relevant cholesterol disease states would exhibit concomitant signaling changes that correlate with circulating cholesterol levels and parallel the data presented here.
Our data demonstrate human eosinophil signaling pathways are selectively sensitive to the levels of cellular membrane cholesterol, likely altering membrane architecture. Numerous diseases linked to loss of optimal cholesterol homeostasis involve eosinophils as part of the inflammatory response and disease progression, including atherosclerosis and Alzheimer's [1]–[10], and recent studies have assessed the relationship between hypercholesterolemia and low levels of serum high density lipoprotein (HDL) and risk for asthma [35], [37], [40], [41]. Future studies will define how the eosinophil responses to cholesterol levels relate to human disease states.
Supporting Information
Cholesterol manipulation did not alter eosinophil size or density as determined via flow cytometry. Scatter plots of side scatter versus forward scatter of eosinophils pretreated 1 hour with i. media, ii. 5 mg/mL MβCD, or iii. 5 mg/mL MβCD+2%Chol (3–5×105 per treatment).
(TIF)
Pretreatment with reduced percentage of cholesterol in MβCD+Chol (1% cholesterol) resulted in no net-change in PBEos cellular membrane cholesterol. (A–B) Eosinophils were treated 1 hour with media, 5 mg/mL MβCD, 5 mg/mL MβCD+2%Chol, or 5 mg/mL MβCD+1%Chol (3–5×105 per treatment). Cells were fixed, stained with 50 µg/mL Filipin III for cholesterol, then analyzed via flow cytometry. (A) Representative histogram from n = 5 experiments of cells stained with FIII after treatment with i. Media, ii. MβCD, iii. MβCD+2%Chol, or iv. MβCD+1%Chol. (B) Quantification of 5 independent experiments of FIII-stained eosinophils after treatment with concentrations indicated above. Error bars indicate SEM, p-values from one-way ANOVA. (C–D) One million eosinophils per treatment were pretreated 1 hour with media, 5 mg/mL MβCD +2%Chol, or 5 mg/mL MβCD +1%Chol, then stimulated +/− IL-5 for 15 min. (C) Samples were immunoblotted for active p38. (D) Pooled data (n = 5) normalized to actin loading, error bars indicate SEM, p-values from non-parametric one-way ANOVA. Irrelevant lanes were digitally removed from between lanes 2 and 3. Unmarked comparisons were non-significant.
(TIF)
EPA treatment increases basal p38 phosphorylation, eliminating an IL-5-stimulated increase. One million eosinophils per treatment pretreated 18 hours with media or 33 uM EPA were stimulated +/− IL-5 for 15 min. (A) Western blots were probed for phosphorylated STAT5 or p38, and reprobed with actin antibodies as a loading control. Example blot is representative of n = 3 experiments. (B) Graph plots p-p38 band density normalized to actin as mean +/− SE for each treatment. Data are pooled from 3 experiments, and ctl + IL-5 samples were set as 1. (C) Graph plots p-STAT5 band density normalized to actin as mean +/− SE for each treatment. Data are pooled from 3 experiments, and only IL-5-stimulated bands were quantified as p-STAT5 was undetectable in lysates from unstimulated cells.
(TIF)
Acknowledgments
The authors are grateful to our blood donors, and we also thank Sameer Mathur, M.D., Ph.D., for oversight of the Laboratory Core that recruited and screened subjects and purified blood eosinophils; Elizabeth Schwantes, B.S., and Paul Fichtinger, B.S., for eosinophil purification; Larissa DeLain, B.S., for her technical support. We would also like to thank Dr. Monica Gavala for invaluable editorial input. This work is dedicated to the memory of Dr. Paul J. Bertics, who died December 22, 2011.
Funding Statement
This work was supported by the National Institutes of Health (grants P01HL088594, 5T32DK007665, and UL1TR000427). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Aceves SS, Broide DH (2008) Airway fibrosis and angiogenesis due to eosinophil trafficking in chronic asthma. Current molecular medicine 8: 350–358. [DOI] [PubMed] [Google Scholar]
- 2. Bochner BS, Busse WW (2005) Allergy and asthma. The Journal of allergy and clinical immunology 115: 953–959. [DOI] [PubMed] [Google Scholar]
- 3. Busse WW, Lemanske RF Jr (2001) Asthma. The New England journal of medicine 344: 350–362. [DOI] [PubMed] [Google Scholar]
- 4. Rothenberg ME, Hogan SP (2006) The eosinophil. Annual review of immunology 24: 147–174. [DOI] [PubMed] [Google Scholar]
- 5. Hou LF, Lloyd-Jones DM, Ning HY, Huffman MD, Fornage M, et al. (2013) White blood cell count in young adulthood and coronary artery calcification in early middle age: coronary artery risk development in young adults (CARDIA) study. European Journal of Epidemiology 28: 735–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oda E, Kawai R, Aizawa Y (2012) Lymphocyte count was significantly associated with hyper-LDL cholesterolemia independently of high-sensitivity C-reactive protein in apparently healthy Japanese. Heart and Vessels 27: 377–383. [DOI] [PubMed] [Google Scholar]
- 7. Siddiqui S, MacGregor MS, Glynn C, Roditi G, Deighan CJ (2005) Factors predicting outcome in a cohort of patients with atherosclerotic renal artery disease diagnosed by magnetic resonance angiography. American Journal of Kidney Diseases 46: 1065–1073. [DOI] [PubMed] [Google Scholar]
- 8. Shim WS, Kim HJ, Kang ES, Ahn CW, Lim SK, et al. (2006) The association of total and differential white blood cell count with metabolic syndrome in type 2 diabetic patients. Diabetes Research and Clinical Practice 73: 284–291. [DOI] [PubMed] [Google Scholar]
- 9. Liu GT, Hwang CS, Hsieh CH, Lu CH, Chang SLY, et al. (2013) Eosinophil-Derived Neurotoxin Is Elevated in Patients with Amyotrophic Lateral Sclerosis. Mediators of inflammation [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vidal R, Ghetti B (2011) Characterization of Amyloid Deposits in Neurodegenerative Diseases. In: Manfredi G, Kawamata H, editors. Neurodegeneration: Methods and Protocols. pp. 241–258. [DOI] [PubMed] [Google Scholar]
- 11. Sedgwick JB, Calhoun WJ, Vrtis RF, Bates ME, McAllister PK, et al. (1992) Comparison of airway and blood eosinophil function after in vivo antigen challenge. Journal of immunology 149: 3710–3718. [PubMed] [Google Scholar]
- 12. Sedgwick JB, Calhoun WJ, Gleich GJ, Kita H, Abrams JS, et al. (1991) Immediate and late airway response of allergic rhinitis patients to segmental antigen challenge. Characterization of eosinophil and mast cell mediators. The American review of respiratory disease 144: 1274–1281. [DOI] [PubMed] [Google Scholar]
- 13. Pazdrak K, Olszewska-Pazdrak B, Stafford S, Garofalo RP, Alam R (1998) Lyn, Jak2, and Raf-1 kinases are critical for the antiapoptotic effect of interleukin 5, whereas only Raf-1 kinase is essential for eosinophil activation and degranulation. The Journal of experimental medicine 188: 421–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Monahan J, Siegel N, Keith R, Caparon M, Christine L, et al. (1997) Attenuation of IL-5-mediated signal transduction, eosinophil survival, and inflammatory mediator release by a soluble human IL-5 receptor. Journal of immunology 159: 4024–4034. [PubMed] [Google Scholar]
- 15. Bates ME, Liu LY, Esnault S, Stout BA, Fonkem E, et al. (2004) Expression of interleukin-5- and granulocyte macrophage-colony-stimulating factor-responsive genes in blood and airway eosinophils. American journal of respiratory cell and molecular biology 30: 736–743. [DOI] [PubMed] [Google Scholar]
- 16. Bates ME, Green VL, Bertics PJ (2000) ERK1 and ERK2 activation by chemotactic factors in human eosinophils is interleukin 5-dependent and contributes to leukotriene C(4) biosynthesis. The Journal of biological chemistry 275: 10968–10975. [DOI] [PubMed] [Google Scholar]
- 17. Ip WK, Wong CK, Wang CB, Tian YP, Lam CW (2005) Interleukin-3, -5, and granulocyte macrophage colony-stimulating factor induce adhesion and chemotaxis of human eosinophils via p38 mitogen-activated protein kinase and nuclear factor kappaB. Immunopharmacology and immunotoxicology 27: 371–393. [DOI] [PubMed] [Google Scholar]
- 18. Kankaanranta H, De Souza PM, Barnes PJ, Salmon M, Giembycz MA, et al. (1999) SB 203580, an inhibitor of p38 mitogen-activated protein kinase, enhances constitutive apoptosis of cytokine-deprived human eosinophils. The Journal of pharmacology and experimental therapeutics 290: 621–628. [PubMed] [Google Scholar]
- 19. Stout BA, Bates ME, Liu LY, Farrington NN, Bertics PJ (2004) IL-5 and granulocyte-macrophage colony-stimulating factor activate STAT3 and STAT5 and promote Pim-1 and cyclin D3 protein expression in human eosinophils. Journal of immunology 173: 6409–6417. [DOI] [PubMed] [Google Scholar]
- 20. Wong CK, Zhang JP, Ip WK, Lam CW (2002) Activation of p38 mitogen-activated protein kinase and nuclear factor-kappaB in tumour necrosis factor-induced eotaxin release of human eosinophils. Clinical and experimental immunology 128: 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhu X, Jacobs B, Boetticher E, Myou S, Meliton A, et al. (2002) IL-5-induced integrin adhesion of human eosinophils caused by ERK1/2-mediated activation of cPLA2. Journal of leukocyte biology 72: 1046–1053. [PubMed] [Google Scholar]
- 22. Zhu Y, Bertics PJ (2011) Chemoattractant-induced signaling via the Ras-ERK and PI3K-Akt networks, along with leukotriene C4 release, is dependent on the tyrosine kinase Lyn in IL-5- and IL-3-primed human blood eosinophils. Journal of immunology 186: 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Andina N, Didichenko S, Schmidt-Mende J, Dahinden CA, Simon HU (2009) Proviral integration site for Moloney murine leukemia virus 1, but not phosphatidylinositol-3 kinase, is essential in the antiapoptotic signaling cascade initiated by IL-5 in eosinophils. The Journal of allergy and clinical immunology 123: 603–611. [DOI] [PubMed] [Google Scholar]
- 24. Gozgit JM, Bebernitz G, Patil P, Ye M, Parmentier J, et al. (2008) Effects of the JAK2 inhibitor, AZ960, on Pim/BAD/BCL-xL survival signaling in the human JAK2 V617F cell line SET-2. The Journal of biological chemistry 283: 32334–32343. [DOI] [PubMed] [Google Scholar]
- 25. Ge B, Xiong X, Jing Q, Mosley JL, Filose A, et al. (2003) TAB1beta (transforming growth factor-beta-activated protein kinase 1-binding protein 1beta), a novel splicing variant of TAB1 that interacts with p38alpha but not TAK1. The Journal of biological chemistry 278: 2286–2293. [DOI] [PubMed] [Google Scholar]
- 26. McDermott EP, O'Neill LA (2002) Ras participates in the activation of p38 MAPK by interleukin-1 by associating with IRAK, IRAK2, TRAF6, and TAK-1. The Journal of biological chemistry 277: 7808–7815. [DOI] [PubMed] [Google Scholar]
- 27. Wang W, Chen JX, Liao R, Deng Q, Zhou JJ, et al. (2002) Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Molecular and cellular biology 22: 3389–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mendelsohn AR, Hamer JD, Wang ZB, Brent R (2002) Cyclin D3 activates Caspase 2, connecting cell proliferation with cell death. Proceedings of the National Academy of Sciences of the United States of America 99: 6871–6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Janes PW, Ley SC, Magee AI (1999) Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. The Journal of cell biology 147: 447–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kranenburg O, Verlaan I, Moolenaar WH (2001) Regulating c-Ras function. cholesterol depletion affects caveolin association, GTP loading, and signaling. Current biology : CB 11: 1880–1884. [DOI] [PubMed] [Google Scholar]
- 31. Leitinger B, Hogg N (2002) The involvement of lipid rafts in the regulation of integrin function. Journal of cell science 115: 963–972. [DOI] [PubMed] [Google Scholar]
- 32. Szabo G, Dolganiuc A, Dai Q, Pruett SB (2007) TLR4, ethanol, and lipid rafts: a new mechanism of ethanol action with implications for other receptor-mediated effects. Journal of immunology 178: 1243–1249. [DOI] [PubMed] [Google Scholar]
- 33. Tuluc F, Meshki J, Kunapuli SP (2003) Membrane lipid microdomains differentially regulate intracellular signaling events in human neutrophils. International immunopharmacology 3: 1775–1790. [DOI] [PubMed] [Google Scholar]
- 34. Lucero HA, Robbins PW (2004) Lipid rafts-protein association and the regulation of protein activity. Archives of biochemistry and biophysics 426: 208–224. [DOI] [PubMed] [Google Scholar]
- 35. Al-Shawwa B, Al-Huniti N, Titus G, Abu-Hasan M (2006) Hypercholesterolemia is a potential risk factor for asthma. The Journal of asthma : official journal of the Association for the Care of Asthma 43: 231–233. [DOI] [PubMed] [Google Scholar]
- 36. Liu LY, Sedgwick JB, Bates ME, Vrtis RF, Gern JE, et al. (2002) Decreased expression of membrane IL-5 receptor alpha on human eosinophils: I. Loss of membrane IL-5 receptor alpha on airway eosinophils and increased soluble IL-5 receptor alpha in the airway after allergen challenge. Journal of immunology 169: 6452–6458. [DOI] [PubMed] [Google Scholar]
- 37. Yao X, Dai C, Fredriksson K, Dagur PK, McCoy JP, et al. (2011) 5A, an apolipoprotein A-I mimetic peptide, attenuates the induction of house dust mite-induced asthma. Journal of immunology 186: 576–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yao X, Dai C, Fredriksson K, Lam J, Gao M, et al. (2012) Human apolipoprotein E genotypes differentially modify house dust mite-induced airway disease in mice. American journal of physiology Lung cellular and molecular physiology 302: L206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yao X, Fredriksson K, Yu ZX, Xu X, Raghavachari N, et al. (2010) Apolipoprotein E negatively regulates house dust mite-induced asthma via a low-density lipoprotein receptor-mediated pathway. American journal of respiratory and critical care medicine 182: 1228–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yao X, Vitek MP, Remaley AT, Levine SJ (2012) Apolipoprotein mimetic peptides: a new approach for the treatment of asthma. Frontiers in pharmacology 3: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yiallouros PK, Savva SC, Kolokotroni O, Behbod B, Zeniou M, et al. (2012) Low serum high-density lipoprotein cholesterol in childhood is associated with adolescent asthma. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology 42: 423–432. [DOI] [PubMed] [Google Scholar]
- 42. Akuthota P, Melo RC, Spencer LA, Weller PF (2012) MHC Class II and CD9 in human eosinophils localize to detergent-resistant membrane microdomains. American journal of respiratory cell and molecular biology 46: 188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lei JT, Martinez-Moczygemba M (2008) Separate endocytic pathways regulate IL-5 receptor internalization and signaling. Journal of leukocyte biology 84: 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Burnham ME, Koziol-White CJ, Esnault S, Bates ME, Evans MD, et al. (2013) Human Airway Eosinophils Exhibit Preferential Reduction in STAT Signaling Capacity and Increased CISH Expression. Journal of Immunology In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Esnault S, Kelly EA, Nettenstrom LM, Cook EB, Seroogy CM, et al. (2012) Human eosinophils release IL-1β and increase expression of IL-17A in activated CD4(+) T lymphocytes. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology 42: 1756–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Milhaud J (1992) Permeabilizing action of filipin III on model membranes through a filipin-phospholipid binding. Biochimica et biophysica acta 1105: 307–318. [DOI] [PubMed] [Google Scholar]
- 47. Bittman R, Fischkoff SA (1972) Fluorescence studies of the binding of the polyene antibiotics filipin 3, amphotericin B, nystatin, and lagosin to cholesterol. Proceedings of the National Academy of Sciences of the United States of America 69: 3795–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McGookey DJ, Fagerberg K, Anderson RG (1983) Filipin-cholesterol complexes form in uncoated vesicle membrane derived from coated vesicles during receptor-mediated endocytosis of low density lipoprotein. The Journal of cell biology 96: 1273–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hassall DG (1992) Three probe flow cytometry of a human foam-cell forming macrophage. Cytometry 13: 381–388. [DOI] [PubMed] [Google Scholar]
- 50. Muller CP, Stephany DA, Winkler DF, Hoeg JM, Demosky SJ Jr, et al. (1984) Filipin as a flow microfluorometry probe for cellular cholesterol. Cytometry 5: 42–54. [DOI] [PubMed] [Google Scholar]
- 51. Schley PD, Brindley DN, Field CJ (2007) (n-3) PUFA alter raft lipid composition and decrease epidermal growth factor receptor levels in lipid rafts of human breast cancer cells. Journal of Nutrition 137: 548–553. [DOI] [PubMed] [Google Scholar]
- 52. Stulnig TM, Huber J, Leitinger N, Imre EM, Angelisova P, et al. (2001) Polyunsaturated eicosapentaenoic acid displaces proteins from membrane rafts by altering raft lipid composition. Journal of Biological Chemistry 276: 37335–37340. [DOI] [PubMed] [Google Scholar]
- 53. Stulnig TM (2003) Immunomodulation by polyunsaturated fatty acids: Mechanisms and effects. International archives of allergy and immunology 132: 310–321. [DOI] [PubMed] [Google Scholar]
- 54. Dendele B, Tekpli X, Hardonniere K, Holme JA, Debure L, et al. (2014) Protective action of n-3 fatty acids on benzo a pyrene-induced apoptosis through the plasma membrane remodeling-dependent NHE1 pathway. Chemico-Biological Interactions 207: 41–51. [DOI] [PubMed] [Google Scholar]
- 55. Cheung PF, Wong CK, Lam CW (2008) Molecular mechanisms of cytokine and chemokine release from eosinophils activated by IL-17A, IL-17F, and IL-23: implication for Th17 lymphocytes-mediated allergic inflammation. Journal of immunology 180: 5625–5635. [DOI] [PubMed] [Google Scholar]
- 56. Weller PF (1997) Human eosinophils. The Journal of allergy and clinical immunology 100: 283–287. [DOI] [PubMed] [Google Scholar]
- 57. Shaw G, Kamen R (1986) A CONSERVED AU SEQUENCE FROM THE 3′ UNTRANSLATED REGION OF GM-CSF MESSENGER-RNA MEDIATES SELECTIVE MESSENGER-RNA DEGRADATION. Cell 46: 659–667. [DOI] [PubMed] [Google Scholar]
- 58. Esnault S, Malter JS (2002) Extracellular signal-regulated kinase mediates granulocyte-macrophage colony-stimulating factor messenger RNA stabilization in tumor necrosis factor-alpha plus fibronectin-activated peripheral blood eosinophils. Blood 99: 4048–4052. [DOI] [PubMed] [Google Scholar]
- 59. Brooks AR, Shiffman D, Chan CS, Brooks EE, Milner PG (1996) Functional analysis of the human cyclin D2 and cyclin D3 promoters. The Journal of biological chemistry 271: 9090–9099. [DOI] [PubMed] [Google Scholar]
- 60. Yamaguchi Y, Suda T, Ohta S, Tominaga K, Miura Y, et al. (1991) Analysis of the survival of mature human eosinophils: interleukin-5 prevents apoptosis in mature human eosinophils. Blood 78: 2542–2547. [PubMed] [Google Scholar]
- 61. Zhuang S, Demirs JT, Kochevar IE (2000) p38 mitogen-activated protein kinase mediates bid cleavage, mitochondrial dysfunction, and caspase-3 activation during apoptosis induced by singlet oxygen but not by hydrogen peroxide. The Journal of biological chemistry 275: 25939–25948. [DOI] [PubMed] [Google Scholar]
- 62. ten Hove W, Houben LA, Raaijmakers JA, Bracke M, Koenderman L (2007) Differential regulation of TNFalpha and GM-CSF induced activation of P38 MAPK in neutrophils and eosinophils. Molecular immunology 44: 2492–2496. [DOI] [PubMed] [Google Scholar]
- 63. Ferreiro I, Joaquin M, Islam A, Gomez-Lopez G, Barragan M, et al. (2010) Whole genome analysis of p38 SAPK-mediated gene expression upon stress. BMC genomics 11: 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen YL, Huang YL, Lin NY, Chen HC, Chiu WC, et al. (2006) Differential regulation of ARE-mediated TNF alpha and IL-1 beta mRNA stability by lipopolysaccharide in RAW264.7 cells. Biochemical and biophysical research communications 346: 160–168. [DOI] [PubMed] [Google Scholar]
- 65. Pelicci G, Dente L, De Giuseppe A, Verducci-Galletti B, Giuli S, et al. (1996) A family of Shc related proteins with conserved PTB, CH1 and SH2 regions. Oncogene 13: 633–641. [PubMed] [Google Scholar]
- 66. Rockett BD, Teague H, Harris M, Melton M, Williams J, et al. (2012) Fish oil increases raft size and membrane order of B cells accompanied by differential effects on function. Journal of lipid research 53: 674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hooper NM (1999) Detergent-insoluble glycosphingolipid/cholesterol-rich membrane domains, lipid rafts and caveolae (review). Molecular Membrane Biology 16: 145–156. [DOI] [PubMed] [Google Scholar]
- 68. Brown DA, London E (1998) Functions of lipid rafts in biological membranes. Annual review of cell and developmental biology 14: 111–136. [DOI] [PubMed] [Google Scholar]
- 69. Brown DA (2006) Lipid rafts, detergent-resistant membranes, and raft targeting signals. Physiology 21: 430–439. [DOI] [PubMed] [Google Scholar]
- 70. Pike LJ (2004) Lipid rafts: heterogeneity on the high seas. The Biochemical journal 378: 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dahl ME, Arai KI, Watanabe S (2000) Association of Lyn tyrosine kinase to the GM-CSF and IL-3 receptor common betac subunit and role of Src tyrosine kinases in DNA synthesis and anti-apoptosis. Genes to cells : devoted to molecular & cellular mechanisms 5: 143–153. [DOI] [PubMed] [Google Scholar]
- 72. Brewer CF, Miceli MC, Baum LG (2002) Clusters, bundles, arrays and lattices: novel mechanisms for lectin-saccharide-mediated cellular interactions. Curr Opin Struct Biol 12: 616–623. [DOI] [PubMed] [Google Scholar]
- 73. Zhuang LY, Kim J, Adam RM, Solomon KR, Freeman MR (2005) Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. Journal of Clinical Investigation 115: 959–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cholesterol manipulation did not alter eosinophil size or density as determined via flow cytometry. Scatter plots of side scatter versus forward scatter of eosinophils pretreated 1 hour with i. media, ii. 5 mg/mL MβCD, or iii. 5 mg/mL MβCD+2%Chol (3–5×105 per treatment).
(TIF)
Pretreatment with reduced percentage of cholesterol in MβCD+Chol (1% cholesterol) resulted in no net-change in PBEos cellular membrane cholesterol. (A–B) Eosinophils were treated 1 hour with media, 5 mg/mL MβCD, 5 mg/mL MβCD+2%Chol, or 5 mg/mL MβCD+1%Chol (3–5×105 per treatment). Cells were fixed, stained with 50 µg/mL Filipin III for cholesterol, then analyzed via flow cytometry. (A) Representative histogram from n = 5 experiments of cells stained with FIII after treatment with i. Media, ii. MβCD, iii. MβCD+2%Chol, or iv. MβCD+1%Chol. (B) Quantification of 5 independent experiments of FIII-stained eosinophils after treatment with concentrations indicated above. Error bars indicate SEM, p-values from one-way ANOVA. (C–D) One million eosinophils per treatment were pretreated 1 hour with media, 5 mg/mL MβCD +2%Chol, or 5 mg/mL MβCD +1%Chol, then stimulated +/− IL-5 for 15 min. (C) Samples were immunoblotted for active p38. (D) Pooled data (n = 5) normalized to actin loading, error bars indicate SEM, p-values from non-parametric one-way ANOVA. Irrelevant lanes were digitally removed from between lanes 2 and 3. Unmarked comparisons were non-significant.
(TIF)
EPA treatment increases basal p38 phosphorylation, eliminating an IL-5-stimulated increase. One million eosinophils per treatment pretreated 18 hours with media or 33 uM EPA were stimulated +/− IL-5 for 15 min. (A) Western blots were probed for phosphorylated STAT5 or p38, and reprobed with actin antibodies as a loading control. Example blot is representative of n = 3 experiments. (B) Graph plots p-p38 band density normalized to actin as mean +/− SE for each treatment. Data are pooled from 3 experiments, and ctl + IL-5 samples were set as 1. (C) Graph plots p-STAT5 band density normalized to actin as mean +/− SE for each treatment. Data are pooled from 3 experiments, and only IL-5-stimulated bands were quantified as p-STAT5 was undetectable in lysates from unstimulated cells.
(TIF)